

ABSTRACT
Retroviruses spread more efficiently when infected and uninfected cells form tight, physical interfaces known as virological synapses (VSs). VS formation is initiated by adhesive interactions between viral Envelope (Env) glycoproteins on the infected cell and CD4 receptor molecules on the uninfected cell. How high-avidity Env-CD4 linkages are resolved over time is unknown. We describe here a tractable two-color, long-term (>24 h) live cell imaging strategy to study VS turnover in the context of a large cell population, quantitatively. We show that Env's conserved cytoplasmic tail (CT) can potently signal the recruitment of Gag capsid proteins to the VS, a process also dependent on residues within Gag's N-terminal matrix (MA) domain. Additionally, we demonstrate that Env's CT and Gag's MA domain both regulate the duration of interactions between viral donor and target cells, as well as the stability of this interaction over time (i.e., its capacity to resolve or form a syncytium). Finally, we report the unexpected finding that modulating extracellular fluid viscosity markedly impacts target T cell trafficking and thus affects the duration, stability, and turnover of virus-induced cell-cell contacts. Combined, these results suggest a stepwise model for viral cell-to-cell transmission wherein (i) Env-receptor interactions anchor target cells to infected cells, (ii) Env signals Gag's recruitment to the cell-cell contact dependent on an intact Env CT and Gag MA, and (iii) Env CT and Gag MA, in conjunction with extracellular forces, combine to regulate VS stability and infectious outcomes.
IMPORTANCE HIV-1 spreads efficiently at physical, cell-cell interfaces known as virological synapses (VSs). The VS provides for spatiotemporal coupling of virus assembly and entry into new host cells and may transmit signals relevant to pathogenesis. Disrupting this mode of transmission may be critical to the goal of abolishing viral persistence in infected individuals. We describe here a long-term live cell imaging strategy for studying virus-induced effects on cell behavior in the context of a large cell population. We demonstrate cooperative roles for viral Gag capsid proteins and Envelope glycoproteins in regulating VS formation and turnover. We also show that modulating fluid viscosity markedly affects T cell trafficking and VS stability. Thus, extracellular factors also play an important role in modulating the nature of infectious cell-cell interactions. In sum, our study provides new tools and insights relevant to exposing vulnerabilities in how HIV-1 and other viruses spread infection among cells, tissues, and people.
INTRODUCTION
Retroviruses encode transmembrane Envelope (Env) glycoproteins that regulate virion-receptor binding and mediate the fusion of viral and cellular membranes necessary to deliver viral capsids to the cytoplasm of uninfected target cells (1–4). Human immunodeficiency virus type 1 (HIV-1) Env is translated as a 160-kDa polyprotein (also known as gp160) that self-interacts and is cleaved in the secretory pathway to generate a trimer of noncovalently linked surface (SU; gp120) and transmembrane (TM; gp41) subunits. During entry, HIV-1 SU/gp120 binds CD4 (cluster of differentiation 4) receptor molecules and a chemokine coreceptor (CXCR4 or CCR5) found on the surfaces of T cells and macrophages. Conformational changes in SU/gp120 expose the TM/gp41 fusion peptide that subsequently drives virion-cell membrane fusion. During viral budding, the incorporation of Env trimers into virions involves interactions between Env's 150 amino acid cytoplasmic tail (CT) domain (found in TM/gp41) and the N-terminal matrix (MA) domain of viral Gag capsid proteins (5–7). In vivo, Env incorporation is inefficient, yielding only 10 to 17 trimers per >1,500 Gag proteins in each virion (8). Thus, Env-MA interactions appear to regulate a “just enough” mechanism that ensures Env incorporation into virions while limiting its net exposure to the host immune system.
Env plays additional, nonfusogenic roles in the infected cell, including driving the formation of specialized, virus-induced cell-cell contacts known as virological synapses (VSs), named for their morphological similarity to the immunological synapse formed between T cells and antigen-presenting cells (9, 10). VS formation among T cells is driven by high-avidity binding interactions between Env and CD4 on the donor and target cell, respectively, with Env, Gag, CD4, F-actin, and other cell factors accumulating to form a stable, long-term interface (9, 11–13). The transfer of infection can be up to 10,000-fold more efficient when cells can form these physical contacts (or “VS-like” structures, e.g., filopodial bridges or nanotubes [14, 15]) than for cell-free virions (12, 16–20), reflecting the ability of the VS to directly couple the processes of viral egress and entry in space and time (9, 12, 21–23). Contact-mediated spread can increase the frequency of multiploid (i.e., multigenome) inheritance (20, 24–26) and protect transmission from some antiviral drugs, neutralizing antibodies, or host restriction factors (20, 27–40). Furthermore, Env-dependent intercellular signaling is implicated in potentiating cell responses relevant to the efficiency of viral replication (41) and/or immune dysregulation linked to HIV/AIDS pathogenesis (42, 43).
Remaining questions for VS function pertain to the mechanics of virus-induced Env-receptor interactions at the VS and how they are maintained and resolved over time. Recent live cell imaging studies have confirmed retroviral Env's capacity to lengthen the duration of donor-target cell interactions both in culture (12, 15) and in vivo (44, 45). Intravital imaging of HIV-1-infected cells in a humanized mouse model recently revealed Env-dependent peripheral cell extensions and instances of Env-dependent cell-cell fusion (syncytium formation) (44, 46–48). Additional studies have clarified roles for the cytoskeleton and immune signaling factors (e.g., LFA-1, ICAM-1, and the tetraspannins CD9, CD63, and CD81) in regulating VS stability (9, 11, 13, 20, 49–56). HIV-1 Env's conserved CT domain is also relevant to VS stability, considering that it is required for spreading infection in most T cell lines, (57–61) can regulate Env's capacity to form syncytia (54, 62), and can, in some instances, direct Gag's trafficking to sites of cell-cell contact (58).
Here, we established a long-term (>24 h) live cell imaging strategy with the goal of monitoring cell-cell contact formation, duration, and stability within the context of a large cell population quantitatively. We show that, in nonpolarized cells, HIV-1 Env actively recruits large quantities of Gag to sites of cell-cell contact, requiring cooperation between Env's CT domain and Gag's N-terminal MA domain. Gag's capacity to accumulate at the contact significantly modulated the duration and stability of interactions between cells, a finding consistent with one or more functions for Gag at the contact extending beyond its role in virus particle assembly. We further illustrate that MA's effects on donor-target cell interactions can be abrogated by a single amino acid substitution at leucine-12 (L12E), a mutation previously reported to reduce Gag-Env convergence at the plasma membrane during virus particle assembly (63–65). In addition, we report the surprising finding that increasing extracellular viscosity enhances Jurkat target T cell motility on physical substrates, reducing the duration of donor-target cell interactions but, unexpectedly, triggering a higher frequency of cell-cell fusion events. Thus, both viral and extracellular determinants contribute to the outcome of infectious cell-cell interactions.
MATERIALS AND METHODS
Plasmids and cell lines.
Plasmids encoding HIV-1NL4-3 Env and HIV-1SF2 Env were a kind gift of Robert Doms, University of Pennsylvania, Philadelphia, PA. The ΔCT Env mutant construct was generated using overlapping PCR to delete sequences encoding the Env CT (amino acids 703 to 847), prior to reinsertion into the parental plasmid using EcoRI- and XhoI-cut sites. Gag-mCherry and mutants (S-ΔMA, ΔNC-zip, and Δp6) were generated by fusing gag and mCherry cDNAs separated by a flexible glycine linker sequence (encoding PGISGGGGGILD) using overlapping PCR prior to replacing the native gag reading frame in a pcDNA3.1-based Gag-Pol-Vif (GPV) construct encoding the first 5,297 nucleotides of the HIV-1NL4-3 RNA genome upstream of a Rev response element (RRE) and a polyadenylation signal (66). S-ΔMA Gag bears a replacement of the native MA sequence (Gag amino acids 1 to 126) with the myristoylation signal from the Src kinase (MGSSKSKPK) coupled to a short linker peptide (TVSFNF) upstream of the native capsid (CA) sequence. Δp6 Gag lacked Gag's C-terminal amino acids 453 to 504. Plasmids encoding cDNAs for generating ΔNC-zip Gag were kindly provided by David Ott (National Cancer Institute, Frederick, MD) (67). Plasmids encoding Rev have been previously described (68). Retroviral vectors encoding green fluorescent protein (GFP) and CD4-YFP (yellow fluorescent protein) were generated by inserting the relevant cDNA into the MIGR1-based retroviral vector pCMS28 using BglII- and EcoRI-cut sites upstream of an encephalomyocarditis virus internal ribosomal entry site that drives expression of puromycin N-acetyltransferase (69). For CD4-YFP, cd4 and yfp cDNAs were fused in-frame using overlapping PCR and sequence encoding the same flexible glycine linker (PGISGGGGGILD) described for Gag-mCherry above.
NIH 3T3 murine fibroblasts, Cos7 African green monkey osteosarcoma cells, and Jurkat T cells were obtained from the American Type Culture Collection. 3T3 and Jurkat cells stably expressing CD4-YFP and GFP were generated using retroviral transduction as previously described (70). Cos7 and 3T3 cell lines were maintained in Dulbecco modified Eagle medium (DMEM; Life Technologies) containing 10% fetal bovine serum (FBS), 1% penicillin-streptomycin, and 1% l-glutamine. Jurkat cell lines were maintained in complete RPMI culture medium (Life Technologies) containing 10% FBS, 1% penicillin-streptomycin and 1% l-glutamine.
Microscopy and image analysis.
For live cell imaging, donor and target cells were cocultured in 8-well μ-Slides (iBiDi) maintained at 37°C with 5% CO2 and 50% relative humidity using a LiveCell incubator (Pathology Devices). At 21 h prior to coculture, donor cells were transfected with 200 ng of total plasmid DNA (85 ng of Gag-mCherry, 85 ng of Env, and 30 ng of Rev) mixed with 0.45 μg of polyethylenimine (Sigma) in 20 μl of Opti-MEM (Life Technologies). The cells were washed, and target cells were added to donor cells at a 1:1 ratio and allowed to interact for 3 h prior to fixation or live cell imaging. Donor and target cells were cocultured at a 1:1 DMEM/RPMI ratio with, in some experiments, media supplemented with up to 2% (wt/vol) methylcellulose (MeC) to increase extracellular viscosity. Images were acquired using the CFI S Fluor 10× (N.A. 0.5) or CFI Plan Apo Lambda 60× Oil (N.A. 1.40) objective lenses of an Eclipse Ti automated epifluorescence microscope (Nikon). Movies were postprocessed and analyzed using NIS Elements (Nikon) and FIJI/ImageJ2 (NIH) software packages. For Gag VS targeting (see Fig. 2 and 3) analyses, cell-cell contacts were determined from the overlap or juxtaposition of cell boundaries, as determined by fluorescent signal and only for CD4-YFP puncta that were enriched ≥5-fold in fluorescence intensity over a length of at least 0.2 μm. Approximately 50 cell-cell contacts were measured per condition for each experiment (see Fig. 1C). Gag-mCherry polarization was indicated by an overlap of CD4-YFP and Gag-mCherry fluorescence intensity profiles, adapted from Li et al. (71). The duration of cell-cell interactions (see Fig. 4) was calculated by tracking the overlap of donor cell Gag-mCherry and target cell GFP/YFP signals over time for ≥50 cells per condition per experiment. Cell fusion was similarly monitored using live cell imaging (see Fig. 4B) or measured by fixing cells in 4% paraformaldehyde prior to staining cell nuclei with DAPI (4′,6′-diamidino-2-phenylindole) and counting Jurkat nuclei per donor cell for >50 donor cells per condition (see Fig. 5).
FIG 2.

Gag's recruitment to the cell-cell contact is Env CT-dependent. (A) Depiction of Env's TM/gp41 subunit and the Env CT truncation mutant (ΔCT Env). ED, ectodomain; MSD, membrane-spanning domain. (B) Coculture assay between Cos7 donor cells and target 3T3.CD4-YFP fibroblasts as for Fig. 1B demonstrating Gag-mCherry (red) clustering with CD4-YFP (green) for wild-type Env (WT Env, blue) but not for ΔCT Env (blue). (C) Quantification of the experiment depicted in panel B. Gag-mCherry clustering to the VS was scored for >50 cells per condition expressing the indicated Env variants in three independent experiments. Error bars represent the standard deviations from the mean. (D) Quantification of synapse length for >200 Cos7 donor and 3T3 target contacts from four independent experiments, demonstrating that Gag-mCherry/CD4-YFP-enriched contact zones range in length from <0.5 μm to >10 μm. Error bars represent standard deviations from the mean.
FIG 3.

Gag's recruitment to the cell-cell contact is Gag MA dependent. (A) Depiction of Gag-mCherry and mutants lacking matrix (MA), nucleocapsid (NC), and p6. NC was replaced with a leucine zipper motif, providing for Gag capsid assembly in the absence of NC-dependent RNA binding (67). (B) Gag variants depicted in panel A assemble virus-like particles (VLPs). Donor cells were transfected with plasmids encoding each protein, and lysates and supernatants were harvested at 48 h as described in Materials and Methods. Cell-associated Gag (lysates) or VLP-associated Gag was detected by immunoblotting with anti-p24Gag antisera and secondary antibodies conjugated to infrared fluorescent molecules. HSP90 was detected from cell lysates as a loading control. (C) Imaging of the coculture assay, performed as for Fig. 2B, demonstrating the loss of Gag-mCherry clustering to the VS when MA is deleted but not for the ΔNC or Δp6 mutants. (D) Quantification of Gag-mCherry/CD4-YFP coclustering for the experiment depicted in panel C. Error bars represent the standard deviations from the mean for 50 contacts per condition for three independent experiments. The P values were calculated by using the Fisher exact test in MStat (University of Wisconsin—Madison). ***, P < 0.0001; n.s., not significant. (E) Quantification of synapse length for >100 Cos7 donor–3T3.CD4-YFP fibroblast contacts from three independent experiments per condition, demonstrating that contact size is smaller when S-ΔMA is present. (F) Model for CT- and MA-dependent interactions at the VS.
FIG 1.

Monitoring VS formation using live cell imaging. (A) Diagram of the coculture assay and visual outcome when Gag-mCherry (red) and CD4-YFP (green) converge at sites of cell-cell contact. (B) VS formation for Cos7 donor cells cocultured with 3T3 fibroblasts (top) or Jurkat T cells (bottom). Donor cells expressing Gag-mCherry (red) with or without coexpression of Env were cocultured with the indicated target cell for 3 h prior to imaging. Note that CD4-YFP enrichment was Env dependent and used as a marker for contact formation. A dashed line indicates the boundary of the donor cell. Scale bars, 10 μm. (C) Quantification of the fold enhancement for CD4-YFP and Gag-mCherry at cell-cell contacts based on the relative mean fluorescence intensity (MFI). Error bars represent the standard deviations from the mean for 70 contacts derived from two independent experiments. (D) Env coclustering with CD4-YFP in the presence or absence of cyan fluorescent protein-tagged Gag (Gag-CFP) at cell-cell contacts, detected using indirect immunofluorescence and anti-gp120 antisera. Scale bars represent 5 μm for the top image panels and 10 μm for the bottom image panels. (E) Time-lapse imaging of donor and fibroblast target cells demonstrating coincident Gag-mCherry/CD4-YFP enrichment and the lateral clustering of individual punctae, or microsynapses, to form larger contacts over time. Images correspond to Movie S2 in the supplemental material. Scale bars, 10 μm.
FIG 4.

Gag's MA domain increases the duration of donor-target cell interactions. (A) Cell-cell interaction tracking assay using adherent Cos7 donor cells (red) and nonadherent GFP-expressing Jurkat T cell targets (green). Donor cells were transfected to express Gag-mCherry (red) and Env 24 h prior to coculture with target cells. Individual T cells exhibiting GFP signal overlapping the Gag-mCherry donor signal were tracked for 8 h. The figure presents examples of T cells that interacted transiently after contacting a donor cell (top panels, cells A, B, and C) and T cells that were immobilized with donor cells (bottom panels, cells A and B). Tracks for individual cells over 8 h are presented on the right. Scale bar, 25 μm. (B) More than 1,000 cells from three independent experiments were tracked for each indicated condition and scored for the time of donor-target cell interaction. The black line represents the median time of interaction for each condition. Black dots represent interactions that terminated due to cell-cell fusion. Statistical analysis was performed in GraphPad Prism 6 using the Kruskal-Wallis test, followed by a Dunn's multiple-comparison posttest. ***, P < 0.0001. (C) Representative images of cells expressing Gag-mCherry (red) and stained for surface level expression (top panels) or total expression (bottom panels) of Env gp120 (green) by indirect immunofluorescence. Scale bars, 50 μm (except for the WT Env, WT Gag and ΔCT Env, and WT Gag total expression conditions, which are 25 μm in length). (D) Quantification of single cell Env surface expression, based on the mean fluorescence intensity (MFI) for 50 cells for the conditions presented in panel C. The black line represents the MFI; error bars represent the standard deviations from the mean.
FIG 5.

Env and Gag cooperate to regulate Env's fusogenic potential. (A) Cos7 donor cells and target Jurkat T cells were cocultured and fixed 5 h later prior to scoring for syncytium formation, as indicated by the overlap between donor cell Gag-mCherry (red) and target cell GFP (green) fluorescence. Scale bars, 500 μm. (B) Percent donor cells imaged that formed syncytia (determined by multinuclear DAPI staining) for Cos7 donor cells transfected with subgenomic plasmids (trans constructs, left) or 293T donor cells transfected with full-length proviral DNA (cis constructs, right). Error bars represent the standard deviations of the mean for three independent experiments. (C) VS collapse assay. 293T donor cells were cocultured with Jurkat.GFP cells and processed as for panel A. Cells were then stained with DAPI to visualize nuclei prior to scoring total cell-cell fusion events based on the number of T cell nuclei per syncytium. The mean was calculated from three replicates for two independent experiments. Error bars represent the standard deviations of the mean, and P values were calculated by using the Fisher exact test in MStat. ***, P < 0.0001. (D) Heat map representation of 293T donor cell size after coculturing with Jurkat.GFP target T cells. Cell area was determined for the conditions represented in panel C and then sorted and colored based on size (as indicated).
Immunofluorescence and immunoblotting.
Immunofluorescence detection of Env was performed as previously described (72), with cells plated on glass coverslips, transfected as described above, and fixed at 24 h posttransfection in 4% paraformaldehyde prior to permeabilization using 0.2% Triton X-100 to determine the total protein signal. To measure Env surface expression, antibodies were added to nonpermeabilized cells in order to only stain the cell surface. Cells were incubated with human anti-Env antiserum (2G12) obtained through the NIH AIDS Research and Reference Reagent Program (Bethesda, MD) for 1 h prior to washing and addition of anti-human secondary antibodies conjugated to Alexa Fluor 488 (Life Technologies) or Dylight405 (Jackson Immunoresearch). Immunoblots were performed as previously described (73) using mouse anti-p24Gag antiserum (HIV-1 p24Gag hybridoma 183-H12-5C from Bruce Chesebro) also obtained from the NIH AIDS Research and Reference Reagent Program and rabbit anti-Hsp90 (Santa Cruz Biotechnology) antisera prior to incubation with anti-mouse or anti-rabbit secondary antibodies conjugated to infrared fluorophores IRDye800 or IRDye680 (Li-Cor Biosciences).
RESULTS
Modeling HIV-1-induced cell-cell interactions using long-term live cell imaging.
To establish a minimal visual system for studying Env-receptor interactions in the context of a cell population over time, we plated HIV-1 donor cells coexpressing Gag-mCherry and Env on glass coverslips prior to coculture with target cells engineered to stably express CD4-YFP (Fig. 1A). Cos7 cells were chosen as “infected” donors because they exhibit expansive lamellipodia ideal for visualizing plasma membrane events. Critically, Cos7 cells are also nonmotile, thus providing us with a means to monitor hundreds of single donor cells concurrently with subcellular resolution over many hours of time-lapse video microscopy. As target cells, we selected either Jurkat T cells (Jurkat.CD4-YFP) or, in a subset of experiments (Fig. 1 to 3), murine 3T3 fibroblasts (3T3.CD4-YFP) as a model for donor interactions with stromal cells. Murine fibroblasts also lack functional HIV-1 coreceptor molecules, thus allowing us to visualize Env-CD4 interactions independently of CXCR4/CCR5 and cell-cell fusion events.
In donor cells expressing either R5- or X4-tropic Env, we observed remarkable accumulations of Gag-mCherry at cell-cell contacts colocalizing with CD4-YFP within 3 h of coculture (Fig. 1B). Gag and CD4 coclustering was Env dependent (Fig. 1B), with Gag-mCherry enriched at the site of contact 200-fold and CD4-YFP enriched ∼7-fold on average, based on the relative mean fluorescence intensities (Fig. 1C). gp120Env was also enriched at these clusters, as visualized using indirect immunofluorescence, and consistent with the formation of tripartite Gag/Env/CD4 cell-cell adhesion domains (Fig. 1D). Taken together, these experiments confirmed that our system recapitulates anticipated features of VS formation at sites of cell-cell contact in nonpolarized cells.
Interestingly, live cell imaging revealed VS formation in these cell types to be initiated by small Gag/CD4 punctae, or “microsynapses,” prior to their coalescing over time to form larger zones of contact (Fig. 1E and see Movies S1 to S4 in the supplemental material). In fibroblast coculture, larger contacts were remarkably stable and could be maintained for several hours, only disrupted due to mechanical cell-cell pulling forces (e.g., see Movie S3 in the supplemental material). Cell-cell interactions were typically more transient for target T cells (see Movies S5 and S6 in the supplemental material), reflecting the tendency of these cells to scan the donor cell surface using dynamic actin-rich lamellipodial protrusions. For either target cell type, we observed instances of membrane exchange as demarcated by the transfer of either Gag-mCherry or CD4-YFP fluorescent signal to the conjugate cell, illustrating that Gag/Env/receptor complexes are linked to endocytic uptake pathways (see Movies S1 to S4 in the supplemental material) as previously suggested (14, 21). All together, these experiments demonstrate that (i) Env-receptor interactions at cell-cell interfaces are sufficient to drive the formation of VS-like structures between cells, (ii) Gag-mCherry and CD4-YFP are enriched at these contacts, and (iii) the contact is a site of bidirectional cell-cell membrane exchange. Observations of microsynapses converging over time (Fig. 1E and see Movies S1 to S4 in the supplemental material) suggest that VS formation is a cooperative process, i.e., many discrete Gag/Env/CD4 punctae nucleate single sites of cell-cell interaction prior to their lateral accumulation and the formation of larger adhesive structures.
Gag's targeting to cell-cell contacts is Env CT and Gag MA dependent.
Because Gag/CD4 enrichment served as a robust marker for VS-like contact formation, we next explored the viral determinants that underpin VS formation over time. We first tested the effects of deleting the gp41Env cytoplasmic tail (CT) domain based on prior work demonstrating that the CT domain can regulate Gag subcellular trafficking for HIV-1 and other retroviruses (58, 74–76) (Fig. 2A). As expected, Gag-mCherry accumulated at the strong majority of CD4-YFP-enriched cell-cell contacts (81% ± 6%, n = 4 independent experiments, 212 total contacts) when coexpressed with wild-type (WT) Env (Fig. 2B and C). In contrast, deleting Env amino acids 703 to 847 (ΔCT Env; retaining only the first six amino acids of the CT) severely reduced Gag-mCherry's recruitment to cell-cell contacts (16% ± 2% of cells, n = 4 independent experiments, 260 total contacts), despite having no apparent defects to Env-CD4-YFP contact formation (Fig. 2B, note CD4-YFP enrichment in lower panels) or size (Fig. 2D). Rapid acquisition live cell imaging, however, indicated that ΔCT Env synapses might be less stable over time (see Movie S4 in the supplemental material). These results confirmed an important role for one or more CT-encoded signaling determinants in regulating Gag's trafficking to and/or retention at cell-cell contacts in nonpolarized cells.
The processes of virus particle assembly and VS formation both require a convergence of Env and Gag at the plasma membrane. Because Env incorporation during virion assembly is regulated by Gag's N-terminal matrix (MA) domain, we next tested the ability of wild-type Env to recruit a Gag mutant, wherein the native MA domain was replaced with a heterologous membrane-targeting signal (derived from the Src oncogene; S-ΔMA Gag) (Fig. 3A). This mutation had no deleterious effects on the capacity of Gag to form virus-like particles compared to wild-type Gag (Fig. 3B) but severely impacted Gag's ability to accumulate at the contact (Fig. 3C and D). In contrast, other Gag mutants where (i) the nucleocapsid domain was replaced with a leucine zipper motif, thus providing for Gag multimerization in the absence of RNA binding (ΔNCzip Gag), or (ii) the C-terminal p6 domain essential for efficient virus particle release mediated through interactions with the cellular endosomal sorting complexes required for transport (ESCRT) machinery was deleted (Δp6 Gag) (Fig. 3A), localized to VS-like contacts to similar levels as wild-type Gag (Fig. 3C and D).
Interestingly, VS contacts formed with S-ΔMA Gag were smaller than those formed in the presence of WT Gag (Fig. 3E). More than 50% of the S-ΔMA Gag contacts were 0.5 to 0.99 μm in length, while the majority of WT Gag contacts fell between 1 and 2.99 μm (compare Fig. 2D and 3E), a range of sizes similar to those previously described for T cell donors (21). Since ΔCT Env maintained a VS size distribution similar to WT (Fig. 2D), this demonstrates that while both CT and MA are necessary for Gag's recruitment to the VS, these conditions do not result in phenocopies. Taken together, our data illustrate that Env's CT regulates Gag's clustering to the cell-cell contact, requiring Gag's native MA membrane targeting domain but not Gag nucleocapsid domain (NC)-regulated RNA binding or p6-dependent ESCRT recruitment (Fig. 3F).
Env and Gag cooperate to regulate the duration and stability of cell-cell contacts.
Based on the above results, we tested the roles of Env's CT and Gag's MA domain on long-term cell-cell interactions by coculturing Cos7 donor cells with Jurkat T cells stably expressing GFP (Jurkat.GFP) and tracking >300 donor-target cell interactions over 8 h (Fig. 4; see also Movie S5 in the supplemental material). We hypothesized that for successful virion transfer between cells, VS stability should be balanced with a mechanism to resolve the contact, thereby releasing newly infected cells to traffic and thus spread infection (44, 77). Other possible outcomes include contact-induced cell death or cell-cell fusion events resulting in syncytia (42, 44, 48, 78).
For the WT Env and Gag conditions, many T cell-donor cell interactions were transient, enduring for <1 h (Fig. 4A, top panels, and Fig. 4B). However, for >78% (±7%, n = 3, 442 total contacts) of the interactions, the T cells remained associated with donor cells for extended periods of time (>1 h) and, in several instances, for the entire imaging period (Fig. 4A, lower panels, and Fig. 4B). Short-term video microscopy revealed that these immobilized cells exhibited either of two behaviors. In the first, T cells appeared anchored to donor cells and exhibited dynamic interactions mediated by peripheral lamellipodial protrusions (see Movie S6, left panel, in the supplemental material). For the second, we observed more extensive interactions, wherein T cells were practically engulfed by the donor cell (see Movie S6, right panel, in the supplemental material). Additionally, 21% (±4%, n = 3 independent experiments, 442 total interactions) of longer-term cell-cell interactions terminated due to membrane fusion resulting in syncytium formation (Fig. 4B, black dots).
As expected, the expression of Env in donor cells increased the median time of interaction, ∼2-fold relative to the Env-minus control. Removal of Env's CT domain markedly reduced the duration of cell-cell interaction time. However, this result largely reflected the highly fusogenic nature of this mutant and an increase to the frequency of syncytialization events (Fig. 4B, black dots, and see Movie S7 in the supplemental material). Remarkably, however, expression of the S-ΔMA Gag mutant reduced the time of interaction to near baseline levels even in the presence of wild-type Env, suggesting that Env is unable to stabilize VS-like contacts in the absence of Gag's MA domain (Fig. 4B) and despite only minor effects on the Env surface expression levels (Fig. 4C and D). Taken together, these data suggested that Gag's MA domain plays a stabilizing role at the cell-cell contact that extends beyond its role driving virus particle production.
Differences in the extent of cell-cell fusion events over time provided an additional metric by which to evaluate the outcomes of Env-receptor interactions (Fig. 5). Interestingly, we observed ∼10-fold more cell-cell fusion events over 8 h for WT Gag-mCherry compared to either a no Gag control or Gag-mCherry expressed in the form of the S-ΔMA Gag mutant (Fig. 5A and B, trans constructs). This result provided further evidence that an intact MA domain plays a role in regulating Env stability and/or fusogenicity, either through modulating Env's trafficking or conformation. To ensure that the effects observed were not due to overexpressing viral components from non-native promoters (i.e., trans constructs) or were cell type specific, we also tested identical mutations engineered into plasmids encoding full-length Gag-mCherry NL4-3 reporter viruses (cis constructs). HIV-1 proteins driven off native LTRs in human 293T cells exhibited very similar trends both in the percentage of cells undergoing syncytiation (Fig. 5B), as well as the total number of fusion events measured either as the number of nuclei per heterokaryon (Fig. 5C) or the total cell area (Fig. 5D) relative to the trans constructs. Interestingly, ΔNCzip Gag-mCherry also yielded a moderate increase to cell-cell fusion events relative to WT Gag, suggesting that NC may also play a minor role in regulating cell-cell contact stability.
Increases to extracellular viscosity markedly reduce the stability of Env-dependent cell-cell interactions.
In the course of these experiments, we also tested the effects of modulating extracellular viscosity on Env/CD4 stability using the thickening agent methylcellulose (MeC) (Fig. 6). Our initial rationale for adding MeC to our cocultures was to force virus particle transmission to only occur through a cell-cell mode of spread, since cell-free virus particles would be unable to freely diffuse through the culture media (79). However, we also recognized that, in vivo, cells exist in blood plasma or dense tissue microenvironments with greater viscosity than typical cell culture conditions (80, 81). Surprisingly, we found that increasing the levels of MeC from 0 to 2% enhanced the frequency of Env-dependent cell-cell fusion events up to 3-fold, with 2% MeC causing nearly as much syncytiation for WT Env as for the ΔCT Env condition in the absence of MeC (Fig. 6A). To explain this increase in fusion, we measured both Jurkat T cell motility and the duration of donor-target interactions for 8 h for the 0, 1, and 2% MeC conditions (Fig. 6B and C). In contrast to our expectations, higher levels of MeC led to a dose-dependent decrease to the median duration of donor-target cell-cell interactions (Fig. 6B), corresponding to an increase to T cell trafficking on the glass substrate (Fig. 6C and see Movie S8 in the supplemental material). Thus, increasing MeC reduces cell-cell interaction times through apparently activating Jurkat T cell motility but markedly enhances cell-cell fusion events likely due to an increased frequency of cell-cell collisions.
FIG 6.
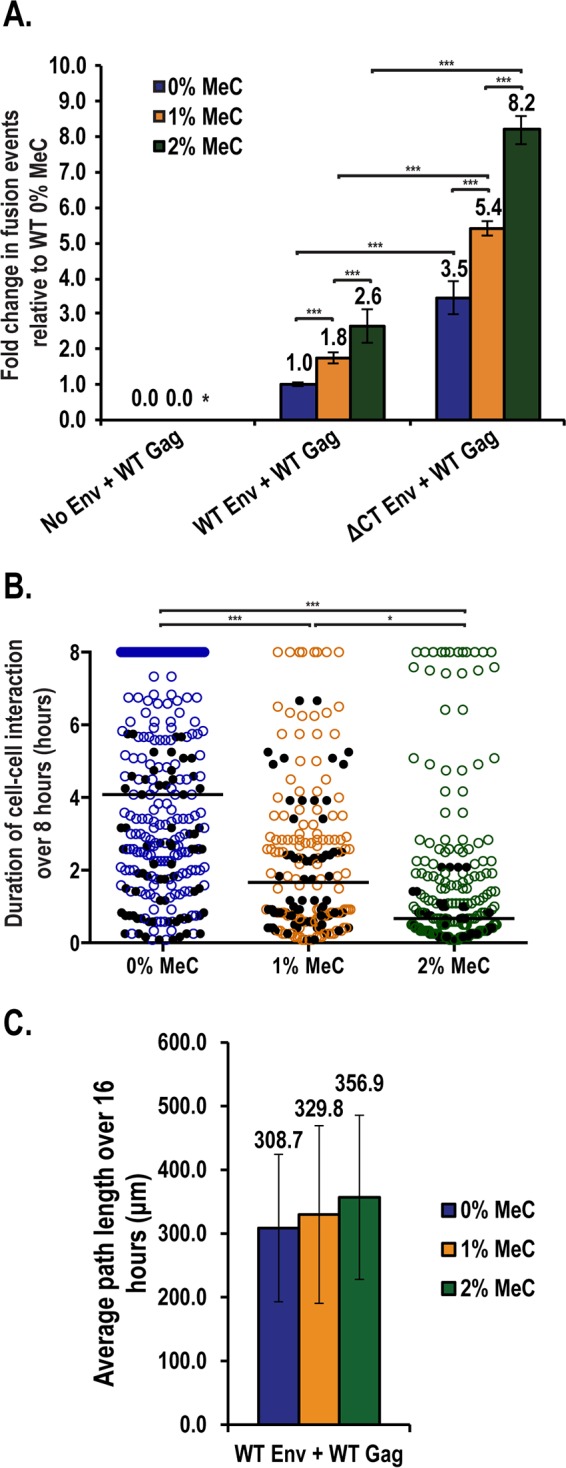
Extracellular viscosity impacts the stability of Env-dependent cell-cell contacts. (A) Cos7 donor cells expressing the indicated viral proteins were cocultured with Jurkat.GFP target T cells for 3 h in standard culture media and then switched to media supplemented with increasing amounts of methylcellulose (MeC), as indicated, prior to live-cell imaging. The cells were scored for the total number of donor-target fusion events observed over 8 to 16 h of imaging. Error bars represent the standard deviations of the mean for two independent experiments. An asterisk (*) denotes condition not measured. ***, P < 0.0001. (B) Duration of Cos7 donor-Jurkat.GFP interactions for movies taken from panel A were measured as described in Fig. 4B. The black line represents the median time of interaction for each condition. Black dots represent interactions that terminated due to cell-cell fusion. Statistical analysis was performed in GraphPad Prism 6 using the Kruskal-Wallis test, followed by a Dunn's multiple-comparison posttest. ***, P < 0.0001; *, P < 0.01. (C) All Jurkat.GFP cells imaged in panel A were tracked for net distance traversed (path length) over the course of imaging. The mean path length was calculated from two independent experiments. Error bars represent the standard deviations of the mean.
Further evidence for a functional link between Gag MA and Env CT in regulating VS stability and turnover.
Taken together, the results presented above suggested that VS stabilization or turnover is a function of the capacity of Gag, Env, and CD4 to form long-lived cell-cell interfaces, combined with the nature of donor-target cell migratory behaviors within the context of the extracellular environment. To further explore the hypothesis that cooperative interplay between Gag and Env stabilizes the VS, we tested the effects of additional Gag MA and Env mutations previously implicated in regulating Gag/Env cotrafficking during virus particle assembly (Fig. 7A and B). Replacing a single Gag MA leucine residue at position 12 to glutamic acid (L12E Gag) was previously shown by Freed and colleagues to reduce Env incorporation into assembling virions (Fig. 7A) (63–65). In our assays, this mutant yielded a pronounced reduction in the duration in donor-target cell interactions relative to WT Gag, very similar to the effects of the S-ΔMA Gag condition (Fig. 7C). Importantly, this mutant had minimal impact on Env surface levels relative to WT Gag (Fig. 7D). Expression of the L12E mutation also markedly reduced the frequency of cell-cell fusion events at all levels of MeC tested (Fig. 7E). However, a second-site suppressor mutant (L12E/Q62R Gag) reported to rescue Env incorporation during virion assembly (63) triggered only a very minor increase to donor-target cell interactions and cell-cell fusion events relative to L12E mutant Gag (Fig. 7D and E).
FIG 7.

Donor-target cell contact duration and outcome altered by a single point mutation in MA. (A) Depiction of Gag-mCherry mutants bearing the incorporation point mutation (L12E) and the secondary compensator mutation (L12E/Q62R) (63–65). (B) Depiction of Env's TM/gp41 subunit and the Env/CD4 chimera mutant (CD4 CT Env). ED, ectodomain; MSD, membrane-spanning domain. (C) Duration of Cos7 donor-Jurkat.GFP interactions for movies taken from panel A were measured as described for Fig. 4B. The black line represents the median time of interaction for each condition. Black dots represent interactions that were terminated due to cell-cell fusion. Statistical analysis was performed in GraphPad Prism 6 using a Kruskal-Wallis test, followed by a Dunn's multiple-comparison posttest. ***, P < 0.0001; n.s., not significant. (D) Quantification of Env surface expression, based on the mean fluorescent intensity (MFI) for 50 cells for the conditions presented in panel C. A black line represents the MFI; error bars represent the standard deviations of the mean. (E) VS collapse assay. Cos7 donor cells expressing the indicated viral proteins were cocultured with Jurkat.GFP target T cells for 2 h in standard culture media and then switched to media supplemented with increasing amounts (0, 1, or 2%) of methylcellulose (MeC) for 3 h prior to fixing. A total of 50 donor cells per condition were scored for the total number of donor-target fusion events observed based on the number of extraneous nuclei, as in Fig. 5C. Means were calculated from three independent experiments; error bars represent the standard deviations from the mean. P values were calculated by using the Fisher exact test in MStat. ***, P < 0.0001; **, P < 0.001; n.s., not significant. (F) Quantification of the number of donor cells that underwent syncytiation in panel E.
In this context, we also tested an Env chimeric protein wherein the CT of Env was swapped with that of CD4 (CD4 CT Env) (Fig. 7B) because, interestingly, CD4 has been shown to be actively copackaged and incorporated into budding HIV-1 particles in a CT-dependent manner (i.e., is competent for viral “pseudotyping”) (82, 83). CD4 CT Env increased the duration of donor-target interactions relative to ΔCT Env (Fig. 7C, P = 0.0963) despite exhibiting a similar level of enhanced fusogenicity (Fig. 7E). However, CD4 CT Env did not restore contact duration times to WT levels (Fig. 7C). Combined, these results were more consistent with a model wherein donor-target cell contact stability is regulated by a mechanism of Gag/Env convergence related to pseudotyping, rather than the mechanism by which L12E/Q62R mediates efficient Env incorporation into budding virions. Similar dose-dependent cell-cell fusion trends for most mutants at various MeC levels (Fig. 7E and F, the exception being L12E/Q62R) served as further evidence that the extracellular environment and MA-CT interplay affect Env's fusogenic potential through different but apparently synergistic mechanisms.
DISCUSSION
In this study, we describe a quantitative imaging approach for measuring viral and cellular contributions to the formation, stabilization, and net outcome (e.g., resolution or syncytium formation) of HIV-1 Env-induced cell-cell contacts. As summarized in Fig. 8, our long-term system allows us to integrate measurements of at least five stages of viral cell-to-cell transmission over time in a single experiment, using only two fluorescently tagged proteins. These measurements include (i) the extent of initial probing interactions between donor cells and CD4-expressing target cells, (ii) CD4 clustering indicative of VS formation, (iii) Gag's recruitment to the cell-cell contact, (iv) the duration of cell-cell interactions and extent of virion/membrane exchange among cells, and (v) contact resolution or collapse to form a syncytium.
FIG 8.

Model for Env-induced cell-cell contact formation, turnover, and/or collapse. Our imaging strategy allowed us to measure viral, cellular, and extracellular determinants affecting the stepwise progression of Env-CD4 interactions leading to transient cell-cell contacts (step 1), Env-dependent recruitment of Gag to the VS (step 2), Gag-dependent stabilization of the contact and lateral accumulation of microsynapses (step 3), virion transmission (step 4), and resolution of the contact due to mechanical disengagement or, alternatively, collapse to form a syncytium (step 5).
To establish our system, we exploited fluorescent viruses and adherent, nonmotile Cos7 donor cells cocultured with target 3T3 fibroblasts expressing human CD4-YFP (Fig. 1 to 3) or Jurkat T cells (Fig. 4 to 7). The power of this approach is the capacity to modulate and quantitatively access viral and cellular determinants under tightly controlled conditions, as well as to measure hundreds of cell-cell interactions over time in a single experiment. Moreover, Cos7 donor cells were ideal for quantitative imaging due to their expansive lamellipodia and flat cell morphology. However, we emphasize that, in vivo, HIV-1 replicates predominantly in human primary T cells and macrophages. Multiple studies have suggested important roles for Env in cell trafficking both in culture (12, 61) and in vivo using humanized mouse models (46, 48). However, Gag and Env expression levels are likely to vary markedly depending on the stage of infection, cell type, or cell activation status. Thus, an important goal for the near term is to adapt these quantitative, long-term strategies for studying HIV-1 in primary cell systems in ex vivo or in vivo models that more accurately replicate spreading infection in complex tissues.
To test our system, we examined the role of Env's conserved CT domain in signaling Gag to accumulate at the zone of cell-cell contact. As shown in Fig. 2 and 3, Gag's accumulation at the contact requires both an intact Env CT domain and residues within Gag's MA domain in this cell coculture system. A parsimonious explanation is that Gag is targeted to the VS by Env in the same way that Env is incorporated into virus particles during virion assembly, i.e., through interactions between Env and the immature Gag capsid lattice (5–7). Alternatively, Env's clustering with CD4 at the cell-cell contact could signal Gag's targeting to a specialized membrane microdomain, as demonstrated for the model retrovirus murine leukemia virus (MLV) (71, 74). In this light, we tested a Gag mutant bearing a single amino acid change in MA (L12E) known to abrogate Env incorporation during budding (63–65), and found that it reduced cell contact duration and also affected contact stability to levels equivalent to deleting MA completely (Fig. 7). However, the L12E/Q62R compensatory Gag mutant, which rescues Env incorporation during budding, had no substantial effect in our system relative to L12E Gag. Thus, the L12E Gag mutation may impact Env conformation and fusogenicity at the VS in a way that is independent of its role in the Env incorporation pathway. In contrast, our observation that the CD4 CT Env chimeric proteins moderately enhanced contact duration relative to ΔCT Env (Fig. 7C) may indicate that a more general mechanism is involved, perhaps akin to the more nebulous “pseudotyping” pathway by which various transmembrane proteins, including several viral glycoproteins (e.g., VSV-G), are preferentially incorporated into HIV-1 budding particles (2, 84).
In this context, we also note prior work by Li et al. showing that, similar to HIV-1, deletion of MLV Gag's MA domain abrogated Gag targeting to the VS; however this could be rescued by replacing MLV MA with the myristoylation signal from the Src oncogene provided that it contained multiple basic residues (MGSSKSKPK, where basic lysine residues are highlighted in bold) (71). In our experiments, we replaced HIV-1 MA with the identical Src-derived membrane-targeting signal but did not detect Gag targeting to the cell-cell contact (Fig. 3). Thus, we confirm that the mechanisms by which MLV and HIV-1 Gag are trafficked to the VS are fundamentally different, as previously suggested (71, 74). Also, whereas MLV can bias virus particle assembly to occur preferentially at the VS contact zone (79), we and others have not detected individual HIV-1 budding events at the VS using video microscopy (14, 21, 74). Instead, HIV-1 Gag tends to form surface-associated assemblages that migrate to the cell-cell contact after Env-CD4 binding (e.g., see Fig. 1 and see Movies S1 to S4 in the supplemental material). Similarly, Chen and coworkers demonstrated surface-linked Gag assemblages (viral buttons) undergoing lateral trafficking to the VS prior to cell-cell transfer in T cells (21, 85). Here, we also demonstrated that tripartite CD4/Env/Gag puncta, or “microsynapses,” appear to nucleate VS linkages prior to coalescing to form larger, and often remarkably stable structures (Fig. 1E and see Movies S3 and S4 in the supplemental material). Combined, these observations suggest that HIV-1 Gag/Env complexes form at the cell surface prior to CD4 engagement.
We also report the unexpected finding that Gag's MA domain plays a role in prolonging Env-dependent interactions between cells (Fig. 4). The VS is so named due to its similarity to the immunological synapse that forms between T cells and antigen-presenting cells expressing major histocompatibility (MHC) complexes (10, 49). Immunological synapse formation requires stabilizing interactions between the donor cell surface protein lymphocyte function-associated antigen 1 and the T cell receptor (TCR) with the target cell surface protein intercellular adhesion molecule 1 and MHC, respectively (86). In T cells, Env and CD4 are thought to mimic TCR-MHC binding and thus provide a stop signal to the otherwise mobile target T cell that culminates in stable VS formation (52). Our data demonstrate that Env can arrest target cell trafficking even in the absence of other T cell factors (Fig. 4). However, this activity requires coexpression of wild-type Gag. Interestingly, Kräusslich and coworkers demonstrated large accumulations of Env, greater than what is incorporated into particles, forming a ring around non-VS virion bud sites, and speculated that Gag is capable of recruiting excess Env in order to promote the formation of cell-cell contacts (87). Thus, in conjunction with Gag's tendency to accumulate at the VS for the purpose of virion production, we propose that a subset of cell surface Gag-Env complexes are not involved in budding but rather in stabilizing the cell-cell contact (e.g., see Movies S2 and S3 in the supplemental material).
In vivo, such a role for Gag may ensure sufficient duration of cell-cell interactions necessary for the transfer of infection. However, a conundrum for Env's fusogenic potential is that whereas prolonged cell-cell interactions may be beneficial to viral transmission, they also increase the odds that two cells will fuse together to form a syncytium. Our study illustrates that MA contributes both to the duration of cell-cell interactions and also to the frequency of syncytiation. This observation is in conflict with recent models suggesting that Gag plays an inhibitory role in regulating Env fusogenicity at the cell surface (54) and, to an extent, data suggesting that the immature capsid lattice in virions restricts Env membrane diffusion until proteolytic maturation, thereby providing a trigger to activate Env's fusion peptide (88, 89). This reduced frequency of Env-dependent membrane fusion events may be explained by the fact that ΔMA Gag does not accumulate at and stabilize the VS, thereby reducing the duration of cell-cell interactions (Fig. 4). Alternatively, MA's presence at the VS may regulate the recruitment of cellular factors that modulate Env fusogenicity. Consistent with this hypothesis, MA can remodel the composition of the plasma membrane at sites of Gag multimerization (56, 90), and Thali and coworkers have demonstrated a correlation between Env's capacity to drive syncytium formation and MA-dependent coclustering of Gag with the tetraspanin molecules CD9 and CD63 (56).
We also made the interesting observation that increases to extracellular viscosity, an inducer of cell surface tension and membrane stress linked to cortical actin dynamics (91), dramatically enhances the frequency of Env-driven cell-cell fusion events. We note that, in vivo, T cells experience viscosities substantially higher than those typically encountered in cell culture (80, 81, 92). Although the relevance of syncytium formation to HIV-1 replication or pathogenesis in vivo is still debated (93, 94), HIV-induced syncytia have been observed in patients (95, 96) and were recently directly visualized in the lymph nodes of a humanized mouse model using intravital imaging (44, 47).
As for the mechanism(s) by which viscosity potentiates membrane fusion, a reasonable explanation would be that increased viscosity reduces cell migration and thus prolongs the cell-cell interaction. We were surprised, however, to find that higher percentages of MeC actually decreased the duration of cell-cell interactions due to increasing the motility and migration of Jurkat target T cells (Fig. 6B and C). Thus, a more plausible explanation is that increases to cell-cell fusion events reflect a higher frequency of cell-cell collisions. An alternative explanation is that the increased viscosity increases the external pressure applied to the cell surface, thereby decreasing the net tension required to drive membrane fusion. This could potentially be facilitated by the recruitment of myosin II to the cell surface since its mechanosensing ability has been implicated in driving nonviral syncytiation events in response to membrane tension (97). Thus, further studies investigating the role of the extracellular environment in the context of HIV-1 replication may provide insight into how Env strikes the delicate balance between fusion and adhesion at the VS, as well as the mechanisms by which cells fuse during development and disease.
In contrast to these models suggesting important roles for Env/Gag interactions at the VS, it was recently demonstrated that both HIV-1 and MLV Gag proteins are targeted to the uropod at the rear of polarized lymphocytes in an Env-independent manner. The uropod was shown to direct donor cell binding to uninfected target cells (90, 98). In addtion, Gag's targeting to the VS formed between infected macrophages and uninfected T cells has been reported to be Env independent (99). Hence, there appear to be fundamental differences to how Gag and Env are trafficked to cell-cell contacts depending on the identity of the cell and its polarization status. We think it likely that these differences reflect the existence of multiple pathways exploited by the virus to spread and persist in diverse cell types and tissues. Consistent with this view, Env's CT domain is essential for replication in most but not all T cell types (e.g., MT4 cells) (57, 59–61). Importantly, Bosch and coworkers recently correlated this activity to the CT's capacity to recruit Gag to the VS (58). These results, combined with the results from our present study, emphasize the multifactorial nature of viral dissemination among cells, wherein viral, cellular, and extracellular determinants combine to determine the net outcome of spreading infection.
Supplementary Material
ACKNOWLEDGMENTS
We thank Bill Sugden and the NIH AIDS Research and Reference Reagent Program for generously supplying vital cells and reagents. We thank Laraine Zimdars, Jordan Becker, Halena VanDeusen, Eric Britigan, and Yousra Mohamoud for technical advice and support.
This study was supported in part by funding from the NIH, National Institute of Allergy and Infectious Diseases (RO1AI110221A1), the Wisconsin Partnership Program New Investigator Program (ID 2830), and the Greater Milwaukee Foundation's Shaw Scientist Program to N.M.S. J.C.G. received training support from the University of Wisconsin—Madison Science and Medicine Graduate Research Scholars (SciMed GRS) program and National Science Foundation Graduate Research Fellowship Program grant DGE-1256259. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
Funding Statement
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00600-16.
REFERENCES
- 1.Adamson CS, Freed EO. 2007. Human immunodeficiency virus type 1 assembly, release, and maturation. Adv Pharmacol 55:347–387. doi: 10.1016/S1054-3589(07)55010-6. [DOI] [PubMed] [Google Scholar]
- 2.Checkley MA, Luttge BG, Freed EO. 2011. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J Mol Biol 410:582–608. doi: 10.1016/j.jmb.2011.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klatzmann D, Champagne E, Chamaret S, Gruest J, Guetard D, Hercend T, Gluckman J-C, Montagnier L. 1984. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature 312:767–768. doi: 10.1038/312767a0. [DOI] [PubMed] [Google Scholar]
- 4.Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. 1996. CC CKR5: a RANTES, MIP-1α, MIP-1α receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 5.Da Silva ES, Mulinge M, Bercoff DP. 2013. The frantic play of the concealed HIV envelope cytoplasmic tail. Retrovirology 10:54. doi: 10.1186/1742-4690-10-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steckbeck JD, Kuhlmann A-S, Montelaro RC. 2014. Structural and functional comparisons of retroviral envelope protein C-terminal domains: still much to learn. Viruses 6:284–300. doi: 10.3390/v6010284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tedbury PR, Freed EO. 2014. The role of matrix in HIV-1 envelope glycoprotein incorporation. Trends Microbiol 22:372–378. doi: 10.1016/j.tim.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu P, Chertova E, Bess J, Lifson JD, Arthur LO, Liu J, Taylor KA, Roux KH. 2003. Electron tomography analysis of envelope glycoprotein trimers on HIV and simian immunodeficiency virus virions. Proc Natl Acad Sci U S A 100:15812–15817. doi: 10.1073/pnas.2634931100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jolly C, Kashefi K, Hollinshead M, Sattentau QJ. 2004. HIV-1 cell-to-cell transfer across an Env-induced, actin-dependent synapse. J Exp Med 199:283–293. doi: 10.1084/jem.20030648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Igakura T, Stinchcombe JC, Goon PKC, Taylor GP, Weber JN, Griffiths GM, Tanaka Y, Osame M, Bangham CRM. 2003. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 299:1713–1716. doi: 10.1126/science.1080115. [DOI] [PubMed] [Google Scholar]
- 11.Jolly C, Mitar I, Sattentau QJ. 2007. Requirement for an intact T-cell actin and tubulin cytoskeleton for efficient assembly and spread of human immunodeficiency virus type 1. J Virol 81:5547–5560. doi: 10.1128/JVI.01469-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen P, Hübner W, Spinelli MA, Chen BK. 2007. Predominant mode of human immunodeficiency virus transfer between T cells is mediated by sustained Env-dependent neutralization-resistant virological synapses. J Virol 81:12582–12595. doi: 10.1128/JVI.00381-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rudnicka D, Feldmann J, Porrot F, Wietgrefe S, Guadagnini S, Prévost M-C, Estaquier J, Haase AT, Sol-Foulon N, Schwartz O. 2009. Simultaneous cell-to-cell transmission of human immunodeficiency virus to multiple targets through polysynapses. J Virol 83:6234–6246. doi: 10.1128/JVI.00282-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sherer NM, Lehmann MJ, Jimenez-Soto LF, Horensavitz C, Pypaert M, Mothes W. 2007. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat Cell Biol 9:310–315. doi: 10.1038/ncb1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sowinski S, Jolly C, Berninghausen O, Purbhoo MA, Chauveau A, Köhler K, Oddos S, Eissmann P, Brodsky FM, Hopkins C, & Ouml;nfelt B, Sattentau Q, Davis DM. 2008. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat Cell Biol 10:211–219. doi: 10.1038/ncb1682. [DOI] [PubMed] [Google Scholar]
- 16.Carr JM, Hocking H, Li P, Burrell CJ. 1999. Rapid and efficient cell-to-cell transmission of human immunodeficiency virus infection from monocyte-derived macrophages to peripheral blood lymphocytes. Virology 265:319–329. doi: 10.1006/viro.1999.0047. [DOI] [PubMed] [Google Scholar]
- 17.Dimitrov DS, Willey RL, Sato H, Chang LJ, Blumenthal R, Martin MA. 1993. Quantitation of human immunodeficiency virus type 1 infection kinetics. J Virol 67:2182–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin N, Welsch S, Jolly C, Briggs JAG, Vaux D, Sattentau QJ. 2010. Virological synapse-mediated spread of human immunodeficiency virus type 1 between T cells is sensitive to entry inhibition. J Virol 84:3516–3527. doi: 10.1128/JVI.02651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sourisseau M, Sol-Foulon N, Porrot F, Blanchet F, Schwartz O. 2007. Inefficient human immunodeficiency virus replication in mobile lymphocytes. J Virol 81:1000–1012. doi: 10.1128/JVI.01629-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhong P, Agosto LM, Ilinskaya A, Dorjbal B, Truong R, Derse D, Uchil PD, Heidecker G, Mothes W. 2013. Cell-to-cell transmission can overcome multiple donor and target cell barriers imposed on cell-free HIV. PLoS One 8:e53138. doi: 10.1371/journal.pone.0053138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hübner W, McNerney GP, Chen P, Dale BM, Gordon RE, Chuang FYS, Li X-D, Asmuth DM, Huser T, Chen BK. 2009. Quantitative 3D video microscopy of HIV transfer across T cell virological synapses. Science 323:1743–1747. doi: 10.1126/science.1167525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mothes W, Sherer NM, Jin J, Zhong P. 2010. Virus cell-to-cell transmission. J Virol 84:8360–8368. doi: 10.1128/JVI.00443-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sattentau QJ. 2010. Cell-to-cell spread of retroviruses. Viruses 2:1306–1321. doi: 10.3390/v2061306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duncan CJA, Williams JP, Schiffner T, Gärtner K, Ochsenbauer C, Kappes J, Russell RA, Frater J, Sattentau QJ. 2014. High-multiplicity HIV-1 infection and neutralizing antibody evasion mediated by the macrophage-T cell virological synapse. J Virol 88:2025–2034. doi: 10.1128/JVI.03245-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Del Portillo A, Tripodi J, Najfeld V, Wodarz D, Levy DN, Chen BK. 2011. Multiploid inheritance of HIV-1 during cell-to-cell infection. J Virol 85:7169–7176. doi: 10.1128/JVI.00231-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Russell RA, Martin N, Mitar I, Jones E, Sattentau QJ. 2013. Multiple proviral integration events after virological synapse-mediated HIV-1 spread. Virology 443:143–149. doi: 10.1016/j.virol.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 27.Abela IA, Berlinger L, Schanz M, Reynell L, Günthard HF, Rusert P, Trkola A. 2012. Cell-cell transmission enables HIV-1 to evade inhibition by potent CD4bs directed antibodies. PLoS Pathog 8:e1002634. doi: 10.1371/journal.ppat.1002634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agosto LM, Zhong P, Munro J, Mothes W. 2014. Highly active antiretroviral therapies are effective against HIV-1 cell-to-cell transmission. PLoS Pathog 10:e1003982. doi: 10.1371/journal.ppat.1003982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casartelli N, Sourisseau M, Feldmann J, Guivel-Benhassine F, Mallet A, Marcelin A-G, Guatelli J, Schwartz O. 2010. Tetherin restricts productive HIV-1 cell-to-cell transmission. PLoS Pathog 6:e1000955. doi: 10.1371/journal.ppat.1000955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coleman CM, Spearman P, Wu L. 2011. Tetherin does not significantly restrict dendritic cell-mediated HIV-1 transmission and its expression is upregulated by newly synthesized HIV-1 Nef. Retrovirology 8:26. doi: 10.1186/1742-4690-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ganesh L, Leung K, Loré K, Levin R, Panet A, Schwartz O, Koup RA, Nabel GJ. 2004. Infection of specific dendritic cells by CCR5-tropic human immunodeficiency virus type 1 promotes cell-mediated transmission of virus resistant to broadly neutralizing antibodies. J Virol 78:11980–11987. doi: 10.1128/JVI.78.21.11980-11987.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giese S, Marsh M. 2014. Tetherin can restrict cell-free and cell-cell transmission of HIV from primary macrophages to T cells. PLoS Pathog 10:e1004189. doi: 10.1371/journal.ppat.1004189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gupta P, Balachandran R, Ho M, Enrico A, Rinaldo C. 1989. Cell-to-cell transmission of human immunodeficiency virus type 1 in the presence of azidothymidine and neutralizing antibody. J Virol 63:2361–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jolly C, Booth NJ, Neil SJD. 2010. Cell-cell spread of human immunodeficiency virus type 1 overcomes tetherin/BST-2-mediated restriction in T cells. J Virol 84:12185–12199. doi: 10.1128/JVI.01447-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuhl BD, Sloan RD, Donahue DA, Bar-Magen T, Liang C, Wainberg MA. 2010. Tetherin restricts direct cell-to-cell infection of HIV-1. Retrovirology 7:115. doi: 10.1186/1742-4690-7-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malbec M, Porrot F, Rua R, Horwitz J, Klein F, Halper-Stromberg A, Scheid JF, Eden C, Mouquet H, Nussenzweig MC, Schwartz O. 2013. Broadly neutralizing antibodies that inhibit HIV-1 cell to cell transmission. J Exp Med 210:2813–2821. doi: 10.1084/jem.20131244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Massanella M, Puigdomènech I, Cabrera C, Fernandez-Figueras MT, Aucher A, Gaibelet G, Hudrisier D, García E, Bofill M, Clotet B, Blanco J. 2009. Antigp41 antibodies fail to block early events of virological synapses but inhibit HIV spread between T cells. AIDS Lond Engl 23:183–188. doi: 10.1097/QAD.0b013e32831ef1a3. [DOI] [PubMed] [Google Scholar]
- 38.McCoy LE, Groppelli E, Blanchetot C, de Haard H, Verrips T, Rutten L, Weiss RA, Jolly C. 2014. Neutralisation of HIV-1 cell-cell spread by human and llama antibodies. Retrovirology 11:83. doi: 10.1186/s12977-014-0083-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richardson MW, Carroll RG, Stremlau M, Korokhov N, Humeau LM, Silvestri G, Sodroski J, Riley JL. 2008. Mode of transmission affects the sensitivity of human immunodeficiency virus type 1 to restriction by rhesus TRIM5alpha. J Virol 82:11117–11128. doi: 10.1128/JVI.01046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, Milo R, Baltimore D. 2011. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature 477:95–98. doi: 10.1038/nature10347. [DOI] [PubMed] [Google Scholar]
- 41.Yoder A, Yu D, Dong L, Iyer SR, Xu X, Kelly J, Liu J, Wang W, Vorster PJ, Agulto L, Stephany DA, Cooper JN, Marsh JW, Wu Y. 2008. HIV envelope-CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell 134:782–792. doi: 10.1016/j.cell.2008.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Doitsh G, Cavrois M, Lassen KG, Zepeda O, Yang Z, Santiago ML, Hebbeler AM, Greene WC. 2010. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell 143:789–801. doi: 10.1016/j.cell.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, Hunt PW, Hatano H, Sowinski S, Muñoz-Arias I, Greene WC. 2014. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 505:509–514. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murooka TT, Deruaz M, Marangoni F, Vrbanac VD, Seung E, von Andrian UH, Tager AM, Luster AD, Mempel TR. 2012. HIV-infected T cells are migratory vehicles for viral dissemination. Nature 490:283–287. doi: 10.1038/nature11398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sewald X, Gonzalez DG, Haberman AM, Mothes W. 2012. In vivo imaging of virological synapses. Nat Commun 3:1320. doi: 10.1038/ncomms2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murooka TT, Mempel TR. 2013. Intravital miroscopy in BLT-humanized mice to study cellular dynamics in HIV infection. J Infect Dis 208:S137–S144. doi: 10.1093/infdis/jit447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murooka TT, Sharaf RR, Mempel TR. 2015. Large syncytia in lymph nodes induced by CCR5-tropic HIV-1. AIDS Res Hum Retroviruses 31:471–472. doi: 10.1089/aid.2014.0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Symeonides M, Murooka TT, Bellfy LN, Roy NH, Mempel TR, Thali M. 2015. HIV-1-induced small T cell syncytia can transfer virus particles to target cells through transient contacts. Viruses 7:6590–6603. doi: 10.3390/v7122959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jolly C, Mitar I, Sattentau QJ. 2007. Adhesion molecule interactions facilitate human immunodeficiency virus type 1-induced virological synapse formation between T cells. J Virol 81:13916–13921. doi: 10.1128/JVI.01585-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fackler OT, Kräusslich H-G. 2006. Interactions of human retroviruses with the host cell cytoskeleton. Curr Opin Microbiol 9:409–415. doi: 10.1016/j.mib.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 51.Lehmann M, Nikolic DS, Piguet V. 2011. How HIV-1 takes advantage of the cytoskeleton during replication and cell-to-cell transmission. Viruses 3:1757–1776. doi: 10.3390/v3091757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vasiliver-Shamis G, Tuen M, Wu TW, Starr T, Cameron TO, Thomson R, Kaur G, Liu J, Visciano ML, Li H, Kumar R, Ansari R, Han DP, Cho MW, Dustin ML, Hioe CE. 2008. Human immunodeficiency virus type 1 envelope gp120 induces a stop signal and virological synapse formation in noninfected CD4+ T cells. J Virol 82:9445–9457. doi: 10.1128/JVI.00835-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vasiliver-Shamis G, Cho MW, Hioe CE, Dustin ML. 2009. Human immunodeficiency virus type 1 envelope gp120-induced partial T-cell receptor signaling creates an F-actin-depleted zone in the virological synapse. J Virol 83:11341–11355. doi: 10.1128/JVI.01440-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roy NH, Chan J, Lambelé M, Thali M. 2013. Clustering and mobility of HIV-1 Env at viral assembly sites predict its propensity to induce cell-cell fusion. J Virol 87:7516–7525. doi: 10.1128/JVI.00790-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krementsov DN, Weng J, Lambelé M, Roy NH, Thali M. 2009. Tetraspanins regulate cell-to-cell transmission of HIV-1. Retrovirology 6:64. doi: 10.1186/1742-4690-6-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weng J, Krementsov DN, Khurana S, Roy NH, Thali M. 2009. Formation of syncytia is repressed by tetraspanins in human immunodeficiency virus type 1-producing cells. J Virol 83:7467–7474. doi: 10.1128/JVI.00163-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murakami T, Freed EO. 2000. The long cytoplasmic tail of gp41 is required in a cell type-dependent manner for HIV-1 envelope glycoprotein incorporation into virions. Proc Natl Acad Sci U S A 97:343–348. doi: 10.1073/pnas.97.1.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emerson V, Haller C, Pfeiffer T, Fackler OT, Bosch V. 2010. Role of the C-terminal domain of the HIV-1 glycoprotein in cell-to-cell viral transmission between T lymphocytes. Retrovirology 7:43. doi: 10.1186/1742-4690-7-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Akari H, Fukumori T, Adachi A. 2000. Cell-dependent requirement of human immunodeficiency virus type 1 gp41 cytoplasmic tail for Env incorporation into virions. J Virol 74:4891–4893. doi: 10.1128/JVI.74.10.4891-4893.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dubay JW, Roberts SJ, Hahn BH, Hunter E. 1992. Truncation of the human immunodeficiency virus type 1 transmembrane glycoprotein cytoplasmic domain blocks virus infectivity. J Virol 66:6616–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Durham ND, Chen BK. 2015. HIV-1 cell-free and cell-to-cell infection are differentially regulated by distinct determinants in the Env gp41 cytoplasmic tail. J Virol 89:9324–9337. doi: 10.1128/JVI.00655-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kalia V, Sarkar S, Gupta P, Montelaro RC. 2003. Rational site-directed mutations of the LLP-1 and LLP-2 lentivirus lytic peptide domains in the intracytoplasmic tail of human immunodeficiency virus type 1 gp41 indicate common functions in cell-cell fusion but distinct roles in virion envelope incorporation. J Virol 77:3634–3646. doi: 10.1128/JVI.77.6.3634-3646.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tedbury PR, Ablan SD, Freed EO. 2013. Global rescue of defects in HIV-1 envelope glycoprotein incorporation: implications for matrix structure. PLoS Pathog 9:e1003739. doi: 10.1371/journal.ppat.1003739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tedbury PR, Novikova M, Ablan SD, Freed EO. 2016. Biochemical evidence of a role for matrix trimerization in HIV-1 envelope glycoprotein incorporation. Proc Natl Acad Sci U S A 113:E182–E190. doi: 10.1073/pnas.1516618113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Freed EO, Martin MA. 1996. Domains of the human immunodeficiency virus type 1 matrix and gp41 cytoplasmic tail required for envelope incorporation into virions. J Virol 70:341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Swanson CM, Sherer NM, Malim MH. 2010. SRp40 and SRp55 promote the translation of unspliced human immunodeficiency virus type 1 RNA. J Virol 84:6748–6759. doi: 10.1128/JVI.02526-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ott DE, Coren LV, Gagliardi TD. 2005. Redundant roles for nucleocapsid and matrix RNA-binding sequences in human immunodeficiency virus type 1 assembly. J Virol 79:13839–13847. doi: 10.1128/JVI.79.22.13839-13847.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Swanson CM, Puffer BA, Ahmad KM, Doms RW, Malim MH. 2004. Retroviral mRNA nuclear export elements regulate protein function and virion assembly. EMBO J 23:2632–2640. doi: 10.1038/sj.emboj.7600270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gallois-Montbrun S, Kramer B, Swanson CM, Byers H, Lynham S, Ward M, Malim MH. 2007. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J Virol 81:2165–2178. doi: 10.1128/JVI.02287-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sherer NM, Lehmann MJ, Jimenez-Soto LF, Ingmundson A, Horner SM, Cicchetti G, Allen PG, Pypaert M, Cunningham JM, Mothes W. 2003. Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic 4:785–801. doi: 10.1034/j.1600-0854.2003.00135.x. [DOI] [PubMed] [Google Scholar]
- 71.Li F, Jin J, Herrmann C, Mothes W. 2013. Basic residues in the matrix domain and multimerization target murine leukemia virus gag to the virological synapse. J Virol 87:7113–7126. doi: 10.1128/JVI.03263-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aligeti M, Behrens RT, Pocock GM, Schindelin J, Dietz C, Eliceiri KW, Swanson CM, Malim MH, Ahlquist P, Sherer NM. 2014. Cooperativity among Rev-associated nuclear export signals regulates HIV-1 gene expression and is a determinant of virus species tropism. J Virol 88:14207–14221. doi: 10.1128/JVI.01897-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sherer NM, Swanson CM, Papaioannou S, Malim MH. 2009. Matrix mediates the functional link between human immunodeficiency virus type 1 RNA nuclear export elements and the assembly competency of Gag in murine cells. J Virol 83:8525–8535. doi: 10.1128/JVI.00699-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jin J, Li F, Mothes W. 2011. Viral determinants of polarized assembly for the murine leukemia virus. J Virol 85:7672–7682. doi: 10.1128/JVI.00409-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lodge R, Gottlinger H, Gabuzda D, Cohen EA, Lemay G. 1994. The intracytoplasmic domain of gp41 mediates polarized budding of human immunodeficiency virus type 1 in MDCK cells. J Virol 68:4857–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lodge R, Lalonde JP, Lemay G, Cohen EA. 1997. The membrane-proximal intracytoplasmic tyrosine residue of HIV-1 envelope glycoprotein is critical for basolateral targeting of viral budding in MDCK cells. EMBO J 16:695–705. doi: 10.1093/emboj/16.4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fackler OT, Murooka TT, Imle A, Mempel TR. 2014. Adding new dimensions: towards an integrative understanding of HIV-1 spread. Nat Rev Microbiol 12:563–574. doi: 10.1038/nrmicro3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Blanco J, Barretina J, Ferri KF, Jacotot E, Gutiérrez A, Armand-Ugón M, Cabrera C, Kroemer G, Clotet B, Esté JA. 2003. Cell-surface-expressed HIV-1 envelope induces the death of CD4 T cells during GP41-mediated hemifusion-like events. Virology 305:318–329. doi: 10.1006/viro.2002.1764. [DOI] [PubMed] [Google Scholar]
- 79.Jin J, Sherer NM, Heidecker G, Derse D, Mothes W. 2009. Assembly of the murine leukemia virus is directed towards sites of cell-cell contact. PLoS Biol 7:e1000163. doi: 10.1371/journal.pbio.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bonithon-Kopp C, Levenson J, Scarabin P-Y, Guillanneuf M-T, Kirzin J-M, Malmejac A, Guize L. 1993. Longitudinal associations between plasma viscosity and cardiovascular risk factors in a middle-aged French population. Atherosclerosis 104:173–182. doi: 10.1016/0021-9150(93)90188-Z. [DOI] [PubMed] [Google Scholar]
- 81.Fröhlich E, Bonstingl G, Höfler A, Meindl C, Leitinger G, Pieber TR, Roblegg E. 2013. Comparison of two in vitro systems to assess cellular effects of nanoparticles-containing aerosols. Toxicol In Vitro 27:409–417. doi: 10.1016/j.tiv.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gregory DA, Olinger GY, Lucas TM, Johnson MC. 2014. Diverse viral glycoproteins as well as CD4 copackage into the same human immunodeficiency virus (HIV-1) particles. Retrovirology 11:28. doi: 10.1186/1742-4690-11-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Henriksson P, Bosch V. 1998. Inhibition of cellular glycoprotein incorporation into human immunodeficiency virus-like particles by coexpression of additional cellular interaction partner. Virology 251:16–21. doi: 10.1006/viro.1998.9403. [DOI] [PubMed] [Google Scholar]
- 84.Johnson MC. 2011. Mechanisms for Env glycoprotein acquisition by retroviruses. AIDS Res Hum Retroviruses 27:239–247. doi: 10.1089/aid.2010.0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dale BM, McNerney GP, Thompson DL, Hubner W, de Los Reyes K, Chuang FYS, Huser T, Chen BK. 2011. Cell-to-cell transfer of HIV-1 via virological synapses leads to endosomal virion maturation that activates viral membrane fusion. Cell Host Microbe 10:551–562. doi: 10.1016/j.chom.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dustin ML, Chakraborty AK, Shaw AS. 2010. Understanding the structure and function of the immunological synapse. Cold Spring Harb Perspect Biol 2:a002311. doi: 10.1101/cshperspect.a002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Muranyi W, Malkusch S, Müller B, Heilemann M, Kräusslich H-G. 2013. Super-resolution microscopy reveals specific recruitment of HIV-1 envelope proteins to viral assembly sites dependent on the envelope C-terminal tail. PLoS Pathog 9:e1003198. doi: 10.1371/journal.ppat.1003198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wyma DJ, Jiang J, Shi J, Zhou J, Lineberger JE, Miller MD, Aiken C. 2004. Coupling of human immunodeficiency virus type 1 fusion to virion maturation: a novel role of the gp41 cytoplasmic tail. J Virol 78:3429–3435. doi: 10.1128/JVI.78.7.3429-3435.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Murakami T, Ablan S, Freed EO, Tanaka Y. 2004. Regulation of human immunodeficiency virus type 1 Env-mediated membrane fusion by viral protease activity. J Virol 78:1026–1031. doi: 10.1128/JVI.78.2.1026-1031.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Llewellyn GN, Grover JR, Olety B, Ono A. 2013. HIV-1 Gag associates with specific uropod-directed microdomains in a manner dependent on its MA highly basic region. J Virol 87:6441–6454. doi: 10.1128/JVI.00040-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Makino A, Shin HY, Komai Y, Fukuda S, Coughlin M, Sugihara-Seki M, Schmid-Schönbein GW. 2007. Mechanotransduction in leukocyte activation: a review. Biorheology 44:221–249. [PubMed] [Google Scholar]
- 92.Jain RK. 1988. Determinants of tumor blood flow: a review. Cancer Res 48:2641–2658. [PubMed] [Google Scholar]
- 93.Thali M. 2011. Tetraspanin functions during HIV-1 and influenza virus replication. Biochem Soc Trans 39:529–531. doi: 10.1042/BST0390529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gallo RC, Jay G. 2012. The human retroviruses. Elsevier, New York, NY. [Google Scholar]
- 95.Frankel SS, Wenig BM, Burke AP, Mannan P, Thompson LDR, Abbondanzo SL, Nelson AM, Pope M, Steinman RM. 1996. Replication of HIV-1 in dendritic cell-derived syncytia at the mucosal surface of the adenoid. Science 272:115–117. doi: 10.1126/science.272.5258.115. [DOI] [PubMed] [Google Scholar]
- 96.Anderson JM. 2000. Multinucleated giant cells. Curr Opin Hematol 7:40–47. doi: 10.1097/00062752-200001000-00008. [DOI] [PubMed] [Google Scholar]
- 97.Kim JH, Ren Y, Li S, Kee Y, Zhang G, Robinson D, Chen E. 2014. Myosin II functions as a direct mechanosensor for intercellular invasion during cell-cell fusion. Biophys J 106:574a. doi: 10.1016/j.bpj.2013.11.3182. [DOI] [Google Scholar]
- 98.Li F, Sewald X, Jin J, Sherer NM, Mothes W. 2014. Murine leukemia virus Gag localizes to the uropod of migrating primary lymphocytes. J Virol 88:10541–10555. doi: 10.1128/JVI.01104-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gousset K, Ablan SD, Coren LV, Ono A, Soheilian F, Nagashima K, Ott DE, Freed EO. 2008. Real-time visualization of HIV-1 GAG trafficking in infected macrophages. PLoS Pathog 4:e1000015. doi: 10.1371/journal.ppat.1000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.