

ABSTRACT
Conserved genes essential to sulfur assimilation and trafficking in aerobic organisms are missing in many methanogens, most of which inhabit highly sulfidic, anaerobic environmental niches. This suggests that methanogens possess distinct pathways for the synthesis of key metabolites and intermediates, including cysteine, homocysteine, and protein persulfide groups. Prior work identified a novel tRNA-dependent two-step pathway for cysteine biosynthesis and a new metabolic transformation by which sulfur is inserted into aspartate semialdehyde to produce homocysteine. Homocysteine biosynthesis requires two of the three proteins previously identified in our laboratory by a comprehensive bioinformatics approach. Here, we show that the third protein identified in silico, the ApbE-like protein COG2122, facilitates sulfide assimilation in Methanosarcina acetivorans. Knockout strains lacking the gene encoding COG2122 are severely impaired for growth when sulfide is provided as the sole sulfur source. However, rapid growth is recovered upon supplementation with cysteine, homocysteine, or cystathionine, suggesting that COG2122 is required for efficient biosynthesis of both cysteine and homocysteine. Deletion of the gene encoding COG2122 does not influence the extent of sulfur modifications in tRNA or the prevalence of iron-sulfur clusters, indicating that the function of COG2122 could be limited to sulfide assimilation for cysteine and homocysteine biosynthesis alone.
IMPORTANCE We have found that the conserved M. acetivorans ma1715 gene, which encodes an ApbE-like protein, is required for optimal growth with sulfide as the sole sulfur source and supports both cysteine and homocysteine biosynthesis in vivo. Together with related functional-genomics studies in methanogens, these findings make a key contribution to elucidating the novel pathways of sulfide assimilation and sulfur trafficking in anaerobic microorganisms that existed before the advent of oxygenic photosynthesis. The data suggest that the MA1715 protein is particularly important to sustaining robust physiological function when ambient sulfide concentrations are low. Phylogenetic analysis shows that MA1715 and other recently discovered methanogen sulfur-trafficking proteins share an evolutionary history with enzymes in the methanogenesis pathway. The newly characterized genes thus likely formed an essential part of the core metabolic machinery of the ancestral euryarchaeote.
INTRODUCTION
Genes that function in sulfate assimilation and sulfur trafficking in aerobic organisms are often not identifiable in the genomes of methanogens and other anaerobes, suggesting that these ancient organisms may retain metabolic vestiges characteristic of life on the early earth (1). In aerobes, sulfur is imported into the cell as sulfate, which is reduced to sulfide in an ATP-dependent pathway (2). In turn, sulfide is incorporated directly into activated serine and homoserine derivatives to yield cysteine (Cys) and homocysteine (Hcy), respectively (Fig. 1, left) (3). Cys and Hcy can also be interconverted via the metabolic intermediate cystathionine in a bidirectional transsulfuration pathway (Fig. 1, right). The metabolic role of Hcy is limited to its use as the precursor to methionine (Met) (Fig. 1, left), which is required for protein synthesis and incorporation into S-adenosylmethionine. In contrast, Cys is required for protein synthesis, plays a key role in the biosynthesis of coenzyme A and glutathione, and functions as a sulfur donor for the synthesis of a wide range of sulfur-containing compounds (1). Sulfur is mobilized from Cys by the activity of cysteine desulfurase (CD), which removes sulfane (S°) from Cys to yield a persulfide-modified Cys residue (Cβ-S-SH) in the active site (Fig. 1) (4). The terminal sulfur atom of the persulfide modification is then relayed as sulfane between Cys residues of different proteins until it is incorporated into cofactors (thiamine, biotin, molybdopterin, and iron-sulfur clusters), tRNA modifications, or the C termini of ubiquitin-like proteins, which can serve as intermediate sulfur carriers (4).
FIG 1.
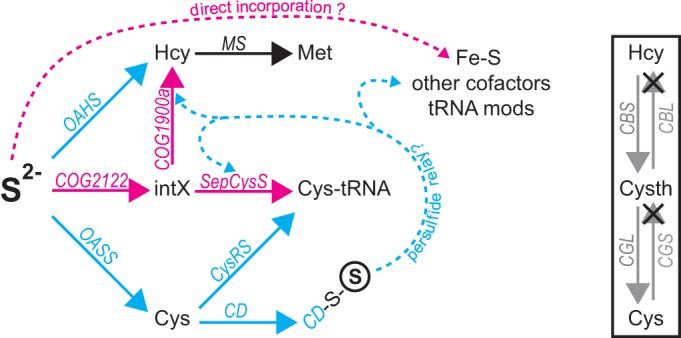
(Left) Model for sulfur assimilation. In M. acetivorans, the biosynthesis of cysteine and homocysteine is facilitated by a combination of methanogen-type (pink), universal (black), and conventional-type (blue) enzymes. Although the conventional-type enzymes found in most aerobes are often absent from other methanogens, those depicted here were presumably acquired by lateral gene transfer and are present in M. acetivorans. Sulfane (circled S) is relayed as persulfide (dashed arrows) between proteins to support the biosynthesis of cofactors and tRNA modifications. Catalysis by SepCysS and COG1900a may also proceed by a persulfide-dependent mechanism. COG2122 may be required for the biosynthesis of a yet-unidentified sulfur-containing metabolic intermediate (intX) of Cys-tRNA and homocysteine biosynthesis (see the text). MS, methionine synthase. (Right) Schematic depiction of transsulfuration pathways for interconversion of Cys and Hcy through the cystathionine (Cysth) intermediate. CBS, cystathionine β-synthase; CBL, cystathionine β-lyase; CGL, cystathionine γ-lyase; CGS, cystathionine γ-synthase. × indicates that the Cys-to-Hcy pathway is not present in M. acetivorans. The Hcy-to-Cys pathway may be present. See the text for details.
Many methanogens lack the pathway by which sulfate is reduced to sulfide, as well as the genes encoding O-acetylserine sulfhydrylase (OASS), O-acetylhomoserine sulfhydrylase (OAHS), and CD. Further, methanogens also often lack the gene encoding cysteinyl-tRNA synthetase (CysRS), which ligates tRNACys with Cys to yield Cys-tRNACys for protein synthesis (Fig. 1) (1, 5, 6). Cys-tRNACys biosynthesis in these methanogens is accomplished by a two-step pathway in which tRNACys is first ligated to phosphoserine (Sep) by phosphoseryl-tRNA synthetase (SepRS), and the resulting Sep-tRNACys intermediate is then converted to Cys-tRNACys by Sep-tRNA:Cys-tRNA synthase (SepCysS) (5). It has also been shown that free Cys is not synthesized independently of tRNACys in Methanococcus maripaludis (5), consistent with later findings showing that, in contrast to aerobic cells, neither Hcy nor iron-sulfur clusters are synthesized with sulfur originating from Cys in that organism (6). Studies of SepCysS expressed in Escherichia coli suggested that the enzyme does not efficiently use sulfide as a sulfur source in vitro (7) but instead consumes sulfane from persulfide to form Cys-tRNACys (7, 8) (Fig. 1). These discoveries provided key insights into the methanogen-type strategy for sulfur assimilation. However, the genes responsible for Hcy and persulfide biosynthesis, and the source of sulfur provided to SepCysS in native methanogen cells, remained elusive.
We took a bioinformatics approach to identify new genes for Hcy and persulfide synthesis in methanogens (9). Genotype-dependent growth and metabolite-labeling experiments revealed that the Methanosarcina acetivorans genes ma1821 and ma1822 encode proteins (COG1900a and a genomically linked ferredoxin) that together catalyze sulfur insertion into Hcy using aspartate-4-semialdehyde as a precursor (9, 10) (Fig. 1). A third M. acetivorans gene, ma1715, was also identified in silico (9). This gene encodes the uncharacterized protein domain COG2122, a distant homolog of the ApbE enzyme, which cleaves flavin adenine dinucleotide (FAD) to flavin mononucleotide (FMN) and AMP for thiamine biosynthesis and iron-sulfur cluster maintenance (11). Alignment of COG2122 proteins from methanogens revealed the presence of a single conserved Cys residue, but the portion of ApbE that binds FAD is not conserved in the protein encoded by ma1715, suggesting that it may have a different function (see Fig. S4 in the supplemental material) (12, 13). Genes encoding COG2122 are exclusively present in strict anaerobes, occur in all genomes that encode SepCysS and/or COG1900a, share an evolutionary history with methanogenesis enzymes, and are often genomically linked with ma1715 and genes dedicated to Met biosynthesis (9). These observations suggest that COG2122, COG1900a, and SepCysS may perform closely related functions in methanogens.
Here, the physiological role of COG2122 in M. acetivorans is investigated using genotype-dependent growth experiments. We establish that its gene, ma1715, is required for efficient growth with sulfide as the sole sulfur source and provide further evidence suggesting that it may mobilize sulfur for the reactions catalyzed by SepCysS and COG1900a. However, ma1715 is not required to maintain cellular levels of iron-sulfur clusters or sulfur modifications in tRNA, suggesting that its function may be limited to amino acid biosynthesis. Finally, the evolutionary history of COG2122 is revisited in light of the finding that methanogens are also present in the Bathyarchaeota (14). This further supports the notion that COG2122 shares an evolutionary history with methanogenesis.
MATERIALS AND METHODS
Strains and media.
All M. acetivorans strains were derived from the wild-type-like strain WWM75 (Table 1) (15). Anaerobic high-salt media based on formulations of Sowers et al. and Metcalf et al. (16, 17) were employed throughout, as described previously (9). Methanol was used universally as the methanogenesis substrate.
TABLE 1.
M. acetivorans strains and plasmids used in this study
| Strain or plasmid | Description | Parent strain | Manipulation | Relevant characteristics | Source or reference |
|---|---|---|---|---|---|
| Strains | |||||
| WWM75 | Wild typea | M. acetivorans C2A | Δhpt::(PmcrB Tetr ΦC31 int attB) | 19 | |
| BJR01 | Δma1821-Δma1822b | WWM75 | Δma1821-Δma1822 | 15 | |
| BJR02 | Δma1715 | WWM75 | Δma1715 | This study | |
| BJR09 | OASSc knockout | WWM75 | OASS knockout | 15 | |
| BJR22 | OASS-OAHSd double knockout | BJR09 | OAHS knockout | 15 | |
| BJR23 | OASS-OAHS-MA1715 triple knockout | BJR22 | Δma1715 | This study | |
| BJR24 | OASS-MA1715 double knockout | BJR09 | Δma1715 | This study | |
| Plasmids | |||||
| pMP44 | Facilitates markerless exchange in M. acetivorans | 22 | |||
| pBR23 | Fused ma1715-flanking sequences inserted into pMP44 via SpeI/KpnI; used to construct BJR02, BJR23, and BJR24 | This study | |||
| pET22b | E. coli expression vector for C-terminal 6×His fusion proteins | Novagen | |||
| pBR06 | ma1715 gene sequence inserted into pET22b via NdeI/XhoI | This study | |||
| pBR31 | M. acetivorans expression vector; tetracycline inducible | (15) | |||
| pBR60 | COG2122-6×His coding sequence from pBR06 inserted into pBR31 via SphI/BamHI | This study |
The strain contains modifications from the original M. acetivorans isolate but is referred to as wild type for the purposes of this study.
Encodes COG1900a and its associated ferredoxin.
OASS is an E. coli-like cysteine biosynthesis enzyme.
OAHS is a yeast-like homocysteine biosynthesis enzyme.
Plasmid construction.
The knockout vector for ma1715 (pBR23) was constructed by ligating a 2-kb fusion of the genomic DNA sequences that flank ma1715 into the SpeI and KpnI restriction sites of pMP44 (Table 1). Upstream and downstream flanking regions were amplified separately by PCR using primers 1 and 2 and primers 3 and 4, respectively. Subsequently, they were fused by overlap extension PCR using primers 1 and 4 (see Table S2 in the supplemental material for a list of primers).
The MA1715 expression plasmid (pBR60) was constructed by ligating the ma1715 gene, augmented with the coding sequence for a polyhistidine tag, into the tetracycline-inducible autonomously replicating plasmid pBR31 (Table 1). The ma1715 gene was PCR amplified from genomic DNA using primers 5 and 6 and subsequently ligated into the NdeI and XhoI sties of pET-22b(+) (Novagen) to yield plasmid pBR6. Using primers 7 and 8, the augmented ma1715 gene was PCR amplified from pBR6 and ligated into the SphI and ApaI sites of pBR31 (see Table S2 in the supplemental material for a list of primers).
Construction of knockout strains.
pBR23 was employed to excise the ma1715 gene from M. acetivorans strains WWM75, BJR09, and BJR22 to yield the markerless deletion strains BJR02, BJR24, and BJR23, respectively (Table 1). Transformants undergoing recombination were selected for puromycin resistance as described previously (18, 19). The resulting merodiploid intermediate strains were resolved by counterselective plating and purification in the presence of 0.2 mg/ml 8-aza-2,6-diaminopurine (8-ADP). Knockout strains were first identified by PCR and later confirmed by DNA-DNA hybridization directly targeting the ma1715 gene. The DNA probe was synthesized by PCR using primers 9 and 10 (see Table S2 in the supplemental material). In anticipation of Cys or Hcy auxotrophy, HSMet medium, a minimal medium supplemented with 0.4 mM sodium sulfide, 3 mM Cys, and 3 mM Met, was employed as needed (9).
Sulfur-source-dependent growth experiments.
M. acetivorans strains were grown anaerobically on sulfur-source-free HSDTT medium supplemented with various sulfur sources that were added after autoclaving, immediately prior to inoculation, as described previously (9). Growth (10 ml) was monitored by determining the A600, measured after inserting sealed culture tubes directly into a spectrophotometer (9).
Detection of thiolated tRNAs in the cellular lysates of stationary-phase M. acetivorans cultures.
The strains of interest were grown anaerobically to saturation on 10 ml HSDTT medium supplemented with various sulfur sources (9). All subsequent manipulations were performed aerobically. Cells were first harvested by centrifugation (1 min; 16,000 relative centrifugal force [RCF]) and subsequently lysed by resuspension to a 400-μl mixture containing 5 mM Tris · HCl (pH 7.5), 5 mM MgCl2, and 50% Tris-saturated phenol (pH 8.0). The resuspensions were vortexed for 1 min. The organic and aqueous phases were then separated by centrifugation (2 min; 16,000 RCF). Nucleic acids were precipitated from the aqueous layer over the course of a 1-h incubation on ice in the presence of 1.25 M ammonium acetate (pH 5.2) and 50% isopropanol. The precipitated nucleic acids were recovered by centrifugation (30 min; 16,000 RCF; 4°C). The pellets were washed with 500 μl ice-cold 70% ethanol and recovered by centrifugation (10 min; 16,000 RCF; 4°C). The washed pellets were dried under vacuum and then resuspended in 40 μl of a solution containing 10 mM Tris·HCl (pH 7.5), 10 mM MgCl2, 1 mM CaCl2, and 10 μg/ml DNase I. The solutions were incubated for 30 min at room temperature and then stored at −20°C. To specifically label tRNAs at the 3′ internucleotide linkage, aliquots (5 μl from each crude RNA solution) were reacted with tRNA nucleotidyltransferase in the presence of [α-32P]ATP in a reaction volume of 80 μl, essentially as described previously (20). The labeled tRNAs were gel purified and subsequently subjected to electrophoresis through a matrix containing 10% polyacrylamide (19:1 acrylamide-bisacrylamide), 8 M urea, 1× Tris-borate-EDTA (TBE), and (N-acryloylamino)phenyl mercury (APM), which specifically retards the migration of sulfur-containing compounds (21, 22). The labeled tRNAs were then visualized by phosphorimaging.
Preparation of cellular lysates from actively dividing M. acetivorans cultures.
All manipulations were carried out anaerobically. Strains were grown to late exponential phase (0.5 < A600 < 0.7) in duplicate on 1,000 ml HSDTT medium supplemented with 0.2 mM sodium sulfide. Cells were harvested by centrifugation (20 min; 10,000 RCF; 4°C) and subsequently washed three times with 10 ml high-salt wash buffer, which contained 100 mM Tris · HCl (pH 8.0) and 1,000 mM KCl. The buffer was purged with anaerobic N2 prior to use. The washed cell pellets (0.4 to 0.5 ml per strain) were pooled and resuspended in 1 ml lysis buffer (100 mM ammonium acetate [pH 8.5] purged with anaerobic N2-CO2 [80%/20%] prior to use). The resuspended cells were lysed by sonication (0.5 min) and then cleared by centrifugation (30 min; 16,000 RCF). Approximately 0.9 ml of cleared lysate was recovered from each strain. The lysates were then dialyzed three times for 4 h each time against 300 ml lysis buffer. The protein content was assessed by bicinchoninic acid (BCA) assay (Sigma). The lysates contained approximately 15 mg/ml protein and were stored anaerobically at −20°C as 0.1-mg protein aliquots.
Quantitation of acid-labile sulfide.
Dialyzed lysates of BJR22 and BJR23 (see above) were processed as described previously with 0.3 to 0.6 mg of protein per sample (6). Three independent experiments were performed for each strain.
Measurement of tRNA thiolation activity.
tRNATyr from Methanocaldococcus jannaschii was transcribed in vitro with T7 RNA polymerase as described previously (23). After gel purification, the tRNA was radiolabeled at the 3′ internucleotide linkage with 32P using tRNA nucleotidyltransferase (see above). The labeled transcript (<150 nM) was incubated at room temperature in 20-μl reaction mixtures containing 20 mM HEPES · KOH (pH 7.5), 10 mM MgCl2, 75 mM KCl, 2.5 mM MgCl2 · ATP, and dialyzed cellular lysate (5 mg/ml protein). The reactions were quenched by 2-fold dilution in a solution containing 94% formamide, 1% SDS, 0.025% bromophenol blue, 0.025% xylene cyanol FF, and 5 mM EDTA (pH 8.0). The quenched samples were subjected to APM gel electrophoresis (see above) and analyzed by phosphorimaging.
Phylogenetic reconstruction of COG2122.
Protein sequences were acquired following a blastp search of all translated archaeal genomes using the MA1715 protein sequence as a query. All search results below the expectation threshold of 10−20 were downloaded. After truncated sequences (<230 amino acids [aa]) were discarded, a multiple-sequence alignment was generated with ClustalW, using default parameters. A neighbor-joining tree was then constructed using the Poisson model, a uniform distribution of rates, and complete deletion of gaps, as directed within the graphic user interface of MEGA 6. Bootstrapping support values, which reflect a percentage of 1,000 replicates, are shown adjacent to selected nodes.
RESULTS
COG2122 is required for optimal growth of M. acetivorans when sulfide is the sole sulfur source.
Strong genomic cooccurrence suggests that the function of COG2122 is related to SepCysS and COG1900a. All genomes that encode SepCysS and/or COG1900a also encode COG2122, while fewer than 2% of genomes that encode COG2122 lack genes for either SepCysS or COG1900a (9). In part because sulfide-dependent SepCysS activity is inefficient in vitro (5, 24), we postulated that COG2122 could play a role in converting sulfide to metabolic intermediates for incorporation into Cys and Hcy. To test this hypothesis, we took a reverse-genetics approach, using M. acetivorans as a model organism. In contrast with more streamlined hydrogenotrophic methanogens, M. acetivorans possesses an apparently redundant full set of acquired, nonancestral sulfide assimilation genes (Fig. 1). We posited that the presence of these genes, which encode OASS, OAHS, CD, and CysRS, might be necessary to obtain a viable strain after deleting the gene encoding COG2122. Consistent with this notion, a genome-wide transposon mutagenesis analysis of M. maripaludis has suggested that COG2122 is essential (25).
We first constructed a single-deletion strain lacking the ma1715 gene, which encodes COG2122 in M. acetivorans (Table 1). We used an established markerless approach to excise the coding region of ma1715 from the chromosome and demonstrated that the desired manipulation had occurred using PCR and hybridization analyses (Fig. 2). As a precautionary measure to overcome potential growth defects associated with deleting ma1715, manipulations to construct the deletion strain were performed with a sulfur-rich growth medium that contained sodium sulfide, Cys, and Met.
FIG 2.
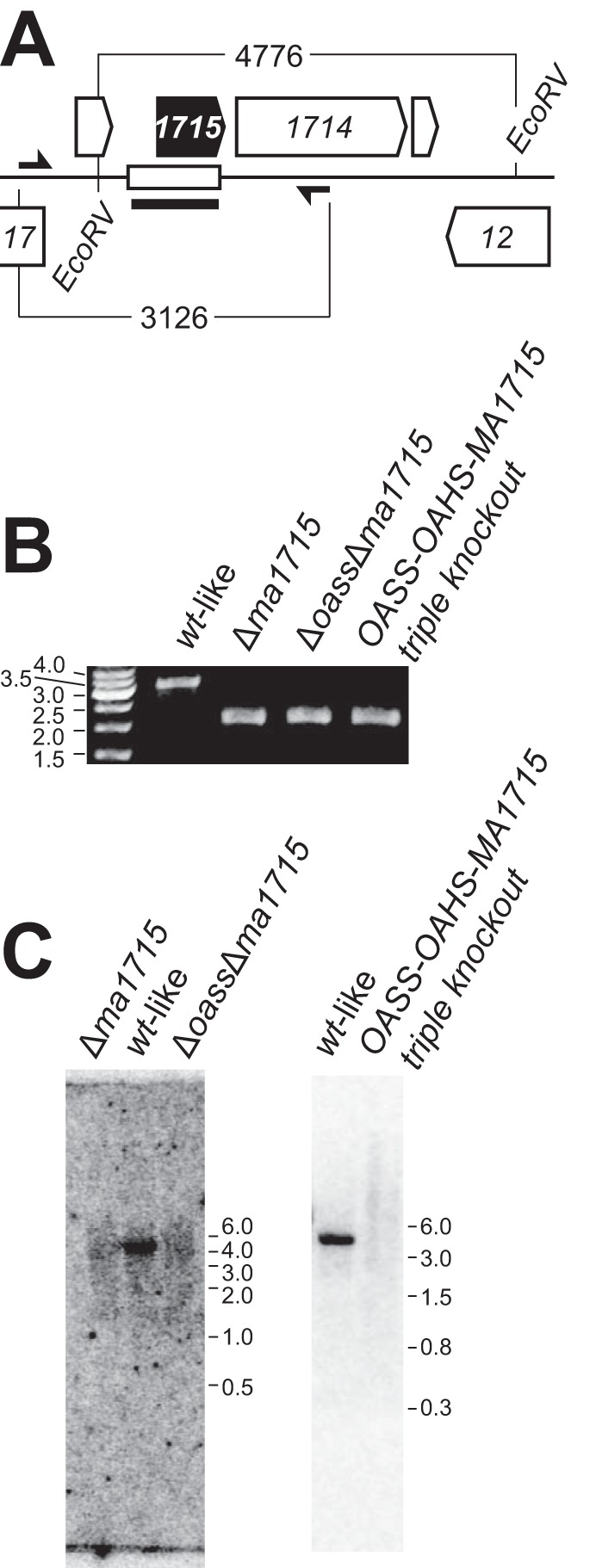
Deletion of ma1715 from M. acetivorans. (A) Reference map for the genomic neighborhood of ma1715 featuring the deleted chromosomal segment (open rectangle) and its flanking EcoRV sites, in addition to the PCR primers (half-arrows) and ma1715-hybridizing probe (thick line segment) that were used to confirm the deletions. (B) Genotyping PCR products for knockout strains BJR02 (Δma1715), BJR24 (OASS-MA1715 double knockout), and BJR23 (OASS-OAHS-MA1715 triple knockout) were resolved on an agarose gel. A wild-type (wt)-like control strain (WWM75) was also analyzed. (C) Gene deletions were confirmed by Southern blotting. The blots were visualized by phosphorimaging.
To establish a link between COG2122 and sulfide assimilation, we tested the Δma1715 single-deletion strain for its ability to utilize sodium sulfide as its sole sulfur source. Relative to the wild-type parent strain, the Δma1715 strain was severely impaired for sulfide-dependent growth. This was most apparent at lower sulfide concentrations (0.05 to 0.2 mM), which did not support any growth of the deletion strain (Fig. 3). In comparison, wild-type M. acetivorans was capable of robust growth at both 0.05 mM and 0.2 mM sulfide, although the growth yield was attenuated at the lower concentration. Growth of the Δma1715 strain was partially restored at 0.8 mM sulfide and nearly fully restored when 3.2 mM sulfide was present in the growth medium.
FIG 3.

Growth of M. acetivorans strains with different concentrations of sodium sulfide present as the sole sulfur source. The wild-type-like and Δma1715 strains are compared across the top row, whereas the OASS-OAHS double-knockout and OASS-OAHS-MA1715 triple-knockout strains are compared across the bottom row. All the data points represent averages of the results of at least four independent experiments. The error bars reflect standard deviations. Prior to each experiment, the strains were grown for at least 12 generations with 0.8 mM sodium sulfide as the sole sulfur source.
To confirm that the observed growth phenotype arose from deleting the ma1715 gene, we tested whether growth of the Δma1715 strain could be recovered at low sulfide concentrations by reintroducing the ma1715 gene on an autonomously replicating plasmid. Transformants were then assayed for growth in the presence of 0.05 mM sulfide. The transformed cells indeed exhibited substantially recovered growth, indicating that deletion of ma1715 was responsible for the sulfide concentration dependence phenotype (see Fig. S1 in the supplemental material).
The inability of the Δma1715 strain to grow at low sulfide concentrations suggests that the gene may indeed encode a sulfide assimilation protein of critical importance. The phenotype of the Δma1715 strain is particularly striking, considering that this single-deletion strain still retains the genes coding for OAHS and OASS, which insert sulfide into activated precursors for biosynthesis of Hcy and Cys, respectively (9). OAHS and OASS are the only two known proteins able to incorporate sulfide into these metabolites, which then serve as precursors for the biosynthesis of Met, protein persulfides, and Cys-tRNACys through pathways facilitated by enzymes known to be present in M. acetivorans (Fig. 1). It appears that either OAHS or OASS, or both, cannot function efficiently at ambient sulfide concentrations of 0.2 mM or lower in a cellular background lacking the ma1715 gene product. This would lead to deficient Hcy and/or Cys levels, blocking the growth of the Δma1715 strain at these low sulfide concentrations.
To further explore the interplay of ma1715 and the OASS and OAHS genes, we next characterized the growth phenotype of an OASS-OAHS double-knockout strain that we had previously constructed (9). In sharp contrast to the behavior of the Δma1715 strain, the OASS-OAHS double-knockout strain was unimpaired for growth at all sulfide concentrations tested (Fig. 3). Therefore, the ma1715 gene product alone is capable of channeling sulfide for the biosynthesis of all sulfur-containing cellular metabolites, including iron-sulfur clusters, tRNA modifications, and cofactors, such as coenzyme M and coenzyme B (Fig. 1). The ability of the MA1715 protein to fully sustain cellular function is consistent with its ubiquitous presence and with the absence of OAHS and OASS in many methanogens (9).
We next constructed new M. acetivorans strains containing knockouts of the sulfide assimilation genes in other combinations. As expected, we found that an OASS-MA1715 double-deletion strain, like the Δma1715 strain, also could not be propagated at ambient sulfide concentrations of 0.2 mM or lower (see Fig. S2 in the supplemental material). This suggests that the presence of OASS in the Δma1715 strain does not exert an inhibitory effect. Strikingly, however, further deletion of the OAHS gene to produce the OASS-OAHS-MA1715 triple knockout allowed growth even at sulfide concentrations as low as 0.05 mM (Fig. 3). The OASS-OAHS-MA1715 triple-knockout strain exhibited a doubling time 2- to 3-fold longer than those of the wild-type and OASS-OAHS double-deletion strains, indicating significant impairment. Nonetheless, it is remarkable that deletion of all three genes still generated a viable strain. These findings show that ma1715 is not essential in M. acetivorans and demonstrate that other mechanisms for assimilating sulfide must exist (see Discussion).
The ability of the OASS-OAHS-MA1715 triple-knockout strain to grow at 0.05 mM and 0.2 mM sulfide suggests that OAHS may play an inhibitory role in the Δma1715 and OASS-MA1715 double-knockout strains, explaining why these deletion strains are not viable at low sulfide concentrations. To study this further, we analyzed the capacity of a Δma1821-Δma1822 double-knockout strain to grow with sulfide as the sole sulfur source. MA1821 and MA1822 together catalyze the conversion of aspartate semialdehyde to Hcy and are essential for Hcy biosynthesis in an M. acetivorans strain that lacks the OAHS gene (9, 10). We found that the Δma1821-Δma1822 strain grew comparably to wild-type-like M. acetivorans at 0.05 mM sulfide (see Fig. S1 in the supplemental material). Since OAHS activity is required for sulfide-dependent homocysteine biosynthesis (and growth) in the Δma1821-Δma1822 strain, we infer that the enzyme is capable of converting sulfide to Hcy at these low concentrations. Possibly, the appropriation of sulfide for Hcy biosynthesis by OAHS in the Δma1715 and OASS-MA1715 double-deletion strains precludes its availability for the alternate assimilation mechanisms that must be present in the OASS-OAHS-MA1715 triple-knockout strain. Such alternate mechanisms may not be able to mobilize sulfide as efficiently as OAHS, explaining the growth-inhibitory effect of the enzyme.
COG2122 is not implicated in the biosynthesis of iron-sulfur clusters or thiolated tRNA.
To examine whether the COG2122 protein has a role in the biosynthesis of downstream sulfur-containing metabolites, we measured iron cluster formation and tRNA thiolation in several of the knockout strains. The abundance of acid-labile sulfide, which approximates the sulfur content of iron-sulfur clusters, was determined in dialyzed cell extracts of the OASS-OAHS double-knockout and OASS-OAHS-MA1715 triple-knockout strains that were grown using 0.2 mM sulfide as the sole sulfur source, using the methylene blue method (6, 26). We observed similar acid-labile sulfide contents in each strain (see Table S1 in the supplemental material), suggesting that ma1715 does not play a crucial role in iron-sulfur cluster biosynthesis.
To detect sulfur modifications in tRNA, we performed electrophoresis experiments using APM-containing polyacrylamide gels. APM gels retard the migration of sulfur-containing compounds, allowing direct comparison of the abundances of modified and unmodified tRNAs. To simultaneously monitor all tRNAs and limit the required amount of tRNA to microgram quantities, we specifically radiolabeled bulk tRNA from crude nucleic acid extracts at the 3′ internucleotide linkage using tRNA nucleotidyltransferase. These experiments were conducted using tRNA extracted from stationary-phase cultures grown with 3.2 mM sulfide as the sole sulfur source. They reveal the presence of similarly high levels of tRNA thiolation in wild-type M. acetivorans, Δma1715, OASS-OAHS double-knockout, and OASS-OAHS-MA1715 triple-knockout strains (Fig. 4A; see Table S1 in the supplemental material). Supplementing the media used to grow these cultures with 3 mM Cys and 3 mM Met produced identical results. We also directly assayed cell extracts from actively growing cultures of OASS-OAHS double-knockout and OASS-OAHS-MA1715 triple-knockout strains for tRNA thiolation activity. Dialyzed extracts from strains grown with 0.2 mM sulfide as the sole sulfur source were incubated with a radiolabeled, in vitro-transcribed tRNA lacking posttranscriptional modifications. Comparable levels of tRNA thiolation were produced in each extract. Thus, both sets of experiments suggest that COG2122 is not required for tRNA thiolation in M. acetivorans (Fig. 4B).
FIG 4.

Deletion of ma1715 does not impair tRNA thiolation in M. acetivorans. (A) Radiolabeled tRNAs from M. acetivorans cells grown to saturation were analyzed by APM gel electrophoresis. Strains were cultured to saturation on sulfide-containing medium with (+) or without (−) added cysteine and methionine. Thiolated (E. coli bulk tRNA) and unthiolated (in vitro transcript tRNA) control samples were also analyzed. The gels were visualized by phosphorimaging. (B) tRNA thiolation activities of M. acetivorans cellular lysates, obtained from actively growing cultures, were compared using APM gel electrophoresis. A radiolabeled, unmodified tRNA transcript was incubated for 60 s with either dialyzed cellular lysate (+) or buffer (−). Reactions were performed anaerobically and in the absence of sulfide or any other exogenously added sulfur source. The gels were visualized by phosphorimaging.
Growth of the OASS-OAHS-MA1715 triple-knockout strain in media with sulfur-containing amino acids.
We next compared the growth of the OASS-OAHS-MA1715 triple-knockout strain with that of its parent OASS-OAHS double-knockout strain under conditions in which 0.2 mM sulfide was supplemented with 0.75 mM Hcy, 2 mM Cys, 1 mM cystathionine, or 2 mM Met (Fig. 5). The OASS-OAHS double-knockout strain already grew robustly with 0.2 mM sulfide alone, and so, growth was only slightly improved by further addition of these amino acids. In contrast, the relatively weak growth of the OASS-OAHS-MA1715 triple-knockout strain at 0.2 mM sulfide was greatly accelerated by supplementation with either Hcy or Cys (Fig. 5). These data are consistent with the notion that COG2122 is required for the efficient biosynthesis of both Cys and Hcy. The triple knockout was also fully reconstituted by supplementation with cystathionine. Robust growth with either Hcy or cystathionine supplementation is consistent with the existence of a pathway from Hcy through cystathionine to Cys, as in other microbes, despite the apparent absence in M. acetivorans of all the genes involved in the transsulfuration pathways that interconvert Hcy and Cys (1). It is important to note, however, that cystathionine supplementation does not complement Hcy auxotrophy in an MA1821-MA1822-OAHS triple-knockout M. acetivorans strain (9). Thus, Cys-to-Hcy transulfuration does not function in this organism. The robust growth from Cys supplementation in the OASS-OAHS-MA1715 triple-knockout strain may instead arise because cysteine desulfurases direct sulfur to MA1821-MA1822 for Hcy synthesis (see Discussion).
FIG 5.

Growth of M. acetivorans strains in the presence of 0.2 mM sodium sulfide and amino acid supplements. Cultures of the OASS-OAHS-MA1715 triple-knockout (left) and OASS-OAHS double-knockout (right) strains were supplemented with 0.75 mM homocysteine (+Hcy), 2 mM cysteine (+Cys), 1 mM cystathionine (+cysth), or 2 mM methionine (+Met). Unsupplemented cultures (no supp.) are also shown. All the data points represent averages of the results of at least four independent experiments. The error bars reflect standard deviations. Prior to each experiment, the strains were grown for at least 12 generations under the reported assay conditions. The data for both unsupplemented growth experiments were replotted from Fig. 3.
Unlike with the other three amino acids, Met supplementation of the OASS-OAHS-MA1715 triple-knockout strain growing at 0.2 mM sulfide did not reconstitute robust growth (Fig. 5). This suggests that Met recycling (through S-adenosylmethionine and S-adenosylhomocysteine) is inadequate to meet cellular demand for Hcy and that the metabolic utility of Hcy extends beyond Met biosynthesis in M. acetivorans. In particular, the absence of OASS in the OASS-OAHS-MA1715 triple-knockout strain suggests that Hcy provides sulfur for Cys biosynthesis.
Finally, we characterized the growth of the OASS-OAHS-MA1715 triple-knockout strain with Hcy as the sole sulfur source. While 0.8 mM sulfide promoted growth of the triple knockout to levels comparable to that observed in the wild-type strain (albeit with nearly a 3-fold increase in the doubling time), 0.8 mM Hcy as the sole sulfur source supported only very weak growth (see Fig. S3 in the supplemental material). Therefore, sulfide appears to function more efficiently than Hcy in promoting sulfur trafficking by mechanisms independent of OAHS, OASS, and MA1715. The advantage provided by sulfide, however, disappears at higher concentrations of about 3 mM Hcy (Fig. 3; see Fig. S3 in the supplemental material).
We also noted that the slow growth of the triple knockout observed with 0.2 mM sulfide (Fig. 5) was restored to wild-type levels when 0.75 mM Hcy was added, even though 0.8 mM Hcy supports only very weak growth when it is the sole sulfur source (Fig. 5; see Fig. S3 in the supplemental material). One interpretation of this finding is that there are multiple pathways by which sulfur can be incorporated into downstream metabolites. Employing more than one such pathway through the use of multiple sulfur sources has synergistic effects compared to those of either individual pathway alone.
Revisiting the evolutionary history of COG2122.
The recent finding that the phylum Bathyarchaeota also contains methanogens allows phylogenetic reconstructions that provide new insights into the evolution of ancient anaerobic biochemical processes. Recent analysis of methanogenesis protein phylogeny, including sequence information from Bathyarchaeota, suggested vertical inheritance of genes encoding methanogenesis enzymes, allowing the inference that methanogenesis emerged prior to the divergence of the Bathyarchaeota and Euryarchaeota (27).
Previously, we generated phylogenetic reconstructions of COG2122 and COG1900a suggesting that the genes encoding both proteins were vertically inherited from the ancestral euryarchaeote (9). To determine whether COG2122 is ancestral to the Bathyarchaeota and Euryarchaeota, we generated a new phylogenetic reconstruction of COG2122 that included all the archaeal homologs. An unrooted tree clearly distinguished bathyarchaeal sequences from euryarchaeal sequences (Fig. 6). Moreover, its topology was suggestive of vertical inheritance, since it resembled those from reported reconstructions of mcrA and organismal phylogenies (14, 27). Thus, we infer that COG2122 shares an evolutionary history with methanogenesis dating back to the common ancestor of the Euryarchaeota and Bathyarchaeota.
FIG 6.

Phylogenetic reconstruction of archaeal COG2122 proteins. Shown is a neighbor-joining tree representing COG2122 phylogeny. Collapsed braches are shown as triangles, and the corresponding taxonomic designations are shown in boldface. Bootstrapping support is shown adjacent to selected nodes as a percentage of 1,000 replicates.
DISCUSSION
We have demonstrated that the conserved protein MA1715 of M. acetivorans, a member of the COG2122 family, plays a key role in sulfide assimilation and trafficking in that organism. The most likely role of MA1715 is to directly bind and mobilize sulfide for insertion into metabolic precursors of both Cys and Hcy (Fig. 1). This proposed function is supported by three lines of experimental evidence. First, the Δma1715 and OASS-MA1715 double-deletion strains do not grow at submillimolar sulfide concentrations, while the OASS-OAHS-MA1715 triple-knockout strain exhibits weak growth under these conditions (Fig. 3; see Fig. S2 in the supplemental material). Thus, while COG2122 is not always essential for cell growth, it is required for optimal growth with sulfide as the sole sulfur source in all the genetic backgrounds tested. Next, the OASS-OAHS double-deletion strain exhibited wild-type levels of growth at all sulfide concentrations tested (Fig. 3). OASS and OAHS are the only enzymes in M. acetivorans previously known to assimilate sulfide for Cys and Hcy biosynthesis. Therefore, unless other sulfide-mobilizing enzymes for Cys and Hcy biosynthesis remain undiscovered, it appears that COG2122 is capable of replacing the functions of both of these enzymes. Finally, supplementing cultures of the OASS-OAHS-MA1715 triple-knockout strain grown at low sulfide concentrations with either Cys or Hcy fully restored growth to wild-type levels (Fig. 5). Together with the wild-type growth properties of the OASS-OAHS double-deletion strain, this suggests that COG2122 plays an important role in both Cys and Hcy biosynthesis in M. acetivorans.
The data show that M. acetivorans harbors at least three distinct pathways by which sulfur is incorporated into Cys and Hcy. In addition to the acquired OASS and OAHS enzymes (pathway 1) and the anaerobe-specific MA1715 protein (pathway 2), a third pathway must operate under low sulfide concentrations when all three of these activities are absent (Fig. 3). It is likely that in wild-type cells this additional pathway operates only at high ambient sulfide concentrations. Sulfide oxidation and incorporation into Cys and Hcy in the OASS-OAHS-MA1715 triple-knockout strain might be enabled by nonenzymatic processes or by as yet undiscovered enzymatic activities (6). Further, while efficient SepCysS function likely requires persulfide formation (7), the extent to which protein persulfidation and relay are essential in methanogens remains largely unknown. It is possible that pathways of sulfur incorporation into Cys, Hcy, cofactors, and tRNA exhibit varied dependencies on persulfide formation, depending on the ambient sulfide concentrations. Further, some enzymes that incorporate sulfur into products may be able to function either through persulfide formation or via direct binding and oxidation of sulfide (10, 28). While MA1715 may promote persulfide formation on SepCysS and MA1821 for Cys and Hcy biosynthesis, its dispensability for iron-sulfur cluster formation and tRNA thiolation suggests that persulfide intermediates are less important to these processes (Fig. 4) (6, 28).
Sequence homology searches did not detect genes in M. acetivorans that interconvert Cys and Hcy through cystathionine, which are prevalent in many other organisms (Fig. 1, right). However, our findings that robust growth of the OASS-OAHS-MA1715 triple-knockout strain can be recovered by supplementation with either Hcy or cystathionine nonetheless suggest the presence of still-undiscovered enzymes that can convert these metabolites to Cys. Although robust growth recovery of the OASS-OAHS-MA1715 triple-knockout strain is also achieved by supplementing the growth medium with Cys, we previously found that cystathionine did not relieve Hcy auxotrophy in an M. acetivorans strain lacking both COG1900a and OAHS (9). Therefore, enzymes that convert Cys to cystathionine and subsequently to Hcy appear not to be present. Growth recovery by Cys supplementation may instead be due to cysteine-dependent persulfide relay by cysteine desulfurases, possibly resulting in the transfer of sulfane sulfur to COG1900a for Hcy biosynthesis.
COG2122 is a member of the large ApbE-like protein family, many of whose members bind FAD (12, 13). Interestingly, structure-based sequence alignments suggest that the COG2122 protein in M. acetivorans and other methanogens lacks the FAD-binding portion of the structure (see Fig. S4 in the supplemental material). Since sulfide activation does not require electron transfer, we speculate that COG2122 may facilitate the nucleophilic attack of sulfide on a different metabolite. Interestingly, in the Archaea and Eukarya, selenocysteine is biosynthesized as selenocysteinyl-tRNASec (Sec-tRNASec) in a pathway that involves initial formation of Sep-tRNASec from Ser-tRNASec, followed by conversion to Sec-tRNASec via attack of selenophosphate (29). By analogy, thiophosphate formed by COG2122 (perhaps via reaction of sulfide with a compound containing a high-energy phosphate linkage) could promote conversion of Sep-tRNACys to Cys-tRNACys. An indirect role for COG2122 as an activator of SepCysS and COG1900a is also plausible but seems unlikely considering that Cys supplementation restores rapid growth of the OASS-OAHS-MA1715 triple-knockout strain (Fig. 5). Since this strain lacks OAHS, rapid growth implies that COG1900a is highly active for Hcy biosynthesis, even though COG2122 is absent.
Finally, since an updated phylogenetic reconstruction of archaeal COG2122 proteins is congruous with reported organismal phylogenies, COG2122 proteins are likely to share an evolutionary history with methanogenesis and may thus participate in an ancient strategy for sulfide assimilation that emerged prior to the divergence of the Bathyarchaeota and Euryarchaeota. Although the precise mechanism by which COG2122 enhances sulfide assimilation remains to be elucidated, the experiments reported here nonetheless significantly advance our understanding of how anaerobes possessing SepCysS and COG1900a assimilate sulfide into Cys and Hcy.
Supplementary Material
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00141-16.
REFERENCES
- 1.Liu Y, Beer LL, Whitman WB. 2012. Methanogens: a window into ancient sulfur metabolism. Trends Microbiol 20:251–258. doi: 10.1016/j.tim.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 2.Sekowska A, Kung HF, Danchin A. 2000. Sulfur metabolism in Escherichia coli and related bacteria: facts and fiction. J Mol Microbiol Biotechnol 2:145–177. [PubMed] [Google Scholar]
- 3.Kessler D. 2006. Enzymatic activation of sulfur for incorporation into biomolecules in prokaryotes. FEMS Microbiol Rev 30:825–840. doi: 10.1111/j.1574-6976.2006.00036.x. [DOI] [PubMed] [Google Scholar]
- 4.Mueller EG. 2006. Trafficking in persulfides: delivering sulfur in biosynthetic pathways. Nat Chem Biol 2:185–194. doi: 10.1038/nchembio779. [DOI] [PubMed] [Google Scholar]
- 5.Sauerwald A, Zhu W, Major TA, Roy H, Palioura S, Jahn D, Whitman WB, Yates JR III, Ibba M, Söll D. 2005. RNA-dependent cysteine biosynthesis in archaea. Science 307:1969–1972. doi: 10.1126/science.1108329. [DOI] [PubMed] [Google Scholar]
- 6.Liu Y, Sieprawska-Lupa M, Whitman WB, White RH. 2010. Cysteine is not the sulfur source for iron-sulfur cluster and methionine biosynthesis in the methanogenic archaeon Methanococcus maripaludis. J Biol Chem 285:31923–31929. doi: 10.1074/jbc.M110.152447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y, Dos Santos PC, Zhu X, Orlando R, Dean DR, Söll D, Yuan J. 2012. Catalytic mechanism of Sep-tRNA:Cys-tRNA synthase: sulfur transfer is mediated by disulfide and persulfide. J Biol Chem 287:5426–5433. doi: 10.1074/jbc.M111.313700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Helgadottir S, Sinapah S, Söll D, Ling J. 2012. Mutational analysis of Sep-tRNA:Cys-tRNA synthase reveals critical residues for tRNA-dependent cysteine formation. FEBS Lett 586:60–63. doi: 10.1016/j.febslet.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rauch BJ, Gustafson A, Perona JJ. 2014. Novel proteins for homocysteine biosynthesis in anaerobic microorganisms. Mol Microbiol 94:1330–1342. doi: 10.1111/mmi.12832. [DOI] [PubMed] [Google Scholar]
- 10.Allen KD, Miller DV, Rauch BJ, Perona JJ, White RH. 2015. Homocysteine is biosynthesized from aspartate semialdehyde and hydrogen sulfide in methanogenic archaea. Biochemistry 54:3129–3132. doi: 10.1021/acs.biochem.5b00118. [DOI] [PubMed] [Google Scholar]
- 11.Boyd JM, Endrizzi JA, Hamilton TL, Christopherson MR, Mulder DW, Downs DM, Peters JW. 2011. FAD binding by ApbE protein from Salmonella enterica: a new class of FAD-binding proteins. J Bacteriol 193:887–895. doi: 10.1128/JB.00730-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bertsova YV, Fadeeva MS, Kostyrko VA, Serebryakova MV, Baykov AA, Bogachev AV. 2013. Alternative pyrimidine biosynthesis protein ApbE is a flavin transferase catalyzing covalent attachment of FMN to a threonine residue in bacterial flavoproteins. J Biol Chem 288:14276–14286. doi: 10.1074/jbc.M113.455402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deka RK, Brautigam CA, Liu WZ, Tomchick DR, Norgard MV. 2013. The TP0796 lipoprotein of Treponema pallidum is a bimetal-dependent FAD pyrophosphatase with a potential role in flavin homeostasis. J Biol Chem 288:11106–11121. doi: 10.1074/jbc.M113.449975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans PN, Parks DH, Chadwick GL, Robbins SJ, Orphan VJ, Golding SD, Tyson GW. 2015. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science 350:434–438. doi: 10.1126/science.aac7745. [DOI] [PubMed] [Google Scholar]
- 15.Guss AM, Rother M, Zhang JK, Kulkarni G, Metcalf WW. 2008. New methods for tightly regulated gene expression and highly efficient chromosomal integration of cloned genes for Methanosarcina species. Archaea 2:193–203. doi: 10.1155/2008/534081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sowers KR, Boone JE, Gunsalus RP. 1993. Disaggregation of Methanosarcina spp. and growth as single cells at elevated osmolarity. Appl Environ Microbiol 59:3832–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Metcalf WW, Zhang JK, Shi X, Wolfe RS. 1996. Molecular, genetic, and biochemical characterization of the serC gene of Methanosarcina barkeri Fusaro. J Bacteriol 178:5797–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pritchett MA, Zhang JK, Metcalf WW. 2004. Development of a markerless genetic exchange method for Methanosarcina acetivorans C2A and its use in construction of new genetic tools for methanogenic archaea. Appl Environ Microbiol 70:1425–1433. doi: 10.1128/AEM.70.3.1425-1433.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buan N, Kulkarni G, Metcalf WW. 2011. Genetic methods for methanosarcina species. Methods Enzymol 494:23–42. doi: 10.1016/B978-0-12-385112-3.00002-0. [DOI] [PubMed] [Google Scholar]
- 20.Wolfson AD, Uhlenbeck OC. 2002. Modulation of tRNAAla identity by inorganic pyrophosphatase. Proc Natl Acad Sci U S A 99:5965–5970. doi: 10.1073/pnas.092152799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Igloi GL. 1988. Interaction of tRNAs and of phosphorothioate-substituted nucleic acids with an organomercurial. Probing the chemical environment of thiolated residues by affinity electrophoresis. Biochemistry 27:3842–3849. [DOI] [PubMed] [Google Scholar]
- 22.Biondi E, Burke DH. 2012. Separating and analyzing sulfur-containing RNAs with organomercury gels. Methods Mol Biol 883:111–120. doi: 10.1007/978-1-61779-839-9_8. [DOI] [PubMed] [Google Scholar]
- 23.Sherlin LD, Bullock TL, Nissan TA, Perona JJ, Lariviere FJ, Uhlenbeck OC, Scaringe SA. 2001. Chemical and enzymatic synthesis of tRNAs for high-throughput crystallization. RNA 7:1671–1678. [PMC free article] [PubMed] [Google Scholar]
- 24.Hauenstein SI, Perona JJ. 2008. Redundant synthesis of cysteinyl-tRNACys in Methanosarcina mazei. J Biol Chem 283:22007–22017. doi: 10.1074/jbc.M801839200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarmiento F, Mrazek J, Whitman WB. 2013. Genome-scale analysis of gene function in the hydrogenotrophic methanogenic archaeon Methanococcus maripaludis. Proc Natl Acad Sci U S A 110:4726–4731. doi: 10.1073/pnas.1220225110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moest R. 1975. Hydrogen sulfide determination by the methylene blue method. Anal Chem 47:1204–1205. doi: 10.1021/ac60357a008. [DOI] [Google Scholar]
- 27.Blank CE. 2009. Phylogenomic dating—the relative antiquity of archaeal metabolic and physiological traits. Astrobiology 9:193–219. doi: 10.1089/ast.2008.0248. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Zhu X, Nakamura A, Orlando R, Söll D, Whitman WB. 2012. Biosynthesis of 4-thiouridine in tRNA in the methanogenic archaeon Methanococcus maripaludis. J Biol Chem 287:36683–36692. doi: 10.1074/jbc.M112.405688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan J, Palioura S, Salazar JC, Su D, O'Donoghue P, Hohn MJ, Cardoso AM, Whitman WB, Söll D. 2006. RNA-dependent conversion of phosphoserine forms selenocysteine in eukaryotes and archaea. Proc Natl Acad Sci U S A 103:18923–18927. doi: 10.1073/pnas.0609703104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.