

ABSTRACT
Next-generation sequencing technology is now being increasingly applied to study the within- and between-host population dynamics of viruses. However, information on avian influenza virus evolution and transmission during a naturally occurring epidemic is still limited. Here, we use deep-sequencing data obtained from clinical samples collected from five industrial holdings and a backyard farm infected during the 2013 highly pathogenic avian influenza (HPAI) H7N7 epidemic in Italy to unravel (i) the epidemic virus population diversity, (ii) the evolution of virus pathogenicity, and (iii) the pathways of viral transmission between different holdings and sheds. We show a high level of genetic diversity of the HPAI H7N7 viruses within a single farm as a consequence of separate bottlenecks and founder effects. In particular, we identified the cocirculation in the index case of two viral strains showing a different insertion at the hemagglutinin cleavage site, as well as nine nucleotide differences at the consensus level and 92 minority variants. To assess interfarm transmission, we combined epidemiological and genetic data and identified the index case as the major source of the virus, suggesting the spread of different viral haplotypes from the index farm to the other industrial holdings, probably at different time points. Our results revealed interfarm transmission dynamics that the epidemiological data alone could not unravel and demonstrated that delay in the disease detection and stamping out was the major cause of the emergence and the spread of the HPAI strain.
IMPORTANCE The within- and between-host evolutionary dynamics of a highly pathogenic avian influenza (HPAI) strain during a naturally occurring epidemic is currently poorly understood. Here, we perform for the first time an in-depth sequence analysis of all the samples collected during a HPAI epidemic and demonstrate the importance to complement outbreak investigations with genetic data to reconstruct the transmission dynamics of the viruses and to evaluate the within- and between-farm genetic diversity of the viral population. We show that the evolutionary transition from the low pathogenic form to the highly pathogenic form occurred within the first infected flock, where we identified haplotypes with hemagglutinin cleavage site of different lengths. We also identify the index case as the major source of virus, indicating that prompt application of depopulation measures is essential to limit virus spread to other farms.
INTRODUCTION
Today, next-generation sequencing (NGS) techniques allow the investigation of viral population dynamics at any level (from within host to the epidemiological scale) with high resolution. In addition, NGS can be used to identify low-frequency variants, which may be selected for and transmitted to other hosts. Avian influenza viruses exist in the host as populations of genetically related variants (1). The rate at which genetic diversity is generated within the host, the competitive replication ability of each variant, and the occurrence of genetic drift and of bottleneck events are some of the processes that drive virus evolution.
NGS has been applied to avian influenza virus (i) to characterize the emergence of mutations in the viral subpopulations associated with an increased virulence (2, 3) or with adaptation to new hosts, (4, 5), (ii) to study genetic bottlenecks upon transmission events (6, 7), (iii) to investigate the dynamics of virus evolution during outbreaks in poultry (8), and (iv) to identify coinfection with different subtypes (9). However, application of high-throughput sequencing for the exploration of avian influenza virus evolution and transmission during a naturally occurring epidemic is still limited, making the interpretation of genomic data collected from outbreaks far from straightforward.
Between 13 August and 3 September 2013, 13 years after the last highly pathogenic avian influenza (HPAI) outbreak, Italy experienced a new avian influenza epidemic caused by an HPAI virus of the H7N7 subtype, which infected five industrial poultry holdings, four of which belonged to a large vertically integrated layer company, and one backyard flock (10). Detailed information on these outbreaks has been provided in a previous study (10). The epidemiological investigation indicated that the contact between free-range hens and wild waterfowl in the first affected holding may have favored the introduction of a low-pathogenicity avian influenza (LPAI) virus, which rapidly mutated into a highly pathogenic form within the infected sheds (10) through the acquisition of multiple basic amino acids at the hemagglutinin (HA) cleavage site, which is considered being the major molecular determinant of an HPAI virus (11).
We used NGS to unravel the virus population diversity and the evolution of virus pathogenicity within the affected poultry farms. We also determined the transmission pathways of the H7N7 virus between different holdings and sheds during the course of the epidemic by combining deep-sequencing and epidemiological data.
MATERIALS AND METHODS
Viruses.
Fourteen positive clinical samples (organs and swabs) were collected between 13 August and 3 September 2013 from each infected shed of the five industrial farms and a backyard flock, counting all the sheds infected during the epidemic (10). The epidemiological data, including the collection date, the sample type (swabs, organs), the farm and shed of origin, and the number of birds present at each farm at the time of the forfeiture and depopulation date, are presented in Table 1. The viral RNA copy numbers (Table 1) were determined for each sample using a quantitative real-time RT-PCR assay with a standard curve targeting the M gene of influenza A virus, using the published probes and primers from Spackman et al. (12).
TABLE 1.
Epidemiological information for 14 samples collected during HPAI H7N7 outbreak
| Site | Sample | RNA copies/μl | Mean depth of coverage | Sample typea | Animal (farm type) | Collection date | Province | No. of birds | Depopulation date |
|---|---|---|---|---|---|---|---|---|---|
| Farm 1, shed 2 | 4527-11 | 1.24E+05 | 19,354 | Pool of 10 TS | Laying hen (industrial farm) | 13 Aug 2013 | Ferrara | 128,000 | 27 Aug 2013 |
| 4527-12 | 9.22E+07 | 36,772 | Pool of 10 TS | Laying hen (industrial farm) | 13 Aug 2013 | Ferrara | 128,000 | 27 Aug 2013 | |
| 4541-7 | 1.04E+06 | 24,696 | Organ pool* | Laying hen (industrial farm) | 13 Aug 2013 | Ferrara | 128,000 | 27 Aug 2013 | |
| 4541-32 | 2.49E+07 | 53,292 | Kidney | Laying hen (industrial farm) | 13 Aug 2013 | Ferrara | 128,000 | 27 Aug 2013 | |
| Farm 1, shed 4 | 4541-8 | 5.24E+05 | 34,018 | Organ pool* | Laying hen (industrial farm) | 13 Aug 2013 | Ferrara | 128,000 | 27 Aug 2013 |
| 4541-33 | 4.98E+03 | 42,661 | Kidney | Laying hen (industrial farm) | 13 Aug 2013 | Ferrara | 128,000 | 27 Aug 2013 | |
| Farm 1, shed 5 | 4541-9 | 3.93E+04 | 23,390 | Organ pool* | Laying hen (industrial farm) | 13 Aug 2013 | Ferrara | 128,000 | 27 Aug 2013 |
| 4541-34 | 4.32E+04 | 58,893 | Kidney | Laying hen (industrial farm) | 13 Aug 2013 | Ferrara | 128,000 | 27 Aug 2013 | |
| Farm 2 | 4603-1 | 2.89E+05 | 43,810 | Pool of 10 TS | Laying hen (industrial farm) | 19 Aug 2013 | Bologna | 584,900 | 8 Sept 2013 |
| Farm 3 | 4678 | 7.88E+05 | 19,893 | Organ pool* | Meat turkey (industrial farm) | 21 Aug 2013 | Ferrara | 19,850 | 27 Aug 2013 |
| Farm 4 | 4774 | 2.30E+08 | 31,804 | Organ pool** | Laying hen (industrial farm) | 27 Aug 2013 | Bologna | 121,705 | 8 Sept 2013 |
| Farm 5 | 5091 | 1.21E+07 | 24,510 | Organ pool* | Backyard flock | 2 Sept 2013 | Ferrara | 3 | 5 Sept 2013 |
| Farm 6 | 5051-1 | 5.27E+07 | 46,615 | Trachea | Pullets (industrial farm) | 3 Sept 2013 | Bologna | 98,200 | 8 Sept 2013 |
| 5051-3 | 1.02E+08 | 48,562 | Trachea | Pullets (industrial farm) | 3 Sept 2013 | Bologna | 98,200 | 8 Sept 2013 |
TS, tracheal swabs; *, pool of organs from two animals; **, pool of organs from three animals.
Generation of viral sequence data.
Total RNA was purified from 14 infected clinical samples using a Nucleospin RNA kit (Macherey-Nagel, Duren, Germany). Complete influenza A virus genomes were amplified with a SuperScript III One-Step RT-PCR system with Platinum Taq High Fidelity (Invitrogen, Carlsbad, CA) using one pair of primers complementary to the conserved elements of the influenza A virus promoter as described previously (13). PCR products were visualized on a 0.7% agarose gel. Sequencing libraries were obtained using Nextera DNA XT sample preparation kit (Illumina) according to the manufacturer's instructions and quantified using a Qubit dsDNA high-sensitivity kit (Invitrogen). The average fragment length was determined using an Agilent high-sensitivity bioanalyzer kit. Finally, the indexed libraries were pooled in equimolar concentrations and sequenced in multiplex for 250-bp paired-end Illumina MiSeq analysis according to the manufacturer's instructions.
Quality trimming, assembly, and single-nucleotide polymorphism detection.
Illumina MiSeq reads were inspected using FASTQC to assess the quality of data. Fastq files were cleaned with PRINSEQ and Trim Galore to remove low-quality bases at the 5′ and 3′ ends of each read and to exclude reads with a Phred quality score below 30 and shorter than 80 nucleotides. The filtered, trimmed reads were aligned to the eight gene segments of A/chicken/Italy/13VIR4727-11/2013, for which the consensus genome had been previously obtained using the Sanger method (data not shown) with BWA-MEM (v.0.7.5a; http://arxiv.org/abs/1303.3997v2). The BAM alignment files were parsed using the diversiTools program (http://josephhughes.github.io/btctools/) to determine the average base-calling error probability and to identify the frequency of polymorphisms at each site relative to the reference used for the alignment. In order to minimize artifacts introduced through reverse transcription-PCR and sequencing errors for all the analyses conducted here, we considered only polymorphisms with a frequency of >2% identified in positions with a minimum coverage of 500. This choice was based on a comparison of data obtained from two technical replicates of three samples (4541-8, 4541-9, and 4541-34) sequenced on two different Illumina sequencing machines (MiSeq), starting from two separate libraries obtained from the same extracted RNA. This threshold should guarantee the exclusion of 99.6% of the errors from our deep-sequencing data (Fig. 1). For each replicate, only the aligned genome with the highest coverage was used in the following analyses.
FIG 1.
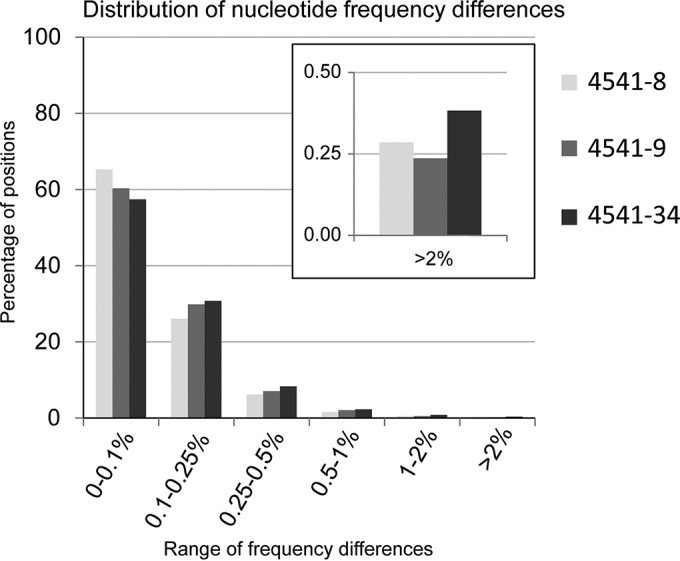
Distribution of nucleotide frequency differences between three technical replicates. For each genome position with coverage of >500, the frequency differences between the four bases (A, C, T, and G) were obtained from a comparison of the replicates of three samples: 4541-8 in yellow, 4541-9 in violet, and 4541-34 in blue. The y axis represents the percentage of nucleotide positions where the highest frequency differences fall within the ranges 0 to 0.1%, 0.1 to 0.25%, 0.25 to 0.5%, 0.5 to 1%, 1 to 2% and >2% (x axis). Frequency differences higher than 2% were observed in only 0.3 to 0.4% of all the analyzed positions (11,501 to 13,308) for all the replicates. Thus, a 2% threshold allows the exclusion of 99.6% of the possible errors.
For each gene, we calculated the number of synonymous and nonsynonymous polymorphisms present either at a consensus level or as subpopulations and normalized to the number of synonymous and nonsynonymous sites in the coding regions. Significant differences between the frequencies of the two types of mutation in the different genes were calculated using a two-way analysis of variance (ANOVA). A P value of <0.05 was considered significant.
Genetic distance, entropy, and transmission tree.
We computed the genetic distance between the complete genome of all pairs of individuals (S1 and S2) using the following formula:
where fAiS1, fCiS1, fTiS1, and fGiS1 are the frequencies of nucleotide A, C, T, and G at position i in the two samples, and N is the length of the sequence. This matrix was used to compute a neighbor-joining phylogenetic tree using the web server T-REX (14). In addition, we combined the distance matrix and the collection dates to reconstruct the transmission tree of the H7N7 during the Italian outbreak, using SeqTrack (15), a graph-based approach particularly suitable to infer maximum-parsimony genealogies of viruses in a densely sampled disease outbreak. The adegenet (16) and igraph packages (17) for the R software were used to perform the analysis and to draw the network.
To measure the complexity of the viral populations within a sample, we calculated the Shannon entropy of each sample and each gene using the following equation:
where fi is the frequency of the nucleotide A, T, G, or C at position i, and N is the total length of the gene segment (average entropy per gene) or of the genome (average entropy per sample). Only nucleotides with a frequency above the 2% threshold identified in positions with a minimum coverage of 500 were included in this calculation. We used one-way ANOVA to determine significant differences between the entropies of each gene for each sample. A P value of <0.05 was considered significant.
Phylogenetic analyses.
Consensus sequences of the complete genome of the 14 samples were aligned using MAFFT v.7 (18) and compared to the most related sequences available in GenBank and in GISAID (accessed on May 2015). In addition, representative H7 viruses circulating in wild and domestic birds in Europe and H7 viruses responsible of important epidemics were included in the alignment. Maximum-likelihood phylogenetic trees were obtained for each gene segment using the best-fit general-time-reversible (GTR) model of nucleotide substitution with gamma-distributed rate variation among sites (with four rate categories, Γ4) available in RAxML-MPI v8.1.7 (19). To assess the robustness of individual nodes of the phylogeny, 100 bootstrap replicates were performed. Phylogenetic trees were visualized with the program FigTree v1.4 (http://tree.bio.ed.ac.uk/software/figtree/).
The eight gene segments of the influenza virus genome were manually concatenated, and the alignment was used to construct a phylogenetic network using the median joining method implemented in the program NETWORK 4.5 (20). This method uses a parsimony approach to reconstruct the relationships between highly similar sequences and allows the creation of “median vectors,” which represent unsampled sequences, that are used to connect the existing genotypes in the most parsimonious way. The parameter “epsilon” was set to 10, and the transition/transversion ratio was set to 3:1. A bootstrap resampling process (1,000 replicates) using a distance-based method (NeighborNet) implemented in SplitsTree4 v4.14.2 (21) was used to assess the robustness of the network edges.
Nucleotide sequence accession numbers.
MiSeq sequences were submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/Traces/sra/) under accession numbers SRR3036850, SRR3036852, SRR3036854, SRR3036856, SRR3036860, SRR3036864, SRR3036910, SRR3036911, SRR3036914, SRR3036916, SRR3036917, SRR3036919, SRR3036920, and SRR3036945. Consensus sequences of the 14 H7N7 viruses were submitted to GISAID under accession numbers EPI677984 to EPI678095.
RESULTS
Phylogenetic analysis of consensus sequences.
To investigate influenza virus variation during the HPAI H7N7 epidemic, we sequenced the eight genomic segments for all the clinical samples received from each infected farm. The highest number of positive samples (8) was submitted from the three infected sheds (sheds 2, 4, and 5) of the index case, whereas only one sample per infected shed was received from the remaining five outbreak sites, for a total of one or two samples per farm. Farms are labeled from 1 to 6, according to the collection date of the samples. Details of the location, date of sample collection, farm characteristics, sample type, and mean depth of coverage are reported in Table 1.
Our maximum-likelihood phylogenetic analyses of the consensus sequences show that the 14 HPAI H7N7 viruses form a distinct genetic group, defined by high bootstrap values (>96%) and long branches in all the eight phylogenies, suggesting the occurrence of a single viral introduction (Fig. 2). This group includes also the sequences of the complete genome available for one of the three poultry workers involved in the depopulation, who developed conjunctivitis due to HPAI H7N7 infection, suggesting a direct transmission of the virus from poultry to humans (22). In the HA and NA phylogenetic trees, the Italian H7N7 HPAI cluster with H7 viruses collected in Europe between 2009 and 2014. In particular, the HA gene segment of the Italian samples show the highest similarity (99.1 to 99.3%) with an LPAI H7N7 virus collected from a wild bird in Italy in 2014, for which only the HA sequence is available (Fig. 2), whereas the NA gene segment displays the highest identity (99 to 99.1%) with an H7N7 virus collected from chicken in the Netherlands (phylogenetic tree is available upon request). In the phylogenies of the internal gene segments, the Italian samples group with viruses of different subtypes circulating mainly among wild birds in Eurasian countries (phylogenetic trees are available upon request).
FIG 2.

Maximum-likelihood phylogenetic tree of the HA gene segment of 172 H7 avian influenza viruses. HPAI H7N7 viruses collected during Italian epidemic are colored according to the farm of collection: gray for farm 1, purple for farm 2, light blue for farm 3, yellow for farm 4, green for farm 5, and orange for farm 6. The numbers at the nodes represent bootstrap values (>70%), whereas the branch lengths are scaled according to the numbers of nucleotide substitutions per site. The tree is midpoint rooted for clarity only.
High genetic variability of the first infected flock.
Surprisingly, molecular analysis of the eight viruses collected from the index case shows the cocirculation of two highly pathogenic strains with a different insertion at the HA cleavage site compared to a H7 LP virus. Specifically, sequences of the two viruses from shed 5 (4541-9 and 4541-34) show an insertion of 6 nucleotides, whereas the remaining samples identified in sheds 2 and 4 possess a longer cleavage site with a 9-nucleotide insertion (Fig. 3).
FIG 3.

Consensus level nucleotide and amino acid differences among the complete genome of the 14 Italian H7N7 viruses. Each sample (column) is colored according to the farm of collection: gray for farm 1, purple for farm 2, light blue for farm 3, yellow for farm 4, green for farm 5, and orange for farm 6. The farm and shed of belonging (i.e., “1-shed 5” represents “farm 1, shed 5”), and the sample type is indicated above the sample name. The nucleotide (NT) differences identified between each sample and the viruses from shed 5 of the index case (samples 4541-9 and 4541-34, columns 1 and 2) are reported. Amino acid mutations (AA) are highlighted in red, while silent mutations are in black. The 11 different genomes identified during this epidemic are indicated in the last row (A to K).
To better understand the evolution of the pathogenicity of the H7N7 viruses within the first infected flock, we focused our analysis on the deep-sequencing data of the HA cleavage site. The sequencing coverage in this genetic region ranges from 4445 for sample 4541-7 to 23511 for sample 4541-34. We did not identify any reads showing the cleavage site typical of a LPAI strain. A total of 99.9% of the reads of the two samples from shed 5 (named for clarity V+6) possess a cleavage site with an insertion of 6 nucleotides, with only a few reads containing an insertion of 3, 5, and 9 nucleotides (Table 2). A total of 99.7 to 99.9% of the reads of viruses from shed 2 (named V+9) had an insertion of 9 nucleotides, with only a few minority variants showing an insertion of 6, 7, or 8 nucleotides (Table 2). On the other hand, in one of the samples from shed 4 (4541-33) we identified a mixed population with both types of cleavage sites displaying an insertion of 9 (95.7%) and 6 (4.1%) nucleotides. Similarly to the samples from shed 2, the majority (from 99.9 to 100%) of the viral population of the subsequent outbreaks possesses the longer cleavage site, suggesting that this variant (V+9) may have a higher fitness advantage (Fig. 3).
TABLE 2.
Number of reads showing a 0- to 9-nucleotide insertiona
| Nucleotide insertion size (no. of nucleotides) | No. of reads |
|||||||
|---|---|---|---|---|---|---|---|---|
| Shed 5 |
Shed 4 |
Shed 2 |
||||||
| 4541-34 | 4541-9 | 4541-8 | 4541-33 | 4527-11 | 4527-12 | 4541-7 | 4541-32 | |
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 3 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| 4 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| 5 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| 6 | 23,509 | 14,861 | 0 | 591 | 11 | 0 | 1 | 4 |
| 7 | 0 | 0 | 4 | 18 | 5 | 3 | 5 | 27 |
| 8 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 5 |
| 9 | 2 | 1 | 16,587 | 13,700 | 6,660 | 13,725 | 4,439 | 22,929 |
Compared to the sequence of an H7 LPAI strain (CCAAAGAGAAGA) at the HA cleavage site of eight samples collected from three different sheds of the index case.
In addition to the cleavage site, sample V+9 collected from sheds 2 and 4 of the first infected farm can be distinguished from the two samples from shed 5 by 9-nucleotide signatures (HA G471A, PB2 A347G, T1891G, PA A347G, T1891G, NP G219A, C316A, NS1 G353A, and G378A; Fig. 3), which resulted in three amino acid changes (PA Q116R, PA C631G, and NS1 R118K). These signatures are maintained in all samples identified in the subsequent outbreaks, suggesting that only viruses from sheds 2 and 4 of the index case were transmitted to the other five farms (Fig. 3). In addition, we identified one nonsynonymous mutation at position 130 of the M2 gene, responsible of the amino acid substitution D44N, which is shared between the V+6 viruses and sample 4527-11 from shed 2 of farm 1, sample 4603 from farm 2, sample 4678 from farm 3, and sample 5091 from farm 5 (Fig. 3). However, whether this mutation emerged by chance in the four viruses or arose in the shed 2 virus of the index case and was then transmitted to the other outbreaks or was acquired by the V+9 samples through a reassortment event cannot be assessed.
To determine whether the shed 5 viruses (V+6) were the progenitors of the variant V+9, we examined the presence of the nine signature mutations (Fig. 3) as minority variants in the analyzed samples. None of the mutations typical of the V+9 viruses were already present in shed 5 viruses (V+6) with a frequency higher than 2% (the frequency threshold used in this study [see Materials and Methods for details]). Similarly, none of the mutations characteristic of V+6 (Fig. 3) was identified in subpopulations of the V+9 samples, except for the virus from shed 4 of the index case (sample 4541-33), which, besides the shorter cleavage site, possessed subpopulations containing all the mutations distinctive of V+6 variant, with a frequency ranging from 3 to 9%, confirming the presence of a mixed population (V+6 and V+9).
Genetic diversity of H7N7 viruses.
Overall, we observed mutations at 185 sites (excluding the HA cleavage site) distributed among the eight gene segments, of which 111 are nonsynonymous and 74 synonymous. Specifically, a total of 35 consensus-level nucleotide substitutions were recovered along the entire genome, defining 11 different genomes (named A to K in Fig. 3), 5 of which were identified within the first infected farm (A to E). The PB2 gene, with a total of 10 nucleotide variants (8 synonymous and 2 nonsynonymous), is the segment showing the highest number of mutations at the consensus level. The nucleotide distance among the 14 viruses ranged from 0 to 0.1% for the PA, HA, and NA genes to 0 to 0.2% for the PB2, PB1, and NP genes and 0 to 0.4% for the NS gene. Notably, 13 of 35 mutations distributed along 12 proteins (HA, NA, PB2, PB1, PB1-F2, PA, PA-X, NP, M1, M2, NS1, and NS2) are nonsynonymous, with the PA protein showing the highest number of amino acid variations (4) (Fig. 3).
In addition to these consensus-level variant sites, our deep-sequencing analysis identified 209 minority variants in 151 sites (97 nonsynonymous and 54 synonymous) with a frequency ranging from 2 to 49.8% (Fig. 4). The virus collected from shed 4 of the index case (sample 4541-33), which displayed a mixed population of V+6 and V+9, and sample 4603 collected from farm 2, comprised the highest number of minority variants (respectively, 40 and 41). In contrast, we did not detect any subpopulations in samples 4541-34 and 4541-9. No correlation between the number of variants and the type of samples used for the analysis (pool, organs, or swab) was observed (Pearson test, P = 0.254; r = 0.33).
FIG 4.

Heatmap of the nucleotide frequency. The horizontal axis represents the samples, colored according to the farm of collection, while the vertical axis display only the variable nucleotide positions showing nucleotide differences compared to samples 4541-9 and 4541-34. The color scale represents the nucleotide frequency according to the scale bar at the top of the figure. White spaces represent positions for which deep-sequencing data were not available (coverage < 500). Black arrows indicate the variants that are fixed in the viral population of at least one sample of the first infected farm. The dendrogram above the heatmap represents the neighbor-joining tree obtained from the distance matrix calculated from the deep-sequencing data.
We measured the complexity of the viral population of each sample using Shannon entropy (represented by the size of the circles in Fig. 5). In the first infected flock, the entropy measures fluctuate considerably: the lowest values are observed for the two viruses from the shed 5 (V+6) (Wilcoxon rank-sum test, with P values ranging from 9.37 × 10−14 to 4.7 × 10−3), suggesting that these samples (4541-9 and 4541-34) had recently experienced a narrow bottleneck and had not recovered from the loss of complexity. Conversely, viruses from shed 2 show intermediate values of entropy, whereas sample 4541-33 from shed 4 of the index case, sample 4603 from farm 2, and sample 4678 from farm 3 displayed entropy levels significantly higher than the other samples (Wilcoxon rank-sum test, with P values ranging from 9.37 × 10−14 to 1.49 × 10−3), an observation consistent with the high genetic diversity observed across their genomes. There is no significant Pearson correlation between within-host virus diversity and viral RNA content (P = 0.487; r = −0.2; the numbers of RNA copies are reported in Table 1). Thus, the significantly different entropies between the analyzed samples may simply be a bias associated with the time elapsed between infection and sampling or, alternatively, they may be due to the occurrence of random or selective bottleneck events of different intensity within separate sheds or farms.
FIG 5.

Transmission tree obtained from deep-sequencing data. Each circle represents an individual sample, colored according to the farm of collection. The size of the circles is proportional to the mean entropy value. The vertical axis represents the time of collection of each sample (samples in the same row belong to the same farm). Circle outlines are assigned according to the owner of the farm as indicated in the inset. Connecting arrows correspond to the results obtained from SeqTrack, whereas dashed lines are alternative hypotheses of transmission events formulated based on the number of shared mutations. Numbers over the lines are the genetic distances calculated from the deep-sequencing data between the samples. Colored area represents genetic groups identified based on the number of shared mutations and the results of both the neighbor-joining phylogenetic tree (Fig. 4) and the network analysis (Fig. 6).
To evaluate whether a selective-bottleneck or random-founder effect is the major force driving virus evolution, we compared the relative diversity changes on a gene-by-gene basis (see Table S1 in the supplemental material). We would not expect selective bottlenecks to affect all the genes in the same way, and thus the entropy and the number of nonsynonymous mutations found in the genes should vary. To this aim, we calculated the mean entropy and the number of synonymous and nonsynonymous normalized mutations separately for all of the genes in each sample (see Table S1 in the supplemental material). There was no significant difference in the entropy between the genes (one-way ANOVA, P = 0.196) or between the number of nonsynonymous and synonymous mutations (two-way ANOVA, P = 0.249), suggesting that the reduction in diversity observed in the analyzed samples was probably due to founder effects rather than to selective bottlenecks.
Minority variants transmitted between sheds and farms.
Focusing our analysis of the first infected flock, we observed that only a few mutations were shared at a shed and farm level, whereas the majority of the minor changes were unique to individual samples. Specifically, at the shed level we detected 44 minority changes in viruses from shed 2; of these changes, 22 were found in individual samples and were not shared with others, and 48 were found in viruses from shed 4, of which 37 were identified in single samples. Similarly, at the farm scale we counted 92 mutations, of which 12 were shared between two and four samples, whereas 59 minority variants were identified in single individuals (Fig. 4).
Interestingly, five of these variants, shared between viruses of the first infected farm, were fixed in the viral population of at least one sample (variants highlighted with black arrows in Fig. 4), and three of them were also transmitted or independently acquired by viruses collected from the other premises. Four of these were nonsynonymous mutations fixed in the viral population, which cause changes at the protein level (NS M119T, M2 D44N, PA V100I, and PB2 K574R) (Fig. 3).
We detected only seven minority variants (HA 1351A, M 942G, M 955G, PA 1251 G, PA 1748A, PB2 981G, and NA 390A) shared between two or three farms. Interestingly, five of them resulted in amino acid mutations (HA D451N, M2 D85G, PA R583Q, PB2 G327G, and NA M130I). These nonsynonymous mutations may be advantageous variants, associated with changes in viral fitness or due to adaptive evolution of the virus, or they may be neutral or deleterious polymorphisms, which occurred because of random genetic drift or hitchhiking.
Transmission dynamics of the H7N7 virus.
To assess the interfarm transmission, a median joining phylogenetic network was inferred using concatenated consensus sequences of the eight gene segments of the 14 analyzed viruses (Fig. 6). At the first infected farm we identified five sequence genotypes (gray circles in Fig. 6): one within shed 5, two within shed 2, and two in shed 4. Viruses from sheds 2 and 4 appear to be at the origin of the infection to the other farms, although one or two median vectors (red circles), which represent the lost ancestral sequences, separate them from viruses of the other holdings, except for sample 5051-3 from farm 6, which appears to be a direct descendant of shed 2 viruses (bootstrap value 85.5). Sequences from farms 2 to 6 grouped within two main clusters which shared a common ancestor (c1 and c2): c1 includes viruses collected from farms 2 (sample 4603), 3 (sample 4778), and 5 (sample 5091), whereas c2 contains virus sequences from farms 4 (sample 4774) and 6 (sample 5051-1). Sequences within these two clusters were separated by 6- to 10-nucleotide differences, whereas 9- to 13-nucleotide differences were observed between viruses of the two clusters. Therefore, the high number of mutations and median vectors identified between the analyzed samples and the low number of viruses available for the analysis makes the relationship between sequences hard to determine, and we cannot exclude that sampling bias may have affected the results. Our deep-sequencing data may help to elucidate this relationship. To this aim, we first inferred a neighbor-joining phylogenetic tree based on the distance matrix calculated from our NGS data, which confirmed the clustering identified by our network analysis (Fig. 4). We then used the distance matrix and the collection dates to reconstruct a transmission tree using the graph-based algorithm SeqTrack. This approach, which considers the sampled viruses as a fraction of the genealogy, is particularly suitable to infer the transmission pathway during disease outbreaks, where one strain can be the ancestor of another strain (Fig. 5).
FIG 6.

Median-joining phylogenetic network. (A) The network was constructed from the consensus sequences of the eight concatenated gene segments. Each unique sequence genotype is represented by a circle sized relatively to its frequency in the data set. Numbers next to the circles correspond to the samples showing that particular genotype, while the number within the circle represents the shed where the genotype was identified. Genotypes are colored according to farm. Branches represent the shortest trees, and black circles represent the numbers of nucleotide mutations that separate each node. Median vectors are indicated as red circles. The violet and yellow shading represent the two identified genetic groups C1 and C2. The numbers at each branch represent bootstrap values. (B) Map showing the geographic locations of the six infected farms.
Despite 21 days passing from the first to the last outbreak, the inferred genealogy suggests that all but one of the outbreaks descend directly from shed 2 (V+9) of the index case. The only exception is represented by the virus sample 5091, collected from the backyard farm on September 2 (farm 5), which appears to have been infected directly by farm 3.
However, based on the number of shared mutations between the analyzed sequences, we may speculate further scenarios. For example, sample 4603 from farm 2 shared two fixed mutations with sample 4678 (farm 3) and sample 5091 (farm 5) (group c2 of the network analysis); thus, a transmission event from farm 2 to farm 3 cannot be excluded. Similarly, virus samples 4774 and 5051-1 share three unique minority variants and one unique fixed mutation, making a transmission event between these two farms highly plausible. In addition, samples 4774 and 5051-1 share one fixed mutations and three minority variants (group c1 of the network analysis) and, in turn, they share two fixed mutations with sample 5051-3. Although virus samples 5051-1 and 5051-3 were collected from two different sheds of farm 6, we observed a relatively high nucleotide distance between them. Specifically, they show 7- and 14-nucleotide differences at the population and subpopulation levels, respectively, although all of the consensus-level mutations were present as minority variants in the other sample (Fig. 4). Thus, the occurrence of two separate introductions in farm 6 from the index case and/or farm 4 cannot be excluded.
Overall, these analyses indicate that shed 2 of the index case is the major source of the virus. An early strain (c1) appears to have spread from the first infected flock to farm 2 (19 August) and farm 3 (21 August) and then from farm 3 to the backyard farm 5 (2 September). Since farms 2 and 3 belong to different companies (circle outlines in Fig. 5) and are located 50 km apart (see map in Fig. 6), it is more plausible that viruses with similar genetic characteristics were transmitted from the index case to both holdings. A later spread with a slightly different strain (c2) may have occurred from the first infected flock to farm 4 (27 August) and farm 6 (3 September). These two farms are located in the same area, at a distance of 3 km, and belong to the same layer company as the first infected holding; thus, an exchange of virus between them cannot be ruled out.
DISCUSSION
Acquisition of a virulent phenotype by H7 avian influenza viruses may have devastating consequences to the poultry industry and, in some instances, can create major human health issues, including the risk of generating a new pandemic strain (23). Despite the identification of multiple basic amino acids at the HA cleavage site as one of the most important molecular markers of virus pathogenicity, the mechanisms underlying the emergence, spread, and evolution of HPAI during an epidemic are poorly understood and limited to a few studies (3, 24). We performed here for the first time a deep-sequencing analysis of all the samples collected during an HPAI epidemic to evaluate the transmission dynamics and the within- and between-farm genetic diversity of the viral population.
We showed that the 14 H7N7 Italian samples collected from six different farms form a cluster distinct to other Eurasian sequences for all the eight gene segments, suggesting the occurrence in the poultry population of a single viral introduction. The high similarity of the HA gene segment to a virus collected from a wild bird in Italy and the contact between free-range hens and wild waterfowl in the first infected farm (10) indicates that the LPAI progenitor strain may have been introduced from the wild bird population into the first infected holding, where it rapidly mutated into a highly pathogenic form.
Despite our phylogenies suggesting a single viral introduction, we observed a high genetic variability of H7N7 between the different sheds of the first infected flock. In particular, at the consensus level, viruses collected from shed 5 possessed a shorter HA cleavage site and 9-nucleotide differences compared to the viruses from shed 2. None of the fixed mutations had ever been described in previous HPAI H7 outbreaks or recognized as associated with a specific phenotypic effect. Further studies will be necessary to evaluate their possible impact on virus fitness, host range, and virulence. This number of nucleotide substitutions (9) is not compatible with the occurrence of different introductions, when a higher number of mutations is usually observed (25). Moreover, we noticed that the highest genetic distance (means ± the standard error) between the two groups of H7N7 viruses (V+6 and V+9) ranged from 0.1% ± 0.1% for the HA, NA, and PA genes to 0.4% ± 0.2% for the NS gene, whereas the overall mean distance among the sequences included in our phylogenies ranged from 1.8% ± 0.2% for the M gene to 5.5% ± 0.3% for the NA gene. This evidence indicates that the Italian H7N7 sequences are significantly closer to each other than any other random sequences in their tree seem to be, which supports the hypothesis that the two variants V+6 and V+9 had very likely derived from a single introduction. Hence, this high genetic variability can be explained by (i) a rapid evolution of the virus following some bottleneck events or a strong selection, (ii) independent evolution of the same virus within two separate sheds, or (iii) the establishment of the infection starting from two different seeding variants from the same progenitor viral population comprising a cloud of diverse viruses. Nevertheless, our analysis of the mutation spectra of viral populations suggests that the two variants arose as a consequence of a founder event or a narrow population bottleneck. Indeed, the haplotype V+6, circulating in shed 5, was not identified in the viral subpopulations of shed 2 and similarly haplotype V+9, identified in shed 2, was not detected as a minority population in shed 5 animals. In addition, at the HA cleavage site of viruses from sheds 2 (V+9) and 5 (V+6) we identified only a total of 16 and 3 reads with insertions, respectively, of 6 and 9 nucleotides.
Entropy values obtained for the two viruses from shed 5 further supports this hypothesis. Samples founded by few viral particles should have low entropy, since the strong bottleneck/founder effect drastically reduce the diversity of the viral population. On the other hand, samples that experienced relatively loose bottlenecks should display higher entropy. Therefore, the low entropy values of the viruses from shed 5 may indicate that they had recently experienced a narrow bottleneck/founder effect or that they had been subjected to a strong selection which had reduced the within-host diversity and fixed adaptive mutations. To distinguish between selective and random bottlenecks/founder effects, we compared the relative diversity changes on a gene-by-gene basis. We showed that there were no significant differences between the entropy values and the number of nonsynonymous mutations for the different genes, suggesting that founder effects caused by the transmission bottlenecks are a major driving force of virus evolution during this epidemic.
On the other hand, viruses from shed 2 show intermediate entropy values, suggesting that (i) they were founded by a larger seeding population, (ii) they experienced a high-level of replication, or (iii) they had circulated within the shed for a longer period of time. The latter suggestion is supported by the identification of H7-specific antibodies in animals from this shed but not in animals from sheds 4 and 5 (10), whereas the second hypothesis may be supported by the high number of dead birds found in shed 2 compared to the other sheds, considering the virulence of the two variants were equal (intravenous pathogenicity index of 3 for both variants [data not shown]).
However, we cannot exclude that difference in the within-host genetic diversity between the analyzed samples could be associated with a different time of sampling since infection. Unfortunately, the lack of information on the exact time of entrance of the virus in each farm and shed makes it impossible to exclude this possible bias and to ascertain the process responsible of the reduction of the genetic variability, which may be caused both by bottlenecks of different sizes that occurred around the same time since infection or by similar bottlenecks that occurred at different time points relative to sampling.
Surely, sequences of early viruses might have helped us to provide a better characterization of the evolution of this strain within the index case. Indeed, the identification of H7-specific antibodies in animals from shed 2 and from the outer sheds 1 and 7, where no viruses were isolated (10), indicates that the virus had been circulating undetected within the farm before its identification, likely with a low-pathogenicity phenotype.
Moreover, the identification of three human infections during this epidemic highlights the need for constant monitoring during avian influenza outbreaks for the emergence in poultry of amino acid signatures associated with interspecies transmission to provide early warning of pandemic potential.
Our analysis of the transmission dynamics indicates that only one of the two variants (V+9), probably the one with the highest fitness advantage, was transmitted from the index case to the other farms. Four of the six infected farms (farms 1, 2, 4, and 6) belong to one large vertically integrated layer company (Fig. 5); therefore, virus dissemination might have occurred through shared equipment, human-mediated mechanical transport, and also through infected workers, since H7N7 virus was diagnosed for three humans involved in the control of the epidemic (22). The low number of shared mutations between farms (seven) suggests that the transmission depended on the dissemination of a few viral particles. However, the high frequency threshold (2%) used in this study to identify the minority variants and the scarce number of analyzed samples for each farm need to be taken into consideration.
In the farms for which it was possible to sequence more than one sample (eight for farm 1 and two for farm 6), we identified the cocirculation in the same premise of different related variants and the possible occurrence of multiple introductions in the same holdings (i.e., farm 6), which can be detected only through the sequencing of a larger number of samples. Moreover, the high number of median vectors identified between the analyzed samples in our phylogenetic network reveals missing ancestral sequences from our analyses, which might have been detected with increased sampling. As a consequence, increasing the number of viruses sampled from each farm and also from the environment could increase the resolution of our interfarm transmission dynamic.
We identify farm 1 as the major source for the spread of the virus to the other four industrial holdings, while the rural farm (farm 5) appears to have received the virus from the turkey farm (farm 3). Interestingly, this finding allowed the national authorities to demonstrate the occurrence of uncontrolled movements of birds from the infected turkey flock (farm 3), underlining the importance of genetic data to complement the outbreak investigation. Despite 21 days elapsing from the index case (August 13) to the last outbreak (September 3), the late depopulation date of the first infected flock (August 27) and the ability of the avian virus to persist in the environment (26) might explain the virus spread between these two holdings (farms 1 and 6).
In addition, results of our analysis of the transmission dynamics suggests that, though farm 2 is located in close proximity to farms 4 and 6, transmission links are absent between these two premises. In contrast, the virus sampled from this farm appears to be more related to the virus from farms 1 and 3, which are located, respectively, 38 and 36 km from farm 2.
This finding suggests that multiple introductions of different viral haplotypes occurred from farm 1 to the other farms, probably at different time points and with different transmission modes, i.e., neighborhood spread (i.e., farms 1 and 3), human-mediated transport among farms of the same company (i.e., farms 1 and 2 or farms 1 and 4). These different means of viral diffusion have been observed also during other HPAI epidemics (24, 27), suggesting that long-distance transmission events may play an important role in virus dissemination into new areas.
Overall, the present study shows that analysis of deep-sequencing data can complement epidemiological investigations, providing important insights and revealing unexpected dynamics on the interfarm transmission network. Specifically, we demonstrated that the delay in the disease detection and stamping out in the index case might have been the major cause of the emergence and the spread of the HPAI strain. Epidemiological investigations did not recognize the central role of the first infected flock in the diffusion of the virus to most of the farms and suggested an epidemiological link between farms 2, 5, and 6, which has not been confirmed by our data. In addition, the epidemiological data alone were not sufficient to trace back the source of the virus detected in the rural farm (farm 5), which we demonstrated to be linked to the turkey farm (farm 3).
Moreover, we show that a farm can harbor a high level of heterogeneity, potentially caused either by separate bottlenecks and founder effects in the different sheds, or by multiple viral introductions from different sources. Hence, it is important during control activities to collect and analyze several samples from each infected farm to provide a complete picture of the evolutionary process during an avian influenza epidemic.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the authors and the originating and submitting laboratories of the sequences from the GISAID's EpiFlu Database discussed here. A detailed list is found in Table S2 in the supplemental material.
This study was financially supported by the European projects Epi-SEQ (research project supported under the Second Joint Call for Transnational Research Projects by EMIDA ERA-NET [FP7 project no. 219235]) and PREDEMICS (research project supported by the European Community's Seventh Framework Programme [FP7/2007-2013] under grant agreement 278433). P.R.M. and J.H. are supported by the Medical Research Council of the United Kingdom (grant number G0801822).
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00538-16.
REFERENCES
- 1.Manrubia SC, Escarmís C, Domingo E, Lázaro E. 2005. High mutation rates, bottlenecks, and robustness of RNA viral quasispecies. Gene 347:273–282. doi: 10.1016/j.gene.2004.12.033. [DOI] [PubMed] [Google Scholar]
- 2.Iqbal M, Reddy KB, Brookes SM, Essen SC, Brown IH, McCauley JW. 2014. Virus pathotype and deep sequencing of the HA gene of a low pathogenicity H7N1 avian influenza virus causing mortality in turkeys. PLoS One 9:e87076. doi: 10.1371/journal.pone.0087076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Monne I, Fusaro A, Nelson MI, Bonfanti L, Mulatti P, Hughes J, Murcia PR, Schivo A, Valastro V, Moreno A, Holmes EC, Cattoli G. 2014. Emergence of a highly pathogenic avian influenza virus from a low-pathogenic progenitor. J Virol 88:4375–4388. doi: 10.1128/JVI.03181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jonges M, Welkers MRA, Jeeninga RE, Meijer A, Schneeberger P, Fouchier RAM, de Jong MD, Koopmans M. 2014. Emergence of the virulence-associated PB2 E627K substitution in a fatal human case of highly pathogenic avian influenza virus A(H7N7) infection as determined by Illumina ultra-deep sequencing. J Virol 88:1694–1702. doi: 10.1128/JVI.02044-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poole DS, Yú S, Ca Y, Dinis JM, Müller MA, Jordan I, Friedrich TC, Kuhn JH, Mehle A. 2014. Influenza A virus polymerase is a site for adaptive changes during experimental evolution in bat cells. J Virol 88:12572–12585. doi: 10.1128/JVI.01857-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Varble A, Albrecht RA, Backes S, Crumiller M, Bouvier NM, Sachs D, García-Sastre A, ten Oever BR. 2014. Influenza A virus transmission bottlenecks are defined by infection route and recipient host. Cell Host Microbe 16:691–700. doi: 10.1016/j.chom.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilker PR, Dinis JM, Starrett G, Imai M, Hatta M, Nelson CW, O'Connor DH, Hughes AL, Neumann G, Kawaoka Y, Friedrich TC. 2013. Selection on hemagglutinin imposes a bottleneck during mammalian transmission of reassortant H5N1 influenza viruses. Nat Commun 4:2636. doi: 10.1038/ncomms3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fusaro A, Tassoni L, Hughes J, Milani A, Salviato A, Schivo A, Murcia PR, Bonfanti L, Cattoli G, Monne I. 2015. Evolutionary trajectories of two distinct avian influenza epidemics: parallelisms and divergences. Infect Genet Evol 34:457–466. doi: 10.1016/j.meegid.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 9.Yu X, Jin T, Cui Y, Pu X, Li J, Xu J, Liu G, Jia H, Liu D, Song S, Yu Y, Xie L, Huang R, Ding H, Kou Y, Zhou Y, Wang Y, Xu X, Yin Y, Wang J, Guo C, Yang X, Hu L, Wu X, Wang H, Liu J, Zhao G, Zhou J, Pan J, Gao GF, Yang R, Wang J. 2014. Influenza H7N9 and H9N2 viruses: coexistence in poultry linked to human H7N9 infection and genome characteristics. J Virol 88:3423–3431. doi: 10.1128/JVI.02059-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonfanti L, Monne I, Tamba M, Santucci U, Massi P, Patregnani T, Loli Piccolomini L, Natalini S, Ferri G, Cattoli G, Marangon S. 2014. Highly pathogenic H7N7 avian influenza in Italy. Vet Rec 174:382–382. doi: 10.1136/vr.102202. [DOI] [PubMed] [Google Scholar]
- 11.Bosch FX, Garten W, Klenk HD, Rott R. 1981. Proteolytic cleavage of influenza virus hemagglutinins: primary structure of the connecting peptide between HA1 and HA2 determines proteolytic cleavability and pathogenicity of avian influenza viruses. Virology 113:725–735. doi: 10.1016/0042-6822(81)90201-4. [DOI] [PubMed] [Google Scholar]
- 12.Spackman E, Senne DA, Myers TJ, Bulaga LL, Garber LP, Perdue ML, Lohman K, Daum LT, Suarez DL. 2002. Development of a real-time reverse transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin subtypes development of a real-time reverse transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin. J Clin Microbiol 40:3256–3260. doi: 10.1128/JCM.40.9.3256-3260.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou B, Donnelly ME, Scholes DT, St George K, Hatta M, Kawaoka Y, Wentworth DE. 2009. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and swine origin human influenza A viruses. J Virol 83:10309–10313. doi: 10.1128/JVI.01109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boc A, Diallo AB, Makarenkov V. 2012. T-REX: a web server for inferring, validating and visualizing phylogenetic trees and networks. Nucleic Acids Res 40:W573–W579. doi: 10.1093/nar/gks485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jombart T, Eggo RM, Dodd PJ, Balloux F. 2011. Reconstructing disease outbreaks from genetic data: a graph approach. Heredity 106:383–390. doi: 10.1038/hdy.2010.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jombart T. 2008. adegenet: an R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. doi: 10.1093/bioinformatics/btn129. [DOI] [PubMed] [Google Scholar]
- 17.Csárdi G, Nepusz T. 2006. The igraph software package for complex network research. InterJournal Complex Systems 1695. [Google Scholar]
- 18.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bandelt HJ, Forster P, Röhl A. 1999. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 21.Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267. [DOI] [PubMed] [Google Scholar]
- 22.Puzelli S, Rossini G, Facchini M, Vaccari G, Di Trani L, Di Martino A, Gaibani P, Vocale C, Cattoli G, Bennett M, McCauley JW, Rezza G, Moro ML, Rangoni R, Finarelli AC, Landini MP, Castrucci MR, Donatelli I. 2014. Human infection with highly pathogenic A(H7N7) avian influenza virus, Italy, 2013. Emerg Infect Dis 20:1745–1749. doi: 10.3201/eid2010.140512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Capua I, Marangon S. 2006. Control of avian influenza in poultry. Emerg Infect Dis 12:1319–1324. doi: 10.3201/eid1209.060430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bataille A, van der Meer F, Stegeman A, Koch G. 2011. Evolutionary analysis of inter-farm transmission dynamics in a highly pathogenic avian influenza epidemic. PLoS Pathog 7:e1002094. doi: 10.1371/journal.ppat.1002094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bouwstra R, Koch G, Heutink R, Harders F, van der Spek A, Elbers A, Bossers A. 2015. Phylogenetic analysis of highly pathogenic avian influenza A(H5N8) virus outbreak strains provides evidence for four separate introductions and one between-poultry farm transmission in the Netherlands, November 2014. Eurosurveillance 20:21174. doi: 10.2807/1560-7917.ES2015.20.26.21174. [DOI] [PubMed] [Google Scholar]
- 26.Brown JD, Swayne DE, Cooper RJ, Burns RE, Stallknecht DE. 2007. Persistence of H5 and H7 avian influenza viruses in water. Avian Dis 51:285–289. doi: 10.1637/7636-042806R.1. [DOI] [PubMed] [Google Scholar]
- 27.Souris M, Gonzalez J-P, Shanmugasundaram J, Corvest V, Kittayapong P. 2010. Retrospective space-time analysis of H5N1 Avian Influenza emergence in Thailand. Int J Health Geogr 9:3. doi: 10.1186/1476-072X-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.