

ABSTRACT
The structural proteins of flaviviruses carry a unique set of transmembrane domains (TMDs) at their C termini that are derived from the mode of viral polyprotein processing. They function as internal signal and stop-transfer sequences during protein translation, but possible additional roles in protein interactions required during assembly and maturation of viral particles are ill defined. To shed light on the role of TMDs in these processes, we engineered a set of tick-borne encephalitis virus mutants in which these structural elements were replaced in different combinations by the homologous sequences of a distantly related flavivirus (Japanese encephalitis virus). The effects of these modifications were analyzed with respect to protein synthesis, viral particle secretion, specific infectivity, and acidic-pH-induced maturation processes. We provide evidence that interactions involving the double-membrane anchor of the envelope protein E (a unique feature compared to other viral fusion proteins) contribute substantially to particle assembly, stability, and maturation. Disturbances of the inter- and intra-TMD interactions of E resulted in the secretion of a larger proportion of capsidless subviral particles at the expense of whole virions, suggesting a possible role in the still incompletely understood mechanism of capsid integration during virus budding. In contrast, the TMD initially anchoring the C protein to the endoplasmic reticulum membrane does not appear to take part in envelope protein interactions. We also show that E TMDs are involved in the envelope protein rearrangements that are triggered by acidic pH in the trans-Golgi network and represent a hallmark of virus maturation.
IMPORTANCE The assembly of flaviviruses occurs in the endoplasmic reticulum and leads to the formation of immature, noninfectious particles composed of an RNA-containing capsid surrounded by a lipid membrane, with the two integrated envelope proteins, prM and E, arranged in an icosahedral lattice. The mechanism by which the capsid is formed and integrated into the budding viral envelope is currently unknown. We provide evidence that the transmembrane domains (TMDs) of E are essential for the formation of capsid-containing particles and that disturbances of these interactions lead to the preferential formation of capsidless subviral particles at the expense of whole virions. E TMD interactions also appear to be essential for the envelope protein rearrangements required for virus maturation and for the generation of infectious virions. Our data thus provide new insights into the biological functions of E TMDs and extend their role during viral polyprotein processing to additional functions in particle assembly and maturation.
INTRODUCTION
The genus Flavivirus in the family Flaviviridae comprises 53 taxonomically recognized species (1), including the human-pathogenic mosquito-borne dengue, Zika, yellow fever, Japanese encephalitis, and West Nile viruses as well as tick-borne encephalitis virus (TBEV) (2). Flaviviruses are small enveloped positive-strand RNA viruses with two membrane-associated proteins (prM/M and E) arranged in specific icosahedral lattices at the virion surface that differ between immature and mature forms of the virion. Immature viruses carry 60 spikes of trimers of prM-E heterodimers (3, 4) and assemble by budding into the lumen of the endoplasmic reticulum (ER) (5). This process is driven by lateral interactions between prM-E oligomers and as yet undefined interactions with the capsid (6). How the nucleocapsid is formed and integrated into flavivirus particles is still unresolved. As a by-product, the envelope protein interactions also lead to the generation of subviral particles that contain only prM/M and E associated with a lipid membrane (reviewed in reference 7). Virus maturation occurs during the exocytic transit of immature particles and is triggered by the slightly acidic pH of the trans-Golgi network (TGN). Here the 60 trimeric spikes of prM-E heterodimers rearrange into a herringbone-like lattice of 90 E protein dimers characteristic of mature virions (8). These structural changes result in the exposure of a previously cryptic protease cleavage site in prM, which is cleaved by the TGN protease furin to yield pr and M. The cleaved pr fragment remains associated with E at the virion surface in the TGN at acidic pH, but it falls off when particles encounter neutral pH upon release from the cell by exocytosis (9). In its mature conformation, E is metastable and mediates the viral entry functions of cell attachment as well as low-pH-triggered fusion after endocytic uptake into cells (10).
Both envelope proteins (prM and E) have double-membrane anchors that are remnants of flavivirus polyprotein processing which occurs at the ER membrane (Fig. 1A and B) (5). The first protein to be translated is the C protein, which is initially (before cleavage by the viral NS2B-NS3 protease) anchored to the ER membrane (anchC) by a C-terminal transmembrane domain (TMD) that serves as a signal sequence for the translocation of prM into the lumen of the ER (Fig. 1B). The two TMDs of prM and E serve as stop-transfer signals (TMD1) and as internal signal sequences (TMD2) for the following glycoproteins (Fig. 1B) (5). The presence of such a double-TMD structure at its C terminus makes the flavivirus E protein (a class II viral fusion protein) unique among viral fusion proteins, all of which are anchored to their membranes by single membrane-spanning domains only (11, 12). This difference even holds true for the structurally closely related class II fusion proteins of alpha- and bunyaviruses (13, 14). Cryo-electron microscopy (cryo-EM) structures of immature and mature dengue viruses revealed that the prM and E TMDs form hairpins of antiparallel helices that penetrate the outer membrane leaflet and dip into the inner leaflet but do not reach the intraparticle space (3, 4, 15, 16). Structurally, interactions between the membrane anchors of prM and E have been visualized only in mature, not immature, virions (4). Importantly, the positions of the TMDs are different in the icosahedral envelope protein lattices of immature and mature viruses, indicating the occurrence of lateral movements in the viral membrane during the maturation process (4).
FIG 1.

Representations of the flavivirus genome and polyprotein (A), processing of the structural proteins (B), modifications engineered into the transmembrane domains (TMDs) of prM and E (C), and sequence alignments of the TM regions of prM and E (D). (A) Schematic diagrams of the flavivirus genome and the derived polyprotein, with protease cleavage sites indicated. (B) Membrane topology of the structural proteins and NS1 during polyprotein processing at the ER membrane. The N-terminal C protein is cytosolic and anchored in the ER membrane by a signal sequence (anchC TMD). On the cytosolic side, C is cleaved by the viral NS2B-NS3 protease (open arrow), and on the luminal side, it is cleaved by the host signal peptidase (solid arrow). The signal peptidase also cleaves at the prM-E as well as E-NS1 junctions. (C) Schematic diagrams of the C-terminal modifications of prM and E in the TBEV infectious clone used to produce mutant viruses. (D) Alignments of the amino acid sequences of the prM and E membrane anchor regions of the following flaviviruses (TBEV numbering): TBEV (GenBank accession number U27495), JEV (GenBank accession number EF571853), West Nile virus (WNV) (GenBank accession number DQ211652), dengue type 2 virus (DENV2) (GenBank accession number NC_001474), and yellow fever virus (YFV) (GenBank accession number AY640589). Identical amino acid residues are highlighted in gray. The TMD1 and TMD2 helices are indicated at the bottom. NCR, noncoding region; N-ter, N terminus; TMD, transmembrane domain; WT, wild type.
Using recombinant subviral particles (RSPs) as a model system, previous studies indicated the requirement of E TMD interactions for efficient membrane fusion during virus entry (17), a process mediated by acidic-pH-induced conformational and oligomeric rearrangements of E in the endosome. Preliminary experiments to elucidate similar effects in a whole-virus system were largely unsuccessful because of insufficient recovery of infectious virus (17), suggesting that E TMD interactions might also play a role in virus assembly and/or maturation. Because subviral particles are smaller and have a more pronounced membrane curvature and a different icosahedral arrangement of the E proteins (18), unequivocal conclusions related to the role of E TMDs in the assembly and maturation process of whole virions cannot be drawn from results obtained with this model system. We therefore replaced the TMDs of E as well as prM and anchC—all of which are present at the assembly site—with the functionally homologous but substantially sequence-divergent TMDs of Japanese encephalitis virus (JEV) in an infectious clone of TBEV and studied the effects of these modifications on the generation of infectious particles. Our data provide evidence that interactions involving the TMDs of E contribute to the efficient production and release of infectious virus particles and might be involved in the recruitment of the nucleocapsid. We did not, however, find evidence for an involvement of specific interactions of the anchC TMD (Fig. 1B) in this process. Our study also suggests that the rearrangements occurring on the virus surface during maturation are facilitated by TMD interactions of E.
MATERIALS AND METHODS
Virus sequences.
The different constructs used in this study were based on Western subtype TBEV strain Neudoerfl (GenBank accession number U27495) and JEV strain Nakayama (GenBank accession number EF571853).
Virus plasmids and cloning procedures.
Plasmid pTNd/c contains a full-length genomic cDNA insert of TBEV strain Neudoerfl cloned into the vector pBR322 under the control of a T7 transcription promoter (19). The membrane anchor region (Fig. 1B and C) of wild-type (WT) TBEV E [E(T1-T2)] or prM [prM(T1-T2)] in the pTNd/c vector was replaced by a chemically synthesized DNA fragment (GeneArt AG, Germany) containing the heterologous membrane anchor of JEV [E(J1-J2) or prM(J1-J2)] or a shuffled membrane anchor [E(J1-T2), E(T1-J2), prM(J1-T2), or prM(T1-J2)] by using unique restriction sites. In addition, we constructed a clone in which both the prM and E TMD regions were exchanged with the corresponding elements of JEV [prM(J1-J2)+E(J1-J2)]. The TMD of anchC was replaced in the WT and E(J1-J2) clones, resulting in the C(J) and C(J)+E(J1-J2) mutants. The amino acid sequences of the membrane anchors of the WT and the mutants are displayed in detail in Table 1.
TABLE 1.
Amino acid sequences of the membrane anchors (underlined) of the structural proteins E, prM/M, and anchC present in the TBEV infectious WT and mutant clones used in this study
| Protein and clone | Sequencea |
|---|---|
| E | |
| WT | VGFLPKLLLGVALAWLGLNMRNPTMSMSFLLAGGLVLAMTLGVGA (TBEV E aa 452–496) |
| E(J1-J2) | MSWITQGLMGALLLWMGVNARDRSIALAFLATGGVLVFLATNVHA |
| E(J1-T2) | MSWITQGLMGALLLWMGVNARNPTMSMSFLLAGGLVLAMTLGVGA |
| E(T1-J2) | VGFLPKLLLGVALAWLGLNMRDRSIALAFLATGGVLVFLATNVHA |
| prM/M | |
| WT | KLLALAMVTVVWLTLESVVTRVAVLVVLLCLAPVYA (TBEV prM aa 129–164) |
| prM(J1-J2) | PGYAFLAAILGWMLGSNNGQRVVFTILLLLVAPAYS |
| prM(J1-T2) | PGYAFLAAILGWMLGESVVTRVAVLVVLLCLAPVYA |
| prM(T1-J2) | KLLALAMVTVVWLTLENNGQRVVFTILLLLVAPAYS |
| anchC | |
| WT | KRRSATDWMSWLLVITLLGMTLA (TBEV anchC aa 94–116) |
| C(J) | KRRGGNEGSIMWLASLAVIIACAGA |
TBEV sequences are indicated in regular type, and JEV sequences are shown in bold.
Full-length DNA templates for in vitro transcription of TBEV plasmids were generated as described previously (19). The correct sequences were confirmed by next-generation sequencing of the whole clones.
Generation of virus.
In vitro transcription and transfection of BHK-21 (C-13) cells (ATCC CCL-10) by electroporation were performed as described previously (17). Briefly, RNAs were synthesized from full-length cDNA clones by use of a T7 Megascript kit (Ambion, Thermo Fisher) according to the manufacturer's instructions. Each template DNA was digested with DNase I, and the quality of the RNA was checked by electrophoresis on a 1% agarose gel containing 6% formalin. RNA was purified with an RNeasy minikit (Qiagen) and quantified spectrophotometrically, and equimolar amounts of the corresponding RNAs were used for transfection of BHK-21 cells. At 30 h posttransfection, the cell culture supernatant was harvested and clarified by low-speed centrifugation.
For blocking of virus maturation by increasing the pH in the TGN, NH4Cl was added to the medium at 4 h postelectroporation, to a final concentration of 10 mM.
Concentration of viral and subviral particles.
The cell culture supernatant obtained 30 h after transfection was clarified, layered onto a 10% sucrose cushion in TAN buffer, pH 8.0 (50 mM triethanolamine, 100 mM NaCl), and centrifuged for 2 h at 50,000 rpm and 4°C (Beckman Ti 90 rotor). Control experiments revealed that both virions and subviral particles are pelleted through the sucrose cushion under these conditions. The pellet (containing virions and subviral particles) was then resuspended in TAN buffer, pH 8.0, supplemented with 0.1% bovine serum albumin (BSA).
Lysis of transfected cells.
Cells were lysed using RIPA cell lysis buffer (containing 1% Nonidet P-40 substitute, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate; Amresco, VWR) supplemented with mammalian protease inhibitor cocktail (Amresco, VWR) according to the manufacturer's instructions.
Quantification of viral components (E protein and vRNA).
Genomic RNA (RNA copies) in the cell culture supernatant was measured by quantitative PCR (qPCR) after reverse transcription (RT) of the viral RNA. The cell culture supernatant was treated with RLN lysis buffer (250 mM Tris-HCl, pH 8.0, 700 mM NaCl, 7.5 mM MgCl2, 2.5% [vol/vol] Igepal, 5 mM dithiothreitol [DTT]) supplemented with 1,000 U RNase inhibitor/ml (Roche) for 1 min on ice. Viral RNA (vRNA) was isolated using an RNeasy minikit (Qiagen) according to the manufacturer's instructions. The vRNA was then subjected to RT by use of an iScript cDNA synthesis kit (Bio-Rad) following the manufacturer's instructions. An aliquot corresponding to 7 μl of the original supernatant was used for qRT-PCR using TaqMan Universal PCR master mix (Applied Biosystems). TBEV NS5-specific primers were used for qRT-PCR, as described previously (20). Serial 10-fold dilutions of the plasmid pTNd/c, containing the full-length genomic cDNA insert of TBEV strain Neudoerfl (19), were used to generate a standard curve for quantification.
The amount of E protein in the cell culture supernatants, lysates, and pellets was quantified by enzyme-linked immunosorbent assay (ELISA) after solubilization with 0.4% sodium dodecyl sulfate (SDS) at 65°C for 30 min as described previously (21).
Focus assay.
Virus samples were added to confluent cell monolayers in serial 10-fold dilutions in medium 199 (supplemented with 0.1% BSA, 15 mM HEPES, and 15 mM HEPPS [4-(2-hydroxyethyl)piperazine-1-propanesulfonic acid], pH 7.6). After 30 min of incubation at 37°C, cells were covered with a 3% carboxymethyl cellulose overlay in the same medium. Two days after infection, cells were fixed with 4% paraformaldehyde for 20 min at room temperature and treated with a Tris buffer (50 mM Tris, 150 mM NaCl, pH 7.6) containing 3% nonfat dry milk powder, 0.5% Triton X-100, and 0.05% Tween 20 for 30 min at 37°C. A virus-specific polyclonal rabbit anti-TBEV serum was then added and incubated with the fixed cells for 90 min at 37°C. Bound antibodies were detected with alkaline phosphatase-labeled goat anti-rabbit immunoglobulin G (Sigma-Aldrich), with SigmaFast Fast Red TR/naphthol AS-MX used as the substrate.
Immunofluorescence test (IFT).
RNA-transfected BHK-21 cells were seeded into 24-well tissue culture plates containing microscope coverslips and incubated for 24 h at 37°C. Cells were fixed and permeabilized with acetone-methanol (1:1) as described previously (22). Staining was performed by successive incubations with a rabbit polyclonal anti-TBEV serum recognizing the prM/M and E proteins or an anti-NS1 mouse monoclonal antibody (MAb) (E611-1-1) (23) and an Alexa Fluor 488-conjugated goat anti-rabbit antibody (Thermo Fisher Scientific) or a rhodamine Red-X-conjugated goat anti-mouse antibody (Jackson Immune Research Laboratory). Hoechst 33342 (Thermo Fisher Scientific) was used for nuclear DNA staining.
NS1 quantification.
RNA-transfected BHK-21 cells were seeded into 96-well tissue culture plates and incubated for 30 h at 37°C. Cells were fixed with 4% paraformaldehyde for 20 min at room temperature and treated with a Tris buffer (50 mM Tris, 150 mM NaCl, pH 7.6) containing 3% nonfat dry milk, 0.5% Triton X-100, and 0.05% Tween 20 for 30 min at 37°C. An NS1-specific monoclonal antibody (5D9-1-1) (23) or an anti-mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) monoclonal antibody (Pierce) was added to the fixed cells for 90 min at 37°C. Bound antibodies were detected with alkaline phosphatase-labeled goat anti-mouse immunoglobulin G (Sigma-Aldrich), with SigmaFast pNPP used as the substrate. The reaction was stopped by addition of 1.5 N NaOH, and the absorbance at 405 nm was measured. To obtain a measure of the relative amount of NS1 produced in the cell, the ratio of NS1 absorbance to GAPDH absorbance was determined. The ratio obtained with the wild type was set to 100.
HA assay.
Hemagglutination (HA) activity was determined in microtiter plates at pH 6.4 by use of goose erythrocytes as described by Clarke and Casals (24).
ELISA for determining virus maturation.
Samples (0.25 μg/ml E protein) in phosphate-buffered saline (PBS), pH 7.4, containing 2% lamb serum and 2% Tween 20 were captured by incubation with a polyclonal guinea pig anti-TBEV serum for 1 h at 37°C as described previously (21). After blocking with PBS, pH 7.4, containing 1% BSA for 1 h at 37°C, serial 10-fold dilutions of monoclonal antibodies 8H1 (prM specific) and B4 (E specific) were added. Bound antibodies were detected by use of a horseradish peroxidase-conjugated rabbit anti-mouse antibody (Nordic Immunology) and o-phenylenediamine (OPD) substrate (Sigma). To evaluate the maturation state of particles, the ratios of 8H1 to B4 absorbance values were calculated.
Rate zonal sucrose density gradient centrifugation.
For the separation of virions and subviral particles, samples were applied to continuous 5 to 30% (wt/wt) sucrose gradients in TAN buffer, pH 8.0. Centrifugation was carried out for 70 min at 38,000 rpm at 4°C in a Beckmann SW40 rotor. Twenty-one fractions (0.6 ml) were collected by upward displacement, and the pellets were resuspended in TAN buffer, pH 8.0 (fraction 22). The amount of E in each fraction was determined by an E-specific four-layer ELISA after treatment with 0.4% SDS at 65°C for 30 min (21).
Production of purified mature/immature virus and RSPs.
TBEV purification was carried out essentially as described previously (25). In brief, supernatants from infected primary chicken embryo cells were subjected to ultracentrifugation, and the virus in the resuspended pellet was purified by rate zonal followed by equilibrium sucrose density gradient centrifugation. For the production of immature virus, NH4Cl was added to the medium at 24 h postinfection, to a final concentration of 20 mM (21).
For the production of RSPs, COS-1 cells were transfected with recombinant plasmids by electroporation as described previously (26). RSPs were harvested from cell culture supernatants 48 h after transfection and were pelleted by ultracentrifugation (26).
Detection of anchC TMD in virus particles.
Fifty micrograms of purified TBEV was subjected to SDS-PAGE by use of a Tris-boric acid-EDTA Ready Gel 4 to 20% system (Bio-Rad) according to the manufacturer's instructions. The proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad) and stained with Ponceau Red (0.25% Ponceau S in 1% acetic acid). Fifty micrograms of TBEV spiked with 1.4 μg of a synthesized peptide corresponding to the TMD of the TBEV C protein (JPT Peptide Technologies, Berlin, Germany) served as a positive control (molar ratio of C to E = 1:1). In parallel, 10 μg of the peptide was applied to the gel to identify the corresponding bands, which were cut from the blot and sent for automated N-terminal protein sequencing by Edman degradation (Toplab GmbH, Martinsried, Germany).
Statistical analyses.
Statistical significances of the results were determined by one-way analysis of variance (ANOVA) followed by Dunnett's (comparison to the WT) or Tukey's (comparison of all groups) multiple-comparison test, using logarithmically transformed values (Graph Pad Prism 5). P values of <0.05 were regarded as statistically significant.
RESULTS
Generation of prM and E TMD mutants and intracellular expression of viral proteins.
To investigate the influence of TMD interactions on the production of virus particles, we generated different TBEV mutants in which we replaced either both or individual TMDs of E and prM with those of JEV [E(J1-J2), E(J1-T2), E(T1-J2), prM(J1-J2), prM(J1-T2), and prM(T1-J2)] (Fig. 1C). In addition, we combined all of the JEV prM and E TMDs in one construct [prM(J1-J2)+E(J1-J2)] (Fig. 1C). This strategy should maintain the principal functions of the TMDs for polyprotein processing, i.e., the stop-transfer signal (TMD1) for prM or E and the internal signal sequence for the synthesis of E or NS1 (TMD2) (Fig. 1A and B). Due to the low sequence conservation between TBEV and JEV TMDs (only about 17%) (Fig. 1D), however, specific interactions between the different TMDs as well as with other TBEV proteins would be expected to be disturbed.
We first verified that the TMD replacements did not impair polyprotein processing. For this purpose, WT and mutant RNAs were in vitro transcribed from infectious cDNA clones and transfected into BHK cells. Twenty-four hours after electroporation, immunofluorescence staining was carried out by using a polyclonal serum recognizing prM/M and E as well as a monoclonal antibody (MAb) specific for the first NS protein (NS1). As shown in Fig. 2A, WT-like expression of structural proteins and NS1 was observed for each of the E TMD mutants, but the substitutions of TMDs in prM—alone and in combination with those of E—abolished protein synthesis.
FIG 2.

Intracellular expression of viral proteins of prM and E TMD mutants. (A) Immunofluorescence staining of BHK cells transfected with mutant or WT viral RNAs, as indicated in the left panels. Staining was carried out using a polyclonal serum recognizing the prM/M and E proteins of TBEV and a monoclonal antibody recognizing NS1. Nuclear DNA was stained with Hoechst dye. The data are representative examples of three or more independent experiments. (B) Quantification of E in cell lysates by ELISA at 30 h posttransfection. (C) Quantification of NS1 in fixed cells by ELISA at 30 h posttransfection. The absorbance ratio of NS1 to GAPDH (cellular protein control) was determined, and the results are expressed as percentages of the ratio obtained with the WT. The data are presented as means ± standard errors for at least four independent experiments. Asterisks indicate significant differences relative to the WT (ANOVA and Dunnett's multiple-comparison test).
For a more quantitative analysis of protein expression of the E TMD mutants, we determined (i) the amounts of E protein in transfected cell lysates by SDS ELISA and (ii) the efficiency of NS1 expression by an ELISA using fixed transfected cells (see Materials and Methods). As shown in Fig. 2B and C, the intracellular expression of both viral proteins was not affected in the case of the E(J1-J2) and E(J1-T2) mutants, but in the case of the mutant in which only the second TMD was replaced [E(T1-J2)], a significant reduction occurred.
Taken together, the results indicate that the 2nd E TMD of JEV can function as an internal signal sequence in the TBEV context, but its efficiency appears to be affected by the interaction with TMD1 in the E membrane anchor hairpin. Since the replacement of the prM TMDs abolished the expression of both structural and nonstructural proteins (Fig. 2A), further analyses of these mutants were not possible.
Release of E protein and viral RNA.
To obtain information on the effects of E TMD replacements on the production and release of viral particles, we first quantified the amounts of E protein (by ELISA) and genomic RNA (by qPCR) in the cell culture supernatant 30 h after electroporation of in vitro-transcribed RNA into BHK cells. Compared to that in the WT, the secretion of E and vRNA was slightly but not significantly reduced in the case of the E(J1-J2) and E(J1-T2) mutants, but a strong and significant decrease was observed for the E(T1-J2) mutant (Fig. 3A and B). Similarly, the ratio of RNA copies to E protein was moderately reduced for the E(J1-J2) and E(J1-T2) mutants (Fig. 3C), although statistical significance was not reached at this point. A significant reduction, however, was observed for the E(T1-J2) mutant (Fig. 3C).
FIG 3.

Release of E and viral RNA (vRNA) from E TMD mutants. (A) Quantification of E in transfected cell culture supernatants by ELISA at 30 h posttransfection. (B) Quantification of vRNA in transfected cell culture supernatants by qPCR at 30 h posttransfection. (C) Ratios of vRNA to E (number of RNA copies per picogram of E). The data are presented as means ± standard errors for at least three independent experiments. Asterisks indicate significant differences relative to the WT (ANOVA and Dunnett's multiple-comparison test).
These data suggest that certain E TMD modifications affect the formation and/or release of RNA-containing particles, possibly by favoring the formation of capsidless subviral particles without RNA.
Characterization of released E protein.
To characterize the form of E secreted into the cell culture supernatant (soluble versus particulate), we first determined the amount of E protein incorporated into particles. For this purpose, the supernatants of the WT and mutant strains were subjected to ultracentrifugation under conditions which resulted in the pelleting of both viral and subviral particles (see Materials and Methods). The amounts of E in the supernatant and pellet fractions were quantified by ELISA. As shown in Fig. 4A, the pellets contained about 70% of the total E protein in the case of the WT as well as the E(J1-J2) and E(J1-T2) constructs. In contrast, this percentage was significantly lower for the E(T1-J2) mutant, indicating either a lower proportion or reduced stability of E-containing particles. As revealed by an ELISA using prM- and E-specific MAbs (see Materials and Methods), these effects were not due to different extents of prM cleavage, which were similar for the WT and all mutants (Fig. 4B).
FIG 4.

Pelleting efficiencies of secreted E proteins of E TMD mutants and determination of the particle maturation state. (A) Clarified supernatants from transfected cells were subjected to ultracentrifugation under conditions resulting in the pelleting of both virions and subviral particles. The amounts of E protein in the pellet and supernatant fractions after centrifugation were quantified by ELISA. The data are expressed as percentages of E found in the pellet. (B) The resuspended pellets from panel A were analyzed by an ELISA using a prM-specific (8H1) and an E-specific (B4) MAb. An immature preparation of TBEV was included as a control (first bar). The data are expressed as ratios of 8H1 absorbance to B4 absorbance and represent means ± standard errors for at least three independent experiments. Asterisks in panel A indicate a significant difference relative to the WT (ANOVA and Dunnett's multiple-comparison test).
To assess the proportions of virions and subviral particles in the pellets of the different mutants compared to the WT, the samples were subjected to rate zonal sucrose gradient centrifugation, which allowed differentiation between the two particle forms. As shown in Fig. 5A and quantitatively evaluated in Fig. 6, WT particles consisted mostly of virions, and only a small amount of subviral particles could be detected. In all mutants, however, a change of this distribution was observed at the expense of virions (Fig. 5D to F and 6). In the case of the E(J1-J2) mutant, approximately 50% of E sedimented as subviral particles, and this percentage was even higher with the E(J1-T2) mutant (∼80% subviral particles). In the case of the E(T1-J2) mutant, the virion peak had disappeared and the E protein was distributed over many fractions in the first half of the gradient, suggesting that mutant particles disintegrated during centrifugation (Fig. 5F and 6). As expected for the WT as well as the E(J1-J2) and E(J1-T2) mutants, more than 80% of the total vRNA and infectivity was found in the virion-containing fractions, with only a minor proportion in the fractions containing subviral particles. The RNA distribution for the E(T1-J2) mutant is consistent with its disintegration during centrifugation (Table 2), and all of the very small amount of infectivity detected (see the comparison of specific infectivities below) was confined to the position of the virus peak in the control (Table 2).
FIG 5.

Analysis of E TMD mutants by rate zonal centrifugation. Resuspended pellets from Fig. 4A, containing viral as well as subviral particles, were subjected to sedimentation analysis. Centrifugation was carried out on continuous sucrose gradients (5 to 30% [wt/wt]), and the amount of E protein in each fraction was quantified by ELISA. Sedimentation is shown from left to right. Representative data from six independent experiments are shown. (A and D to F) WT and mutant E TMD samples; (B) virion control; (C) RSP control. SP, subviral particles.
FIG 6.
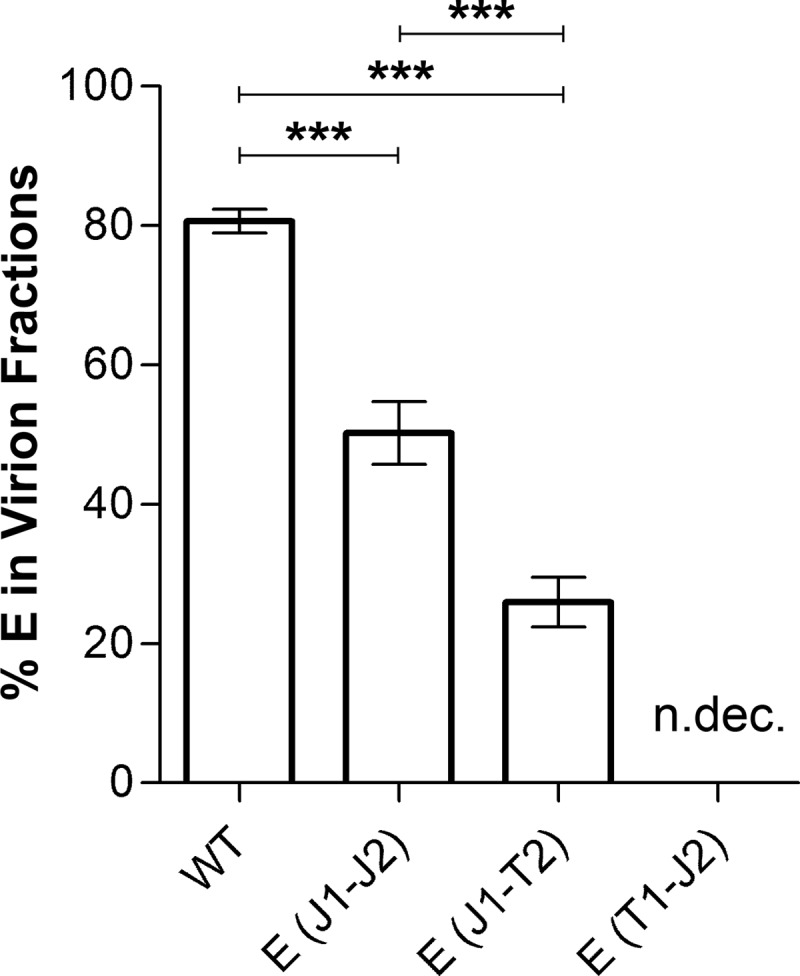
Proportions of E in virions of E TMD mutants. The proportions of viral particles of WT and mutant preparations [E(J1-J2) and E(J1-T2)] in the density gradients shown in Fig. 5 were calculated by determining the area under the curve for each peak as a percentage of the total area under the curve (GraphPad Prism 5). The data are presented as means ± standard errors for six independent experiments. Asterisks indicate significant differences between the samples (ANOVA and Tukey's multiple-comparison test). n.dec., not detectable.
TABLE 2.
Distributions of vRNA and infectivity in different fraction pools of E TMD mutants after rate zonal centrifugationa
| Virus | % of total vRNA |
Infectivity (% of total FFU) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gradient fractions 1–9 | Statistical significance | Gradient fractions 10–14b | Statistical significance | Gradient fractions 15–22 | Statistical significance | Gradient fractions 1–9 | Statistical significance | Gradient fractions 10–14b | Statistical significance | Gradient fractions 15–22 | Statistical significance | |
| WT | 4.6 | 86.8 | 8.6 | 2.6 | 89.8 | 7.6 | ||||||
| E(J1-J2) | 3.7 | n.s. | 87.1 | n.s. | 9.3 | n.s. | 4.6 | n.s. | 86.6 | n.s. | 8.8 | n.s. |
| E(J1-T2) | 5.6 | n.s. | 81.1 | n.s. | 13.2 | n.s. | 3.0 | n.s. | 83.2 | n.s. | 13.8 | n.s. |
| E(T1-J2) | 19.2 | *** | 57.4 | *** | 23.4 | ** | n.dec. | 100 | n.s. | n.dec. | ||
See Fig. 5. n.dec., not detectable; n.s., no significant difference relative to the WT; **, significantly different from the WT (P < 0.01; ANOVA and Dunnett's multiple-comparison test); ***, significantly different from the WT (P < 0.001; ANOVA and Dunnett's multiple-comparison test).
Virus peak.
To verify the nature of the subviral particle fractions shown in Fig. 5, we determined their specific hemagglutination (HA) activities (Fig. 7) and physical properties in comparison to those of recombinant subviral particles (RSPs) as well as virions. As shown in Fig. 7, the subviral particle fractions of our experiments had the same specific HA activity as that of the RSP control (Fig. 7A), similar to virions (Fig. 7B). Like with RSPs, results of detergent solubilization experiments were consistent with the presence of a lipid membrane, and equilibrium sucrose gradient centrifugation revealed the same buoyant density as that of RSPs (1.14 g/ml), in contrast to that of whole virions (1.19 g/ml) (26; data not shown).
FIG 7.

HA activities of subviral particles and virions of WT and E TMD mutant viruses. (A) Specific HA activities of the subviral particle peak fractions from Fig. 5, expressed as HA titers per microgram of E protein. (B) Specific HA activities of the virion peak fractions from Fig. 5, expressed as HA titers per microgram of E protein. The E(T1-J2) mutant was not analyzed due to its significantly lower yields and the absence of defined particle peaks. n.a., not analyzed; pur. virus, purified virus.
Taken together, the data suggest that the modifications of the E TMDs impaired the assembly and/or affected the stability of virions and favored the formation of nucleocapsidless, noninfectious subviral particles.
Specific infectivity of virus E TMD mutants.
To evaluate the effects of the presence of heterologous E TMDs on the infectious properties of virions, we determined the specific infectivities (numbers of focus-forming units relative to numbers of vRNA copies) of the virion peak fractions from the gradients shown in Fig. 5A, D, and E. In the case of the E(T1-J2) mutant, for which a virus peak was not detectable (Fig. 5F and 6), the fractions corresponding to the control virion peak (Fig. 5B) were analyzed. As shown in Fig. 8A, the E(J1-J2) and E(J1-T2) mutants yielded specific infectivities similar to that of the WT, although the sizes of their foci were smaller than that of the WT (Fig. 8B and C). In the case of the E(T1-J2) mutant, both the specific infectivity and the size of the foci were significantly reduced (Fig. 8A to C).
FIG 8.

Infectivities of WT and E TMD mutant viruses. (A) Specific infectivities of the virion peak fractions from Fig. 5, expressed as numbers of focus-forming units relative to numbers of RNA copies (FFU per 107 RNA copies). In the case of the E(T1-J2) mutant, for which a virus peak was not detectable (Fig. 6), the fractions corresponding to the control virion peak (Fig. 5B) were used. (B) Photographs of focus assay wells. (C) Focus sizes. In the case of the WT and two mutants [E(J1-J2) and E(J1-T2)], 39 foci were used for measurements, while 11 foci were used for the E(T1-J2) mutant. The data represent means ± standard errors for at least three independent experiments. Asterisks indicate significant differences relative to the WT (ANOVA and Dunnett's multiple-comparison test).
Since virions produced by the E(J1-J2) and E(J1-T2) mutants exhibited WT-like specific infectivities—despite the smaller focus size—it can be assumed that the first round of infection was not impaired by the modifications of their E membrane anchors.
Characterization of anchC TMD mutants.
The overall results obtained so far with the different E TMD mutants are consistent with defects in the assembly process of capsid-containing particles. The interactions required for RNA/capsid packaging are currently unknown, but they may involve transient interactions of the anchC TMD with the prM and/or E TMDs at the assembly site in the ER (Fig. 1B). We therefore studied the effect of the replacement of the TBEV anchC TMD (prM signal sequence) with the corresponding JEV element in our WT construct [C(J)] (Fig. 9A). Since the sequential cleavages at the luminal side by signalase and at the cytoplasmic side by the viral protease NS2B-NS3 (Fig. 1A and B) are essential for virus morphogenesis (27–29), we left the dibasic motif of the TBEV NS2B-NS3 cleavage site (RR) unchanged in all our clones (Fig. 9A and B and Table 1).
FIG 9.

Analysis of C TMD mutants. (A) Schematic diagrams of the C-terminal modifications of C and E in the TBEV infectious clone used to produce mutant virions. (B) Alignment of the amino acid sequences of the C membrane anchor regions (signal sequences of prM) of TBEV (GenBank accession number U27495) and JEV (GenBank accession number EF571853). The signal sequences are underlined, and the cleavage sites are indicated by diagonal slashes. The single conserved amino acid in the signal sequence is indicated in bold. (C to F) Clarified supernatants from transfected cells of the WT (C) and the C(J) (D), E(J1-J2) (E), and C(J)+E(J1-J2) (F) mutants were pelleted by ultracentrifugation, and resuspended pellets were subjected to rate zonal sucrose gradient centrifugation as described in the legend to Fig. 5. After centrifugation, the amount of E protein in each fraction was quantified by ELISA. Sedimentation is shown from left to right. Representative data from at least two independent experiments are shown. SP, subviral particles.
The replacement of the TBEV anchC TMD with that of JEV [C(J)] did not result in any differences with respect to virus production (secretion of E and vRNA, maturation state of particles, specific infectivity, and focus size) (data not shown) or the distribution of viral and subviral particles in sucrose gradients (Fig. 9C and D). Apparently, the anchC TMD of JEV can function properly as an internal signal sequence for prM in the TBEV backbone. Since there is virtually no sequence conservation between the TBEV and JEV anchC TMDs (Fig. 9B), these data argue against specific interactions between the C and prM/E TMDs. Such a conclusion was further supported by the finding that the inclusion of the JEV anchC TMD in the E(J1-J2) mutant [C(J)+E(J1-J2)] did not result in any compensation of the defects observed with the E TMD mutants (Fig. 9E and F), as well as by the results of an experiment in which we searched for the presence of the anchC TMD in purified virus particles. We hypothesized that one would expect to find the anchC TMD as a remnant in assembled virions if it played an important role in interactions at the virion assembly site. For this analysis, the proteins of a purified TBEV preparation were separated by SDS-PAGE and blotted onto a PVDF membrane, and the region corresponding to the anchC TMD was subjected to N-terminal sequencing by Edman degradation, as described in Materials and Methods. As a control, the virus preparation was spiked with a synthetic peptide corresponding to the TBEV anchC TMD in an amount equimolar to that of E. Only in the spiked control was the anchC peptide identified by its N-terminal sequence. Considering that the C protein is present in at least a 3-fold molar excess over E in virions (30), these data suggest that the TMD of anchC is excluded from virus particles during assembly and/or budding.
Effects on envelope rearrangements in immature virions.
The data so far suggested a possible role for E TMDs in protein interactions involved in virus assembly and the formation of infectious virions. As described in the introduction, the maturation process of immature virions in the TGN includes a dramatic acidic-pH-induced rearrangement of the E protein lattice, which is a hallmark of virus maturation and converts the spiky immature structure to a smooth herringbone-like arrangement of 90 E dimers characteristic of mature virions. Since it is feasible that TMD interactions are involved in these rearrangements, we designed an experiment to assess the effect of E TMD replacements specifically on this step of virus maturation. This was made possible by comparing particles released from cells cultured in the absence and presence of NH4Cl, which raises the pH in the TGN and thus prevents the acidic-pH-induced rearrangements characteristic of virus maturation.
The differential sedimentation analyses of ultracentrifugation pellets obtained from WT and mutant viruses grown in the absence and presence of NH4Cl revealed substantial differences for the E(J1-T2) mutant only (Fig. 10). Specifically, the shift toward subviral particles at the expense of whole virions observed with the E(J1-T2) mutant in the absence of NH4Cl (Fig. 10C) was much less pronounced in its presence (Fig. 10D), suggesting that the defects observed with this mutant were a composite of impairments of TMD interactions during both assembly and maturation rearrangements in the TGN.
FIG 10.

Analysis by rate zonal centrifugation of the WT and the E(J1-T2) mutant grown in the absence (−) (A and C) or presence (+) (B and D) of NH4Cl. Clarified supernatants from transfected cells, with or without NH4Cl, were pelleted by ultracentrifugation, and resuspended pellets were subjected to rate zonal sucrose gradient centrifugation as described in the legend to Fig. 5. After centrifugation, the amount of E protein in each fraction was quantified by ELISA. Sedimentation is shown from left to right. Representative data from at least three independent experiments are shown. SP, subviral particles.
In contrast, no such differences relative to the effects seen in the absence of NH4Cl were observed for the E(J1-J2) and E(T1-J2) mutants (data not shown), indicating that their defects (Fig. 5) were restricted to the stage of virus assembly.
DISCUSSION
The specific structural features of the TMDs of the flavivirus envelope proteins prM/M and E—both of which are alpha-helical hairpins that penetrate only the outer leaflet of the viral membrane (15)—prompted us to assess their role in the assembly and maturation processes of flaviviruses, using TBEV as a model. We chose a mutational approach in which we replaced these TMDs in an infectious TBEV clone with those of the distantly related virus JEV, assuming that the TMDs would maintain their functions in polyprotein processing (serving as stop-transfer and internal signal sequences) but that the changes would reveal effects on interactions required for virus assembly and/or maturation because of the low degree of sequence conservation of the TMDs. Previous studies provided information on the role of TMDs in the assembly of recombinant capsidless subviral particles (RSPs) and indicated that intra- and inter-TMD interactions play a role in the fusion process (17), but conclusions with respect to whole-virion particles were not possible because of architectural differences between RSPs and virions (18, 31).
It is an important finding of our study that the replacements of both E TMDs [E(J1-J2)] and of the first E TMD only [E(J1-T2)] did not affect polyprotein processing but that the particles secreted from cells contained proportionally more subviral particles and fewer virions. The same replacements had no effect on RSP formation (17), suggesting that these elements are not specifically involved in the lateral interactions known to drive the assembly of the viral envelope (reviewed in reference 7). These findings are consistent with the results of an alanine insertion mutagenesis study of E and prM TMD mutants of yellow fever virus in which prM/E heterodimerization at the ER membrane was not disturbed by the introduced alanines (although some of the mutations affected virus growth) (32). TMD interactions are apparently required for steps that contribute to capsid integration for virion formation but not to subviral particle assembly. In addition, the mutant in which only the second E TMD was replaced [E(T1-J2)] appeared to be quite labile, indicating that such interactions are not only required for the formation of virions but also contribute to their stability.
Interestingly, the specific infectivities of gradient-purified virions of the E(J1-J2) and E(J1-T2) mutants were comparable to that of the WT, suggesting that their entry into cells was not impaired, although they produced more subviral particles than the WT did. However, since the sizes of their foci were significantly smaller than that for the WT, it can be assumed that virus spread was decreased due to the reduced release of infectious virus particles. Such an interpretation would be in agreement with the results of liposome fusion assays with the corresponding TBEV RSP mutants (17). The mutant with the complete JEV membrane anchor had fusion levels similar to that of WT RSPs, and the E(J1-T2) RSPs still yielded fusion levels of approximately 60% of the WT level.
Consistent with the apparent instability of the E(T1-J2) mutant virus particles [and different from the E(J1-T2) mutant], the corresponding virion-containing gradient fractions displayed not only a smaller focus size but also a significantly reduced specific infectivity. One possible explanation for the different effects could be that the E(T1-J2) hairpin might be more disordered than the E(J1-T2) hairpin. This could be due to the presence of different numbers of TMD helix dimerization motifs in the E membrane anchors of TBEV and JEV (Fig. 11). The TMD1 regions of JEV and TBEV both contain one SmXXXSm helix dimerization motif, where “Sm” is a small residue (Gly, Ala, Ser, or Thr) (reviewed in reference 33). In TMD2 of TBEV E, however, four overlapping dimerization motifs are present, whereas in JEV TMD2, only two such motifs could be identified (Fig. 11). These differences between the TBEV and JEV E TMDs could lead to an unstable hairpin that might impair the formation of complex capsid-containing virions but might still allow the budding of subviral particles (17).
FIG 11.

Predicted helix dimerization motifs in the E membrane anchors of TBEV and JEV. The positions of SmXXXSm helix dimerization motifs (“Sm” is a small residue, i.e., Gly, Ala, Ser, or Thr) (reviewed in reference 33) are indicated by black lines under the respective membrane anchor sequences. Residues conserved among flavivirus E proteins are shaded in gray, and those conserved between TBEV and JEV are shown in bold. The TMD1 and TMD2 helices are indicated at the top.
Experiments with NH4Cl as an agent to prevent acidic-pH-induced rearrangements in the TGN provided evidence that E TMD interactions are also involved in the maturation-associated envelope rearrangements. Specifically, we found that the shift toward subviral particles was less pronounced with the E(J1-T2) mutant in the presence of NH4Cl, suggesting that part of the mutational effect was caused by an influence on the rearrangements in the TGN. The positions of the E and prM/M transmembrane regions relative to each other differ in the two particle forms (3, 4, 15, 16, 18), and the presence of heterologous TMDs might thus have different effects. In the smaller RSPs, the E dimers are organized in a T=1 icosahedral lattice in the mature form (18), and TMD repositioning in the TGN is probably less extensive, allowing or even favoring the release of simpler subviral particles over whole virions. For virions, in contrast, the acidic-pH-induced rearrangements in the TGN are quite dramatic, and TMD replacements might thus be more prone to interfere with the proper reorganization of the viral envelope, leading to unstable particles that possibly already undergo fusion at the slightly acidic pH of the TGN.
Virion assembly requires specific interactions with the capsid protein or capsid protein-RNA complexes. These are still elusive, and cryo-EM of mature and immature flaviviruses did not reveal any contacts between the capsid and the envelope proteins (3, 4, 15, 16). Surprisingly, despite a lack of sequence conservation (Fig. 9B), the anchC TMD of JEV in the TBEV backbone did not impair virion formation, specific infectivity, or focus size, suggesting that specific interactions of anchC TMDs with the TMDs of other viral proteins are not required for proper virus assembly. This was further supported by the fact that the peptide corresponding to the anchC TMD was not found to be incorporated into virus particles in significant amounts, suggesting an assembly mechanism that specifically excludes this sequence element from budding virus particles. Nevertheless, the anchC TMDs might interact with each other and thus bring together several copies of C for nucleocapsid formation. One may speculate that during assembly, the E and/or prM TMDs interact transiently with the internal hydrophobic domain (IHD) of C, which has the ability to associate with membranes (34, 35). Consistent with such a notion is the finding that the removal of incremental parts of the IHD of the TBEV capsid protein impaired the release of infectious virus particles but still allowed the secretion of subviral particles (36), similar to what we observed with two of our E TMD mutants. Other candidates for transient interactions with E and/or prM TMDs during virus assembly and/or budding are the nonstructural proteins NS1 and NS2A, which have both been proposed to be involved in virus particle morphogenesis (37–43).
It was unexpected that—in contrast to what was observed with the C and E TMDs—the analogous replacement of prM TMDs resulted in the complete abolishment of protein expression, thus making further studies of their roles in assembly/maturation impossible. The molecular basis for these data is unclear, but the defects in the mutants might already occur at the RNA level, causing the RNA to adopt an unfavorable structure for translation and/or replication (44, 45). Alternatively, the extensive sequence divergence of prM TMDs—which is much higher than that of E TMDs (Fig. 1D)—might result in an abrogation of the complex processes required for proper flavivirus polyprotein processing (5).
Taken together, our results indicate that flavivirus assembly and maturation rearrangements are finely tuned processes that involve the coordinated interplay of several viral proteins, including TMD interactions and/or movements of the three structural proteins. Apart from specific contacts within and between the TMD hairpins, interactions of the membrane anchors with other viral proteins, such as the capsid, might be important for virus assembly. This holds especially true for C-prM TMD interactions, which could not be resolved in the present investigation. Future studies might also address the question of whether transient interactions of prM and E TMDs (both buried in the viral membrane and not protruding into the interior of finally assembled immature/mature virus particles [3, 4, 15, 16]) might assist in the assembly of virus components before particle formation.
ACKNOWLEDGMENTS
We thank Cornelia Stöckl, Walter Holzer, and Jutta Hutecek for their excellent technical assistance, as well as Connie Schmaljohn for providing hybridoma cells secreting prM- and NS1-specific monoclonal antibodies.
REFERENCES
- 1.ICTV. 2014. Virus taxonomy: 2014 release. http://www.ictvonline.org/virusTaxonomy.asp.
- 2.Pierson TC, Diamond MS. 2013. Flaviviruses, p 747–794. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Zhang Y, Corver J, Chipman PR, Zhang W, Pletnev SV, Sedlak D, Baker TS, Strauss JH, Kuhn RJ, Rossmann MG. 2003. Structures of immature flavivirus particles. EMBO J 22:2604–2613. doi: 10.1093/emboj/cdg270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kostyuchenko VA, Zhang Q, Tan JL, Ng TS, Lok SM. 2013. Immature and mature dengue serotype 1 virus structures provide insight into the maturation process. J Virol 87:7700–7707. doi: 10.1128/JVI.00197-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindenbach BD, Murray CL, Thiel HJ, Rice CM. 2013. Flaviviridae, p 712–746. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 6.Apte-Sengupta S, Sirohi D, Kuhn RJ. 2014. Coupling of replication and assembly in flaviviruses. Curr Opin Virol 9:134–142. doi: 10.1016/j.coviro.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roby JA, Setoh YX, Hall RA, Khromykh AA. 2015. Post-translational regulation and modifications of flavivirus structural proteins. J Gen Virol 96:1551–1569. doi: 10.1099/vir.0.000097. [DOI] [PubMed] [Google Scholar]
- 8.Yu IM, Zhang W, Holdaway HA, Li L, Kostyuchenko VA, Chipman PR, Kuhn RJ, Rossmann MG, Chen J. 2008. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science 319:1834–1837. doi: 10.1126/science.1153264. [DOI] [PubMed] [Google Scholar]
- 9.Li L, Lok SM, Yu IM, Zhang Y, Kuhn RJ, Chen J, Rossmann MG. 2008. The flavivirus precursor membrane-envelope protein complex: structure and maturation. Science 319:1830–1834. doi: 10.1126/science.1153263. [DOI] [PubMed] [Google Scholar]
- 10.Harrison SC. 2015. Viral membrane fusion. Virology 479–480:498–507. doi: 10.1016/j.virol.2015.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Langosch D, Hofmann M, Ungermann C. 2007. The role of transmembrane domains in membrane fusion. Cell Mol Life Sci 64:850–864. doi: 10.1007/s00018-007-6439-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.White JM, Delos SE, Brecher M, Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol 43:189–219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuhn RJ. 2013. Togaviridae, p 629–650. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 14.Elliott RM, Schmaljohn CS. 2013. Bunyaviridae, p 1244–1282. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 15.Zhang X, Ge P, Yu X, Brannan JM, Bi G, Zhang Q, Schein S, Zhou ZH. 2013. Cryo-EM structure of the mature dengue virus at 3.5-A resolution. Nat Struct Mol Biol 20:105–110. doi: 10.1038/nsmb.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang W, Chipman PR, Corver J, Johnson PR, Zhang Y, Mukhopadhyay S, Baker TS, Strauss JH, Rossmann MG, Kuhn RJ. 2003. Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat Struct Biol 10:907–912. doi: 10.1038/nsb990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fritz R, Blazevic J, Taucher C, Pangerl K, Heinz FX, Stiasny K. 2011. The unique transmembrane hairpin of flavivirus fusion protein E is essential for membrane fusion. J Virol 85:4377–4385. doi: 10.1128/JVI.02458-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferlenghi I, Clarke M, Ruttan T, Allison SL, Schalich J, Heinz FX, Harrison SC, Rey FA, Fuller SD. 2001. Molecular organization of a recombinant subviral particle from tick-borne encephalitis virus. Mol Cell 7:593–602. doi: 10.1016/S1097-2765(01)00206-4. [DOI] [PubMed] [Google Scholar]
- 19.Mandl CW, Ecker M, Holzmann H, Kunz C, Heinz FX. 1997. Infectious cDNA clones of tick-borne encephalitis virus European subtype prototypic strain Neudoerfl and high virulence strain Hypr. J Gen Virol 78:1049–1057. doi: 10.1099/0022-1317-78-5-1049. [DOI] [PubMed] [Google Scholar]
- 20.Taucher C, Berger A, Mandl CW. 2010. A trans-complementing recombination trap demonstrates a low propensity of flaviviruses for intermolecular recombination. J Virol 84:599–611. doi: 10.1128/JVI.01063-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heinz FX, Stiasny K, Puschner-Auer G, Holzmann H, Allison SL, Mandl CW, Kunz C. 1994. Structural changes and functional control of the tick-borne encephalitis virus glycoprotein E by the heterodimeric association with protein prM. Virology 198:109–117. doi: 10.1006/viro.1994.1013. [DOI] [PubMed] [Google Scholar]
- 22.Kofler RM, Hoenninger VM, Thurner C, Mandl CW. 2006. Functional analysis of the tick-borne encephalitis virus cyclization elements indicates major differences between mosquito-borne and tick-borne flaviviruses. J Virol 80:4099–4113. doi: 10.1128/JVI.80.8.4099-4113.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iacono-Connors LC, Smith JF, Ksiazek TG, Kelley CL, Schmaljohn CS. 1996. Characterization of Langat virus antigenic determinants defined by monoclonal antibodies to E, NS1 and preM and identification of a protective, non-neutralizing preM-specific monoclonal antibody. Virus Res 43:125–136. doi: 10.1016/0168-1702(96)01325-1. [DOI] [PubMed] [Google Scholar]
- 24.Clarke DH, Casals J. 1958. Techniques for hemagglutination and hemagglutination-inhibition with arthropod-borne viruses. Am J Trop Med Hyg 7:561–573. [DOI] [PubMed] [Google Scholar]
- 25.Heinz FX, Kunz C. 1981. Homogeneity of the structural glycoprotein from European isolates of tick-borne encephalitis virus: comparison with other flaviviruses. J Gen Virol 57:263–274. doi: 10.1099/0022-1317-57-2-263. [DOI] [PubMed] [Google Scholar]
- 26.Schalich J, Allison SL, Stiasny K, Mandl CW, Kunz C, Heinz FX. 1996. Recombinant subviral particles from tick-borne encephalitis virus are fusogenic and provide a model system for studying flavivirus envelope glycoprotein functions. J Virol 70:4549–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amberg SM, Rice CM. 1999. Mutagenesis of the NS2B-NS3-mediated cleavage site in the flavivirus capsid protein demonstrates a requirement for coordinated processing. J Virol 73:8083–8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lobigs M, Lee E. 2004. Inefficient signalase cleavage promotes efficient nucleocapsid incorporation into budding flavivirus membranes. J Virol 78:178–186. doi: 10.1128/JVI.78.1.178-186.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lobigs M, Lee E, Ng ML, Pavy M, Lobigs P. 2010. A flavivirus signal peptide balances the catalytic activity of two proteases and thereby facilitates virus morphogenesis. Virology 401:80–89. doi: 10.1016/j.virol.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 30.Schwaiger J, Aberle JH, Stiasny K, Knapp B, Schreiner W, Fae I, Fischer G, Scheinost O, Chmelik V, Heinz FX. 2014. Specificities of human CD4+ T cell responses to an inactivated flavivirus vaccine and infection: correlation with structure and epitope prediction. J Virol 88:7828–7842. doi: 10.1128/JVI.00196-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, Lenches E, Jones CT, Mukhopadhyay S, Chipman PR, Strauss EG, Baker TS, Strauss JH. 2002. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell 108:717–725. doi: 10.1016/S0092-8674(02)00660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Op De Beeck A, Molenkamp R, Caron M, Ben Younes A, Bredenbeek P, Dubuisson J. 2003. Role of the transmembrane domains of prM and E proteins in the formation of yellow fever virus envelope. J Virol 77:813–820. doi: 10.1128/JVI.77.2.813-820.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li E, Wimley WC, Hristova K. 2012. Transmembrane helix dimerization: beyond the search for sequence motifs. Biochim Biophys Acta 1818:183–193. doi: 10.1016/j.bbamem.2011.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Markoff L, Falgout B, Chang A. 1997. A conserved internal hydrophobic domain mediates the stable membrane integration of the dengue virus capsid protein. Virology 233:105–117. doi: 10.1006/viro.1997.8608. [DOI] [PubMed] [Google Scholar]
- 35.Samsa MM, Mondotte JA, Iglesias NG, Assunção-Miranda I, Barbosa-Lima G, Da Poian AT, Bozza PT, Gamarnik AV. 2009. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog 5:e1000632. doi: 10.1371/journal.ppat.1000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kofler RM, Heinz FX, Mandl CW. 2002. Capsid protein C of tick-borne encephalitis virus tolerates large internal deletions and is a favorable target for attenuation of virulence. J Virol 76:3534–3543. doi: 10.1128/JVI.76.7.3534-3543.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kümmerer BM, Rice CM. 2002. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J Virol 76:4773–4784. doi: 10.1128/JVI.76.10.4773-4784.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leung JY, Pijlman GP, Kondratieva N, Hyde J, Mackenzie JM, Khromykh AA. 2008. Role of nonstructural protein NS2A in flavivirus assembly. J Virol 82:4731–4741. doi: 10.1128/JVI.00002-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winkelmann ER, Widman DG, Suzuki R, Mason PW. 2011. Analyses of mutations selected by passaging a chimeric flavivirus identify mutations that alter infectivity and reveal an interaction between the structural proteins and the nonstructural glycoprotein NS1. Virology 421:96–104. doi: 10.1016/j.virol.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 40.Wu R-H, Tsai M-H, Chao D-Y, Yueh A. 2015. Scanning mutagenesis studies reveal a potential intramolecular interaction within the C-terminal half of dengue virus NS2A involved in viral RNA replication and virus assembly and secretion. J Virol 89:4281–4295. doi: 10.1128/JVI.03011-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xie X, Zou J, Puttikhunt C, Yuan Z, Shi P-Y. 2015. Two distinct sets of NS2A molecules are responsible for dengue virus RNA synthesis and virion assembly. J Virol 89:1298–1313. doi: 10.1128/JVI.02882-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vossmann S, Wieseler J, Kerber R, Kümmerer BM. 2015. A basic cluster in the N terminus of yellow fever virus NS2A contributes to infectious particle production. J Virol 89:4951–4965. doi: 10.1128/JVI.03351-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scaturro P, Cortese M, Chatel-Chaix L, Fischl W, Bartenschlager R. 2015. Dengue virus non-structural protein 1 modulates infectious particle production via interaction with the structural proteins. PLoS Pathog 11:e1005277. doi: 10.1371/journal.ppat.1005277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tuplin A. 2015. Diverse roles and interactions of RNA structures during the replication of positive-stranded RNA viruses of humans and animals. J Gen Virol 96:1497–1503. doi: 10.1099/vir.0.000066. [DOI] [PubMed] [Google Scholar]
- 45.Mortimer SA, Kidwell MA, Doudna JA. 2014. Insights into RNA structure and function from genome-wide studies. Nat Rev Genet 15:469–479. doi: 10.1038/nrg3681. [DOI] [PubMed] [Google Scholar]