

Abstract
We derived macrocyclic HIV-1 antagonists as a new class of peptidomimetic drug leads. Cyclic peptide triazoles (cPTs) retained the gp120 inhibitory and virus-inactivating signature of parent PTs, arguing that cyclization locked an active conformation. The 6-residue cPT 9 (AAR029b) exhibited sub-micromolar antiviral potencies in inhibiting cell infection and triggering gp120 shedding that causes irreversible virion inactivation. Importantly, cPTs were stable to trypsin and chymotrypsin, compared to substantial susceptibility of corresponding linear PTs.
Keywords: HIV-1 inactivator, gp120, Macrocycles, Peptide triazoles, Peptidomimetics
Table of Contents Graphic
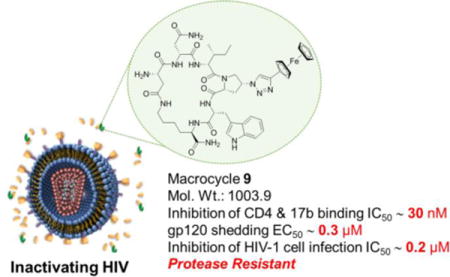
Effective HIV-1 cell entry inhibitors remain a critical unmet need in the effort to prevent virus infection and to intervene with disease progression in infected individuals.1 The virus surface Env spike protein complex composed of gp120 and gp41 is required for cell receptor binding and consequent entry. The gp120 interaction with cell surface receptor CD4 and subsequent interaction with co-receptor CCR5 or CXCR4 are the first events in the entry process.2,3 While its exposure on the virus makes gp120 perhaps the most enticing HIV-1 target for entry inhibitors, discovering clinically useful antagonists targeting gp120 has remained elusive due to such factors as conformational, glycan and mutational masking of conserved binding sites. Some gp120 antagonist candidates are currently under investigation as potential entry inhibitors. BMS class inhibitors, for example, target the unliganded, inactivated state of the viral spike.4 They have been shown to be effective both in vitro and in vivo and currently are in phase IIb clinical trials.5 In addition, both small molecule6,7 and miniprotein8 CD4 mimics, have been identified that target an activated spike state and have in vitro antiviral activity, though their in vivo antiviral efficacies are yet to be evaluated.9 To date, there are no licensed drugs that target the gp120 glycoprotein machinery.9
In the light of a continuing need for effective gp120 antagonists, we have discovered a class of HIV-1 entry inhibitors10, gp120-targeting peptide triazoles (PTs)11, that are able to recognize a non-CD4-bound conformation of HIV-1 Env gp120 from a broad range of virus subtypes with sub-micromolar affinities, to suppress gp120 interactions at both its CD4 and co-receptor binding sites, to cause gp120 shedding12 from the virion particle and as a consequence to inactivate the virus and prevent cell infection. While these functional phenotypes are tantalizing, the peptidic nature of PTs makes them proteolytically susceptible, and hence limits their potential for clinical use. We thus considered it important to identify methods to convert PTs into metabolically stable forms that retain their functional potencies. In the current report, we describe our ability to achieve this goal by PT cyclization. Some macrocyclic HIV-1 inhibitors targeting either HIV-1 protease13 or gp4114 have been previously investigated.15 Others were reported as co-receptor CXCR4 inhibitors.16 However, there are currently no macrocyclic inactivators that target gp120 to suppress receptor binding and inactivate HIV-1.
We recently showed that PTs utilize a conserved two-cavity binding site in gp120 to effect dual receptor site inhibition.10 Modeling studies of the active linear 6-residue PTs bound to gp120 with 2-cavity occupancy that suggested the proximity of the N- and C-termini. In the model of peptide 1, Figure 1, the N terminal Asn side chain, residue 2, could be a solvent exposed moiety. The peptide C terminus extending from a central three-residue primary functional pharmacophore is proximal to the peptide residue Asn-2 side chain amide. The presence of a Pro residue also supports β-turn formation, bringing the peptide termini closer.17 Based on this topology, we investigated a strategy for constraining the active peptide conformation by connecting the C terminus to the exposed Asn-2 side chain (Figure 1). We chose Lys as a possible C terminus extension, as the 4 carbons in its side chain can provide a flexible linker required to reach the Asn-2 side chain at the N terminus. Moreover, the amine group of Lys side chain can be utilized for a covalent bond through amide formation. At the N terminus, residue Asn-2 was initially mutated to Glu (and also to Asp), providing a carboxylic acid functionality for amide formation. After amide coupling of the Glu COOH side chain with the Lys NH2 side chain, the resultant amide (GluCO-LysNH-) would resemble the original Asn-2 amide side chain, therein retaining an electronic environment that could be useful to provide additional stabilizing interactions with the binding site.
Figure 1.
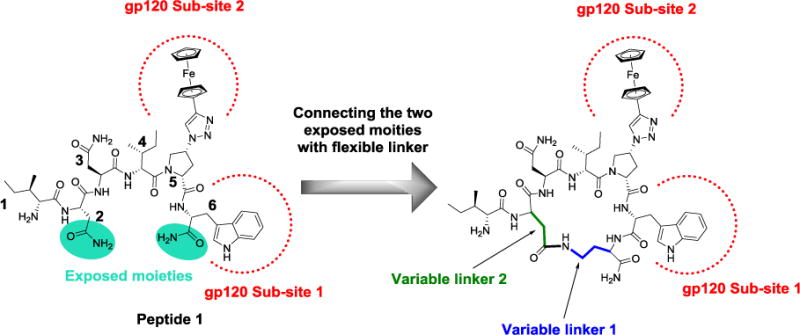
Diagram of the 6-residue peptide 1 depicting the rationale for a side-chain to side-chain cyclization strategy.
For the synthesis of cyclic PT, we initially replaced either the N terminal Asn-2 residue (Supplementary Scheme 1S) or the Ile-1 (Supplementary Scheme 2S) with a Glu residue in which the COOH side chain was protected by ODmab. We also added a Lys (Dde protected side chain NH2) at the C terminal next to the Trp residue. The orthogonal on-resin deprotection of both GluCOOH and LysNH2 with 2% hydrazine in DMF afforded the free COOH and NH2. On-resin side chain to side chain cyclization was successful using microwave synthesizer coupling conditions. On-resin click reaction followed by cleavage/global deprotection and HPLC purification (to ≥ 95% purity) yielded the cyclic PTs. The purity of peptides was confirmed using analytical C18 RP-HPLC column using a gradient of (20 to 95 % ACN)/H2O/0.1% TFA over 42 min run time, with a flow rate of 2 mL min−1 and UV absorbance at 280 nm. Interestingly the cPT 2 (AAR024)18 and 5 (AAR026,18 see supplementary Figure 1S for the chemical structure) not only retained the dual receptor site inhibitory signature of linear PTs, but also displayed enhanced potency compared to the linear analogues (Supplementary Table 1S). Macrocycle 2 was found to be >40 fold more active than the linear 3 (AAR024A,18 see supplementary Figure 1S for the chemical structure). Similarly, the minimized hexapeptide 7 (AAR029)18 was >200 fold more active than 8 (AAR029A)18.
The above observations motivated us to further optimize the cyclic heptapeptide 2 via a series of step-wise modifications (Figure 2 and Supplementary Table 1S). We used SPR competition binding assays to assess the structural elements required for cyclic hexapeptide binding to gp120. As shown in Figure 2, we first trimmed the N terminal Ile to obtain a cyclic hexapeptide 6 (AAR028)18, which surprisingly was found to be inactive at the concentration range used (IC50 > 40 μM). This could be due to the loss of important contacts with the protein upon deletion of the N terminal Ile. In an attempt to restore the lost contact, the length of the two linkers was changed (blue and green linkers in Figure 2). Shortening the blue linker using only two carbon spacers also resulted in inactive macrocycle 10 (AAR030)18. However, incorporating a shorter green linker, by using Asp instead of Glu during the peptide synthesis, resulted in almost two-fold enhanced activity in macrocycle 7 vs 2. This enhancement might be related to the improved positioning of the macrocycle relative to its binding site on gp120. We sought to further optimize 7 by step-wise shortening of the blue linker. Interestingly, incorporating a three-carbon linker, using Orn(Dde), resulted in decreased but not lost activity [11 (AAR031)18, micromolar IC50 value]. However, further shortening with a two carbon linker, using Dab(ivDde), resulted in regaining of activity with almost ten-fold improvement [12 (AAR032)18 vs 11]. These results reflect the significant sensitivity of target binding to changes in the blue linker length. We envision that this sensitivity derives from the different conformations that could be adopted by the constrained sequence and as a consequence could affect the orientations of the PT pharmacophores required for binding.
Figure 2.
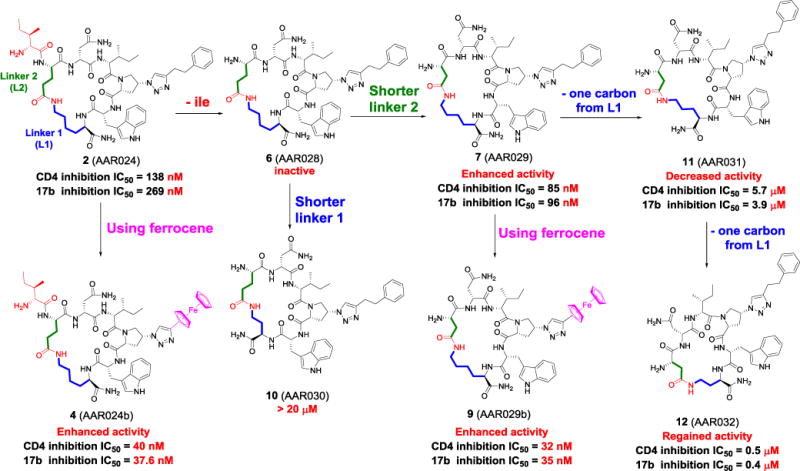
Structure-activity relationship of the synthesized cyclic PTs, starting from macrocycle 2. Each arrow represents a separate synthesis of a subsequent peptide embodying the modifications shown associated with the arrow. The IC50 values shown below each PT reflect the effectiveness of the resulting peptides to inhibit gp120 binding to sCD4 and 17b antibody, using SPR.
We previously showed that introducing the bulky ferrocene moiety during the click conjugation step at the azido-Pro residue in the peptide intermediate results in higher affinities to the gp120 protein.19 Here, we found that ferrocenyl cPTs, 4 (AAR024b)18 and 9 (AAR029b)18 (Supplementary Figure 2S), showed enhanced dual receptor site antagonizing activities compared to their non-ferrocene analogues (Figure 2). The bulky ferrocene moiety would be expected to better stack within the inner domain hydrophobic cavity identified in our previous findings10, therefore enhancing inhibitor binding affinity.
Based on the increased activities of cyclic hexapeptides 7 and 9, we further refined the synthetic route (Scheme 1) using Boc-Asp(Ofm)-OH at the N terminal. After peptide assembly using microwave synthesis, the Asp carboxylic group was deprotected by 20% piperidine in DMF during the final microwave deprotection step. The peptide-bound resin was then subjected to selective deprotection conditions (2% hydrazine in DMF, 3 × 40 mL × 10 min) to deprotect the Lys NH2 group. Cyclization was achieved using two microwave synthesizer coupling steps. Click reaction with the alkyne followed by acidic deprotection/cleavage yielded the crude peptides. Purification (to ≥ 95% purity) of the soluble crude peptide followed procedures similar to those used for macrocycle 2. This modified method (Scheme 1) is cheaper and affords improved yields by at least 3 fold compared to the initially described methods (Supplementary Schemes 1S & 2S), hence representing a simple route to access the highly active class of macrocyclic HIV-1 inhibitors.
Scheme 1.
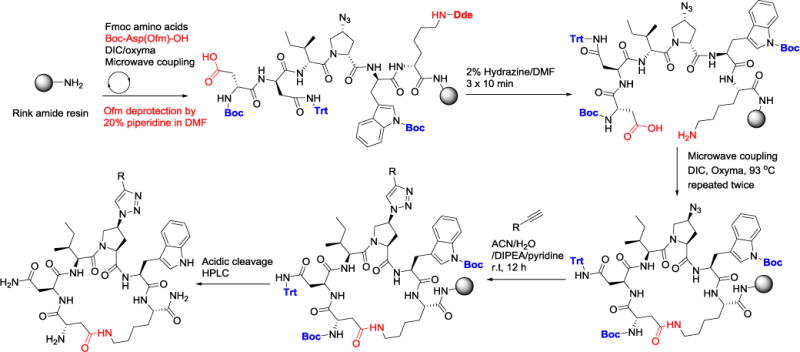
Refined solid-phase synthesis of cyclic peptide triazoles 7 (R = phenylethyl) and 9 (R = ferrocenyl).
The molecular-level inhibitory activities of the cPTs against HIV-1 gp120 glycoproteins (Supplementary Table 1S) argued for their potential use as HIV-1 infection inhibitors. We evaluated the most active cPT derivatives (SPR binding data, Table 1S) against a laboratory adapted strain of HIV-1, Bal.01 and a more resistant strain, JR-FL (Table 1, Supplementary Figure 3S). Importantly, the metallocene containing cPT 9 was able to inhibit the two strains at sub-micromolar concentrations, demonstrating improved potency compared to the parent linear peptide 1. Macrocycle 9 was also found to trigger gp120 shedding, leaving a naked non-infectious virion, with an EC50 value of 316 nM for the JR-FL strain (Supplementary Figure 4S), which is in agreement with its infection inhibition IC50 value (Table 1). As discussed before,10 the shedding function of macrocycle 9 is most likely caused by conformational perturbation of the virus spike Env protein that is triggered when PTs bind in their unique site on the gp120 subunits (Figure 1). This PT-induced conformational perturbation in turn explains suppression of ligand binding at both the CD4 and co-receptor sites of the gp120 protein. The functionality of macrocycle 9 makes it the most potent hexapeptide triazole (linear or cyclic) identified to date. As expected, the non-ferrocenyl cPT 7 was less potent than 9, yet still remained among the most potent hexapeptides10 tested so far. The ability of the cPTs to inhibit both viral strains tested, including the more difficult-to-inhibit tier 2 JR-FL subtype, argues that the cPTs likely retain the breadth of function already observed for linear PTs.20,21,12 Using WST-1 assay, we also observed no significant cell cytotoxicity (Supplementary Figure 3S), even at the highest concentrations used for the infection inhibition analysis. These potency and cellular cytotoxicity results demonstrate the potential use of cPTs as leads for HIV-1 inhibitors.
Table 1.
Infection inhibition against Bal.01 and JR-FL HIV-1 pseudoviruses exhibited by the parent lead peptide 1 and three cPTs, 4, 7 and 9.
| Compound | BaL.01 inhibition IC50 (nM) | JR-FL inhibition IC50 (nM) |
|---|---|---|
| 1 | 850 ± 136 | 453 ± 137 |
| 4 | 650 ± 70 | 603 ± 98 |
| 7 | 2018 ± 95 | 2330 ± 538 |
| 9 | 210 ± 12 | 190 ± 34 |
An important rationale for cyclizing PTs was to eliminate proteolytic susceptibility, which is a major obstacle in developing some peptides into marketed therapeutics despite their high potencies and selectivities.22 Here, we compared the in vitro stability of the cPT 7 to that of its linear analogue 8 against two specific proteases, trypsin and chymotrypsin. The designed cyclic peptides have a trypsin-sensitive Lys amide group incorporated for cyclization, and also have chymotrypsin-sensitive Trp and Ile residues within their sequences. We incubated the two peptides separately with each of the two enzymes at 37 °C and used HPLC (Supplementary Figure 5S) and ESI-MS to monitor the digestion reactions. The results showed striking differences (Figure 3). The macrocycle 7 was completely resistant to both trypsin and chymotrypsin during the 5-hour incubations used. In contrast, almost 60% of the corresponding linear peptide 8 (Figure 3) was cleaved after 5 hours at the C terminal amide as detected by ESI-MS. The linear peptide 8 was even more unstable with chymotrypsin treatment (Figure 3), wherein 100% of the peptide was hydrolyzed after only a 30 minute incubation time compared to almost unchanged cPT 7. Because of the striking difference between the linear and the cyclic peptides, we re-evaluated the intactness of the macrocycle 7 after 20 h and also 40 h of chymotrypsin digestion (Supplementary Figure 6S). We found that the cyclic peptide remained intact even after 40 h incubation as monitored by HPLC and ESI-MS. Overall, those experiments demonstrate the in vitro stability of the cyclized peptide vs the linear analogue, and the consequent ability to greatly reduce the proteolytic susceptibility by macrocycle formation.
Figure 3.
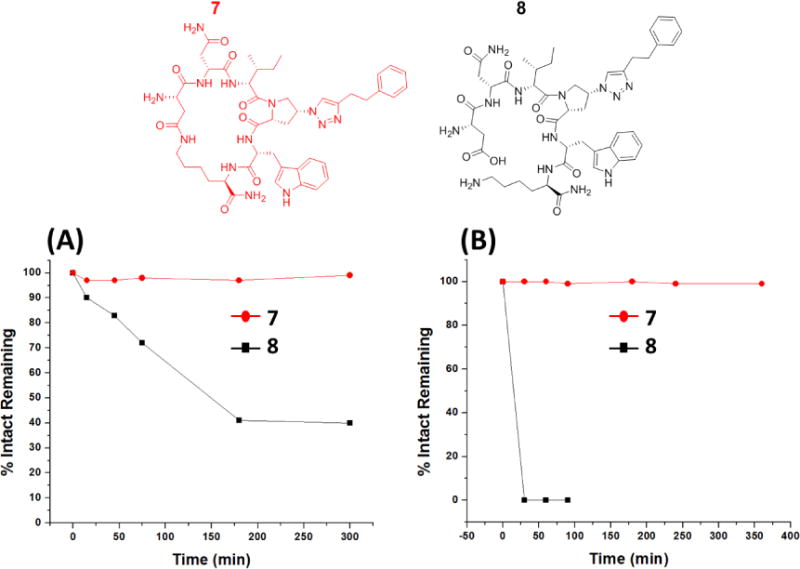
Trypsin (A) and chymotrypsin (B) effects on the cPT 7 (red structure) and the linear analogue 8 (black structure). HPLC peak heights at 0 min were taken as 100% intact. HPLC peak heights at the different time points were used to estimate the % intact peptide remaining compared to the 0 time point.
To rationalize the retention of PT functions by cPTs, we investigated the possible interactions between cPT and envelope HIV-1 gp120 using molecular modeling. Among the gp120 crystal structures available, we chose the recently reported trimeric SOSIP structure (pdb code: 4NCO23), as it represents the gp120 in a non-CD4-bound Env protein in a trimeric form that has been surmised to be close to the structure in the native viral spike.24 We used flexible docking, in which some protein side chains at the binding site along with the peptide were allowed to move during the simulation. Flexible docking can reveal a more plausible binding mode than rigid-docking alone, allowing rearrangements of the protein scaffold in order to better accommodate the docked ligand.25 This docking approach was assessed to be suitable for the highly flexible nature of HIV-1 envelope protein, which can adopt multiple conformations.4 Moreover, our recent work10 showed that PTs require access to binding pocket residues that may be buried in the crystallized protein structure. After the docking simulation (Supplementary Experimental section) of macrocycle 7, the best returned pose (ranked 1 by Glide-Peptide) (Figure 4) had a Glide gscore of −12.04, and a calculated ΔGbinding of −17.64 kcal/mol (as calculated by Szybki 1.8.0.2) indicating stability. This favored binding mode, shown in Figure 4, is consistent with the simulated model for linear PTs,10 where the indole side chain sits in site 1 lined by Ser375 and Thr257 and the triazole substituent sits in site 2 lined by gp120Trp112, gp120Ile109 and gp120Met426. The cPT indole H atom is located at 3.6 Å from gp120Ser375 OH and 3.1 Å from gp120Glu370 COOH group. The solvent exposed C terminal amide NH and Asn-2 side chain NH form 2.1 Å and 2.6 Å H-bonds with the gp120Gly473 backbone carbonyl, respectively. The peptide Pro-4 backbone carbonyl accepts a H-bond (1.93 Å) from the β20/21 loop gp120Trp427 backbone NH. Peptide Ile-3 carbonyl also forms a 1.73 Å H-bond with gp120His105 side chain imidazole NH. The π- π stacking plays a role in complex stabilization, where the peptide phenyl-ethyl stacked with gp120Trp112 and the peptide triazole ring stacked with gp120His105 (Figure 4). This dual site occupancy, along with the constrained peptide conformation, provides an explanation for the functional potency and stability of cPTs.
Figure 4.
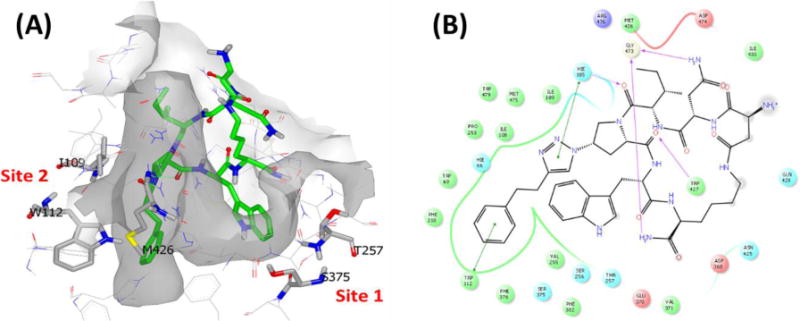
Simulated binding of macrocycle 7 in the HIV-1 envelope crystal structure (pdb code: 4NCO23) using the software Glide. (A) 3D model with peptide carbons colored green and important gp120 interacting residues10 colored in grey (carbons, stick style). (B) 2D representation showing H-bonds and π- π stacking interactions between cPT and gp120 residues.
In summary, we developed a facile five-step synthetic pathway to produce conformationally constrained peptide triazoles that suppress gp120 receptor binding functions and efficiently inhibit HIV-1 cell infection. Unlike linear PTs, cPTs greatly resist in vitro proteolysis as demonstrated by the stability against trypsin and chymotrypsin. The enhanced activity, proteolytic stability and ease of chemical synthesis clearly make this class of cPTs important leads as HIV-1 entry inhibitors. The preliminary SAR data obtained suggest strong opportunities for chemical optimization, which is currently underway. The constrained cPT conformation will also aid crystallization efforts of the highly flexible HIV-1 Env with this class of inhibitors.
Supplementary Material
Acknowledgments
This work was funded by the National Institute of Health through the NIH Program Project GM 56550-17, Structure Based Antagonism of HIV-1 Envelope Function in Cell Entry. We thank Prof. Amos B. Smith III (University of Pennsylvania) for encouragement and helpful discussion during this project. We thank Openeye Scientific Software (Santa Fe, NM. http://www.eyesopen.com) for providing a free academic license of their software package.
ABBREVIATIONS
- ACN
acetonitrile
- Boc
Tertbutyloxycarbonyl
- cPT
Cyclic Peptide triazole
- DIC
N,N′-Diisopropylcarbodiimide
- Dde
N-(1-(4,4-dimethyl-2,6-dioxocyclohexylidene)ethyl)
- ivDde
1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)-3-methylbutyl
- PT
peptide triazole
- ODmab
4-[N-(1-(4,4-dimethyl-2,6-dioxocyclohexylidene)-3-methylbutyl)amino].
- SPR
Surface Plasmon Resonance
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting information including additional synthetic schemes, Surface Plasmon Resonance experimental and sensograms, antiviral functional assays, proteolytic stability experiments with HPLC chromatograms and molecular modelling procedures for this article is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Swindells S, Flexner C, Fletcher CV, Jacobson JM. The Critical Need for Alternative Antiretroviral Formulations, and Obstacles to Their Development. J Infect Dis. 2011;204:669–674. doi: 10.1093/infdis/jir370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doms RW. Beyond Receptor Expression: The Influence of Receptor Conformation, Density, and Affinity in HIV-1 Infection. Virology. 2000;276:229–237. doi: 10.1006/viro.2000.0612. [DOI] [PubMed] [Google Scholar]
- 3.Wyatt R, Kwong PD, Desjardins E, Sweet RW, Robinson J, Hendrickson WA, Sodroski JG. The Antigenic Structure of The HIV gp120 Envelope Glycoprotein. Nature. 1998;393:705–711. doi: 10.1038/31514. [DOI] [PubMed] [Google Scholar]
- 4.Munro JB, Gorman J, Ma X, Zhou Z, Arthos J, Burton DR, Koff WC, Courter JR, Smith AB, 3rd, Kwong PD, Blanchard SC, Mothes W. Conformational Dynamics of Single HIV-1 Envelope Trimers on The Surface of Native Virions. Science. 2014;346:759–763. doi: 10.1126/science.1254426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brinson C, Lalezari J, Gulam LH, Thompson M, Echevarria J, Trevino-Perez S, Stock D, Samit JR, George HJ, Lataillade M. HIV-1 Attachment Inhibitor Prodrug BMS-663068 in Antiretroviral-experienced Subjects: Week 24 Sub-group Analysis. J Int AIDS Soc. 2014;17:19529. doi: 10.7448/IAS.17.4.19529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao Q, Ma LY, Jiang SB, Lu H, Liu SW, He YX, Strick N, Neamati N, Debnath AK. N-phenyl-N′-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a New Class of HIV-1 Entry Inhibitors that Prevent gp120 Binding to CD4. Virology. 2005;339:213–225. doi: 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 7.Courter JR, Madani N, Sodroski J, Schon A, Freire E, Kwong PD, Hendrickson WA, Chaiken IM, LaLonde JM, Smith AB. Structure-Based Design, Synthesis and Validation of CD4-Mimetic Small Molecule Inhibitors of HIV-1 Entry: Conversion of a Viral Entry Agonist to an Antagonist. Acc Chem Res. 2014;47:1228–1237. doi: 10.1021/ar4002735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morellato-Castillo L, Acharya P, Combes O, Michiels J, Descours A, Ramos OHP, Yang YP, Vanham G, Alien KK, Kwong PD, Martin L, Kessler P. Interfacial Cavity Filling To Optimize CD4-Mimetic Miniprotein Interactions with HIV-1 Surface Glycoprotein. J Med Chem. 2013;56:5033–5047. doi: 10.1021/jm4002988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Acharya P, Lusvarghi S, Bewley CA, Kwong PD. HIV-1 gp120 as a Therapeutic Target: Navigating a Moving Labyrinth. Expert Opin Ther Targets. 2015:1–19. doi: 10.1517/14728222.2015.1010513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aneja R, Rashad AA, Li H, Kalyana Sundaram RV, Duffy C, Bailey LD, Chaiken I. Peptide Triazole Inactivators of HIV-1 Utilize a Conserved Two-Cavity Binding Site at the Junction of the Inner Outer Domains of Env gp120. J Med Chem. 2015;58:3843–3858. doi: 10.1021/acs.jmedchem.5b00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biorn AC, Cocklin S, Madani N, Si ZH, Ivanovic T, Samanen J, Van Ryk DI, Pantophlet R, Burton DR, Freire E, Sodroski J, Chaiken IM. Mode of Action for Linear Peptide Inhibitors of HIV-1 gp120 Interactions. Biochemistry. 2004;43:1928–1938. doi: 10.1021/bi035088i. [DOI] [PubMed] [Google Scholar]
- 12.Bastian AR, Contarino M, Bailey LD, Aneja R, Moreira DR, Freedman K, McFadden K, Duffy C, Emileh A, Leslie G, Jacobson JM, Hoxie JA, Chaiken I. Interactions of Peptide Triazole Thiols with Env gp120 Induce Irreversible Breakdown and Inactivation of HIV-1 Virions. Retrovirology. 2013;10 doi: 10.1186/1742-4690-10-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tyndall JD, Reid RC, Tyssen DP, Jardine DK, Todd B, Passmore M, March DR, Pattenden LK, Bergman DA, Alewood D, Hu SH, Alewood PF, Birch CJ, Martin JL, Fairlie DP. Synthesis, Stability, Antiviral Activity, Protease-Bound Structures of Substrate-Mimicking Constrained Macrocyclic Inhibitors of HIV-1 Protease. J Med Chem. 2000;43:3495–3504. doi: 10.1021/jm000013n. [DOI] [PubMed] [Google Scholar]
- 14.Wang D, Lu M, Arora PS. Inhibition of HIV-1 Fusion by Hydrogen-Bond-Surrogate-Based Alpha Helices. Angew Chem. 2008;47:1879–1882. doi: 10.1002/anie.200704227. [DOI] [PubMed] [Google Scholar]
- 15.Hill TA, Shepherd NE, Diness F, Fairlie DP. Constraining Cyclic Peptides To Mimic Protein Structure Motifs. Angew Chem Int Ed. 2014;53:13020–13041. doi: 10.1002/anie.201401058. [DOI] [PubMed] [Google Scholar]
- 16.Demmer O, Frank AO, Hagn F, Schottelius M, Marinelli L, Cosconati S, Brack-Werner R, Kremb S, Wester HJ, Kessler H. A Conformationally Frozen Peptoid Boosts CXCR4 Affinity and Anti-HIV Activity. Angew Chem Int Ed. 2012;51:8110–8113. doi: 10.1002/anie.201202090. [DOI] [PubMed] [Google Scholar]
- 17.Fu H, Grimsley GR, Razvi A, Scholtz JM, Pace CN. Increasing Protein Stability by Improving Beta-Turns. Proteins. 2009;77:491–498. doi: 10.1002/prot.22509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rashad AA, Chaiken I. Novel Cyclic Peptides and Methods Using Same. Provisional US Patent Application Number 62/089,294. 2014
- 19.Gopi H, Cocklin S, Pirrone V, McFadden K, Tuzer F, Zentner I, Ajith S, Baxter S, Jawanda N, Krebs FC, Chaiken IM. Introducing Metallocene Into a Triazole Peptide Conjugate Reduces Its Off-Rate and Enhances Its Affinity and Antiviral Potency For HIV-1 gp120. J Mol Recognit. 2009;22:169–174. doi: 10.1002/jmr.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McFadden K, Fletcher P, Rossi F, Kantharaju, Umashankara M, Pirrone V, Rajagopal S, Gopi H, Krebs FC, Martin-Garcia J, Shattock RJ, Chaiken I. Antiviral Breadth and Combination Potential of Peptide Triazole HIV-1 Entry Inhibitors. Antimicrob Agents Chemother. 2012;56:1073–1080. doi: 10.1128/AAC.05555-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bastian AR, Kantharaju, McFadden K, Duffy C, Rajagopal S, Contarino MR, Papazoglou E, Chaiken I. Free HIV-1 Virucidal Action by Modified Peptide Triazole Inhibitors of Env gp120. Chemmedchem. 2011;6:1335–1339. doi: 10.1002/cmdc.201100177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yudin AK. Macrocycles: Lessons From the Distant Past, Recent Developments, and Future Directions. Chem Sci. 2015;6:30–49. doi: 10.1039/c4sc03089c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Julien JP, Cupo A, Sok D, Stanfield RL, Lyumkis D, Deller MC, Klasse PJ, Burton DR, Sanders RW, Moore JP, Ward AB, Wilson IA. Crystal Structure of a Soluble Cleaved HIV-1 Envelope Trimer. Science. 2013;342:1477–1483. doi: 10.1126/science.1245625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pugach P, Ozorowski G, Cupo A, Ringe R, Yasmeen A, de Val N, Derking R, Kim HJ, Korzun J, Golabek M, de Los Reyes K, Ketas TJ, Julien JP, Burton DR, Wilson IA, Sanders RW, Klasse PJ, Ward AB, Moore JP. A Native-Like SOSIP.664 Trimer Based on an HIV-1 Subtype B env Gene. J Virol. 2015;89:3380–3395. doi: 10.1128/JVI.03473-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma DL, Chan DSH, Leung CH. Molecular Docking for Virtual Screening of Natural Product Databases. Chem Sci. 2011;2:1656–1665. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.