

“I don’t believe medical discoveries are doing much to advance human life. As, fast as we find ways to extend it we are inventing ways to shorten it.”
-Christiaan Barnard.
Many drugs that are relatively new to the market their way out as fast as they entered it.
Is expediting drug discovery really solving the unmet needs of society?
WHAT IS “FAST TRACK” AND WHY WAS IT INTRODUCED?
The United States Food and Drug Administration (US-FDA) defines Fast Track as a process designed to facilitate the development and expedite the review of drugs to treat serious diseases and fill an unmet medical need. Filling an unmet medical need is further defined as providing a therapy where none exists or providing a therapy which may be potentially superior to existing therapy. This was introduced to curtail the duration in drug regulation and approval process, and to facilitate the discovery and marketing of drugs targeted for serious or rare diseases and to accelerate the approval of molecules showing superior efficacy than the existing one. Fast-track approach was introduced in 1988; a sponsor can apply for fast-track drug approval process along with investigational new drug application. The FDA conveys the decision within sixty calendar days to the sponsor about fast-track consideration. When a drug is accredited with the fast-track process, it enables investigators to work along with FDA to conduct the trial and to submit the relevant data. The FDA can approve the drug under fast-track designation after reviewing a single phase 2 study.[1]
FAST-TRACK STATISTICS AND OTHER EXPEDITED DRUG REVIEW PROCESS
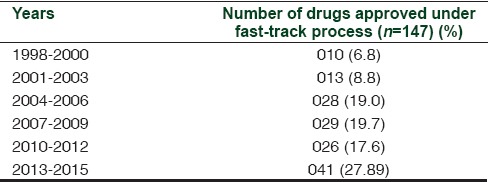
In 1992, the FDA introduced two more drug review processes, namely, “Priority Review” by which a drug review process will be completed within 6 months against the 10 months of the standard review schedule and “Accelerated Approval” of a drug or biologics based on its efficacy shown by surrogate markers. Most of the drugs considered under fast-track process may also be considered under priority review and accelerated approval. According to FDA archival, efavirenz, the anti-HIV drug was the first accepted fast-track product on September 17, 1998. Until now, 147 drugs or biologics have been approved as fast-track products. The approval trend has shown steady increase since 1998 as depicted in Table 1. The number of products utilizing the expedited review processes is about 543. These products undergo scrutiny by the Centre for Drug Evaluation and Research or Centre for Biologics Evaluation and Research before obtaining marketing authorization.[2]
Table 1.
Fast-track designation products statistics since inception
EXPERIENCE FROM THE PAST
Careful interpretation of the preclinical and clinical studies performed for a drug is very crucial. The best example is the thalidomide tragedy in Europe. The drug was not marketed in the USA only because a single drug controller with the US-FDA, Dr. Frances Kelsey, raised concerns regarding the insufficient safety data and risk of peripheral neuropathy. This decision prevented children with phocomelia due to thalidomide being born in the United States. Later, experimental findings provided the scientific basis for the revival of thalidomide based on its immunomodulatory effects and ability to inhibit angiogenesis. In 2006, FDA approved thalidomide for erythema nodosum leprosum and multiple myeloma.
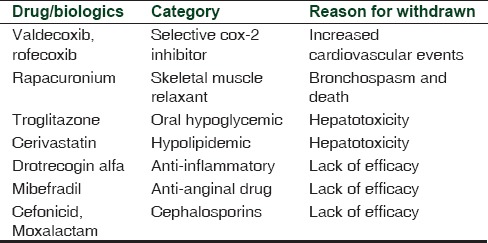
Although fast-track processing of drugs facilitates earlier access of drugs that are urgently needed by society, it also may undermine safety. From 1988 to 2010, some of the fast-track products have gone unpredicted and withdrawn which is mentioned in Table 2. Increased cardiovascular events from selective cox-2 inhibitors valdecoxib and rofecoxib, rapacuronium-induced bronchospasm, alosetron-induced ischemic colitis, hepatotoxicity from cerivastatin, and troglitazone were grievous. Lack of efficacy is also exemplified from some fast-track products such as drotrecogin alfa, mibefradil, moxalactam, and cefonicid.[3]
Table 2.
Fast-track designation products withdrawn from the market
CURRENT SCENARIO
Is it too fast and furious?
There are critics that FDA is liberal and approve the molecules in an accelerated way based on trivial information submitted by marketing authorities. The critic is evidenced by the annual report of FDA approval of 35 new chemical entities in 2011; the review was completed in an average of 8.2 months will make us frown surprisingly. Despite the critics, FDA approved 41 novel compounds in 2014 (15 fast-track products) and 45 novel compounds (14 fast-track products) in 2015.[4]
Dabigatran, an oral direct thrombin inhibitor developed by Boehringer Ingelheim, was projected as superior as conventional warfarin and awarded fast-track process. The FDA reviewed and approved the molecules within 6 months on October 19, 2010. FDA justified for producing fast-track status stating dabigatran is safe and hence monitoring can be omitted, unlike warfarin. This justification is nullified by postmarketing surveillance, which revealed increased gastrointestinal bleeding by dabigatran and safety alert announced by May 13, 2014.[5] The same pharmaceutical Boehringer Ingelheim is now marketing Idarucizumab as an antidote for dabigatran-induced bleeding in 2015. Dabigatran and its antidote are neither cost-effective nor safer than warfarin and its antidote, Vitamin K. Developing and marketing a new molecule out of weak hypothesis and then inventing an antidote by the same pharmaceutical company within 4 years support the pitfalls of inadequate review time and process by FDA.
Ezogabine, a potassium channel blocker, was approved under expedited program for partial seizure has shown severe retinal damage and loss of vision in postmarketing evaluation.[6] Similarly, chronic myeloid leukemia is treated by imatinib and two more molecules, namely, dasatinib and nilotinib in imatinib resistance cases with very high success rate. For the same indication, ponatinib was granted fast-track status and was approved by the FDA on December 2012. Widespread exposure of ponatinib unmasked its propensity to cause fatal veno-occlusive disease and it was withdrawn in the span of 11 months in October 2013. It is totally unconvincing to expose patients to a dangerous molecule arose out of reluctant interpretation of a single Phase II trial.[7]
Is it going on right track?
Most of the drugs for hepatitis C underwent a fast-track process and were approved based on viral load reduction. These molecules produced severe acute liver injury among recipients. Ombitasvir, paritaprevir, and dasabuvir were approved on December 19, 2014, and on January 15, 2016, FDA issued warning of severe hepatic decompensation. Other approved anti-hepatitis C drugs simeprevir and sofosbuvir are also proven to be hepatotoxic. This is justifiable on medical grounds since no specific anti-hepatitis C drugs are available, yet assessing these drugs with greater number of patients might have predicted the toxicity. For diabetes mellitus, two group of drugs, namely, dipeptidyl peptidase-IV (DPP-IV) inhibitors saxagliptin (2009), linagliptin (2011), alogliptin (2013), and sodium glucose co-transporter 2 (SGLT2) inhibitors canagliflozin (2013), dapagliflozin (2014), and empagliflozin (2014) were granted fast-track approval and subsequently marketed worldwide. DPP-IV inhibitors were implicated in severe mouth ulcers, renal impairment, and angioedema. SGLT2 inhibitors are known to precipitate ketoacidosis and urosepsis. Presently, the risk-benefit assessment of almost all DPP-IV inhibitors and SGLT2 inhibitors is being reworked.[8,9,10,11]
Is it leading to unpredictable journey?
Pharmacological effects of cytokine analogues and immunomodulators are complex and underexplored. Assessing the effects of biologics through fast-track processing seems incongruous. All the tumor necrosis factor-alpha (TNF-α) blockers introduced for several autoimmune diseases such as rheumatoid arthritis, inflammatory bowel disease (IBD), and relapsing multiple sclerosis produce serious adverse effects. TNF-α analogs have been shown to induce sarcoidosis and found to induce hepatosplenic lymphoma.[12] Anti-integrin molecule natalizumab used in IBD has been implicated in the development of fatal progressive multifocal leukoencephalopathy and fingolimod produces a rare disorder called hemophagocytic syndrome.[13,14] There is enough evidence to alert us that biologics should have longer review time to predict their complex biological effects. Expedited approval may cure an autoimmune disorder and incite another one.
Is it leading to a blind end?
Anticancer drugs and biologics are being approved based on its effect on surrogate markers of a particular cancer. In reality, the significance in terms of regression or preventing the progression is inconspicuous,[15,16] and hence, adding a new drug or biologics may not actually benefit the patients. The classical example is vemurafenib, approved for advanced melanoma on the basis of increased survival of patients on vemurafenib versus dacarbazine. The median survival increased by about 3.5 months only between the two groups which may gain statistical significance, but not in terms of quality of life. Adding to unclear benefits, vemurafenib has a potential to induce squamous cell carcinoma among receivers.[17,18,19,20]
Is it an effective track or deceptive?
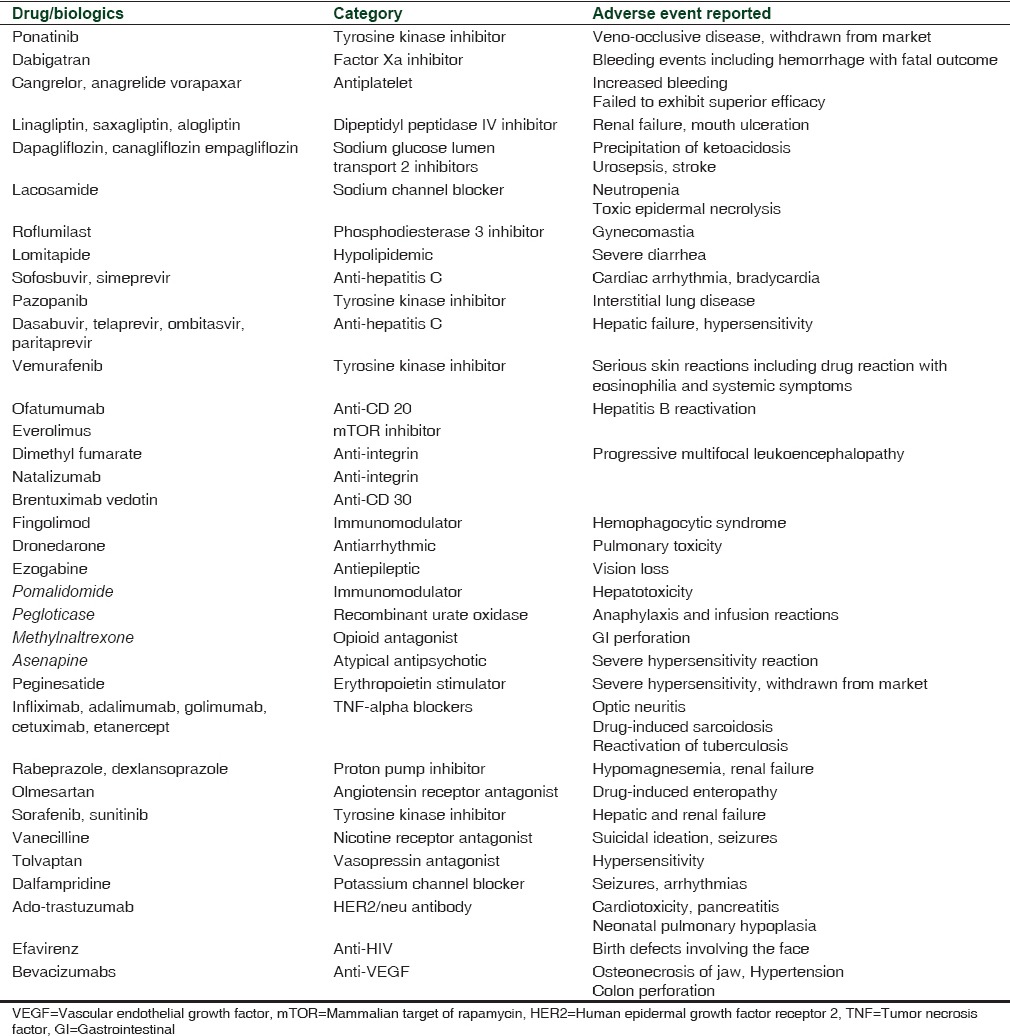
Fast-track processing gave an edge in the 1990s to combat HIV, AIDS, and certain malignancies. Some of the drugs that were developed as fast products such as lamivudine, imatinib, erlotinib, oxaliplatin, and levofloxacin are used extensively even today. At present, fast-track processing should be restricted only to instances when the need for the drug is very crucial. The manner in which these drugs are proven as “non-inferior” to the standard treatment also needs to be modified. New, noninferior molecules may have adverse effects and are usually costlier.[19,20] In the last 5 years, nearly 57% fast-track molecules received a black box warning. Summary of all significant adverse events alerts is shown in Table 3.[21] The most recent issue which prompted this editorial is the fast-track antiplatelet product called cangrelor, which was approved as an adjunct in percutaneous intervention. This drug has shown no additional benefits in trials comparing cangrelor and clopidogrel except for increased bleeding during the procedure.[22,23]
Table 3.
Fast-track designation products received potential adverse effects alert during 2011-2015
CONCLUSION
Fast-track processing will be more effective: (1) When the developing molecule is really expected to treat a disease with no cure. (2) When there is an existing therapy, a new drug or biologic of novel mechanism can be approved by fast-track process. However, the other members of the same group should be considered for fast-track approval only after sufficient postmarketing surveillance supporting the efficacy and safety of the new compound. (3) Approval based only on surrogate markers should be reconsidered. (4) In malignancies with very poor prognosis, the statistical significance of an increase in survival from a fast-track process must be matched with clinical benefits. (5) Regulatory authorities should be more vigilant about undue or fallacious evidence-based information by pharmaceuticals.[24,25,26,27]
Acknowledgment
I acknowledge Dr. S. Manikandan, Department of Pharmacology, JIPMER, Puducherry, for his inputs in the editorial preparation.
REFERENCES
- 1.Fast Track, Breakthrough Therapy, Accelerated Approval, Priority Review. U.S. Food and Drug Administration. [Last updated on 2015 Sep 14; Last cited on 2016 Mar 01]. Available from: http://www.fda.gov/ForPatients/Approvals/Fast/default.htm .
- 2.Guidance for Industry Expedited Programs for Serious Conditions – Drugs and Biologics. U.S. Food and Drug Administration. [Last updated on 2014 May 01; Last cited on 2016 Mar 01]. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm358301.pdf .
- 3.Recalls, Market Withdrawals & Safety Alerts. U.S. Food and Drug Administration. [Last updated on 2016 Feb 29; Last cited on 2016 Mar 01]. Available from: http://www.fda.gov/Safety/Recalls/
- 4.Kesselheim AS, Wang B, Franklin JM, Darrow JJ. Trends in utilization of FDA expedited drug development and approval programs, 1987-2014: Cohort study. BMJ. 2015;351:h4633. doi: 10.1136/bmj.h4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.FDA Drug Safety Communication: Update on the Risk for Serious Bleeding Events with the Anticoagulant Pradaxa (Dabigatran) U.S. Food and Drug Administration. [Last updated on 2014 May 13; Last cited on 2016 Mar 01]. Available from: http://www.fda.gov/Drugs/DrugSafety/ucm326580.htm .
- 6.Potiga (Ezogabine): Drug Safety Communication – Linked to Retinal Abnormalities and Blue Skin Discoloration. U.S. Food and Drug Administration. [Last updated on 2013 Jan 11; Last cited on 2016 Mar 01]. Available from: http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm349847.htm .
- 7.Gainor JF, Chabner BA. Ponatinib: Accelerated disapproval. Oncologist. 2015;20:847–8. doi: 10.1634/theoncologist.2015-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The Novel New Drugs of 2011. U.S. Food and Drug Administration. [Last updated on 2012 Jan 01; Last cited on 2016 Mar 01]. Available from: http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DrugInnovation/UCM289229.pdf .
- 9.The Novel New Drugs of 2013. U.S. Food and Drug Administration. [Last updated on 2014 Jan 01; Last cited on 2016 Mar 01]. Available from: http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DrugInnovation/UCM381803.pdf .
- 10.The Novel New Drugs of 2014. U.S. Food and Drug Administration. [Last updated on 2015 Jan 01; Last cited on 2016 Mar 01]. Available from: http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DrugInnovation/UCM430299.pdf .
- 11.Potential Signals of Serious Risks/New Safety Information Identified by the FDA Adverse Event Reporting System (FAERS) between July – September 2015. U.S. Food and Drug Administration. 2015. [Last updated on 2016 Feb 02; Last cited on 2016 Mar 01]. Available from: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/ucm484294.htm .
- 12.Potential Signals of Serious Risks/New Safety Information Identified by the FDA Adverse Event Reporting System (FAERS) between July – September 2015. U.S. Food and Drug Administration. 2015. [Last updated on 2015 Sep 11; Last cited on 2016 Mar 01]. Available from: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/ucm281856.htm .
- 13.FDA Drug Safety Communication: New Risk Factor for Progressive Multifocal Leukoencephalopathy (PML) Associated with Tysabri (natalizumab) U.S. Food and Drug Administration. [Last updated on 2012 Jan 20; Last cited on 2016 Mar 01]. Available from: http://www.fda.gov/Drugs/DrugSafety/ucm288186.htm .
- 14.Potential Signals of Serious Risks/New Safety Information Identified by the FDA Adverse Event Reporting System (FAERS) between October – December 2012. U.S. Food and Drug Administration. 2012. [Last updated on 2011 Sep 11; Last cited on 2016 Mar 01]. Available from: http://www.fda.gov/Drugs/DrugSafety/ucm278267.htm .
- 15.Zhou S, Huang Y, Wei Y, et al. Goel A, editor. No Survival Benefit from Adding Cetuximab or Panitumumab to Oxaliplatin-Based Chemotherapy in the First-Line Treatment of Metastatic Colorectal Cancer in KRAS Wild Type Patients: A Meta-Analysis. PLoS ONE. 2012;7(11):e50925. doi: 10.1371/journal.pone.0050925. doi: 10.1371/journal.pone. 0050925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grimes DA, Schulz KF. Surrogate end points in clinical research: Hazardous to your health. Obstet Gynecol. 2005;105((5 Pt 1)):1114–8. doi: 10.1097/01.AOG.0000157445.67309.19. [DOI] [PubMed] [Google Scholar]
- 17.FDA Approval for Vemurafenib. U.S. Food and Drug Administration. [Last updated on 2013 Jul 03; Last cited on 2016 Mar 01]. Available from: http://www.cancer.gov/about.cancer/treatment/drugs/fda-vemurafenib .
- 18.Frouin E, Guillot B, Larrieux M, et al. van Steensel MAM, editor. Cutaneous Epithelial Tumors Induced by Vemurafenib Involve the MAPK and Pi3KCA Pathways but Not HPV nor HPyV Viral Infection. PLoS ONE. 2014;9(10):e110478. doi: 10.1371/journal.pone.0110478. doi: 10.1371/journal.pone. 0110478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schumi J, Wittes JT. Through the looking glass: Understanding non-inferiority. Trials. 2011;12:106. doi: 10.1186/1745-6215-12-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hahn S. Understanding noninferiority trials. Korean J Pediatr. 2012;55:403–7. doi: 10.3345/kjp.2012.55.11.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Potential Signals of Serious Risks/New Safety Information Identified from the FDA Adverse Event Reporting System (FAERS) U.S. Food and Drug Administration. [Last updated on 2016 Feb 02; Last cited on 2016 Mar 01]. Available from: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/ucm082196.htm .
- 22.Tang Y, Zhang YC, Chen Y, Xiang Y. Efficacy and safety of cangrelor for patients with coronary artery disease: A meta-analysis of four randomized trials. Int J Clin Exp Med. 2015;8:800–8. [PMC free article] [PubMed] [Google Scholar]
- 23.Serebruany VL, Aradi D, Kim MH, Sibbing D. Cangrelor infusion is associated with an increased risk for bleeding: Meta-analysis of randomized trials. Int J Cardiol. 2013;169:225–8. doi: 10.1016/j.ijcard.2013.08.133. [DOI] [PubMed] [Google Scholar]
- 24.Johnson EP. Pros and cons of the new drug application fast track review process. Clin Res Educ. 2009 Nov 30; [Google Scholar]
- 25.Leonard CE, Freeman CP, MaCurdy T, et al. Data Points Publication Series [Internet] Rockville (MD): Agency for Healthcare Research and Quality (US); 2011. Utilization and cost of anticancer biologic products among Medicare beneficiaries, 2006-2009: Data Points #6. 2011 Aug 26. Available from: http://www.ncbi.nlm.nih.gov/books/NBK65148 . [PubMed] [Google Scholar]
- 26.Osoba D. Health-related quality of life and cancer clinical trials. Ther Adv Med Oncol. 2011;3:57–71. doi: 10.1177/1758834010395342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marx RE. The deception and fallacies of sponsored randomized prospective double-blinded clinical trials: The bisphosphonate research example. Int J Oral Maxillofac Implants. 2014;29:e37–44. doi: 10.11607/jomi.te40. [DOI] [PubMed] [Google Scholar]