

ABSTRACT
Despite an abundance of evidence supporting an important role for the cleavage of minor capsid protein L2 by cellular furin, direct cleavage of capsid-associated L2 during human papillomavirus 16 (HPV16) infection remains poorly characterized. The conserved cleavage site, close to the L2 N terminus, confounds observation and quantification of the small cleavage product by SDS-PAGE. To overcome this difficulty, we increased the size shift by fusing a compact protein domain, the Propionibacterium shermanii transcarboxylase domain (PSTCD), to the N terminus of L2. The infectious PSTCD-L2 virus displayed an appreciable L2 size shift during infection of HaCaT keratinocytes. Cleavage under standard cell culture conditions rarely exceeded 35% of total L2. Cleavage levels were enhanced by the addition of exogenous furin, and the absolute levels of infection correlated to the level of L2 cleavage. Cleavage occurred on both the HaCaT cell surface and extracellular matrix (ECM). Contrary to current models, experiments on the involvement of cyclophilins revealed little, if any, role for these cellular enzymes in the modulation of furin cleavage. HPV16 L2 contains two consensus cleavage sites, Arg5 (2RHKR5) and Arg12 (9RTKR12). Mutant PSTCD-L2 viruses demonstrated that although furin can cleave either site, cleavage must occur at Arg12, as cleavage at Arg5 alone is insufficient for successful infection. Mutation of the conserved cysteine residues revealed that the Cys22-Cys28 disulfide bridge is not required for cleavage. The PSTCD-L2 virus or similar N-terminal fusions will be valuable tools to study additional cellular and viral determinants of furin cleavage.
IMPORTANCE Furin cleavage of minor capsid protein L2 during papillomavirus infection has been difficult to directly visualize and quantify, confounding efforts to study this important step of HPV infection. Fusion of a small protein domain to the N terminus greatly facilitates direct visualization of the cleavage product, revealing important characteristics of this critical process. Contrary to the current model, we found that cleavage is largely independent of cyclophilins, suggesting that cyclophilins act either in parallel to or downstream of furin to trigger exposure of a conserved N-terminal L2 epitope (RG-1) during infection. Based on this finding, we strongly caution against using L2 RG-1 epitope exposure as a convenient but indirect proxy of furin cleavage.
INTRODUCTION
Human papillomaviruses (HPVs) are currently the most common sexually transmitted infection in the United States (1). These viruses infect and replicate in differentiating mucosal and cutaneous epithelia, and a subset of the mucosa-tropic viruses, the high-risk HPVs, cause >99% of cervical cancers in women and are associated with other anogenital and nasopharyngeal cancers in both women and men (2). In all, the high-risk HPVs account for an astounding 5% of total cancers worldwide (3).
HPVs are nonenveloped viruses with a 55-nm icosahedral capsid composed of 72 pentamers of the major capsid protein L1. Encapsidated within the particle are ∼20 to 40 molecules of the minor capsid protein L2 complexed to an ∼8-kb circular double-stranded DNA (dsDNA) genome (vDNA), condensed in a chromatin-like structure (4–6). The initial infection of basal keratinocytes by HPV16 begins with attachment via heparan sulfate proteoglycans (HSPGs) followed by conformational changes and cleavage of L2 by cellular furin, with the virion eventually entering the cell via a micropinocytosis-like process (7–10). Although no sole entry receptor has been reported, entry of HPV16 and other high-risk HPV types appears to involve growth factor receptors, integrins, tetraspanin-enriched membrane microdomains, and the annexin-A2 heterotetramer (11–17).
Internalized virions enter the endosomal pathway, where acidification due to the V-ATPase proton pump triggers L1 uncoating (18) and the L2/vDNA complex separates from the dissociated L1 capsid and retrograde traffics to the trans-Golgi network (TGN) in a furin-, cyclophilin-, γ-secretase-, and retromer-dependent manner (19–24). Transport of L2/vDNA to the TGN is essential, and prolonged residence in the endosomal compartment may be detrimental, as the acid-dependent cathepsins B and L restrict infection (25). Sorting nexin 17 and dynein light chains have also been implicated in the proper subcellular trafficking of virions (26–28). The majority of disassembled L1 capsid remains in the degradative late endosomal/lysosomal compartment (23, 29). Cytoplasmic translocation of the L2/vDNA complex has been suggested to occur at the TGN (19, 30), but few molecular details are known about this process. A recently identified a transmembrane-like hydrophobic domain (residues 45 to 67) is necessary for translocation of L2/vDNA and may mediate an interaction with the limiting membrane (31). The nuclear import of L2/vDNA is believed to depend on mitotic nuclear envelope breakdown (30, 32). Efficient infection requires localization of L2/vDNA to nuclear promyelocytic leukemia protein (PML)/ND10 bodies (33). Notably, inhibition of L2 cleavage results in nonproductive uptake of virions, with L2/vDNA failing to reach the TGN or PML bodies (19).
Furin is a secreted subtilisin-like Ca2+-dependent serine endoprotease that activates a wide variety of immature proprotein substrates through proteolysis at general R-X-X-R or R-X-X-X-K/R-R consensus sites (34, 35). Furin was the first member of the mammalian proprotein convertases (PCs) to be characterized and has a very broad tissue expression pattern. Owing to the enormous diversity of its natural proprotein substrates—hormones, growth factors, clotting factors, cell surface receptors, and proteases—furin is important for an extensive assortment of cellular and physiological processes that underlie development, tissue remodeling, and homeostasis (34, 35). Furin is initially produced in the endoplasmic reticulum (ER) as an inactive transmembrane proprotein with type I topology. Intramolecular autoproteolysis of the propeptide begins in the ER and is completed upon exposure to the mildly acidic environment of the TGN (34, 35). Although the majority of furin is TGN localized at steady state, a trace amount of active furin is present on the cell surface.
Although furin is recognized as a necessary factor for HPV16 infection, cleavage of the L2 capsid protein during infection remains poorly characterized. In this study, we fused a small protein domain to the N terminus of L2 to enable easy measurement and quantification of L2 cleavage during infection. We found that L2 cleavage is incomplete during infection of HaCaT cells, with only ∼25 to 35% of the L2 molecules being cleaved by furin. Levels of L2 cleavage can be boosted through the addition of exogenous furin, resulting in a correlative increase in viral infectivity. Through mutagenesis we showed that cleavage can occur at two different consensus sites in L2, but successful infection requires cleavage at Arg12. Cleavage is also independent of the Cys22-Cys28 disulfide bond. Cleavage can be blocked, unaffected, or potentiated by various neutralizing antibodies against different L1 or L2 epitopes. Finally, cleavage can occur on either the cell surface or the extracellular matrix (ECM) and is largely independent of cyclophilins, contrary to the current model.
MATERIALS AND METHODS
Cell culture.
Cells were cultured at 37°C and 5% CO2. 293TT cells were maintained in high-glucose Dulbecco's modified Eagle's medium (DMEM; Sigma; D5796) with 10% bovine growth serum (BGS, HyClone; SH30541.03), antibiotic/antimycotic (Ab/Am; Sigma; A5955), and 0.4 μg/ml of hygromycin B (Roche; 30-240-CR). HaCaT cells were maintained in high-glucose DMEM with 10% fetal bovine serum (FBS; HyClone; SH30396.03) and Ab/Am. CHO-K1, pgsA-745, CHOfurΔ1 (FurΔ1), and FD11 cells were maintained in high-glucose DMEM with 10% FBS, Ab/Am, and 200 μM l-proline. Conditioned CHO medium (CCM) supernatant from 48-h cultures of FurΔ1 and FD11 cells was collected by centrifugation, concentrated 10-fold with a 30-kDa-molecular-mass-cutoff spin-filter unit (Millipore; UFC903024), aliquoted, and stored at −80°C as a rich source of active furin (FurΔ1) or control supernatant (FD11).
Viruses.
To clone the PSTCD-L2 fusion, the codon-optimized L2 cDNA was PCR amplified from the pXULL expression plasmid, deleting the ATG start codon and introducing unique KpnI and AatII restriction sites for fusion of sequences to the N terminus of L2. The engineered L2 was cloned back into pXULL with NotI and EcoRI, replacing the L2 open reading frame (ORF) in pXULL. The 70-residue PSTCD ORF was then PCR cloned with KpnI and AatII from plasmid pAd-Red-IX-PSTCD (36), introducing a new ATG start codon. The resulting plasmid, pXULL-PSTCD-L2, encodes the PSTCD-L2 fusion in place of the wild-type (wt) L2 ORF. Cloning of mutant viruses was achieved by PCR-based methods or site-directed mutagenesis using the QuikChange-XL II system (Aligent; 200521). All mutations were verified by Sanger sequencing. Virions containing a luciferase expression plasmid (pGL3-basic; Clontech) or the enhanced green fluorescent protein (EGFP) expression plasmid pCIneo-EGFP (Clontech) were generated by CaPO4 transfection of 293TT cells. Purification was done by CsCl density gradient centrifugation as previously described (29, 37). The L1 content of purified virions was determined by SDS-PAGE and Coomassie staining, compared against BSA standards. pGL3 pseudogenome content was determined by SYBR green quantitative PCR (qPCR) using primers specific for the luciferase gene of pGL3, and capsid/genome ratios were all in the ranges typical for wt HPV16 preparations.
Antibodies.
Rabbit anti-HPV16 polyclonal antibody (a gift from M. Ozbun) was used at 1:5,000 for Western blots and live cell immunofluorescence. L2-specific RG-1 monoclonal antibody (MAb) (a gift from R. Roden) was used at 1:100 for live-cell immunofluorescence. The L2-specific K4L220-38 monoclonal antibody (K4, a gift from M. Müller) was used at 1:5,000 for Western blots and 1:200 for neutralization. Rat anti-hemagglutinin (anti-HA) clone 3F10 (Roche; number 1-867-423) was used at 1:1,000 for Western blots. H16.V5 antiserum (V5, a gift from N. Christensen) was used at 1:200 for neutralization unless otherwise specified. H16.U4 antiserum (U4, a gift from N. Christensen) was used at 1:50 for neutralization. Mouse anti-HA (Applied Biological Materials; number G036) was used as a control IgG at 1:200 for neutralization. Rabbit anti-furin (Thermo Scientific; PA1-062) and rabbit anti-CypB (Thermo Scientific; PA1-027A) were both used at 1:1,000 for Western blots. Rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH; Cell Signaling Technology; number 2118) was used at 1:5,000 for Western blots.
Biochemical inhibitors.
Decanoyl-RVKR-chloromethyl ketone (dRVKR; EMD; number 344931) was used at 25 μM unless otherwise indicated. Hexa-d-arginine (6DR; EMD; number 344931) was used at 150 μM. Cyclosporine (CsA; EMD; number 239835) was used at 10 μM unless otherwise indicated. Cytochalasin D (cytoD; Sigma; number C8273) was used at 10 μM. Bafilomycin A (BafA; Sigma; number B1793) was used at 25 nM. γ-Secretase inhibitor XXI (EMD; number 565790) was used at 1 μM. Aphidicolin (Aph; Sigma; number A0781) was used at 6 μM. Bacitracin (Bac; Sigma; number B0125) was prepared as previously described (29) and used at 5 mM.
Infections.
Cells, grown to 50 to 60% confluence in 24-well plates, were infected with 2 × 108 viral genome equivalents of luciferase-expressing HPV16 pseudovirions (PsV) in a volume of 500 μl. At 24 h postinfection, the cells were lysed in 0.1 ml of 1× reporter lysis buffer (Promega; E3971). Luciferase levels were measured with a DTX-800 multimode plate reader (Beckman Coulter) by firefly luciferase assay according to the manufacturer's recommendations (Promega; E4550). Relative light units for each sample were normalized to GAPDH levels by Western blot densitometry and expressed as percent infection. Infections with EGFP-expressing PsV were performed by infecting subconfluent HaCaT cells with 120 ng of L1 PsV in 800 μl. At 18 h postinfection a medium change was performed, keeping the same drug and CCM concentrations on the cells. Cells were trypsinized 48 h postinfection and washed with phosphate-buffered saline (PBS), and EGFP expression was measured by flow cytometry on a BD Biosciences FACSCanto II. Mock-infected cells were used to set the forward scatter (FSC), side scatter (SSC), and EGFP gates. For furin titration experiments, the levels of exogenous furin in the culture were varied by the addition of FurΔ1 and FD11 CCM supernatants. All infections had equivalent final concentrations of CCM, but the absolute levels of furin were modulated by varying the FurΔ1/FD11 ratio. Maximum L2 cleavage was achieved by overnight incubation of purified Propionibacterium shermanii transcarboxylase domain (PSTCD)-L2 viral inoculum in a small volume of FurΔ1 or control FD11 CCM, prior to infection in FurΔ1 or control FD11 CCM.
Furin cleavage assays.
HaCaT cells were cultured to 50 to 60% confluence in 24-well, 12-well, or 6-well plates and infected with 500 to 750 ng of L1 per ml of PsV. Cells were infected for 18 to 24 h unless otherwise specified. Cells were then washed twice with PBS and lysed in RIPA-PAGE cell lysis buffer (800 μl of radioimmunoprecipitation assay [RIPA] buffer, 200 μl of SDS-PAGE loading buffer, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1× protease inhibitors [Sigma; number P1860]). Lysates were heated to 95°C for 5 min and passed through QIAshredder columns (Qiagen; number 79656) to clarify when needed. The samples were run on a 10% acrylamide SDS-PAGE gel, transferred to nitrocellulose membranes, and blocked overnight at 4°C in Tris-buffered saline–Tween (TBST) plus 4% milk, 4% BSA, and 1% goat serum or TBST plus 5% milk. Seven percent gels were used for experiments with L2-HA viruses. Blots were immunostained with the K4 monoclonal antibody (38) diluted 1:5,000 in TBST plus 0.8% milk or rat anti-HA (clone 3F10; Roche) diluted 1:1,000 in TBST plus 0.8% milk. Goat anti-mouse and goat anti-rabbit IR680- and IR800-labeled secondary antibodies were used at 1:10,000 to 1:20,000 in TBST plus 0.8% milk. Dried blots were scanned on an Odyssey instrument (LI-COR), and band intensities were measured by densitometry of high-resolution blot images using ImageJ (39). All cleavage experiments were performed at least two independent times, with average cleavage values shown underneath representative blots below.
siRNA knockdown experiments.
Pools of three small interfering RNA (siRNA) duplexes specific for furin (sc-40595), CypB (sc-35145), or a nonspecific scramble control (sc-37007) were obtained from Santa Cruz Biotechnology. HaCaT cells were transfected with 50 nM siRNAs using Lipofectamine RNAiMax reagent (Invitrogen) in 24-well plates. Twenty-four hours after siRNA transfection, cells (now ∼60% confluent) were rinsed with PBS and infected with PSTCD-L2 virus. Luciferase was quantified 24 h postinfection, and lysates were analyzed for protein knockdown and L2 cleavage by Western blotting. Two independent experiments were performed, each in triplicate.
Live-cell RG-1 immunofluorescence.
Live-cell RG-1 immunofluorescence was performed as described previously (40, 41). Briefly, HaCaT cells, plated to 50% confluence on circular glass coverslips in 24-well plates, were chilled and bound to 1 μg of L1/ml of L2-HA virus in 250 μl of medium containing 0.1% dimethyl sulfoxide (DMSO) at 4°C for 1 h. Some samples were then switched to 37°C for 4 h to allow infection to proceed in the presence of 0.1% the DMSO carrier, 25 μM dRVKR, or 10 μM CsA. After binding and/or infection, cells were chilled to 4°C and washed twice with cold PBS containing 2% FBS and 0.01% NaN3 (PBS-FBS). Live cells were then stained with RG-1 monoclonal antibody (1:100 dilution) and anti-HPV16 polyclonal antibody (1:5,000 dilution) in PBS-FBS for 1 h at 4°C. The cells were washed four times with PBS-FBS and stained with Alexa Fluor 488-conjugated goat anti-mouse and Alexa Fluor 555-conjugated goat anti-rabbit secondary antibodies, each diluted 1:1,000 in PBS-FBS for 1 h at 4°C. The cells were washed five times with PBS-FBS and fixed with 2% paraformaldehyde in PBS for 20 min at 4°C prior to mounting with Prolong Diamond (Life Technologies) containing 4′,6-diamidino-2-phenylindole (DAPI). Images were collected on a Zeiss LSM-510 META confocal microscope and processed with Zeiss LSM Image Browser software. The final images are maximum projection composites of four serial single-plane scans of a z-stack.
Sequence alignment and structure modeling.
Residues 53 to 123 of the 1.3S biotin carboxyl carrier domain from Propionibacterium shermanii transcarboxylase (PDB code 1DCZ) were modeled with backbone cartoon and mesh surface rendering using the MacPyMOL software package. L2 sequence alignments were done using the ClustalW tool within the MacVector software package.
RESULTS
PSTCD-L2 virus is infectious and provides easy visualization of furin cleavage.
Cleavage of L2 by cellular furin is predicted to remove the 12 N-terminal residues of L2. Due to the very low abundance of L2 within purified virions and the aberrant mobility of the protein in SDS-PAGE (L2 is predicted to be 51 kDa but runs at ∼72 kDa), such a small size shift can be difficult to reliably detect in infected cell lysates, confounding efforts to quantitatively visualize furin cleavage of L2 during cellular infection. As the N terminus of L2 has been shown to tolerate a variety of fusions ranging from small epitopes to larger protein fragments (9, 42, 43), we reasoned that addition of a small mass to the N terminus would increase the size shift and greatly facilitate studies of furin cleavage. To generate such a “furin reporter” virus, we fused the 70-residue Propionibacterium shermanii transcarboxylase domain (PSTCD) to the N terminus of the L2 protein (Fig. 1A and B). The PSTCD has been successfully used as a fusion tag within a variety of viral capsid proteins (36, 44, 45), and its size and structure were ideal for our purposes: a small compact β-sandwich fold (46) that is large enough to cause a clear 9-kDa size shift on a polyacrylamide gel when cleaved at the furin consensus site. In addition, the PSTCD is devoid of cysteine, eliminating any potential interference with oxidation of the critical and conserved disulfide bond between Cys22 and Cys28 (37).
FIG 1.

Characterization of the PSTCD-L2 virus. (A) Atomic structure of the 70-residue fragment of the PSTCD domain, modeled with cartoon and mesh surface rendering. The position of the C terminus that is fused to L2 is shown in red. (B) Schematic of the PSTCD-L2 fusion, indicating the positions of the furin cleavage site, the K4 epitope, and predicted cleavage fragment sizes. The asterisk denotes the apparent molecular mass of 67 kDa; the calculated molecular mass is 49 kDa. (C) Western blot of CsCl-purified PSTCD-L2 virions, compared to wt HPV16. L1 and L2 were detected with rabbit anti-HPV16 polyclonal antibody, and L2 was detected with mouse anti-L2 K4 monoclonal antibody. (D and E) Relative infectivity of wt or PSTCD-L2 virus in HaCaT cells (D) or CHO-K1 and HSPG-deficient pgsa-745 cells (E). (F) Representative Western blot of mock- and PSTCD-L2 infected HaCaT cell lysates 24 h postinfection, stained with the K4 monoclonal antibody. Cleavage levels were measured by densitometry. Values are the means from three independent experiments. (G) Representative time course blot of PSTCD-L2 cleavage in HaCaT cells. (H) Plot of PSTCD-L2 cleavage levels from five independent time course experiments. Values are means ± standard deviations.
Purified HPV16 pseudovirions packaged the PSTCD-L2 fusion at levels comparable to that of unmodified wild-type (wt) L2 (Fig. 1C), and there were no defects in levels of reporter vDNA encapsidation or total PsV yield. Infectivity of the PSTCD-L2 virus in HaCaT keratinocytes was comparable to that of wt HPV16 (Fig. 1D). Similar to the wt, PSTCD-L2 infection required HSPGs, as xylosyltransferase-deficient pgsA-745 cells, lacking HSPGs (47), were poorly infected compared to parental CHO-K1 cells (Fig. 1E). This suggests that despite the additional mass on the N terminus, PSTCD-L2 was following the natural HSPG-dependent pathway for infection, rather than an HSPG-independent route utilized by furin-precleaved virions (48).
Furin cleavage of PSTCD-L2 was readily observed by Western blotting of infected cell lysates. Interestingly, 24-h infections typically resulted in cleavage of 25 to 35% of the total cell-associated PSTCD-L2 (Fig. 1F), and complete cleavage was never observed under standard cell culture conditions. Time course experiments revealed that steady-state levels of cleavage in cell-associated virus gradually increased over time, with levels reaching a half-maximal value of 15% by 6 h postinfection (Fig. 1G and H).
Infectivity is directly proportional to furin cleavage.
The incomplete nature of cleavage was unexpected and prompted us to investigate whether furin cleavage is a limiting factor in HaCaT cell infection, and if HPV16 infection could be augmented by the addition of exogenous furin. We utilized cell-free conditioned medium (supernatant) from FurΔ1 cells (FurΔ1 is a CHO-derived clone that constitutively secretes a truncated soluble form of furin lacking the transmembrane domain [49]) as a convenient and affordable source of exogenous furin. Conditioned medium from the furin-deficient CHO-derived clone FD11 (50) was used as a control for conditioned CHO medium (CCM) in these experiments. The furin activity phenotype of FurΔ1 and FD11 cells was verified by cleavage assay with PSTCD-L2 virus (Fig. 2A). HaCaT cell infections were performed in a constant amount of 30% CCM but titrating the ratio of FurΔ1/FD11 from 0 to 30% final medium volume, in increments of 5%. In this way, we were able to vary the amount of extra furin without changing the final concentration of CCM in the infection. Infection was assayed by luciferase assay and cleavage was measured by Western blotting of L2 at 24 h postinfection. When infection is plotted against the percentage of L2 cleavage for each amount of furin, a linear relationship can be observed, with infection increasing as a function of L2 cleavage (Fig. 2B). The highest level of L2 cleavage obtained in this experiment was ∼55%, resulting in about a 5.5-fold increase in infectivity.
FIG 2.
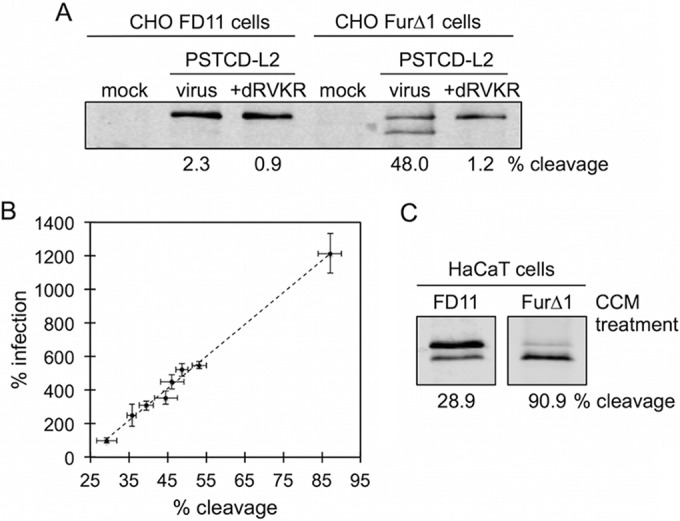
HPV16 Infectivity is proportional to furin cleavage. (A) Cleavage blot from mock- or PSTCD-L2 infected furin-deficient FD11 or furin-secreting FurΔ1 cells in the presence of DMSO or the furin inhibitor dRVKR. (B) PSTCD-L2 infectivity plotted as a function of L2 cleavage. (C) Cleavage blots of PSTCD-L2 virus incubated overnight at 37°C in FD11 or FurΔ1 CCM, followed by infection of HaCaT cells, again in the presence of either FD11 or FurΔ1 CCM.
We empirically determined that maximal cleavage of PSTCD-L2 required overnight pretreatment of purified virus with FurΔ1 CCM, followed by infection of HaCaT cells in 100% FurΔ1 CCM. Under these conditions both infection and cleavage were substantially boosted (1,214% ± 119% infection and 87% ± 3% cleavage) compared to those of appropriate FD11 CCM controls (100% ± 7% infection and 29.2% ± 3% cleavage) (Fig. 2B and C). Additional FurΔ1 CCM supplementation experiments were performed with EGFP-expressing wt or PSTCD-L2 PsV. Similar to the results with luciferase-expressing PSTCD-L2, supplementation with additional furin caused a substantial boost to both the number of EGFP-expressing cells and the mean fluorescence intensity (MFI) compared to those of the proper FD11 controls (Table 1). Increasing the MOI in experiments with EGFP-expressing virus caused a corresponding increase in the percentage of EGFP-positive cells (data not shown). Thus, the additional cleavage essentially increases the effective MOI of the virus, but it is impossible to draw any firm conclusions regarding L2 cleavage levels of individual virions within the population from these experiments.
TABLE 1.
HaCaT cell infections with EGFP-expressing PSTCD-L2 virus
| Condition | % cleavagea | % EGFP positivityb | MFIb |
|---|---|---|---|
| PSTCD-L2 | 35.7 | 100 | 100 |
| FurΔ1 CCM, 50% | 83.9 | 448.3 | 1,018.9 |
| dRVKR, 2.5 μM | 4.4 | 3.2 | 4.2 |
| CsA, 5 μM | 35.6 | 8.1 | 7.6 |
Average of two independent experiments, as measured by densitometry of Western blots.
Average of two independent experiments as measured by fluorescence-activated cell sorting, normalized to the appropriate FD11 or DMSO vehicle control.
Collectively, these findings are in general agreement with other published work reporting a 2- to 20-fold enhancement of infectivity in response to the addition of exogenous furin in a variety of cell types (48, 51). These data suggest that L2 cleavage and subsequent infectivity are quite limited in HaCaT cell culture systems, and this phenomenon is likely true for other cell lines as well. The extent of L2 cleavage during natural infections in vivo remains to be determined.
Cleavage of mutant L2 proteins.
Interestingly, many HPV types have multiple and sometimes overlapping consensus R-X-X-R furin cleavage sites at the N terminus of L2 (Fig. 3A). HPV16 L2 has two consensus sites, 2RHKR5 and 9RTKR12. The latter site is absolutely conserved across all known PV types, suggesting that it is the more relevant site, although the importance of 2RHKR5 has never been formally investigated. We generated R2,5K and R9,12K single-site mutants and the double-site R2,5,9,12K mutant (Fig. 3B) to clarify the role of both consensus sites in cleavage of L2 and HPV16 infection. These mutants were generated in the context of the PSTCD-L2 virus, and lysine was chosen as the substitution to preserve the positive charge of the N terminus, as there is evidence that this region may function as a nuclear localization signal (52) and the positively charged nature of this region has been shown to be essential for infection of bovine papillomavirus 1 (53).
FIG 3.

Cleavage of L2 mutants. (A) Sequence alignment of the N termini from a variety of human and animal PV types. Consensus furin cleavage sites are indicated with red lines, and the pair of conserved cysteines are shaded in yellow. (B) Protein sequences of the L2 mutants used in this work. (C and D) Cleavage blot (C) and infectivity (D) of the furin site PSTCD-L2 mutants. (E) Anti-HA cleavage blot of L2-HA wt and C22,28S mutant viruses.
All three furin site mutants packaged normal levels of luciferase reporter plasmid by qPCR, and no defects in L2 encapsidation were observed by SDS-PAGE of purified virus stocks. Cleavage assays revealed that cellular furin can cleave both sites of L2, as only the double R2,5,9,12K mutant was abrogated for cleavage (Fig. 3C). Infection experiments revealed that mutation of the 9RTKR12 site ablated infectivity, whether the mutation was alone or in combination with the 2RHKR5 site. In contrast, mutation of only the 2RHKR5 site had no effect on viral infectivity (Fig. 3D). These data clearly show that while furin can cleave at either the 2RHKR5 or the 9RTKR12 site, cleavage must occur at the 9RTKR12 site, and that a productive infection requires that the 12 N-terminal positively charged residues of L2 be removed by furin.
We also investigated cleavage of a previously described C22,28S double point mutant of L2 that is devoid of the conserved C22-C28 disulfide bond (37). This mutant is noninfectious and displays a phenotype similar to furin inhibition in that intracellular viral genomes fail to cross the limiting membrane and never localize to PML bodies (unpublished observations). Some furin substrates utilize disulfide bonds for optimal positioning and processing of the cleavage site by furin. For example, HIV gp160 is processed by furin, but efficient cleavage depends on a structural disulfide bond within the gp41 subunit of the protein (54). We therefore wanted to test if the conserved C22-C28 disulfide of L2 is structurally important for accessibility, recognition, or cleavage by furin. All previous experiments with PSTCD-L2 utilized the high-affinity L2-specific K4(20-38) antibody (herein referred to as “K4”) for detection of L2 in whole-cell lysates by Western blotting (see Materials and Methods). The K4 epitope spans from residues 21 to 30, encompassing the Cys22-Cys28 disulfide, and mutation of either Cys22 or Cys28 abrogates K4 reactivity (38), precluding us from studying furin cleavage with this mutant using PSTCD-L2 virus and K4 immunoblotting. To overcome this technical limitation, we measured cleavage of wt and C22,28S mutant L2-HA viruses using an HA-specific antibody. C-terminally HA-tagged L2 has been used previously and does not interfere with infectivity (33). Since L2-HA virus lacks the N-terminal PSTCD tag, the size shift upon cleavage is greatly reduced but can still become apparent when 7% polyacrylamide gels are used and samples are carefully run out for better separation. Results show that mutation of the C22-C28 disulfide had no effect on furin cleavage (Fig. 3E), suggesting that the disulfide is not required to position the N terminus of L2 in a furin-accessible conformation.
Screening of NAbs.
Two L1-specific neutralizing antibodies (NAbs), H16.V5 (V5) and H16.U4 (U4), both block entry of HPV16 but do so through distinct mechanisms (55). V5 binds to a conformational epitope comprised of regions of the BC, DE, FG, and HI loops at the apex of the L1 pentamers (56) and allows virus to bind cell surface HSPGs but prevents virus binding to the underlying extracellular matrix (ECM) (55). U4 recognizes a linear epitope within the C-terminal arm of L1 that reaches across the canyons between L1 pentamers in an extended “suspension bridge” conformation (57, 58) and permits virus binding to the ECM while blocking binding to cell surface HSPGs, similar to inhibition by excess heparin (55). Excess amounts of V5 effectively neutralized the infectivity of PSTCD-L2 and also resulted in strong inhibition of furin cleavage (Fig. 4A and B). Excess U4 was also able to neutralize PSTCD-L2 infection (Fig. 4B) but did so without any effect on furin cleavage (Fig. 4A), suggesting that furin cleavage of L2 can occur on the ECM, where U4-neutralized virions are sequestered (55).
FIG 4.
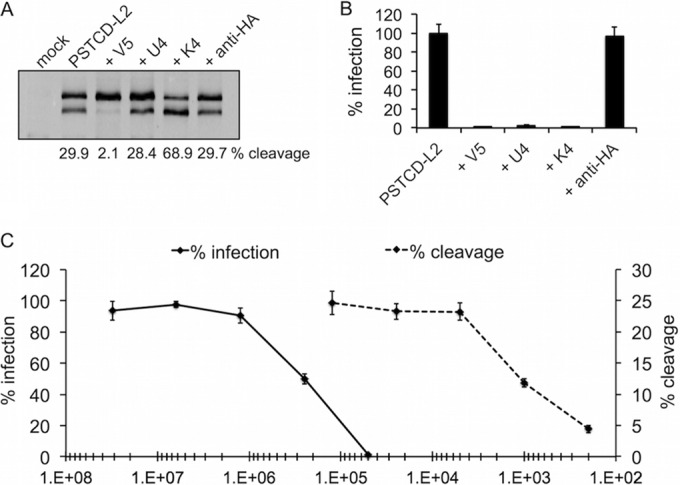
Antibody-mediated inhibition and enhancement of L2 cleavage. (A and B) L2 cleavage blot (A) and infectivity (B) of mock- or PSTCD-L2-infected HaCaT cells in the presence of L1-specific V5 and U4 MAbs, L2-specific K4 MAb, or control HA-specific MAb. (C) Titration curves of V5 neutralization of infection (solid line) and L2 cleavage (dotted line).
The high-affinity L2-specific NAb K4 binds tightly to residues 21 to 30 (38), a region within the previously described “RG-1 epitope” (59). This epitope is normally buried in mature virions and becomes exposed upon furin cleavage of L2 (41). Interestingly, the addition of K4 caused a marked increase in cleavage of PSTCD-L2 while potently neutralizing infection (Fig. 4A and B). Neutralizing antibodies that bind this epitope (including RG-1) cause the relocalization of virus from the cell surface to the ECM in a manner similar to the U4 MAb (41), again suggesting that cleavage can occur on the ECM.
The robust inhibition of L2 cleavage by V5 raised the possibility that the neutralizing activity of V5 itself may be due to inhibition of furin cleavage. To test this, we titrated down the amount of V5 sera by serial dilution and measured both infectivity and furin cleavage of PSTCD-L2 across a wide range of V5 concentrations. We found that the V5 stock was very potent, having a 50% inhibitory concentration (IC50) for infection at a dilution of 1/250,000, with complete neutralization of infection at a dilution of 1/50,000. In contrast, inhibition of furin cleavage by V5 had a very different curve, with an IC50 of 1/1,000 and near complete inhibition of cleavage requiring a 1/200 dilution of NAb (Fig. 4C). In short, blockage of furin cleavage required a much larger amount of V5 than did neutralization of infection, suggesting a steric mode of action whereby excess V5 completely coats the virion to preclude furin cleavage of L2, and one that is not the primary basis for the potent neutralizing activity of V5.
Screening of biochemical inhibitors.
Next we used biochemical inhibitors to verify that the observed cleavage was due to cellular furin and to define other factors important for cleavage. Both the irreversible, cell-permeable furin/PC inhibitor decanoyl-RVKR-chloromethylketone (dRVKR) and the reversible, impermeable peptide inhibitor hexa-d-arginine (6DR) (60) blocked infectivity and cleavage of PSTCD-L2 (Fig. 5). 6DR was less potent and resulted in partial blockage of both infection and furin cleavage.
FIG 5.
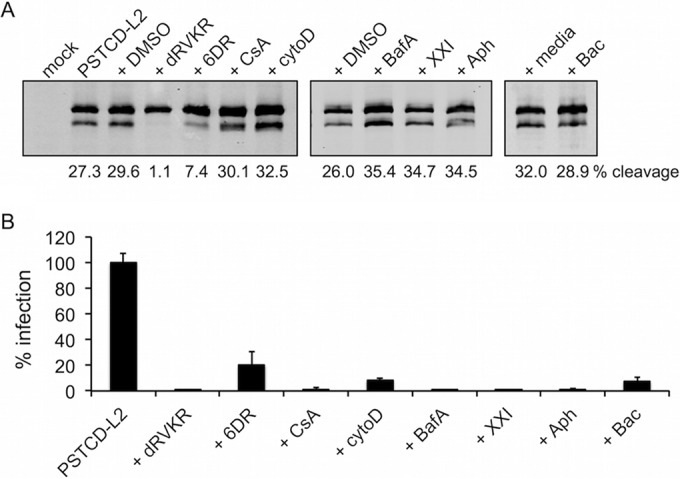
L2 cleavage is extracellular and is blocked by furin but not cyclophilin inhibitors. Shown are L2 cleavage blots (A) and infectivity (B) of mock- or PSTCD-L2-infected HaCaT cells in the presence of carrier controls (DMSO or media), the furin inhibitors dRVKR and 6DR, the cyclophilin inhibitor CsA, the entry inhibitor cytoD, the acidification inhibitor BafA, the γ-secretase inhibitor XXI, the cell cycle blocker Aph, or the PDI inhibitor Bac.
Prior work has suggested a role for cell surface cyclophilins in HPV16 infection (23, 40). Upon HSPG binding, these peptidyl-prolyl isomerases (PPIs) are believed to act upstream of furin by regulating a proline-dependent conformational switch to expose the N terminus of L2 for efficient furin cleavage (13, 61). Surprisingly, the potent and broad PPI inhibitor cyclosporine (CsA) had no effect on furin cleavage of PSTCD-L2, although it potently blocked infection (Fig. 5).
HPV16 entry is dependent on actin dynamics, and treatment with cytochalasin D (cytoD) blocks infection, causing receptor-bound virions to be retained on the surface (10). Although infection was strongly abrogated (Fig. 5B), furin cleavage was unaffected by cytoD (Fig. 5A), suggesting that cleavage is independent of entry. Other postentry inhibitors of HPV infection—the acidification inhibitor bafilomycin A (BafA), γ-secretase inhibitor XXI, and the cell cycle S-phase blocker aphidicolin (Aph)—all had no inhibitory effect on cleavage levels while potently blocking infectivity (Fig. 5). Previously we have shown that infection in the presence of the protein disulfide isomerase (PDI) inhibitor bacitracin (Bac) is blocked postentry, with failure of viral genomes to localize to nuclear PML bodies (29). This inhibitory phenotype is quite similar to that of furin inhibition by dRVKR or mutation of the L2 furin cleavage site (9), raising the possibility that Bac may simply inhibit furin cleavage of L2. We therefore tested Bac and found that it had no direct effect on furin cleavage of PSTCD-L2 (Fig. 5A), suggesting that Bac causes a similar inhibitory phenotype through a different mechanism downstream of furin cleavage. The sensitivity of PSTCD-L2 virus to established biochemical inhibitors and neutralizing antibodies was comparable to that of wt virus, further suggesting that the modified virus behaves like the wt with respect to cellular infection.
Cyclophilins are not essential for furin cleavage.
Drug titration experiments were performed to further confirm and compare the initial findings with dRVKR and CsA, measuring PSTCD-L2 infectivity by luciferase assay and L2 cleavage by Western blotting. Inhibition of furin with increasing concentrations of dRVKR caused a potent block of both L2 cleavage (Fig. 6A) and viral infection (Fig. 6B). When cleavage was measured by densitometry and plotted along with infectivity, the lines were nearly coincident (Fig. 6B). In contrast, no significant inhibition of cleavage was observed across a range of CsA concentrations (Fig. 6C), although infectivity was blocked in a dose-dependent manner, with concentrations at or above 5 μM causing near complete inhibition (Fig. 6D). Infection of EGFP-expressing PSTCD-L2 was also potently blocked using these inhibitory concentrations of dRVKR and CsA, and as before, CsA had a minimal effect on L2 cleavage (Table 1).
FIG 6.
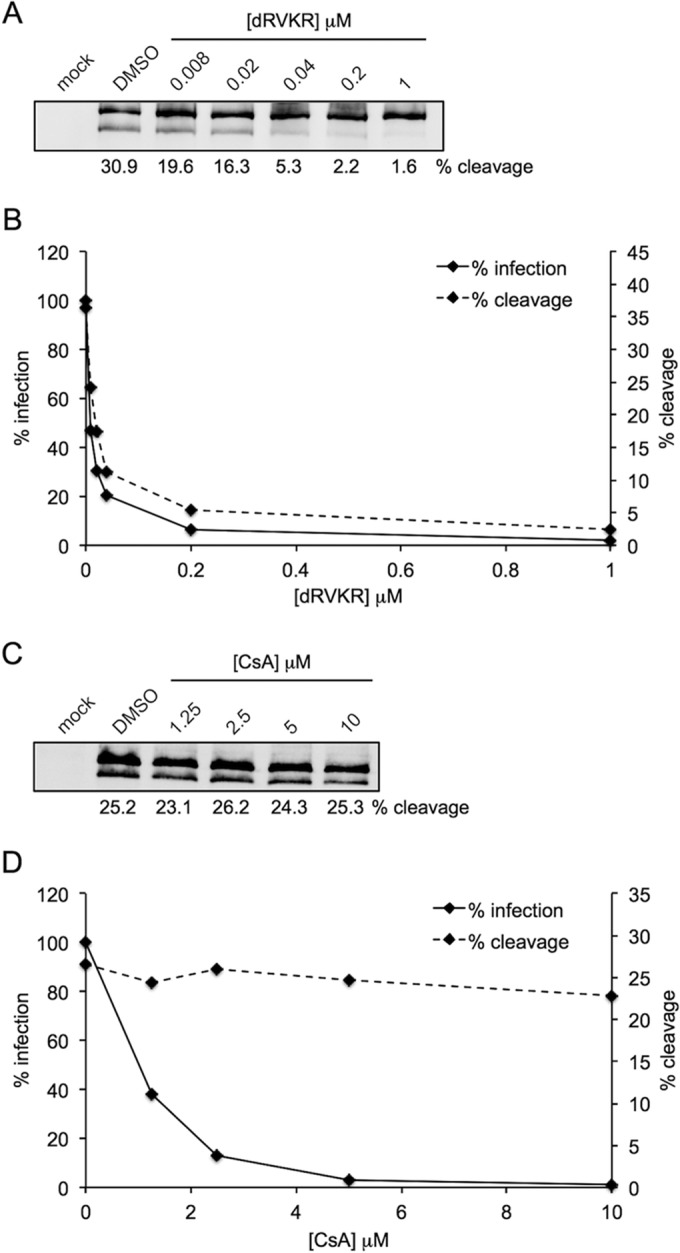
Titration of furin and cyclophilin inhibitors. (A and C) Representative cleavage blots from PSTCD-L2 infected HaCaT cells with increasing concentrations of dRVKR (A) or CsA (C). (B and D) The average cleavage from two independent experiments was measured by densitometry and plotted with infectivity as measured by luciferase assay for titration with dRVKR (B) or CsA (D).
Prior work suggesting a role for cyclophilins in furin cleavage was based largely on indirect immunofluorescence using the L2-specific RG-1 monoclonal antibody, which binds to residues 17 to 36. It has been shown that during infection, this epitope becomes exposed on the surface of cell-associated virions in a time- and furin-dependent manner. This conformational change can be detected by live-cell immunofluorescence with RG-1 antibody, and inhibition of furin with dRVKR causes a potent loss of RG-1 immunoreactivity (41). Thus, live-cell RG-1 immunoreactivity is commonly regarded as a marker for furin cleavage, and the observation that CsA potently blocks RG-1 staining was interpreted as an inhibition of furin cleavage (40). To clarify the effects of CsA on RG-1 epitope exposure, we performed RG-1 live-cell immunofluorescence with PSTCD-L2 virus. As previously reported, RG-1 staining was time dependent and was potently blocked by both dRVKR and CsA (Fig. 7). Similar results were observed for wt virus as well (data not shown). Thus, CsA treatment blocks RG-1 epitope exposure, with minimal effect on furin cleavage (Fig. 5 and 6), suggesting that RG-1 epitope accessibility is not necessarily indicative of L2 cleavage. These data also suggest that like wt HPV16, PSTCD-L2 virions undergo time-, furin-, and cyclophilin-dependent conformational changes to expose the RG-1 epitope.
FIG 7.
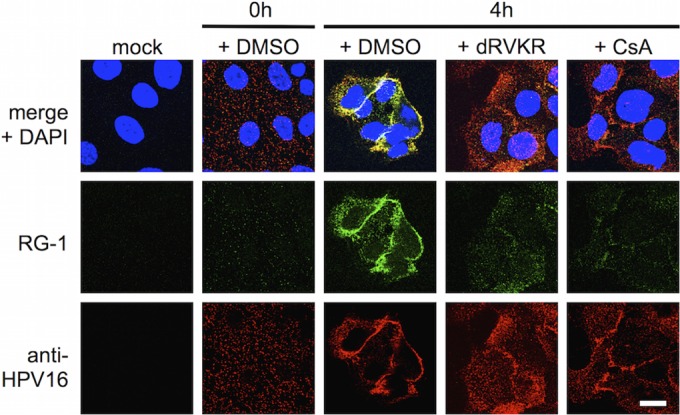
Exposure of the RG-1 epitope during infection. Mock- or PSTCD-L2-infected HaCaT cells were treated with DMSO, dRVKR, or CsA as indicated. Chilled live cells were stained with RG-1 monoclonal antibody and anti-HPV16 polyclonal antibody as described in Materials and Methods at 0 h or 4 h postinfection. Scale bar = 15 μm.
Prior work suggested a role for cyclophilin B in both HPV16 infection and the exposure of the RG-1 epitope on the surface of cell-bound virions (40). To further assess the potential involvement of CypB without using biochemical inhibitors, we performed siRNA knockdown experiments of furin and CypB, looking at both L2 cleavage and infectivity of PSTCD-L2 virus. Specific and strong knockdown was achieved for each siRNA pool compared to the scramble control (Fig. 8A). Only siRNA knockdown of furin had an inhibitory effect on L2 cleavage, strongly suggesting that there is little, if any, role for CypB in modulating furin cleavage (Fig. 8A). Knockdown of furin potently blocked infection, while CypB siRNA had no effect (Fig. 8B). These data suggest that there is no role for CypB in HPV16 infection of HaCaT cells and that the mechanism of CsA inhibition is independent of both furin and CypB.
FIG 8.
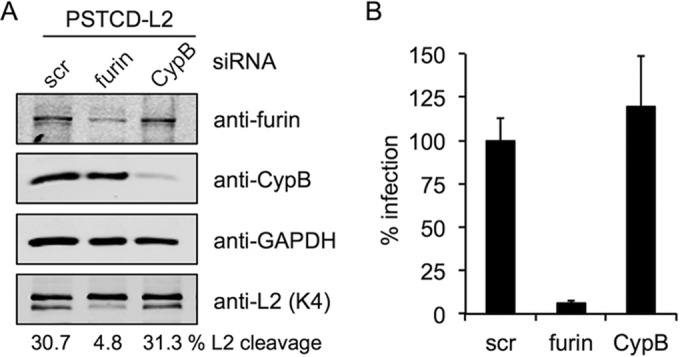
Cyclophilin B is not required for cleavage or infection. Shown are siRNA knockdown and PSTCD-L2 infection of HaCaT cells. Scramble (Scr) control, furin, or CypB specific siRNAs were transfected into HaCaT cells 24 h prior to infection. (A) Immunostaining for furin and CypB shows specific knockdown, GAPDH staining shows equivalent loading, and K4 staining of lysates shows cleavage levels. (B) Infectivity of PSTCD-L2 virus after Scr, furin, or CypB siRNA knockdown.
Given the published work implicating a role for cell surface cyclophilins in furin cleavage (23, 40), we were concerned that our contradicting results with CsA may have been an artifact of the N-terminal PSTCD-L2 fusion. To address this, we compared the effects of furin and cyclophilin inhibitors on cleavage of the L2-HA virus with an unmodified N terminus. Replicates from several independent experiments reveal that cleavage of L2-HA ranged from 24 to 40% in DMSO-treated samples, and treatment with 25 μM dRVKR abrogated cleavage to nearly undetectable levels (Fig. 9). In contrast, the inhibition of cyclophilins with 10 μM CsA caused only a modest change in L2 cleavage levels compared to DMSO (Fig. 9, replicates 1 to 5). Overall L2 cleavage in the presence of CsA ranged from 16 to 36%, and although the average cleavage levels of the CsA-treated groups were sometimes lower than that of DMSO, they were certainly not blocked for furin cleavage like the dRVKR-treated samples. A separate experiment using 2.5 μM dRVKR and 5 μM CsA, concentrations that block infection of wt and PSTCD-L2 viruses, gave similar results, with CsA having a minimal effect on cleavage (Fig. 9, replicate 6). These results clearly show that inhibitory concentrations of CsA have a minimal, if any, effect on L2 cleavage. It is apparent that without the N-terminal PSTCD modification, obtaining quality blots with clear separation of cleaved and uncleaved L2 bands is difficult, with significant variability between experiments. Previously published Western blots also illustrate the difficulty involved with detection and quantification of L2 cleavage, with only small epitope or no N-terminal modification (23, 62). These difficulties and variability were the motivation behind development of the PSTCD-L2 virus.
FIG 9.
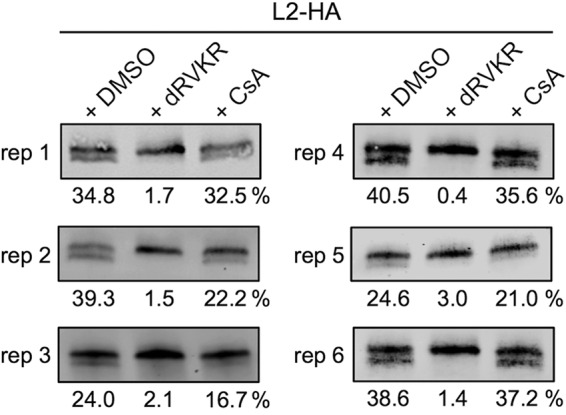
CsA has a minimal effect on cleavage of L2 containing an unmodified N terminus. Shown are representative cleavage blots of L2-HA infected HaCaT cells in the presence of DMSO carrier, furin inhibitor dRVKR, or cyclophilin inhibitor CsA. Replicates (reps) 1 to 5 used dRVKR and CsA at 25 μM and 10 μM, respectively. Replicate 6 used dRVKR and CsA at 2.5 μM and 5 μM, respectively.
DISCUSSION
Furin is a critical cellular factor for HPV16 infection. Although two prior studies have documented cleavage of capsid-associated L2 during cellular entry (23, 62), neither quantitatively measured L2 cleavage. In this study, the N-terminal addition of the small PSTCD domain to L2 greatly facilitated detection and quantification of cleavage, revealing that under normal HaCaT cell culture conditions, L2 cleavage levels plateau at roughly 25 to 35% of the total L2 molecules. The fraction of cleaved L2 could be raised through the addition of extra furin, indicating that viral infectivity is roughly proportional to the level of L2 cleavage.
The measurement of L2 cleavage levels by Western blotting has the inherent limitation that effects can only be observed at the bulk population level. Individual virions contain 20 to 40 molecules of L2. Thus, at two extremes, the cleavage plateau could be a consequence of either complete cleavage of L2 within 30% of the virions of the population or all of the virions being 30% cleaved. Unfortunately, we are unable to make any definitive conclusions in this regard, although we hypothesize that cleavage likely follows a Gaussian distribution, with a peak number of virions being 30% cleaved and decreasing numbers of virions with greater or lesser degrees of cleavage. Boosting the levels of L2 cleavage by furin supplementation of EGFP-expressing PSTCD-L2 caused a marked increase in the number of infected cells (Table 1). This result may be interpreted as raising the effective MOI and could be due to a shifting of the Gaussian distribution above a certain threshold amount that is required for efficient infectivity. We can only speculate until more advanced tools are available to study L2 cleavage at the level of individual virions.
We show that cleavage can occur at one of two N-terminal sites in HPV16 L2, but cleavage must occur at Arg12 for a productive infection. Elimination of the disulfide bond through mutation of Cys22 and Cys28 abrogated infection without any effect on furin cleavage, suggesting no structural role for the C22-C28 disulfide in regard to cleavage. Cleavage occurs extracellularly, as blocking viral entry through biochemical inhibitors or neutralizing antibodies had no effect on cleavage levels. Cleavage was largely unaffected by postentry inhibitors of HPV16, including bafilomycin A, γ-secretase inhibitor XXI, the S-phase blocker aphidicolin, and bacitracin.
Interestingly, two different L1-specific neutralizing antibodies had very different effects on L2 cleavage. V5 was able to completely block cleavage through a dose-dependent steric mechanism, while U4 had no effect on cleavage. Recent cryo-electron microscopy (cryo-EM) studies reveal that while V5 can coat virions by binding to specific loops on the surface of the L1 pentamers (56), U4 preferentially bound to invading arms surrounding the pentavalent L1 pentamers at the 5-fold vertices of the capsid (63). The lack of any observable effect of U4 on furin cleavage suggests that the L2 N terminus may be surface exposed at locations other than the gaps surrounding the 5-fold icosahedral vertices, perhaps through gaps at the 2-fold or 3-fold axes of symmetry.
The L2-specific K4 antibody unexpectedly enhanced L2 cleavage during infection. This puzzling observation could be explained by K4 having a sufficiently high affinity to lock its largely buried (but perhaps transiently exposed or “breathing”) epitope in a conformation that is favorable for furin cleavage. Further experimentation is needed to understand this intriguing phenomenon.
Most surprisingly, cleavage was largely unaffected by the PPI inhibitor CsA or siRNA knockdown of CypB. Furthermore, our results with CypB knockdown suggest no role for this PPI in HPV16 infection. These CypB siRNA findings directly contradict those reported by Bienkowska-Haba and colleagues (23, 40). While no definitive explanation can be provided to reconcile this discrepancy, some noteworthy differences in experimental design should be pointed out. The previous work utilized approximately 10-fold more siRNA duplex than did this work, and 72-h infections were performed at 48 h after siRNA transfection in that study, compared to 24-h infections of cells 24 h after siRNA transfection in this work. More importantly, the siRNA duplexes reported in reference 40 are not specific to CypB. A close look at the PPIB (CypB) locus at 15q21-q22 reveals that the 3′ end of the PPIB mRNA transcript has significant overlap with the 3′ end of the sorting nexin 22 (SNX22) mRNA transcript at 15q22.31. In fact, these transcripts run antisense each other, sharing 1,666 bp of homology. Alignment of the two siRNA duplexes targeting CypB in the work described in reference 40 revealed that they have perfect complementarity to both the PPIB and SNX22 transcripts. Research has shown that both strands of siRNA duplexes can incorporate into the RNA-induced silencing complex (RISC) and target their complementary transcripts in similar situations (64). SNX22 is a cytosolic sorting nexin likely involved with endocytosis and endosomal sorting (65). Importantly, recent work has implicated two other sorting nexins in HPV entry (26, 27). Thus, it is not unreasonable to suggest that knockdown of SNX22 could potentially contribute to the subtle decrease of HPV infection as reported by Bienkowska-Haba and colleagues upon use of those siRNA duplexes. The pool of three CypB-specific siRNA duplexes we used in this work do not share this complementarity with SNX22.
The infectivity of both wt HPV16 or PSTCD-L2 virus was unaffected by CypB knockdown in our hands, yet it was nearly abolished upon CsA treatment. This likely means that other PPIs like CypA contribute elsewhere in the infectious pathway. Unfortunately, we were unable to get consistent and adequate knockdown of CypA to directly assess its involvement, but prior research suggests the involvement of CypA and PPI activity in partitioning of L1/L2 capsid proteins and trafficking of the L2/vDNA complex to the TGN (23, 24).
Furin is a necessary host factor for a variety microbial pathogens. Many enveloped viruses rely on cellular furin for proteolytic activation of their membrane glycoproteins during the initial phases of infection. Cleavage results in conformational changes that enable exposure of their hydrophobic fusion peptides that mediate fusion of the viral and cellular membranes (35, 66). Likewise, a number of membrane-translocating bacterial toxins utilize furin in an analogous manner, with cleavage acting as a conformational switch to promote oligomerization and/or membrane penetration of the toxin (67). It is probable that L2 exploits cell surface furin in an analogous manner. Removal of the highly basic N-terminal 12 residues of HPV16 L2 likely acts as a trigger to induce a structural change necessary for membrane penetration and the proper downstream sorting and translocation of the L2/genome complex from the TGN.
Direct evidence for such a furin-dependent conformational change comes from prior work with the cross-neutralizing RG-1 antibody, which binds to residues 17 to 36 of L2, just downstream of the furin cleavage site (59). Day and colleagues have shown that the epitope at residues 17 to 36 is buried in mature PsV particles, becoming exposed for RG-1 binding upon cleavage of L2 on the cell surface (41), a finding that has been confirmed through immunofluorescence and immunoprecipitation experiments, in cell culture systems, and in an in vivo murine cervicovaginal challenge model (51, 68). RG-1 immunoreactivity has thus become regarded as a convenient proxy for furin cleavage of L2 during infection. Bienkowska-Haba and colleagues found that RG-1 epitope exposure was potently blocked by the PPI inhibitor CsA and concluded that proline isomerization of L2 by cell surface CypB is required for furin cleavage, subsequent furin-dependent RG-1 epitope exposure, and infection (40). Mutagenesis experiments suggested that isomerization of L2 at Pro100 causes a structural change, as a G99A, P100A double mutant was able to expose the RG-1 epitope independently of CypB activity in the presence of CsA and other PPI inhibitors. Notably, the G99A, P100A mutant was fully infectious compared to the wt, but infectivity remained sensitive to CsA, suggesting a role for PPI activity downstream of the early cell surface events. Indeed, cyclophilin activity has been implicated in the partitioning of L1 capsid from L2/vDNA complex later in the endosomal compartment and appears to be necessary for trafficking of vDNA to the TGN and nucleus (23, 24).
The work presented herein is the first to question the relationship between furin cleavage and exposure of the RG-1 epitope. Our data clearly show that unlike RG-1 epitope exposure, furin cleavage occurs independently of PPI or CypB activity, as PPI inhibition with CsA or siRNA knockdown of CypB had little effect on cleavage of L2. Either PPI-mediated isomerization of L2 could be downstream of furin cleavage, with both processes being required for RG-1 epitope exposure, or isomerization and cleavage may occur concomitantly yet independently, with both being required for the structural changes that allow for efficient RG-1 binding. In either case it is unclear if isomerization of L2 and exposure of the RG-1 epitope are even necessary for successful infection. Further work is needed to completely understand the relationship between these two processes, but we urge caution when interpreting RG-1 binding data as a marker of furin cleavage, as has been done in a number of recent studies (8, 29, 40, 69).
ACKNOWLEDGMENTS
This work was supported by grant 1R01AI108751-01 from the National Institute for Allergy and Infectious Diseases and grant RSG 117469 from the American Cancer Society. K.N.B. and A.M.S. were funded in part by a grant to UA from HHMI (52006942) that supports the undergraduate biology research program.
We gratefully acknowledge Martin Müller (DKFZ, Heidelberg, Germany) for the K4L220-38 monoclonal antibody, Richard Roden (Johns Hopkins University, Baltimore, MD) for the RG-1 monoclonal antibody, Neil Christensen (Penn State University, Hershey, PA) for the H16.V5 and H16.U4 monoclonal antibodies, Michelle Ozbun (University of New Mexico School of Medicine, Albuquerque, NM) for the anti-HPV16 polyclonal sera, Patricia Day of the Schiller Group (Laboratory of Cellular Oncology, National Cancer Institute, NIH, Bethesda, MD) for the FurΔ1 furin-secreting CHO cells, and Steven Leppla (Laboratory of Parasitic Diseases, National Institute of Allergy and Infectious Diseases, NIH, Bethesda, MD) for the furin-deficient FD11 CHO cells. We thank Dena Yoder of the UA BIO5 Media Facility, Patty Jansma of the UA ORD Imaging Core-Marley, and Paula Campbell and John Fitch of the UACC/ARL Cytometry Core Facility, which is funded by a UA Cancer Center Support Grant (CCSG-CA 023074).
AM.B.P., C.M.C., and S.K.C. designed and conceived the experiments. M.P.B., C.M.C., S.F.C., S.L., M.L., J.A.C., K.N.B., A.M.S., and S.K.C. performed the experiments and analyzed the data. S.K.C., M.P.B., and C.M.C. wrote the paper.
REFERENCES
- 1.Centers for Disease Control and Prevention. 2015. Genital HPV infection—fact sheet. Centers for Disease Control and Prevention, Atlanta, GA: http://www.cdc.gov/std/hpv/stdfact-hpv.htm. [Google Scholar]
- 2.Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR, Stanley MA. 2012. The biology and life-cycle of human papillomaviruses. Vaccine 30(Suppl 5):F55–F70. [DOI] [PubMed] [Google Scholar]
- 3.Forman D, de Martel C, Lacey CJ, Soerjomataram I, Lortet-Tieulent J, Bruni L, Vignat J, Ferlay J, Bray F, Plummer M, Franceschi S. 2012. Global burden of human papillomavirus and related diseases. Vaccine 30(Suppl 5):F12–F23. [DOI] [PubMed] [Google Scholar]
- 4.Buck CB, Day PM, Trus BL. 2013. The papillomavirus major capsid protein L1. Virology 445:169–174. doi: 10.1016/j.virol.2013.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Favre M, Breitburd F, Croissant O, Orth G. 1977. Chromatin-like structures obtained after alkaline disruption of bovine and human papillomaviruses. J Virol 21:1205–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang JW, Roden RB. 2013. L2, the minor capsid protein of papillomavirus. Virology 445:175–186. doi: 10.1016/j.virol.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cerqueira C, Liu Y, Kuhling L, Chai W, Hafezi W, van Kuppevelt TH, Kuhn JE, Feizi T, Schelhaas M. 2013. Heparin increases the infectivity of human papillomavirus type 16 independent of cell surface proteoglycans and induces L1 epitope exposure. Cell Microbiol 15:1818–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richards KF, Bienkowska-Haba M, Dasgupta J, Chen XS, Sapp M. 2013. Multiple heparan sulfate binding site engagements are required for the infectious entry of human papillomavirus type 16. J Virol 87:11426–11437. doi: 10.1128/JVI.01721-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richards RM, Lowy DR, Schiller JT, Day PM. 2006. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc Natl Acad Sci U S A 103:1522–1527. doi: 10.1073/pnas.0508815103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schelhaas M, Shah B, Holzer M, Blattmann P, Kuhling L, Day PM, Schiller JT, Helenius A. 2012. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog 8:e1002657. doi: 10.1371/journal.ppat.1002657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aksoy P, Abban CY, Kiyashka E, Qiang W, Meneses PI. 2014. HPV16 infection of HaCaTs is dependent on beta4 integrin, and alpha6 integrin processing. Virology 449:45–52. doi: 10.1016/j.virol.2013.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dziduszko A, Ozbun MA. 2013. Annexin A2 and S100A10 regulate human papillomavirus type 16 entry and intracellular trafficking in human keratinocytes. J Virol 87:7502–7515. doi: 10.1128/JVI.00519-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raff AB, Woodham AW, Raff LM, Skeate JG, Yan L, Da Silva DM, Schelhaas M, Kast WM. 2013. The evolving field of human papillomavirus receptor research: a review of binding and entry. J Virol 87:6062–6072. doi: 10.1128/JVI.00330-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheffer KD, Gawlitza A, Spoden GA, Zhang XA, Lambert C, Berditchevski F, Florin L. 2013. Tetraspanin CD151 mediates papillomavirus type 16 endocytosis. J Virol 87:3435–3446. doi: 10.1128/JVI.02906-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spoden G, Freitag K, Husmann M, Boller K, Sapp M, Lambert C, Florin L. 2008. Clathrin- and caveolin-independent entry of human papillomavirus type 16—involvement of tetraspanin-enriched microdomains (TEMs). PLoS One 3:e3313. doi: 10.1371/journal.pone.0003313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spoden G, Kuhling L, Cordes N, Frenzel B, Sapp M, Boller K, Florin L, Schelhaas M. 2013. Human papillomavirus types 16, 18, and 31 share similar endocytic requirements for entry. J Virol 87:7765–7773. doi: 10.1128/JVI.00370-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Surviladze Z, Dziduszko A, Ozbun MA. 2012. Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS Pathog 8:e1002519. doi: 10.1371/journal.ppat.1002519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Müller KH, Spoden GA, Scheffer KD, Brunnhofer R, De Brabander JK, Maier ME, Florin L, Muller CP. 2014. Inhibition by cellular vacuolar ATPase impairs human papillomavirus uncoating and infection. Antimicrob Agents Chemother 58:2905–2911. doi: 10.1128/AAC.02284-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Day PM, Thompson CD, Schowalter RM, Lowy DR, Schiller JT. 2013. Identification of a role for the trans-Golgi network in human papillomavirus 16 pseudovirus infection. J Virol 87:3862–3870. doi: 10.1128/JVI.03222-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lipovsky A, Popa A, Pimienta G, Wyler M, Bhan A, Kuruvilla L, Guie MA, Poffenberger AC, Nelson CD, Atwood WJ, DiMaio D. 2013. Genome-wide siRNA screen identifies the retromer as a cellular entry factor for human papillomavirus. Proc Natl Acad Sci U S A 110:7452–7457. doi: 10.1073/pnas.1302164110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Popa A, Zhang W, Harrison MS, Goodner K, Kazakov T, Goodwin EC, Lipovsky A, Burd CG, DiMaio D. 2015. Direct binding of retromer to human papillomavirus type 16 minor capsid protein L2 mediates endosome exit during viral infection. PLoS Pathog 11:e1004699. doi: 10.1371/journal.ppat.1004699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang W, Kazakov T, Popa A, DiMaio D. 2014. Vesicular trafficking of incoming human papillomavirus 16 to the Golgi apparatus and endoplasmic reticulum requires gamma-secretase activity. mBio 5:e01777-14. doi: 10.1128/mBio.01777-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bienkowska-Haba M, Williams C, Kim SM, Garcea RL, Sapp M. 2012. Cyclophilins facilitate dissociation of the human papillomavirus type 16 capsid protein L1 from the L2/DNA complex following virus entry. J Virol 86:9875–9887. doi: 10.1128/JVI.00980-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DiGiuseppe S, Bienkowska-Haba M, Hilbig L, Sapp M. 2014. The nuclear retention signal of HPV16 L2 protein is essential for incoming viral genome to transverse the trans-Golgi network. Virology 458–459:93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calton CM, Schlegel AM, Chapman JA, Campos SK. 2013. Human papillomavirus type 16 does not require cathepsin L or B for infection. J Gen Virol 94:1865–1869. doi: 10.1099/vir.0.053694-0. [DOI] [PubMed] [Google Scholar]
- 26.Bergant M, Banks L. 2013. SNX17 facilitates infection with diverse papillomavirus types. J Virol 87:1270–1273. doi: 10.1128/JVI.01991-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bergant Marusic M, Ozbun MA, Campos SK, Myers MP, Banks L. 2012. Human papillomavirus L2 facilitates viral escape from late endosomes via sorting nexin 17. Traffic 13:455–467. doi: 10.1111/j.1600-0854.2011.01320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schneider MA, Spoden GA, Florin L, Lambert C. 2011. Identification of the dynein light chains required for human papillomavirus infection. Cell Microbiol 13:32–46. doi: 10.1111/j.1462-5822.2010.01515.x. [DOI] [PubMed] [Google Scholar]
- 29.Campos SK, Chapman JA, Deymier MJ, Bronnimann MP, Ozbun MA. 2012. Opposing effects of bacitracin on human papillomavirus type 16 infection: enhancement of binding and entry and inhibition of endosomal penetration. J Virol 86:4169–4181. doi: 10.1128/JVI.05493-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aydin I, Weber S, Snijder B, Samperio Ventayol P, Kuhbacher A, Becker M, Day PM, Schiller JT, Kann M, Pelkmans L, Helenius A, Schelhaas M. 2014. Large scale RNAi reveals the requirement of nuclear envelope breakdown for nuclear import of human papillomaviruses. PLoS Pathog 10:e1004162. doi: 10.1371/journal.ppat.1004162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bronnimann MP, Chapman JA, Park CK, Campos SK. 2013. A transmembrane domain and GxxxG motifs within L2 are essential for papillomavirus infection. J Virol 87:464–473. doi: 10.1128/JVI.01539-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pyeon D, Pearce SM, Lank SM, Ahlquist P, Lambert PF. 2009. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog 5:e1000318. doi: 10.1371/journal.ppat.1000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Day PM, Baker CC, Lowy DR, Schiller JT. 2004. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc Natl Acad Sci U S A 101:14252–14257. doi: 10.1073/pnas.0404229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas G. 2002. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol 3:753–766. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakayama K. 1997. Furin: a mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem J 327(Part 3):625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campos SK, Barry MA. 2006. Comparison of adenovirus fiber, protein IX, and hexon capsomeres as scaffolds for vector purification and cell targeting. Virology 349:453–462. doi: 10.1016/j.virol.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 37.Campos SK, Ozbun MA. 2009. Two highly conserved cysteine residues in HPV16 L2 form an intramolecular disulfide bond and are critical for infectivity in human keratinocytes. PLoS One 4:e4463. doi: 10.1371/journal.pone.0004463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rubio I, Seitz H, Canali E, Sehr P, Bolchi A, Tommasino M, Ottonello S, Muller M. 2011. The N-terminal region of the human papillomavirus L2 protein contains overlapping binding sites for neutralizing, cross-neutralizing and non-neutralizing antibodies. Virology 409:348–359. doi: 10.1016/j.virol.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 39.Abramoff MD, Magalhaes PJ, Ram SJ. 2004. Image processing with ImageJ. Biophotonics Int 11:36–42. [Google Scholar]
- 40.Bienkowska-Haba M, Patel HD, Sapp M. 2009. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog 5:e1000524. doi: 10.1371/journal.ppat.1000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Day PM, Gambhira R, Roden RB, Lowy DR, Schiller JT. 2008. Mechanisms of human papillomavirus type 16 neutralization by L2 cross-neutralizing and L1 type-specific antibodies. J Virol 82:4638–4646. doi: 10.1128/JVI.00143-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buck CB, Cheng N, Thompson CD, Lowy DR, Steven AC, Schiller JT, Trus BL. 2008. Arrangement of L2 within the papillomavirus capsid. J Virol 82:5190–5197. doi: 10.1128/JVI.02726-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Windram OP, Weber B, Jaffer MA, Rybicki EP, Shepherd DN, Varsani A. 2008. An investigation into the use of human papillomavirus type 16 virus-like particles as a delivery vector system for foreign proteins: N- and C-terminal fusion of GFP to the L1 and L2 capsid proteins. Arch Virol 153:585–589. doi: 10.1007/s00705-007-0025-2. [DOI] [PubMed] [Google Scholar]
- 44.Parrott MB, Adams KE, Mercier GT, Mok H, Campos SK, Barry MA. 2003. Metabolically biotinylated adenovirus for cell targeting, ligand screening, and vector purification. Mol Ther 8:688–700. doi: 10.1016/S1525-0016(03)00213-2. [DOI] [PubMed] [Google Scholar]
- 45.Christakos KJ, Chapman JA, Fane BA, Campos SK. 2016. PhiXing-it, displaying foreign peptides on bacteriophage PhiX174. Virology 488:242–248. doi: 10.1016/j.virol.2015.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reddy DV, Shenoy BC, Carey PR, Sonnichsen FD. 2000. High resolution solution structure of the 1.3S subunit of transcarboxylase from Propionibacterium shermanii. Biochemistry 39:2509–2516. doi: 10.1021/bi9925367. [DOI] [PubMed] [Google Scholar]
- 47.Esko JD, Stewart TE, Taylor WH. 1985. Animal cell mutants defective in glycosaminoglycan biosynthesis. Proc Natl Acad Sci U S A 82:3197–3201. doi: 10.1073/pnas.82.10.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Day PM, Lowy DR, Schiller JT. 2008. Heparan sulfate-independent cell binding and infection with furin-precleaved papillomavirus capsids. J Virol 82:12565–12568. doi: 10.1128/JVI.01631-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chiron MF, Fryling CM, FitzGerald D. 1997. Furin-mediated cleavage of Pseudomonas exotoxin-derived chimeric toxins. J Biol Chem 272:31707–31711. doi: 10.1074/jbc.272.50.31707. [DOI] [PubMed] [Google Scholar]
- 50.Gordon VM, Klimpel KR, Arora N, Henderson MA, Leppla SH. 1995. Proteolytic activation of bacterial toxins by eukaryotic cells is performed by furin and by additional cellular proteases. Infect Immun 63:82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang JW, Jagu S, Kwak K, Wang C, Peng S, Kirnbauer R, Roden RB. 2014. Preparation and properties of a papillomavirus infectious intermediate and its utility for neutralization studies. Virology 449:304–316. doi: 10.1016/j.virol.2013.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mamoor S, Onder Z, Karanam B, Kwak K, Bordeaux J, Crosby L, Roden RB, Moroianu J. 2012. The high risk HPV16 L2 minor capsid protein has multiple transport signals that mediate its nucleocytoplasmic traffic. Virology 422:413–424. doi: 10.1016/j.virol.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roden RB, Day PM, Bronzo BK, Yutzy WHt Yang Y, Lowy DR, Schiller JT. 2001. Positively charged termini of the L2 minor capsid protein are necessary for papillomavirus infection. J Virol 75:10493–10497. doi: 10.1128/JVI.75.21.10493-10497.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sen J, Jacobs A, Jiang H, Rong L, Caffrey M. 2007. The disulfide loop of gp41 is critical to the furin recognition site of HIV gp160. Protein Sci 16:1236–1241. doi: 10.1110/ps.072771407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Day PM, Thompson CD, Buck CB, Pang YY, Lowy DR, Schiller JT. 2007. Neutralization of human papillomavirus with monoclonal antibodies reveals different mechanisms of inhibition. J Virol 81:8784–8792. doi: 10.1128/JVI.00552-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee H, Brendle SA, Bywaters SM, Guan J, Ashley RE, Yoder JD, Makhov AM, Conway JF, Christensen ND, Hafenstein S. 2015. A cryo-electron microscopy study identifies the complete H16.V5 epitope and reveals global conformational changes initiated by binding of the neutralizing antibody fragment. J Virol 89:1428–1438. doi: 10.1128/JVI.02898-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cardone G, Moyer AL, Cheng N, Thompson CD, Dvoretzky I, Lowy DR, Schiller JT, Steven AC, Buck CB, Trus BL. 2014. Maturation of the human papillomavirus 16 capsid. mBio 5:e01104-14. doi: 10.1128/mBio.01104-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carter JJ, Wipf GC, Benki SF, Christensen ND, Galloway DA. 2003. Identification of a human papillomavirus type 16-specific epitope on the C-terminal arm of the major capsid protein L1. J Virol 77:11625–11632. doi: 10.1128/JVI.77.21.11625-11632.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gambhira R, Karanam B, Jagu S, Roberts JN, Buck CB, Bossis I, Alphs HH, Culp T, Christensen ND, Roden RBS. 2007. A protective and broadly cross-neutralizing epitope of human papillomavirus L2. J Virol 81:13927–13931. doi: 10.1128/JVI.00936-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cameron A, Appel J, Houghten RA, Lindberg I. 2000. Polyarginines are potent furin inhibitors. J Biol Chem 275:36741–36749. doi: 10.1074/jbc.M003848200. [DOI] [PubMed] [Google Scholar]
- 61.Sapp M, Bienkowska-Haba M. 2009. Viral entry mechanisms: human papillomavirus and a long journey from extracellular matrix to the nucleus. FEBS J 276:7206–7216. doi: 10.1111/j.1742-4658.2009.07400.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wiens ME, Smith JG. 2015. Alpha-defensin HD5 inhibits furin cleavage of human papillomavirus 16 L2 to block infection. J Virol 89:2866–2874. doi: 10.1128/JVI.02901-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guan J, Bywaters SM, Brendle SA, Lee H, Ashley RE, Christensen ND, Hafenstein S. 2015. The U4 antibody epitope on human papillomavirus 16 identified by cryo-electron microscopy. J Virol 89:12108–12117. doi: 10.1128/JVI.02020-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wei JX, Yang J, Sun JF, Jia LT, Zhang Y, Zhang HZ, Li X, Meng YL, Yao LB, Yang AG. 2009. Both strands of siRNA have potential to guide posttranscriptional gene silencing in mammalian cells. PLoS One 4:e5382. doi: 10.1371/journal.pone.0005382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Worby CA, Dixon JE. 2002. Sorting out the cellular functions of sorting nexins. Nat Rev Mol Cell Biol 3:919–931. doi: 10.1038/nrm974. [DOI] [PubMed] [Google Scholar]
- 66.Schowalter RM, Smith SE, Dutch RE. 2006. Characterization of human metapneumovirus F protein-promoted membrane fusion: critical roles for proteolytic processing and low pH. J Virol 80:10931–10941. doi: 10.1128/JVI.01287-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gordon VM, Leppla SH. 1994. Proteolytic activation of bacterial toxins: role of bacterial and host cell proteases. Infect Immun 62:333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kines RC, Thompson CD, Lowy DR, Schiller JT, Day PM. 2009. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc Natl Acad Sci U S A 106:20458–20463. doi: 10.1073/pnas.0908502106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cerqueira C, Samperio Ventayol P, Vogeley C, Schelhaas M. 2015. Kallikrein-8 proteolytically processes human papillomaviruses in the extracellular space to facilitate entry into host cells. J Virol 89:7038–7052. doi: 10.1128/JVI.00234-15. [DOI] [PMC free article] [PubMed] [Google Scholar]