

ABSTRACT
Influenza viral infections represent a serious public health problem, with influenza virus causing a contagious respiratory disease which is most effectively prevented through vaccination. Segments 7 (M) and 8 (NS) of the influenza virus genome encode mRNA transcripts that are alternatively spliced to express two different viral proteins. This study describes the generation, using reverse genetics, of three different recombinant influenza A/Puerto Rico/8/1934 (PR8) H1N1 viruses containing M or NS viral segments individually or modified M or NS viral segments combined in which the overlapping open reading frames of matrix 1 (M1)/M2 for the modified M segment and the open reading frames of nonstructural protein 1 (NS1)/nuclear export protein (NEP) for the modified NS segment were split by using the porcine teschovirus 1 (PTV-1) 2A autoproteolytic cleavage site. Viruses with an M split segment were impaired in replication at nonpermissive high temperatures, whereas high viral titers could be obtained at permissive low temperatures (33°C). Furthermore, viruses containing the M split segment were highly attenuated in vivo, while they retained their immunogenicity and provided protection against a lethal challenge with wild-type PR8. These results indicate that influenza viruses can be effectively attenuated by the rearrangement of spliced segments and that such attenuated viruses represent an excellent option as safe, immunogenic, and protective live-attenuated vaccines. Moreover, this is the first time in which an influenza virus containing a restructured M segment has been described. Reorganization of the M segment to encode M1 and M2 from two separate, nonoverlapping, independent open reading frames represents a useful tool to independently study mutations in the M1 and M2 viral proteins without affecting the other viral M product.
IMPORTANCE Vaccination represents our best therapeutic option against influenza viral infections. However, the efficacy of current influenza vaccines is suboptimal, and novel approaches are necessary for the prevention of disease caused by this important human respiratory pathogen. In this work, we describe a novel approach to generate safer and more efficient live-attenuated influenza virus vaccines (LAIVs) based on recombinant viruses whose genomes encode nonoverlapping and independent M1/M2 (split M segment [Ms]) or both M1/M2 and NS1/NEP (Ms and split NS segment [NSs]) open reading frames. Viruses containing a modified M segment were highly attenuated in mice but were able to confer, upon a single intranasal immunization, complete protection against a lethal homologous challenge with wild-type virus. Notably, the protection efficacy conferred by our viruses with split M segments was better than that conferred by the current temperature-sensitive LAIV. Altogether, these results open a new avenue for the development of safer and more protective LAIVs on the basis of the reorganization of spliced viral RNA segments in the genome.
INTRODUCTION
Influenza viruses are enveloped pathogens that belong to the Orthomyxoviridae family and contain a segmented genome of eight single-stranded RNA molecules with negative polarity (1). Influenza virus infections cause both seasonal epidemics and occasional pandemics when novel viruses are introduced into humans (2). Despite comprehensive vaccination programs, the World Health Organization (WHO) estimates that the global disease burden from influenza results in 1 billion infections, 3 million to 5 million cases of severe disease, and between 300,000 and 500,000 deaths annually (3). Therefore, infection with influenza virus poses a threat to human health and results in significant negative economic impacts on society every year (4). The public health concerns posed by influenza viruses are aggravated by their efficient transmission and the limited antiviral therapeutic options (5). Hence, vaccination remains our best medical intervention to protect humans against influenza virus (6), even though the effectiveness of current vaccines is suboptimal (7). To date, the U.S. Food and Drug Administration (FDA) approves three types of influenza virus vaccines for human use: inactivated virus, recombinant viral hemagglutinin (HA) protein, and live-attenuated virus vaccines (8, 9). The most widely used influenza vaccine is the inactivated influenza virus vaccine (IIV), which elicits protective humoral immunity by inducing the production of neutralizing antibodies that target epitopes on the viral HA protein and to a lesser extent those on the neuraminidase (NA) protein. The recombinant influenza virus vaccine (RIV), like IIV, is administered intramuscularly and elicits a protective antibody HA-neutralizing response (10). However, these vaccines do not induce a strong cellular response, which is necessary to generate memory against subsequent infections and to protect against heterosubtypic influenza virus infections (8, 9). The remaining option is the live-attenuated influenza virus vaccine (LAIV), which induces better cross-reactive, cell-mediated protection against heterotypic influenza virus infections (11, 12). However, LAIV is recommended only for immunocompetent 2- to 49-year-old persons (13). Moreover, the attenuated phenotype of the virus used in LAIV is conferred by just five point mutations, located in PB2 (N265S), PB1 (K391E, E581G, A661T), and NP (34G) (14–16), that make the virus temperature sensitive (ts). The concern is that reversion of any or a combination of the five mutations could result in a replication-competent and potentially pathogenic virus. Thus, new vaccination strategies that overcome the limitations associated with current influenza vaccination approaches are required for the prevention of viral infections in humans.
At least four of the eight segments of the influenza A virus genome encode more than one polypeptide using alternative splicing mechanisms (M and NS segments) (17, 18), leaky ribosomal scanning (PB1 segment) (19), or ribosomal frame shifting (PA segment) (20). Influenza A virus genome segment 8 encodes the NS mRNA as a continuous primary transcript. Standard processing of this NS mRNA generates nonstructural protein 1 (NS1), whereas alternative processing using a weak 5′ splice site results in a second, less abundant splice product encoding the nuclear export protein (NEP) (21), which accounts for 10 to 15% of the NS-derived mRNA (22). Although both polypeptides are ultimately translated from different open reading frames (ORFs), they still share the first 10 N-terminal amino acids (21). Influenza A virus genome segment 7 (M) uses a similar strategy to produce at least two viral proteins, the primary transcript matrix 1 (M1) protein and the alternatively spliced matrix 2 (M2) protein (18). As with the NS segment, both M1 and M2 share the first 9 N-terminal amino acids and are necessary for the production of replication-competent influenza viruses (23–25).
In the present work, we have engineered the M and NS segments of the influenza A/Puerto Rico/8/1934 (PR8) H1N1 virus genome to encode nonoverlapping independent M1/M2 (split M segment [Ms]), independent NS1/NEP (split NS segment [NSs]), or both independent M1/M2 and independent NS1/NEP (Ms/NSs) ORFs. In vitro characterization of the viruses carrying recombinant Ms and Ms/NSs (referred to here as Ms and Ms/NSs viruses, respectively) showed that they exhibited a ts phenotype. However, recombinant Ms viruses grew at high titers in tissue culture cells at a permissive temperature (33°C). Importantly, the modified Ms or Ms/NSs influenza virus was highly attenuated in a mouse model of infection and able to confer, upon a single intranasal immunization, full protection against a lethal challenge with wild-type (WT) PR8 virus. Notably, mice immunized with the modified Ms or Ms/NSs virus were better protected against subsequent WT PR8 virus lethal challenge than mice immunized with our previously described PR8 ts LAIV (14). In addition, the ability to encode NS1 and NEP (NS segment) and M1 and M2 (M segment) proteins as separate, nonoverlapping transcripts represents an excellent option to independently study mutations in these viral proteins without affecting the viral products encoded by other segments of the viral genome.
MATERIALS AND METHODS
Cell and viruses.
Human embryonic kidney 293T (293T) and Madin-Darby canine kidney (MDCK) cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin (100 units/ml)–streptomycin (100 μg/ml)–2 mM l-glutamine (P-S-G) at 37°C in air enriched with 5% CO2. Influenza A/Puerto Rico/8/1934 (PR8) H1N1 virus (26) was grown in MDCK cells as previously described (27, 28). Viral titers were determined by an immunofocus assay and are presented as fluorescence-forming units (FFU) per milliliter (27, 28).
Plasmids.
To engineer the recombinant PR8 virus NS and M split segments (NSs and Ms, respectively), overlapping PCR and standard molecular biology techniques were used to introduce the modifications into the ambisense pDZ-NS or pDZ-M viral rescue plasmid (26). The modified plasmids, named pDZ-NSs and pDZ-Ms, respectively, contain the NS1 (NS segment) or M1 (M segment) open reading frames (ORFs) without stop codons or splice acceptor sites, followed by the porcine teschovirus 1 (PTV-1) 2A autoproteolytic cleavage site (ATNFSLLKQAGDVEENPGP) and the entire sequence of the NEP (NS segment) or M2 (M segment) ORF (27, 29). The sequences of the modified pDZ-NSs and -Ms plasmids were confirmed by sequencing, and the plasmids were used for virus rescue.
Rescue of recombinant PR8 viruses.
Virus rescues were performed as previously described (27, 28, 30). Briefly, cocultures of 293T and MDCK cells (in a 6-well plate format) were cotransfected in suspension, using the Lipofectamine 2000 reagent, with 1 μg of each of the ambisense plasmids. At 12 h posttransfection, the transfection medium was replaced with DMEM containing 0.3% bovine serum albumin (BSA), 1% P-S-G, and 0.5 μg/ml of tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin. At 48 h posttransfection, tissue culture supernatants were collected, clarified, and used to infect fresh monolayers of MDCK cells (6-well plate format). At 3 to 4 days postinfection, recombinant viruses were plaque purified in 6-well plates and scaled up in MDCK cells (28). Stocks were titrated by the immunofocus assay on MDCK cells (27, 28). Viral rescues and titrations were performed and stocks were produced at 33°C.
Reverse transcription-PCR (RT-PCR).
Total RNA from infected MDCK cells (6-well plate format) was purified using the TRIzol reagent (Invitrogen) according to the manufacturer's specifications. The cDNAs were synthesized with SuperScript II reverse transcriptase, using 1 μg of total RNA as the template and primers specific for the M, NS, and NP viral RNAs. The cDNAs were used as the template to amplify the specific viral RNA segments (M, NS, and NP) using specific primers.
Protein gel electrophoresis and Western blot analysis.
Total proteins from infected MDCK cell lysates (6-well plate format) were separated using 10% or 12% SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Membranes were blocked for 1 h with 5% dried skim milk in phosphate-buffered saline (PBS) containing 0.1% Tween 20 (T-PBS) and incubated overnight at 4°C with the following specific primary monoclonal antibodies (MAbs) or polyclonal antibodies (pAbs): NS1 (MAb 1A7), NEP (pAb a01499; GenScript), M1 (MAb ab22396; Abcam), M2 (MAb ab5416; Abcam), and NP (MAb HB-65). A MAb against actin (MAb A1978; Sigma) was used as an internal loading control. Bound primary antibodies were detected with horseradish peroxidase (HRP)-conjugated antibodies against immunoglobulins of different species (mouse or rabbit). Proteins were detected by quimioluminescence (reagent was from ThermoFisher Scientific) following the manufacturer's recommendations and photographed using a Kodak Image Station. The bands in the Western blots were quantified by densitometry using the software ImageJ (v.1.46). The quantities for the protein bands were normalized to the level of cellular actin. Protein expression in WT-infected cells was considered 100% for comparison with the levels of expression by viruses carrying split mutant sequences.
Virus growth kinetics.
Triplicate wells of confluent MDCK cells (12-well plate format) were infected at a multiplicity of infection (MOI) of 0.001. After 1 h of virus adsorption at room temperature, the cells were washed, overlaid with DMEM containing 0.3% BSA and TPCK-treated trypsin, and incubated at 33°C, 37°C, or 39°C. At the indicated times postinfection, tissue culture supernatants were collected and viral titers were determined by the immunofocus assay (27, 28). The mean value and standard deviation (SD) were calculated using Microsoft Excel software.
Plaque assay and immunostaining.
Confluent monolayers of MDCK cells (6-well plate format) were infected for 1 h at room temperature, overlaid with agar, and incubated at 33°C, 37°C, or 39°C. At 3 days postinfection, the cells were fixed overnight with 4% paraformaldehyde (PFA) and the overlays were removed. Cells were then permeabilized (0.5% Triton X-100 in PBS) for 15 min at room temperature and prepared for immunostaining using NP MAb HB-65 and vector kits (Vectastain ABC vector kits and DAB HRP substrate kit; Vector) according to the manufacturer's specifications (27, 29).
Mouse experiments.
Six- to 8-week-old female C57BL/6 mice were purchased from the National Cancer Institute (NCI) and maintained in the animal care facility at the University of Rochester under specific-pathogen-free conditions. All animal protocols were approved by the University of Rochester Committee of Animal Resources and complied with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Research Council (31). Mice (n = 6) were anesthetized intraperitoneally with tribromoethanol (Avertin) and then inoculated intranasally with 30 μl of a virus preparation containing the number of FFU of the WT, NSs, Ms, or Ms/NSs virus indicated below and monitored daily for body weight loss and mortality. Mice showing a 25% loss of their initial body weight were considered to have reached the experimental endpoint and were humanely euthanized. Virus replication was evaluated by determination of viral titers in the lungs at day 2 postinfection (28). To that end, three mice in each group were sacrificed and their lungs were extracted and homogenized. Virus titers were determined by the immunofocus assay (27, 28). Geometric mean titers (GMTs) and statistical analyses (Mann-Whitney test) were performed using GraphPad Prism software. The 50% mouse lethal dose (MLD50) of WT or recombinant viruses was determined using the method of Reed and Muench (32).
For the vaccination and challenge experiments, 6- to 8-week-old female C57BL/6 mice (n = 6) were inoculated intranasally with PBS or the number of FFU of Ms virus, Ms/NSs virus, or PR8 ts LAIV indicated below. At 2 weeks after priming, the mice were challenged intranasally with 1,000 MLD50s of WT PR8 virus. After challenge, WT PR8 viral replication in the mouse lungs was evaluated at days 2 and 4 postinfection, as described above. Mouse sera were collected by submandibular bleeding 24 h prior to WT PR8 viral challenges and evaluated for the presence of influenza virus antibodies by an enzyme-linked immunosorbent assay (ELISA).
ELISA.
To assess the levels of virus-specific antibodies present in immunized mice, ELISAs were performed as previously described (28). Briefly, 96-well plates were coated with lysates from mock- or WT PR8 virus-infected MDCK cells or with PR8 virus HA (200 ng per well; catalog number NR-19240; BEI Resources) or NP (200 ng per well) recombinant proteins. After washing with PBS, coated wells were blocked with PBS containing 1% BSA overnight and then incubated with 1:2 dilutions (starting dilution, 1:100) of mouse serum at 37°C. After 1 h of incubation, the wells of the plates were washed with H2O and incubated with HRP-conjugated goat anti-mouse IgG for 30 min at 37°C. The reactions were developed with tetramethylbenzidine (TMB) substrate for 10 min at room temperature, quenched with 2 N H2SO4, and read at 450 nm.
HAI assays.
Hemagglutination inhibition (HAI) assays were used to assess the presence of neutralizing antibodies (28). To that end, mouse sera were treated with receptor-destroying enzyme (RDE) and heat inactivated for 30 min at 56°C. The sera were then serially 2-fold diluted (starting dilution, 1:16) in 96-well V-bottom plates and mixed 1:1 with 4 hemagglutinating units (HAU) of WT PR8 virus for 60 min at room temperature. The HAI titers were determined by adding 0.5% turkey red blood cells (RBCs) to the virus-antibody mixtures for 30 min on ice (28).
Antiviral assay.
Antiviral-mediated inhibition of WT and recombinant PR8 viruses was evaluated as previously described (27, 29). Briefly, confluent MDCK cells (24-well plate format, in triplicate) were infected (MOI, 0.001) for 1 h and incubated with infectious medium supplemented with 3-fold serial dilutions of oseltamivir (starting concentration, 10 μM). At 48 h postinfection, the viral titers in tissue culture supernatants were determined by the immunofocus assay (28). The viral titers in virus-infected cells in the absence of drug were used to calculate 100% viral infection. The 50% inhibitory concentration (IC50) was determined by use of a sigmoidal dose-response curve (GraphPad Prism software, v.4.0).
RESULTS
Generation of recombinant PR8 viruses encoding split M1/M2 and NS1/NEP genes.
The regions of the influenza virus genome encoding the NS1 and NEP products in segment 8 (NS) and the M1 and M2 products in segment 7 (M) partially overlap (1), making it difficult to introduce mutations in the overlapping regions in one viral product without affecting the protein sequences in the other viral product. To solve this problem and introduce changes into only one viral protein at a time, we modified the NS and M segments of the viral genome to encode proteins from a single nonoverlapping transcript (Fig. 1). To that end, the overlapping regions in both segments of the viral genome were duplicated and the porcine teschovirus 1 (PTV-1) 2A autoproteolytic cleavage site (33) was inserted between the NS1 and NEP genes (segment 8) or between the M1 and M2 genes (segment 7). This strategy ensured that the viral proteins (the NS1 and NEP proteins and the M1 and M2 proteins, respectively) would be translated as two separate, nonoverlapping, independent ORFs, similar to the strategy that we have previously described for the NS segment (27, 29, 34).
FIG 1.

Schematic representation of WT PR8 H1N1 virus, Ms virus, and NSs virus. Viruses containing wild-type segments (A), Ms (B), NSs (C), and both Ms and NSs (D) are indicated. Black boxes at the end of each viral segment, viral 3′ and 5′ noncoding regions; white boxes, PR8 viral products from the NS (NS1 and NEP) and M (M1 and M2) segments; light gray boxes, the region until the splicing donor in both viral segments (NS and M); dark gray boxes, sequence of the PTV-1 2A autoproteolytic cleavage site. In the virus illustrations, the WT and split NS and M viral segments are indicated with gray and black lines, respectively.
To analyze whether recombinant influenza viruses harboring the modified M and/or NS segments of the viral genome could be rescued and to assess the effect of this new segment organization in the context of viral replication, the modified M and NS segments of the viral genome were cloned into ambisense reverse genetics plasmids and used for viral rescues (35). Three different viruses containing the split M segment (Ms), split NS segment (NSs), or both split segments (Ms/NSs) were rescued (Fig. 1). This is, to our knowledge, the first time that an influenza virus with an Ms has been described.
Characterization of recombinant PR8 viruses with split segments.
The identities of the recombinant Ms, NSs, and Ms/NSs viruses were confirmed by RT-PCR and Western blotting (Fig. 2). By RT-PCR, cDNA products of 895 nucleotides (nt) and 1,150 nt were amplified from the NS viral RNA using parental and NSs viruses, respectively (Fig. 2A). Likewise, DNA band sizes of 1,027 and 1,150 nt were amplified from the M viral RNA using WT and Ms viruses, respectively (Fig. 2A), confirming the identity of the rescued viruses. As expected, a consistent band of 1,565 nt was amplified from the NP viral RNA of all recombinant viruses. To characterize the recombinant Ms, NSs, and Ms/NSs viruses at the protein level, we performed Western blot analyses using antibodies specific for the viral products M1, M2, NS1, NEP, and NP (Fig. 2B). The molecular masses of the modified M1 and NS1 viral proteins were greater for Ms and Ms/NSs virus-infected cells and for NSs and Ms/NSs virus-infected cells, respectively, than for the parental virus-infected cells. This result is consistent with the expression of these proteins fused to the 2A autoproteolytic cleavage site (Fig. 2B). The amounts of processed M1 and M2 proteins were reduced (∼1.5- to 2-fold) in the infections with the Ms and Ms/NSs viruses compared to their amounts in the infections with the parental PR8 virus. Similarly, the amounts of processed NS1 and NEP were lower (∼1.5- to 2-fold) in cells infected with the NSs and Ms/NSs viruses than in cells infected with WT PR8 virus (Fig. 2B), suggesting that M1/M2 and NS1/NEP processing in the viruses with split segments was not 100% efficient.
FIG 2.

Characterization of the influenza PR8 viruses harboring split sequences. MDCK cells were infected (MOI, 3) with WT PR8 virus or PR8 viruses harboring split segments (NSs, Ms, and Ms/NSs), and at 18 h postinfection, cells were harvested to evaluate RNA (A) or protein (B) expression levels. (A) cDNA synthesis and PCRs for NS, M, or NP viral segments were performed using specific primers. (B) Protein expression levels for M1, M2, NS1, NEP, and NP were evaluated using protein-specific antibodies. Actin was used as a loading control. Numbers to the left of the gels indicate the sizes (in nucleotides) of cDNA (A) and the sizes (in kilodaltons) of proteins (B). A schematic representation of the viral segments (A) or protein products (B) is provided on the right of each panel. Western blots were quantified by densitometry using the software ImageJ (v.1.46). Protein expression in WT-infected cells was considered 100% for comparison with the level of expression by the corresponding viruses harboring split segments (indicated by the numbers [in percent] below each blot in panel B).
We next evaluated virus replication by analyzing viral titers and plaque formation in multicycle infections at different temperatures (Fig. 3). The NSs virus grew at slightly lower titers (∼5- to 10-fold) than WT PR8 virus at the three temperatures tested and at all times except 12 h postinfection, at which the differences were greater (Fig. 3A). On the other hand, we observed a ∼100-fold decrease in viral titers for the Ms and Ms/NSs viruses at 33°C at 48 to 96 h postinfection, with no virus being detected at earlier times postinfection (Fig. 3A). Importantly, the Ms and Ms/NSs viruses reached titers close to 107 fluorescence-forming units (FFU)/ml at the peak of infection (72 h). Notably, replication of the Ms and Ms/NSs viruses was significantly impaired at 37°C and was impaired to a greater extent at 39°C. Differences in the titers between the Ms and Ms/NSs viruses and the parental PR8 virus without split sequences reached at least 10,000-fold at 37°C and 100,000-fold at 39°C at the peak of infection (48 h) (Fig. 3A). The ts phenotype of the recombinant modified Ms and Ms/NSs viruses was further confirmed by plaque assay (Fig. 3B). The plaque phenotype of the NSs virus was only slightly smaller than that of the parental PR8 virus at the three temperatures tested (Fig. 3B). As expected, the lysis plaques produced by the Ms and Ms/NSs viruses at 33°C were smaller than those produced by the WT PR8 virus (Fig. 3B). However, these differences were more pronounced at restricted temperatures (37°C and 39°C), where point plaques (37°C) or no plaques (39°C) were observed (Fig. 3B). Altogether, these data indicate that the NSs virus and, to a greater extent, the Ms and Ms/NSs viruses are attenuated in growth in vitro compared to the growth of WT PR8 virus. Importantly, our data also support a temperature-restricted phenotype for the viruses containing the split M segment (Ms and Ms/NSs viruses), suggesting that the modifications introduced in Ms result in viruses with a ts phenotype.
FIG 3.

Viral growth kinetics and plaque morphology. (A) Multicycle growth kinetics. MDCK cells were infected (MOI, 0.001) with the WT, NSs, Ms, or Ms/NSs PR8 virus, and the viral titers in the tissue culture supernatants at the indicated times postinfection (12, 24, 48, 72 and 96 h) were evaluated by the immunofocus assay and are given as the number of FFU per milliliter. The data represent the means and SDs of the results determined for triplicate wells. *, P < 0.05 (WT versus NSs virus, WT versus Ms virus, or WT versus Ms/NSs virus) using Student's t test (n = 3 determinations per time point) from Microsoft Excel software. Dashed lines, the limit of detection (200 FFU/ml). (B) Plaque phenotype. MDCK cells were infected with WT, NSs, Ms, and Ms/NSs PR8 viruses, and viral plaques were assessed at 3 days postinfection using immunostaining with the anti-NP monoclonal antibody HB-65. Schematic representations of the viral segments are provided at the bottom.
Virulence of NSs, Ms, and Ms/NSs PR8 viruses in vivo.
As the PR8 viruses with split segments were attenuated in growth in vitro (Fig. 3), we sought to test whether these viruses were also attenuated in growth in vivo (Fig. 4), as attenuated growth in vivo would support the potential implementation of influenza virus with rearranged spliced segments as live-attenuated vaccine candidates. To that end, groups of mice (n = 6) were inoculated intranasally with different doses of the NSs virus (10, 102, and 103 FFU) or the Ms or Ms/NSs virus (103, 104, and 105 FFU) or with the WT PR8 virus (10, 102, and 103 FFU) for comparison. The mice were monitored for 14 days for signs of morbidity (weight loss) and mortality (survival) (Fig. 4). As expected, the recombinant viruses with split segments showed levels of attenuation and pathogenicity different from those for WT PR8 virus, and the increased morbidity and mortality correlated with the increased virus dose. All mice infected with 103 and 102 FFU of WT PR8 virus rapidly lost weight, and none survived by day 6 or 8 postinfection, respectively, while 66.6% of mice survived after infection with 10 FFU (Fig. 4A). In spite of the limited attenuation observed in vitro (∼5- to 10-fold that of WT PR8 virus; Fig. 3), mice infected with the NSs virus showed less weight loss and mortality than mice infected with the same doses of WT PR8 virus (Fig. 4B). All mice infected with 10 FFU of NSs virus survived the viral infection, 50% of the mice infected with 102 FFU of NSs virus survived the viral infection, and no mice infected with 103 FFU of NSs virus survived the viral infection by day 8 (Fig. 4B). In contrast, the Ms or Ms/NSs virus showed higher levels of attenuation when they were inoculated into the mice (Fig. 4C and D, respectively). Only mice inoculated with 105 FFU of the Ms virus lost weight and succumbed (83.3%) to viral infection (Fig. 4C). Remarkably, none of the mice infected with 104 or 103 FFU of the Ms virus (Fig. 4C) and with up to 105 FFU of the Ms/NSs virus (Fig. 4D) showed weight loss or mortality. From these experiments, the MLD50 (32) for each PR8 virus was ∼17 FFU for WT virus (28), ∼100 FFU for NSs virus, ∼40,000 FFU for Ms virus, and greater than 105 FFU for Ms/NSs virus (Table 1). These data demonstrate that a recombinant virus with a modified split NS segment (NSs) is slightly attenuated (MLD50, ∼5- to 10-fold that of WT PR8 virus) in mice. However, a similar modification in the viral M segment results in a significantly higher level of attenuation (MLD50, ∼2,350-fold that of WT PR8 virus) in vivo. Notably, modification of both the viral M and NS segments resulted in additive attenuation (MLD50, >5,880-fold that of WT PR8 virus) compared to the level of attenuation found for viruses in which a single segment was modified. Importantly, while the MLD50 of the virus containing Ms was similar to that of our recently described PR8 ts LAIV (∼3 × 104 FFU) (14), the MLD50 of the Ms/NSs virus was at least 3-fold higher (>105 FFU) than that of PR8 ts LAIV (Table 1).
FIG 4.

Attenuation of viruses harboring split segments. Six- to 8-week-old female C57BL/6 mice (n = 6) were infected intranasally with the indicated number of FFU of WT (A), NSs (B), Ms (C), and Ms/NSs (D) PR8 viruses and then monitored daily for 2 weeks for body weight loss (left) and survival (right). Mice that lost 25% of their initial body weight were sacrificed. Data represent the means and SDs of the results determined for individual mice (n = 6).
TABLE 1.
MLD50s of PR8 viruses carrying split segments
| Virusa | MLD50 (no. of FFU/mouse) |
|---|---|
| WT | 1.7 × 101 |
| NSs | 10 × 101 |
| Ms | 3.98 × 104 |
| Ms/NSs | >1 × 105 |
| LAIVb | 3.16 × 104 |
Mortality was determined over 2 weeks (n = 6).
The MLD50 of PR8 ts LAIV was previously calculated (14).
Induction of humoral responses in mice infected with PR8 viruses carrying split segments.
Given that the Ms and Ms/NSs viruses were highly attenuated in mice (Fig. 4), we next evaluated the potential of these viruses as vaccine candidates. To this end, groups of mice (n = 3) were mock infected (with PBS) or infected with 0.01 and 0.1 MLD50 of the Ms virus (equal to 400 and 4,000 FFU, respectively), with 10,000 and 100,000 FFU of the Ms/NSs virus (considered 0.01 and 0.1 MLD50, respectively), or with 0.01 and 0.1 MLD50 of our previously described PR8 ts LAIV (equal to 300 and 3,000 FFU, respectively). The humoral immune responses in sera collected 2 weeks after infection were evaluated by ELISA (Fig. 5), using cell extracts from PR8 virus-infected MDCK cells (Fig. 5A) or PR8 virus purified HA (Fig. 5B) or NP (Fig. 5C). Antibodies specific to total viral proteins, viral HA, and viral NP were detected in all infected mice, with the amount of antibodies, as expected, being slightly higher in mice infected with the highest dose of virus (0.1 MLD50). Interestingly, for mice infected with the same MLD50, ELISA titers of antibodies specific for the whole influenza virus-infected cell extracts and NP protein (Fig. 5A and C, respectively) were statistically significantly higher in mice infected with the Ms and Ms/NSs viruses than in those infected with PR8 ts LAIV. These findings suggest that the more attenuated Ms and Ms/NSs viruses induced a better humoral response than PR8 ts LAIV. However, no differences in the titers of antibodies against PR8 HA were observed among the mice infected with the different viruses (Fig. 5B). Additionally, we performed hemagglutination inhibition (HAI) assays to examine the neutralizing activity of the sera from the immunized mice (Table 2). As expected, the HAI titers against WT PR8 virus were 4- to 8-fold higher in animals infected with 0.1 MLD50 than in mice infected with the lower dose of 0.01 MLD50. Serum HAI titers in mice infected with the Ms/NSs virus were 2- and 4-fold lower (MLD50s, 0.01 and 0.1, respectively) than those in mice infected with PR8 ts LAIV. On the other hand, serum HAI titers in mice infected with the Ms virus were the same as or 2-fold less than (MLD50s, 0.01 and 0.1, respectively) those in mice infected with PR8 ts LAIV. Altogether, these data demonstrate that vaccination with the more attenuated Ms or Ms/NSs virus induces humoral neutralizing immune responses similar to those obtained with PR8 ts LAIV.
FIG 5.

Humoral responses to Ms and Ms/NSs virus vaccination. Six- to 8-week-old female C57BL/6 mice (n = 6) were vaccinated intranasally with PBS or the indicated viral MLD50 of the Ms or Ms/NSs PR8 virus or PR8 ts LAIV. Black symbols, 0.1 MLD50; gray symbols, 0.01 MLD50. At 14 days postvaccination, mice were bled and sera were collected, pooled, and evaluated by ELISA for IgG antibodies against total influenza virus proteins using cell extracts of PR8 virus-infected MDCK cells (A) or against recombinant PR8 HA (B) or NP (C) viral proteins. OD, optical density. Since the MLD50 of the Ms/NSs virus was higher than 105 FFU, 105 FFU was defined to be 0.1 MLD50. Likewise, 104 FFU was defined to be 0.01 MLD50 for the Ms/NSs PR8 virus. *, P < 0.05 (when the differences both between the Ms virus and PR8 ts LAIV and between the Ms/NSs virus and PR8 ts LAIV were significant) using Student's t test from Microsoft Excel software.
TABLE 2.
Immunogenicity of PR8 viruses carrying split segments
| Immunization (dose [MLD50])a | Mean HAI titer |
|---|---|
| PBS | ≤16 |
| Ms virus (0.01) | 64 |
| Ms virus (0.1) | 256 |
| Ms/NSs virus (0.01)b | 32 |
| Ms/NSs virus (0.1)b | 128 |
| LAIV (0.01) | 64 |
| LAIV (0.1) | 512 |
Data were calculated for immunized or mock-immunized (n = 6) mice.
Since the MLD50 of the Ms/NSs virus was higher than 105 FFU (Table 1), the mice were vaccinated with 104 FFU (0.01 MLD50) and 105 FFU (0.1 MLD50).
Protection induced by recombinant PR8 viruses with split segments after virus challenge.
To analyze the efficacy of the immune response elicited by vaccination with our modified Ms or Ms/NSs virus against challenge with WT PR8 virus, groups of mice (n = 3) were mock vaccinated (with PBS) or vaccinated with 0.01 and 0.1 MLD50 of the Ms or Ms/NSs virus and PR8 ts LAIV for comparison. Then, at 2 weeks after vaccination, mice were challenged with a lethal dose of 1,000 MLD50s (∼10,000 FFU) of WT PR8 virus and viral titers in mouse lungs, collected at days 2 and 4 after challenge, were evaluated by the immunofocus assay (Fig. 6). As expected, we detected viral titers of ∼5 × 106 FFU/ml in mock-vaccinated mice at both days postchallenge. Compared to the titers in mock-vaccinated mice, animals immunized with 0.01 MLD50 of PR8 ts LAIV had similar viral titers (∼106 FFU/ml) at day 2 postchallenge but reduced viral titers (∼1,000-fold) at day 4 postchallenge. After vaccination with the higher dose of PR8 ts LAIV (0.1 MLD50), we were able to detect WT PR8 virus (3 × 102 FFU/ml) in only 1 of the 3 mice at day 2 postchallenge, whereas no virus was detected at day 4 postchallenge. Remarkably, after vaccination with 0.01 or 0.1 MLD50 of the Ms virus, we were not able to detect WT PR8 virus at day 2 or 4 postchallenge. Likewise, we were not able to detect the challenge WT PR8 virus in mice vaccinated with 0.1 MLD50 of the Ms/NSs virus. Notably, we were able to detect WT PR8 virus in 2 out of 3 mice vaccinated with 0.01 MLD50 of the Ms/NSs virus only at day 2 postinfection, with no WT PR8 virus being detectable at day 4 postchallenge (Fig. 6). Altogether, these data indicate that vaccination with the Ms or Ms/NSs virus induces a strong immune response that prevents the replication of WT PR8 virus and that this protection is greater than that observed with PR8 ts LAIV when comparable MLD50s are used.
FIG 6.
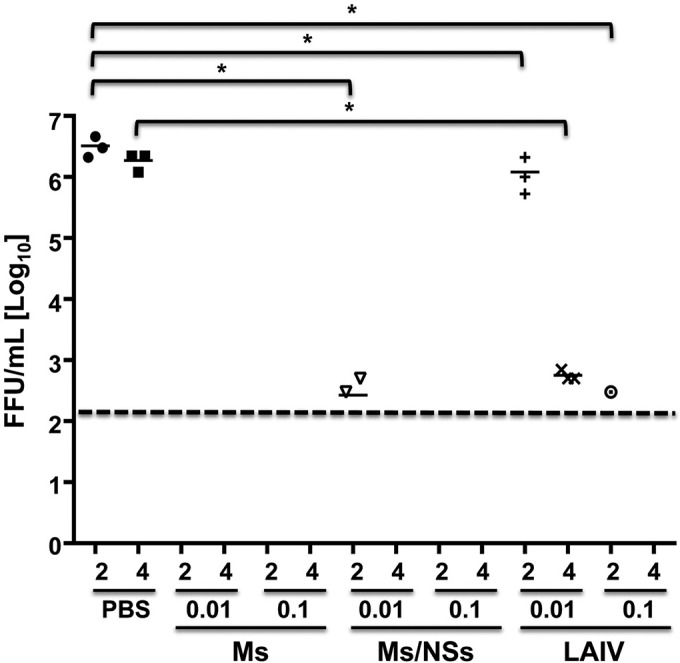
Protection efficacy of Ms, NSs, and M/NSs PR8 viruses in mice. Six- to 8-week-old female C57BL/6 mice (n = 6) were mock vaccinated (with PBS) or vaccinated intranasally with the indicated MLD50 (0.01 or 0.1) of the Ms or Ms/NSs PR8 virus or PR8 ts LAIV. At 2 weeks postvaccination, mice were challenged with 1,000 MLD50 of WT PR8 virus. The replication of WT PR8 virus in the lungs of challenged mice was evaluated at days 2 and 4 postinfection using the immunofocus assay, and titers are given as the number of FFU per milliliter. Symbols represent data for individual mice (n = 3). Bars, geometric mean lung viral titers; dashed line, the limit of detection (200 FFU/ml). Since the MLD50 of the Ms/NSs virus was higher than 105 FFU, 105 FFU was defined to be 0.1 MLD50. Likewise, 104 FFU was defined to be 0.01 MLD50 for the Ms/NSs PR8 virus. *, P < 0.05 using Student's t test from Microsoft Excel software. For the mice vaccinated with 0.01 MLD50 of the Ms/NSs virus, we detected the presence of WT PR8 virus at day 2 postchallenge in only two out of the three mice. Likewise, we detected WT PR8 virus in only one out of the three mice vaccinated with 0.1 MLD50 of PR8 ts LAIV at day 2 postchallenge.
Growth of Ms and Ms/NSs PR8 viruses in vivo and in cell culture.
To further analyze whether the Ms and Ms/NSs viruses replicate in infected mouse lungs, groups of mice (n = 3) were inoculated intranasally with 0.01 and 0.1 MLD50 of the Ms virus (equal to 400 and 4,000 FFU, respectively), with 0.01 and 0.1 MLD50 of the Ms/NSs virus (equal to 10,000 and 100,000 FFU, respectively), or with 0.01 and 0.1 MLD50 of PR8 ts LAIV (equal to 300 and 3,000 FFU, respectively), and the viral titers in the lungs of infected mice were calculated at day 2 postinfection (Fig. 7A). As expected, the viral titers in all cases were greater in mice infected with the higher dose of 0.1 MLD50 than in those infected with the lower dose of 0.01 MLD50 (Fig. 7A). Interestingly, virus replication in the lungs was limited (less than 104 FFU/ml) after infection with all the viruses, correlating with the very low morbidity and mortality observed in infected mice (Fig. 4). However, the viral titers in mice infected with both 0.1 and 0.01 MLD50 were slightly higher in mice infected with the Ms/NSs virus and, more markedly, in mice infected with the Ms virus than in mice infected with PR8 ts LAIV (Fig. 7A). These data suggest that the Ms PR8 viruses replicate better than PR8 ts LAIV in the mouse model used in these studies.
FIG 7.
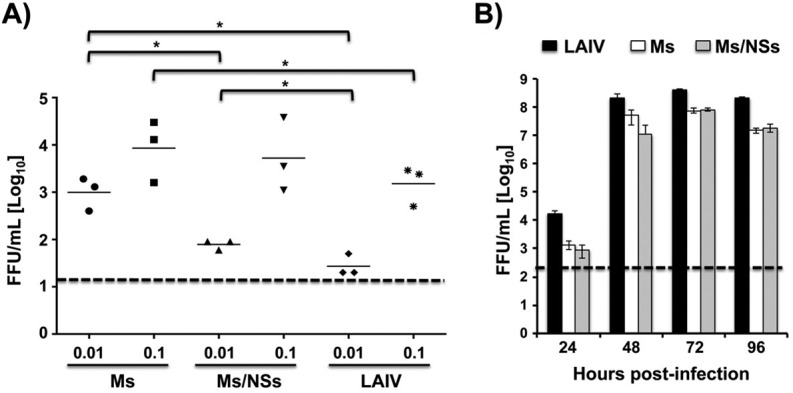
Replication of PR8 Ms and M/NSs viruses in vivo and in cell culture. (A) Viral replication in mice. Six- to-8-week-old female C57BL/6 mice were infected intranasally with the indicated MLD50 (0.01 or 0.1) of the Ms or Ms/NSs PR8 virus or with PR8 ts LAIV as an internal control. Viral replication in the lungs of infected mice was evaluated at day 2 postinfection using the immunofocus assay, and titers are given as the number of FFU per milliliter. Symbols represent data from individual mice (n = 3). Bars, geometric mean lung viral titers; dashed line, the limit of detection (20 FFU/ml). Since the MLD50 of Ms/NSs was higher than 105 FFU, 105 FFU was defined to be 0.1 MLD50. Likewise, 104 FFU was defined to be 0.01 MLD50 for the Ms/NSs PR8 virus. *, P < 0.05 using Student's t test from Microsoft Excel software. (B) Viral replication in vitro. MDCK cells were infected (MOI, 0.001) with the Ms or Ms/NSs virus or PR8 ts LAIV, and the viral titers in the tissue culture supernatants were evaluated at the indicated times postinfection (24, 48, 72, and 96 h) by the immunofocus assay and are given as the number of FFU per milliliter. Data represent the means and SDs of the results determined for triplicate wells. Dashed line, the limit of detection (200 FFU/ml).
To compare the growth kinetics of the Ms and Ms/NSs viruses to those of PR8 ts LAIV in cell culture, MDCK cells were infected (MOI, 0.001) at the permissive temperature (33°C) and the viral titers in the cell culture supernatants were determined at 24, 48, 72, and 96 hpi (Fig. 7B). Maximum viral titers, reached at 72 hpi, were approximately 6-fold lower in cells infected with the Ms and Ms/NSs viruses than in those infected with PR8 ts LAIV (Fig. 7B). These results indicate that, contrary to the situation observed in vivo, the Ms and Ms/NSs viruses grow efficiently in vitro (107 to 108 FFU/ml), demonstrating the feasibility of their use for vaccine production.
Effect of oseltamivir on Ms and Ms/NSs PR8 viral growth.
Despite the extensive efforts invested to control influenza virus infections, only two classes of antivirals are currently clinically available, namely, M2 inhibitors (e.g., amantadine and rimantadine) and NA inhibitors (e.g., zanamivir and oseltamivir). To evaluate the antiviral activity of the FDA-approved agent oseltamivir against our live-attenuated Ms and Ms/NSs viruses, infected MDCK cells were incubated with 3-fold dilutions (starting concentration, 10 μM) of oseltamivir, and the viral titers in tissue culture supernatants were evaluated at 48 h postinfection (Fig. 8A). As internal controls, we used the parental virus, NSs virus, and PR8 ts LAIV. Compared to the growth of virus in the untreated cultures, the lowest drug concentration (0.12 μM) inhibited the growth of the Ms and Ms/NSs viruses and also PR8 ts LAIV by more than 95%, whereas it had no significant effect on NSs and parental PR8 virus growth (Fig. 8A). To determine the active IC50 for all the viruses, lower concentrations (up to 0.0005 μM) of oseltamivir were tested for the more sensitive Ms and Ms/NSs viruses and PR8 ts LAIV (Fig. 8B). The oseltamivir concentration of 0.0015 μM had no effect on Ms virus and PR8 ts LAIV growth, whereas it inhibited (∼80%) the Ms/NSs virus (Fig. 8B). A similar ∼80% inhibition of growth of the Ms virus and PR8 ts LAIV was obtained with a concentration of 0.013 μM oseltamivir (Fig. 8B). The oseltamivir IC50s for the WT, NSs, Ms, and Ms/NSs viruses and PR8 ts LAIV (calculated using sigmoidal dose-response curves) were 0.82 μM, 0.35 μM, 0.0083 μM, 0.00024 μM, and 0.0056 μM, respectively. These results indicate that the Ms and Ms/NSs viruses, as well as PR8 ts LAIV, showed much higher sensitivity than the PR8 WT or NSs virus to inhibition by oseltamivir (27) and that viral replication in people vaccinated with these live-attenuated candidates could easily be controlled in the unlikely event that these viruses induce pathology.
FIG 8.
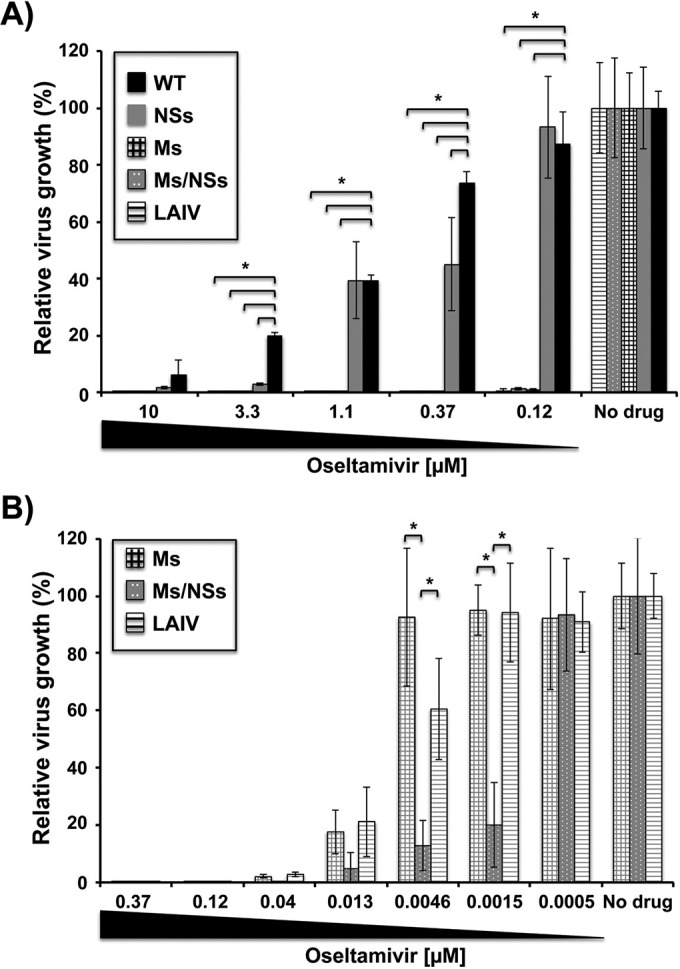
Inhibition of viral replication by oseltamivir treatment. MDCK cells were infected (MOI, 0.001) with the PR8 WT, NSs, Ms, or Ms/NSs virus or PR8 ts LAIV and incubated with 3-fold serial dilutions of oseltamivir starting at a concentration of 10 μM (A) or 0.37 μM (B). Viral titers in the tissue culture supernatants were evaluated at 48 h postinfection by the immunofocus assay using an anti-NP monoclonal antibody (HB-65). Data represent the means and SDs of the results determined for triplicate wells. The viral titers in virus-infected cells in the absence of drug were used to calculate 100% viral infection. *, P < 0.05 (when the differences between WT and NSs viruses, WT and Ms viruses, WT and Ms/NSs viruses, and WT virus and PR8 ts LAIV were significant) using Student's t test from Microsoft Excel software.
DISCUSSION
In this study, we describe a novel approach to generate live-attenuated influenza viruses that would be promising as vaccine candidates that are safer and more protective than those used at present. To that end, the overlapping NS1/NEP (segment NS) and/or M1/M2 (segment M) ORFs were split by using the PTV-1 2A autoproteolytic cleavage site (Fig. 1). Three viruses harboring the modified NS segment, the modified M segment, and both modified NS and M segments (NSs, Ms, and Ms/NSs viruses, respectively) were rescued, and the identity of the modified NS and/or M segments was confirmed by RT-PCR and Western blotting approaches (Fig. 2). In cells infected with the Ms and Ms/NSs viruses, lower levels (∼50 to 75%) of M1 and M2 expression were observed (Fig. 2B). Similarly, in NSs or Ms/NSs virus-infected cells, the amounts of the NS1 and NEP proteins were lower (∼50 to 75%) than those in cells infected with WT PR8 virus (Fig. 2B), suggesting that M1/M2 and NS1/NEP processing may be affected.
In cultured MDCK cells, the NSs virus grew to high titers (∼5- to 10-fold lower than the WT PR8 virus titer) (Fig. 3). However, the expression of processed NS1 and NEP was reduced compared to that in WT PR8 virus infection (Fig. 2B), suggesting that the amount of NS1 and NEP produced in infected cells is not needed for efficient virus growth, at least in MDCK cells. In contrast, the Ms and Ms/NSs viruses were drastically impaired in their replication (Fig. 3). This could be due to altered levels of expression of M1 and M2, which are known to affect viral replication both in cell cultures and in vivo (36); to the M1 protein being fused to the 2A autoproteolytic cleavage site at the C-terminal end (Fig. 1), which could affect M1 functions; or to the presence of alternative proteins encoded by the influenza virus M segment being disrupted during Ms virus generation (18, 37–39). Surprisingly, the defect in replication of the Ms and Ms/NSs viruses was more evident at high temperatures (37°C and 39°C) than at a low temperature (33°C) (Fig. 3). We are currently investigating the molecular mechanism(s) behind the ts phenotype observed in the recombinant PR8 viruses containing the M split viral segment. Remarkably, the Ms and Ms/NSs viruses were highly attenuated in mice (Fig. 4). The MLD50 of the Ms virus (∼40,000 FFU) was ∼2,350-fold higher than the MLD50 of the WT PR8 virus (∼17 FFU) and similar to the MLD50 of PR8 ts LAIV (∼30,000 FFU), whereas the MLD50 of the Ms/NSs virus (>100,000 FFU) was at least ∼5,580-fold higher than that of the WT PR8 virus and ∼3-fold higher than that of PR8 ts LAIV (Table 1).
Because of the safety profiles of the viruses carrying split segments in vivo, we evaluated the immunogenicity and the vaccine potential of the Ms viruses (the Ms and Ms/NSs viruses) after homologous challenge with WT PR8 virus. We found that although the Ms and Ms/NSs viruses were highly attenuated, a single intranasal immunization, using 0.1 or 0.01 MLD50, induced an efficient immune response (Fig. 5), including neutralizing antibodies (Table 2) capable of conferring complete protection against lethal challenge with WT PR8 virus (Fig. 6). Notably, immunization with 0.1 or 0.01 MLD50 of the Ms or Ms/NSs virus inhibited replication of the challenge WT PR8 virus to much greater extents than immunization with the same MLD50 of PR8 ts LAIV (Fig. 6), indicating that these viruses induce better protective immune responses than PR8 ts LAIV. These results suggest that the M split strategy represents an alternative to the current approach of immunization with ts LAIV for immunocompetent 2- to-49-year-old individuals, while the Ms/NSs virus approach represents an improved strategy for vulnerable populations, such as immunocompromised or asthmatic people, who are not currently covered by the marketed ts LAIV (13). Under challenge conditions, the HAI titers of antibodies to the Ms or Ms/NSs virus in sera from infected mice were similar to or slightly lower than those of antibodies to PR8 ts LAIV (Table 2), and the titers of antibodies to NP were higher in sera from mice infected with the Ms or Ms/NSs virus than in those from mice infected with PR8 ts LAIV (Fig. 4). Because of these characteristics of sera and the protection results (Fig. 6), we hypothesize that the Ms and Ms/NSs viruses might induce better cellular immune responses than PR8 ts LAIV. Cellular immune responses have been shown to be required to confer better protection and optimal control of influenza virus replication (40–42). Another possibility is that the Ms and Ms/NSs viruses confer better protection than PR8 ts LAIV because they replicate better in the lower respiratory tract (Fig. 7A).
Importantly, the Ms and Ms/NSs viruses and PR8 ts LAIV were more sensitive to oseltamivir than the PR8 WT or NSs virus (Fig. 8), indicating that in the unlikely event that the vaccine induces illness, virus replication could be easily inhibited by treatment with the FDA-approved NA inhibitor. As the NA proteins of the different recombinant viruses are the same, one explanation for the higher sensitivity to oseltamivir observed in the Ms and Ms/NSs viruses and PR8 ts LAIV is that these viruses replicate less efficiently than the WT counterpart and less drug is required to inhibit viral replication.
LAIVs licensed for human use are recommended only for use by immunocompetent, nonpregnant, 2- to 49-year-old people due to safety issues (13). The attenuated influenza viruses generated by the approach described in this work, based on rearrangement of the viral spliced segments, offer several unique advantages over the currently FDA-approved ts LAIV. First, reorganization of the viral spliced M and/or NS segments makes viral reversion to a virulent phenotype highly unlikely, if not impossible. This approach overcomes safety concerns affecting the current ts LAIV, which is based on a limited number of amino acid substitutions (five) responsible for the ts phenotype. Second, like the current LAIV, our Ms and Ms/NSs viruses have a ts phenotype. However, our data indicate that the ts phenotype of the Ms or Ms/NSs virus results in ts greater than that observed in PR8 ts LAIV, since the viral titers at 37°C or 39°C were higher with PR8 ts LAIV (Fig. 3A) (14). Importantly, the growth of the Ms or Ms/NSs virus is slightly reduced (only 6-fold) compared to that of PR8 ts LAIV at the permissive temperature (33°C) (Fig. 7B), demonstrating the feasibility of using the Ms and Ms/NSs viruses for vaccine production. Third, the Ms virus has an MLD50 (∼40,000 FFU) similar to that of PR8 ts LAIV; however, upon vaccination with the same MLD50 (0.01 or 0.1), we observed greater protection efficacy with our Ms virus than with PR8 ts LAIV (Fig. 6). Moreover, even vaccination with 0.01 MLD50 of the Ms virus conferred better protection efficacy than vaccination with 0.1 MLD50 of PR8 ts LAIV. These results suggest that in order to obtain similar protection efficacy, much less Ms virus than PR8 ts LAIV is required for inoculation. Fourth, the vaccine created by use of the double Ms/NSs approach represents an excellent LAIV for vulnerable people currently excluded from the group eligible to be vaccinated with the currently available LAIV, such as immunocompromised, pregnant, or asthmatic individuals. Therefore, using a similar platform, the Ms or Ms/NSs virus could be used as a safer, more immunogenic, and more robust protective LAIV than the current ts LAIV.
Another advantage of our LAIV approach is that it could be implemented to generate live-attenuated vaccine candidates using the backbone of currently circulating seasonal influenza viruses. This is in contrast to the use of the ts LAIV, which is based on viral reassortants containing the safety backbone (PB2, PB1, PA, NP, M, and NS) of the master donor strain A/Ann Arbor/6/60 virus H2N2 and the two viral RNA segments encoding the viral glycoproteins (HA and NA) from circulating seasonal influenza viruses. This would result in better cellular responses that will assist with the provision of better protection efficacies. Moreover, generation of LAIV on the basis of the rearrangement of the M and/or NS spliced viral segments could be rapidly achieved using state-of-the-art plasmid-based reverse genetics technologies (35). Finally, it is worth noting that since the regions encoding NS1 and NEP (NS segment) and M1 and M2 (M segment) partially overlap, the split virus strategy described here allows introduction of mutations in overlapping regions without affecting the primary amino acid sequence of the NS1 and NEP proteins or M1 and M2 proteins to evaluate individually the contribution of a domain(s) or amino acid residues in one of these viral proteins without affecting the other viral product.
Funding Statement
This research was funded by a 2014 University of Rochester Research Award to Luis Martínez-Sobrido.
REFERENCES
- 1.Palese P, Shaw ML. 2007. Orthomyxoviridae: the viruses and their replication, p 1151–1185. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Li KS, Guan Y, Wang J, Smith GJ, Xu KM, Duan L, Rahardjo AP, Puthavathana P, Buranathai C, Nguyen TD, Estoepangestie AT, Chaisingh A, Auewarakul P, Long HT, Hanh NT, Webby RJ, Poon LL, Chen H, Shortridge KF, Yuen KY, Webster RG, Peiris JS. 2004. Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia. Nature 430:209–213. doi: 10.1038/nature02746. [DOI] [PubMed] [Google Scholar]
- 3.Girard MP, Cherian T, Pervikov Y, Kieny MP. 2005. A review of vaccine research and development: human acute respiratory infections. Vaccine 23:5708–5724. doi: 10.1016/j.vaccine.2005.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molinari NA, Ortega-Sanchez IR, Messonnier ML, Thompson WW, Wortley PM, Weintraub E, Bridges CB. 2007. The annual impact of seasonal influenza in the US: measuring disease burden and costs. Vaccine 25:5086–5096. doi: 10.1016/j.vaccine.2007.03.046. [DOI] [PubMed] [Google Scholar]
- 5.Lynch JP III, Walsh EE. 2007. Influenza: evolving strategies in treatment and prevention. Semin Respir Crit Care Med 28:144–158. doi: 10.1055/s-2007-976487. [DOI] [PubMed] [Google Scholar]
- 6.Anonymous. 2012. Prevention and control of influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP)—United States, 2012-13 influenza season. MMWR Morb Mortal Wkly Rep 61:613–618. [PubMed] [Google Scholar]
- 7.Osterholm MT, Kelley NS, Sommer A, Belongia EA. 2012. Efficacy and effectiveness of influenza vaccines: a systematic review and meta-analysis. Lancet Infect Dis 12:36–44. doi: 10.1016/S1473-3099(11)70295-X. [DOI] [PubMed] [Google Scholar]
- 8.Belshe RB, Edwards KM, Vesikari T, Black SV, Walker RE, Hultquist M, Kemble G, Connor EM, CAIV-T Comparative Efficacy Study Group. 2007. Live attenuated versus inactivated influenza vaccine in infants and young children. N Engl J Med 356:685–696. doi: 10.1056/NEJMoa065368. [DOI] [PubMed] [Google Scholar]
- 9.Belshe RB, Newman FK, Wilkins K, Graham IL, Babusis E, Ewell M, Frey SE. 2007. Comparative immunogenicity of trivalent influenza vaccine administered by intradermal or intramuscular route in healthy adults. Vaccine 25:6755–6763. doi: 10.1016/j.vaccine.2007.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cox MM, Patriarca PA, Treanor J. 2008. FluBlok, a recombinant hemagglutinin influenza vaccine. Influenza Other Respir Viruses 2:211–219. doi: 10.1111/j.1750-2659.2008.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorse GJ, Belshe RB, Munn NJ. 1991. Superiority of live attenuated compared with inactivated influenza A virus vaccines in older, chronically ill adults. Chest 100:977–984. doi: 10.1378/chest.100.4.977. [DOI] [PubMed] [Google Scholar]
- 12.Pica N, Palese P. 2013. Toward a universal influenza virus vaccine: prospects and challenges. Annu Rev Med 64:189–202. doi: 10.1146/annurev-med-120611-145115. [DOI] [PubMed] [Google Scholar]
- 13.De Villiers PJ, Steele AD, Hiemstra LA, Rappaport R, Dunning AJ, Gruber WC, Forrest BD. 2009. Efficacy and safety of a live attenuated influenza vaccine in adults 60 years of age and older. Vaccine 28:228–234. doi: 10.1016/j.vaccine.2009.09.092. [DOI] [PubMed] [Google Scholar]
- 14.Cox A, Baker SF, Nogales A, Martinez-Sobrido L, Dewhurst S. 2015. Development of a mouse-adapted live attenuated influenza virus that permits in vivo analysis of enhancements to the safety of live attenuated influenza virus vaccine. J Virol 89:3421–3426. doi: 10.1128/JVI.02636-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cox NJ, Kitame F, Kendal AP, Maassab HF, Naeve C. 1988. Identification of sequence changes in the cold-adapted, live attenuated influenza vaccine strain, A/Ann Arbor/6/60 (H2N2). Virology 167:554–567. [PubMed] [Google Scholar]
- 16.Snyder MH, Betts RF, DeBorde D, Tierney EL, Clements ML, Herrington D, Sears SD, Dolin R, Maassab HF, Murphy BR. 1988. Four viral genes independently contribute to attenuation of live influenza A/Ann Arbor/6/60 (H2N2) cold-adapted reassortant virus vaccines. J Virol 62:488–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paterson D, Fodor E. 2012. Emerging roles for the influenza A virus nuclear export protein (NEP). PLoS Pathog 8:e1003019. doi: 10.1371/journal.ppat.1003019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wise HM, Hutchinson EC, Jagger BW, Stuart AD, Kang ZH, Robb N, Schwartzman LM, Kash JC, Fodor E, Firth AE, Gog JR, Taubenberger JK, Digard P. 2012. Identification of a novel splice variant form of the influenza A virus M2 ion channel with an antigenically distinct ectodomain. PLoS Pathog 8:e1002998. doi: 10.1371/journal.ppat.1002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hai R, Schmolke M, Varga ZT, Manicassamy B, Wang TT, Belser JA, Pearce MB, Garcia-Sastre A, Tumpey TM, Palese P. 2010. PB1-F2 expression by the 2009 pandemic H1N1 influenza virus has minimal impact on virulence in animal models. J Virol 84:4442–4450. doi: 10.1128/JVI.02717-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jagger BW, Wise HM, Kash JC, Walters KA, Wills NM, Xiao YL, Dunfee RL, Schwartzman LM, Ozinsky A, Bell GL, Dalton RM, Lo A, Efstathiou S, Atkins JF, Firth AE, Taubenberger JK, Digard P. 2012. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 337:199–204. doi: 10.1126/science.1222213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lamb RA, Lai CJ. 1980. Sequence of interrupted and uninterrupted mRNAs and cloned DNA coding for the two overlapping nonstructural proteins of influenza virus. Cell 21:475–485. doi: 10.1016/0092-8674(80)90484-5. [DOI] [PubMed] [Google Scholar]
- 22.Robb NC, Jackson D, Vreede FT, Fodor E. 2010. Splicing of influenza A virus NS1 mRNA is independent of the viral NS1 protein. J Gen Virol 91:2331–2340. doi: 10.1099/vir.0.022004-0. [DOI] [PubMed] [Google Scholar]
- 23.Ali A, Avalos RT, Ponimaskin E, Nayak DP. 2000. Influenza virus assembly: effect of influenza virus glycoproteins on the membrane association of M1 protein. J Virol 74:8709–8719. doi: 10.1128/JVI.74.18.8709-8719.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen BJ, Leser GP, Jackson D, Lamb RA. 2008. The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J Virol 82:10059–10070. doi: 10.1128/JVI.01184-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pinto LH, Holsinger LJ, Lamb RA. 1992. Influenza virus M2 protein has ion channel activity. Cell 69:517–528. doi: 10.1016/0092-8674(92)90452-I. [DOI] [PubMed] [Google Scholar]
- 26.Schickli JH, Flandorfer A, Nakaya T, Martinez-Sobrido L, Garcia-Sastre A, Palese P. 2001. Plasmid-only rescue of influenza A virus vaccine candidates. Philos Trans R Soc Lond B Biol Sci 356:1965–1973. doi: 10.1098/rstb.2001.0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nogales A, Baker SF, Martinez-Sobrido L. 2015. Replication-competent influenza A viruses expressing a red fluorescent protein. Virology 476:206–216. doi: 10.1016/j.virol.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nogales A, Baker SF, Ortiz-Riano E, Dewhurst S, Topham DJ, Martinez-Sobrido L. 2014. Influenza A virus attenuation by codon deoptimization of the NS gene for vaccine development. J Virol 88:10525–10540. doi: 10.1128/JVI.01565-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nogales A, Rodriguez-Sanchez I, Monte K, Lenschow DJ, Perez DR, Martinez-Sobrido L. 2016. Replication-competent fluorescent-expressing influenza B virus. Virus Res 213:69–81. doi: 10.1016/j.virusres.2015.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baker SF, Nogales A, Finch C, Tuffy KM, Domm W, Perez DR, Topham DJ, Martinez-Sobrido L. 2014. Influenza A and B virus intertypic reassortment through compatible viral packaging signals. J Virol 88:10778–10791. doi: 10.1128/JVI.01440-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 32.Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am J Hyg 27:493–497. [Google Scholar]
- 33.Sharma P, Yan F, Doronina VA, Escuin-Ordinas H, Ryan MD, Brown JD. 2012. 2A peptides provide distinct solutions to driving stop-carry on translational recoding. Nucleic Acids Res 40:3143–3151. doi: 10.1093/nar/gkr1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manicassamy B, Manicassamy S, Belicha-Villanueva A, Pisanelli G, Pulendran B, Garcia-Sastre A. 2010. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc Natl Acad Sci U S A 107:11531–11536. doi: 10.1073/pnas.0914994107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinez-Sobrido L, Garcia-Sastre A. 2010. Generation of recombinant influenza virus from plasmid DNA. J Vis Exp 2010:2057. doi: 10.3791/2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chiang C, Chen GW, Shih SR. 2008. Mutations at alternative 5′ splice sites of M1 mRNA negatively affect influenza A virus viability and growth rate. J Virol 82:10873–10886. doi: 10.1128/JVI.00506-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao S, Liu X, Yu M, Li J, Jia X, Bi Y, Sun L, Gao GF, Liu W. 2012. A nuclear export signal in the matrix protein of influenza A virus is required for efficient virus replication. J Virol 86:4883–4891. doi: 10.1128/JVI.06586-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pinto LH, Dieckmann GR, Gandhi CS, Papworth CG, Braman J, Shaughnessy MA, Lear JD, Lamb RA, DeGrado WF. 1997. A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. Proc Natl Acad Sci U S A 94:11301–11306. doi: 10.1073/pnas.94.21.11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rossman JS, Jing X, Leser GP, Balannik V, Pinto LH, Lamb RA. 2010. Influenza virus M2 ion channel protein is necessary for filamentous virion formation. J Virol 84:5078–5088. doi: 10.1128/JVI.00119-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Epstein SL, Lo CY, Misplon JA, Bennink JR. 1998. Mechanism of protective immunity against influenza virus infection in mice without antibodies. J Immunol 160:322–327. [PubMed] [Google Scholar]
- 41.Sridhar S, Begom S, Bermingham A, Hoschler K, Adamson W, Carman W, Bean T, Barclay W, Deeks JJ, Lalvani A. 2013. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat Med 19:1305–1312. doi: 10.1038/nm.3350. [DOI] [PubMed] [Google Scholar]
- 42.Wilkinson TM, Li CK, Chui CS, Huang AK, Perkins M, Liebner JC, Lambkin-Williams R, Gilbert A, Oxford J, Nicholas B, Staples KJ, Dong T, Douek DC, McMichael AJ, Xu XN. 2012. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat Med 18:274–280. doi: 10.1038/nm.2612. [DOI] [PubMed] [Google Scholar]