

Abstract
Background
Lake Magadi and little Magadi are hypersaline, alkaline lakes situated in the southern part of Kenyan Rift Valley. Solutes are supplied mainly by a series of alkaline hot springs with temperatures as high as 86 °C. Previous culture-dependent and culture-independent studies have revealed diverse groups of microorganisms thriving under these conditions. Previous culture independent studies were based on the analysis of 16S rDNA but were done on less saline lakes. For the first time, this study combined illumina sequencing and analysis of amplicons of both total community rDNA and 16S rRNA cDNA to determine the diversity and community structure of bacteria and archaea within 3 hot springs of L. Magadi and little Magadi.
Methods
Water, wet sediments and microbial mats were collected from springs in the main lake at a temperature of 45.1 °C and from Little Magadi “Nasikie eng’ida” (temperature of 81 °C and 83.6 °C). Total community DNA and RNA were extracted from samples using phenol-chloroform and Trizol RNA extraction protocols respectively. The 16S rRNA gene variable region (V4 – V7) of the extracted DNA and RNA were amplified and library construction performed following Illumina sequencing protocol. Sequences were analyzed done using QIIME while calculation of Bray-Curtis dissimilarities between datasets, hierarchical clustering, Non Metric Dimensional Scaling (NMDS) redundancy analysis (RDA) and diversity indices were carried out using the R programming language and the Vegan package.
Results
Three thousand four hundred twenty-six and one thousand nine hundred thirteen OTUs were recovered from 16S rDNA and 16S rRNA cDNA respectively. Uncultured diversity accounted for 89.35 % 16S rDNA and 87.61 % 16S rRNA cDNA reads. The most abundant phyla in both the 16S rDNA and 16S rRNA cDNA datasets included: Proteobacteria (8.33–50 %), Firmicutes 3.52–28.92 %, Bacteroidetes (3.45–26.44 %), Actinobacteria (0.98–28.57 %) and Euryarchaeota (3.55–34.48 %) in all samples. NMDS analyses of taxonomic composition clustered the taxa into three groups according to sample types (i.e. wet sediments, mats and water samples) with evident overlap of clusters between wet sediments and microbial mats from the three sample types in both DNA and cDNA datasets. The hot spring (45.1 °C) contained less diverse populations compared to those in Little Magadi (81–83 °C).
Conclusion
There were significant differences in microbial community structure at 95 % level of confidence for both total diversity (P value, 0.009) based on 16S rDNA analysis and active microbial diversity (P value, 0.01) based on 16S rRNA cDNA analysis, within the three hot springs. Differences in microbial composition and structure were observed as a function of sample type and temperature, with wet sediments harboring the highest diversity.
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-016-0748-x) contains supplementary material, which is available to authorized users.
Keywords: Lake Magadi, Hot springs, 16S rDNA, 16S rRNA cDNA, Microbial diversity
Background
Extreme environment refers to any setting that exhibits life conditions detrimental to living organisms with respect to its physicochemical properties such as pH, temperature, pressure, nutrient and saline concentration [1]. Extreme physicochemical parameters include acidity (pH <5), alkalinity (pH >9), hyper salinity (salinity >35 %), pressure (>0.1 MPa), high temperature (>40 °C), low temperature (<5 °C), water stress (aw <0.80), and high-radiation environments [2]. The extreme environments are inhabited by organisms referred to as extremophiles that are so well-adapted that they readily grow and multiply [3]. In Kenya, the haloalkaline soda lakes are characterized by exceptionally rich productivity rates presumably because of the high ambient temperatures, high light intensities, availability of phosphates and unlimited access to CO2 in these carbonate rich waters [4, 5]. Salinity levels can be as high as 30 % to saturation in Lake Magadi, whereas the pH ranges between 9 and 11.5 [6]. In Lake Magadi, solutes are supplied mainly by a series of alkaline springs with temperatures varying from 33 °C to 86 °C [6, 7].
Previous culture dependent and culture independent studies on Lake Magadi have revealed a dense and diverse population of aerobic, organotropic, halophilic, alkaliphilic, and haloalkaliphilic and alkalitolerant representatives of major bacterial phyla [8–15]. Although conventional microbial cultivation methods have helped shape understanding of physiology and metabolic functions of diverse organisms, they are laborious, time consuming, selective and biased for specific microbial growth. On the other hand, culture - independent studies done on soda lakes in Kenya have been based on the analysis of clone libraries of PCR amplified rDNA. This may not represent an accurate picture of prokaryotic diversity within a given community due to low speed and coverage of a cloning and Sanger-sequencing based approach, which gives a lower number of amplicon sequences compared to the millions of generated by High Throughput Sequencing technologies such as Illumina Sequencing [16]. This is the first culture independent study of the microbial community within the hot springs located around the hypersaline Lakes Magadi and Little Magadi. This study employed Illumina Sequencing of PCR products of both 16S rDNA and 16S rRNA cDNA to obtain a less biased estimation of microbial community within the hot springs’ ecosystem. The main objective of this study was to analyze the targeted total community rDNA and cDNA generated from rRNA so as to compare the total versus active microbial communities within the hot springs of Lake Magadi and Little Magadi in Kenya.
Methods
Research authorization
Research authorization was obtained from National Commission for Science, Technology and Innovation (NACOSTI) on 30th August 2013 in Kenya, and permission to conduct research in Lake Magadi was obtained from Kenya Wildlife Services (KWS) on 24th September 2013.
Study site
Lake Magadi is a hypersaline lake that lies in a naturally formed closed lake basin within the Southern part of the Kenyan Rift Valley. It is approximately 2° 00′ 0″ S and 36° 00′ 0″ E of the Equator at an elevation of about 600 m above sea level [17]. The solutes are supplied mainly by a series of alkaline springs with temperatures as high as 86 °C which are located around the perimeter of the lake. Samples analyzed in this study were collected from three hot springs: one hot spring within the main L. Magadi (02° 00′ 3.7″S 36° 14′ 32″ E) at 45.1 °C and pH 9.8; and two hot springs within Little Magadi “Nasikie eng’ida” (01° 43′ 28″S 36° 16′ 21″E), and (01° 43′ 56″ S 36° 17′ 11″ E) at elevations of 611 and 616 m, temperatures of 81 and 83.6 °C and pH range of 9.2 and 9.4 respectively (Table 1).
Table 1.
Parameters of sampling sites analyzed in this study
| Parameter | Latitude °S | Longitude °E | Elevation (m) | Temperature °C | pH | EC (mS/cm) | TDS (mg/l) | Dissolved Oxygen (mg/l) |
|---|---|---|---|---|---|---|---|---|
| Hot spring 1 | 02° 00′ 3.7″ | 36° 14′ 32″ | 603 | 45.1 | 9.8 | 0.03 | 1 | 12.4 |
| Hot spring 2 | 01° 43′ 28″ | 36° 16′ 21″ | 611 | 83.6 | 9.4 | 1 | 1 | 0.04 |
| Hot spring 3 | 01° 43′ 56″ | 36° 17′ 11″ | 616 | 81 | 9.2 | 1 | 1 | 0.71 |
Measurements of physicochemical parameters
Geographical position of each site in terms of latitude, longitude and elevation was taken using Global Positioning System (GARMIN eTrex 20). The pH for each sampling point was measured with a portable pH-meter (Oakton pH 110, Eutech Instruments Pty. Ltd) and confirmed with indicator strips (Merck, range 5–10). Temperature, Electrical Conductivity (EC), Total dissolved solids (TDS) and dissolved oxygen (DO) were measured on site using Electrical Chemical Analyzer (Jenway - 3405) during sampling. In situ temperature was recorded once for each study site and assigned to all the three sample types for that site.
Sample collection
All samples were collected randomly in triplicates from each hot spring. Water samples were collected using sterile 500 ml plastic containers that had been cleaned with 20 % sodium hypochlorite and UV-sterilized for one hour. Wet sediments were collected by scooping with sterilized hand shovel into sterile 50 ml Falcon tubes. The upper 5 mm from each microbial mat developing on the hot spring water margins was collected into sterile 500 ml plastic jam jars. All samples were preserved on dry ice immediately after sampling, and transported to the laboratory in Jomo Kenyatta University of Agriculture and Technology. Water for nucleic acid extraction (500 ml) was filtered through a 0.22 μM filter membrane (Whatman) and all filter papers containing samples were stored at −80 °C. Pellets were obtained from water samples by re-suspending the filter papers in phosphate buffer solution, and centrifuging 5 ml of the suspension at 13,000 rpm for 10 min. These were used for nucleic acid extraction.
Nucleic acid extraction
Total community DNA was extracted from all the samples in triplicates; pellets from water samples, 0.2 g of sediment samples and 0.4 g of microbial mat samples, as described by as described by Sambrook et al. [18]. Total RNA was extracted from 0.25 g of sediment and mat samples, and pellets obtained from the water samples (described above), in triplicates using Trizol RNA extraction protocol [19]. The respective nucleic acids extracted from triplicate samples were pooled during the precipitation stage, the pellets were air dried and stored at −20 °C. The pellets were lyophilized to protect them from degradation.
Synthesis of cDNA from 16S rRNA
cDNA synthesis, amplification and sequencing were performed at Molecular Research DNA Lab (www.mrdnalab.com, Shallowater, TX, USA). The quality of total RNA was assessed using gel electrophoresis. The extracted RNA was dissolved in RNase-free water and subsequently treated to remove DNA contaminants using the Amplification Grade DNase I Kit (Sigma, MO) according to manufacturer’s instructions. cDNA first-strand and second-strand synthesis was done using the Superscript III First-Strand Synthesis SuperMix (Invitrogen, CA) and the Second-strand cDNA Synthesis Kit (BeyoTime, Jiangsu, China), respectively, following manufacturer’s instructions. Single-strand reverse transcription was done to provide template for amplicon libraries using Superscript III (Invitrogen) according to the manufacturer’s protocol, random hexamer primed and with subsequent RNAse H digestion. The Double stranded cDNA synthesis was carried out as described by as described by Urich et al. [20].
Amplicon library preparation and sequencing
PCR amplification of the 16S rRNA gene V4 variable region was carried out from extracted DNA and cDNA generated from rRNA, using bacteria/archaeal primers 515 F (GTGCCAGCMGCCGCGGTAA) that had barcode and 806R (GGACTACHVGGGTWTCTAAT) according to Caporaso et al. [21]. Amplification proceeded in a 30 cycle PCR using the HotStarTaq Plus Master Mix Kit (Qiagen, USA) with initial denaturation heating at 94 °C for 3 min, followed by 28 cycles of denaturation at 94 °C for 30 s, annealing at 53 °C for 40 s and extension at 72 °C for 1 min, and a final elongation at 72 °C for 5 min. The quality of PCR products was assessed on 2 % agarose gel to determine the success of amplification and the relative intensity of bands. Multiple samples, tagged with different barcodes, were pooled in equimolar ratios based on their DNA concentrations from the gel images. Pooled samples were purified using calibrated Ampure XP beads (Beckman Coulter) for use in library preparation. The pooled and purified PCR products were used to prepare 16S rDNA and cDNA library by following Illumina TruSeq DNA library preparation protocol [22]. Sequencing was performed at MR DNA (www.mrdnalab.com, Shallowater, TX, USA) on a MiSeq 2x300bp Version 3 following the manufacturer’s guidelines.
Sequence analysis, taxonomic classification and data submission
Sequences obtained from the Illumina sequencing platform were depleted of barcodes and primers using a proprietary pipeline (www.mrdnalab.com, MR DNA, Shallowater, TX) developed at the service provider’s laboratory. Low quality sequences were identified by denoising and filtered out of the dataset [23]. Sequences which were <200 base pairs after phred20- based quality trimming, sequences with ambiguous base calls, and those with homopolymer runs exceeding 6 bp were removed. Sequences were analyzed by a script optimized for high-throughput data to identify potential chimeras in the sequence files, and all definite chimeras were depleted as described previously [24]. All this data filtering was done by the service provider using their pipeline. De novo OTU clustering was done with standard UCLUST method using the default settings as implemented in QIIME pipeline Version 1.8.0 at 97 % similarity level. Taxonomy was assigned to each OTU using BLASTn against SILVA SSU Reference 119 database at default e-value threshold of 0.001 in QIIME [25, 26]. Obtained sequences were submitted to the NCBI Sequence Read Archive with SRP# Study accessions: SRP061805. These included SRX1124606: RNA-Seq of Prokaryotes: Alkaline Hot springs and SRX1124607: DNA-Seq of Prokaryotes: Alkaline Hot springs (Additional file 1: Table S1, Additional file 2: Table S2, Additional file 3: Table S3 and Additional file 4: Table S4).
Statistical analysis
Diversity indices (Shannon, Simpson and Evenness) for each sample were calculated using vegan package version 1.16-32 in R software version 3.1.3 [27]. Community and Environmental distances were compared using Analysis of similarity (ANOSIM) test, based upon Bray-Curtis distance measurements with 999 permutations. Significance was determined at 95 % confidence interval (p = 0.05). Calculation of Bray-Curtis dissimilarities between datasets, hierarchical clustering, Non Metric Dimensional Scaling (NMDS), redundancy analysis (RDA) and parameter correlation were carried out using the R programming language [27] and the Vegan package [28]. To support OTU-based analysis, taxonomic groups were derived from the number of reads assigned to each taxon at all ranks from domain to genus using the taxa_summary.txt output from QIIME pipeline Version 1.8.0.
Results and discussion
Sampling
Three hot springs of Lake Magadi and Little Magadi were selected based on different temperature and pH levels. Temperatures ranged from 45.1 to 83.6 °C while pH ranged from 9.2 to 9.8. The TDS was above the measurement range for the Electrical Chemical Analyzer; hence all the readings appeared as one (1). The metadata collected before sampling is summarized in Table 1.
Composition and diversity of the microbial communities
After denoising and demultiplexing, a total of 271,345 and 214,663 sequence reads were generated from 16S rDNA and 16S rRNA cDNA data respectively. Total OTU richness at 3 % distance amounted to 3502 and 1915 OTUs respectively. 85 and 62 OTUs were shared across all hot springs while 82 and 45 OTUs were shared across all sample types in the two data sets respectively. Figure 1a and b shows the distribution of OTUs across hot springs and sample types. For 16S rDNA data, hot spring 83.6 °C and 45.1 °C showed a relatively larger overlap (142 OTUs) than other hot springs, while 45.1 °C harbored most OTUs unique to one hot spring in both datasets.
Fig. 1.
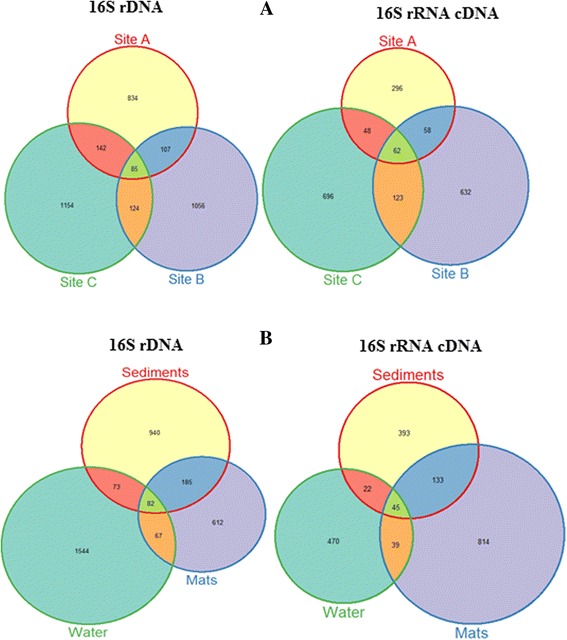
a and b Venn diagrams showing the distribution of unique and shared OTUs within various sample types in the three sampling sites. The number of OTUs in each hot spring is indicated in the respective circle. Site A, B and C represent hot springs 83.6 °C, 81 °C and 45.1 °C respectively
Bacterial taxonomic composition analysis
16S rDNA OTUs comprised of Bacteria (85.55 %) and Archaea (11.27 %) while 16S rRNA cDNA OTUs comprised of Bacteria (83.22 %) and Archaea (13.95 %). These results suggest that bacteria are the most dominant taxa in L. Magadi and Little Magadi hot springs. The groups with highest relative abundances at phylum level belonged to members of Proteobacteria (16–31.4 %), Firmicutes (2.8–28.9 %), Bacteroidetes (3.4–23.8 %), Actinobacteria (1.0–14.4 %), Cyanobacteria (1.1–8.3 %, Chloroflexi (0.3–7.2 %), and Deinococcus-thermus (0.3–5.3 %) (Fig. 2). Other groups scoring high relative abundance in some samples include Gemmatimonadetes (24.1 %) in water samples at 81 °C, Planctomycetes (15.9 %) in water samples at 83.6 °C, Lentisphaerae 5.3 % in mat samples at 81 °C, Spirochaetae 4.5 % in wet sediment samples at 45.1 °C, Thermotogae (1.7 %) in wet sediment samples at 83.6 °C and Verumicrobia (3.9 %) in mat samples at 45.1 °C (Additional file 5: Table S5).
Fig. 2.
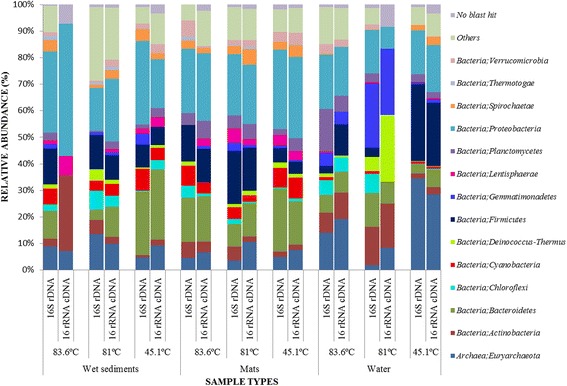
Relative abundance of the most predominant phyla in various samples collected from the hot springs of L. Magadi and Little Magadi. ‘Others’ represent all taxa that scored a relative abundance of below 1 % across all samples in both data sets
At family level, OTUs were distributed in 292 bacterial families with the most abundant belonging to Rhodobacteraceae that scored up to 9.43 % relative abundance in wet sediment samples at 45.1 °C, Cyanobacteria Family I (5.89 %), Desulfonatronaceae (4.55 %), Thermaceae (4.01 %), Ectothiorhodospiraceae (3.71 %), Spirochaetaceae (3.34 %), Nitriliruptoraceae (3.19 %), Anaerolineaceae (3.03 %), Peptococcaceae (3.03 %) and uncultured gamma proteobacterium (3.03 %) in various samples as shown in Additional file 6: Table S6.
Differences in relative abundance were seen as a function of sample type and temperature, with wet sediments harboring the highest taxa. The dominant taxa corresponded with those reported in previous studies conducted on deep sea and marine sediments community composition [29–31]. For example, a review by Brown et al. [30] on microbial life in extreme environments that compared metagenome analyses of different high thermal habitats, observed that microbes adapted to these habitats, are different with respect to species abundance and community structure. However, some bacterial taxa such as Thermotoga, Deinococcus-Thermus and Proteobacteria, were common within the samples under review [30]. These bacterial taxa were also found within the samples from the hot springs of Lakes Magadi and Little Magadi. Species diversity in high temperature environments has been shown to be relatively low [32, 33]. The deep-sea hydrothermal vent chimneys have been found to harbor Proteobacteria [34, 35], Bacteroidetes and Planctomycetes [36].
Archaeal taxonomic composition analysis
The OTUs were distributed among three Archaeal phyla; Euryarchaeota (1.76–34.48 %) across all samples, Crenarchaeota (up to 2.46 %) and Thaumarchaeota (up to 2.78 %) in wet sediment samples at 81 °C (Figs. 2 and 3). At the family level, OTUs were distributed in 24 families with the most abundant belonging to Halobacteriaceae (27.06 %) in water samples at 81 °C, Deep Sea Hydrothermal Vent Gp 6 (DHVEG-6) (0.35–5.15 %) across all samples, South African Goldmine Gp (SAGMEG); Uncultured Archaeon (4.01 %) and Methanocaldococcaceae (2.47 %) in wet sediment samples at 81 °C (Additional file 6: Table S6). Crenarchaeota phyla members identified belonged to the families Desulfurococcaceae (0.93 %), Thermoproteaceae (0.93 %), Thermofilaceae (0.61 %) in wet sediment samples at 81 °C and Sulfolobaceae (0.42 %) in wet sediment samples at 83.6 °C, while Thaumarchaeota were mainly assigned to uncultured archaeon with up to 1.68 % relative abundance in wet sediment samples at 83.6 °C (Additional file 6: Table S6). Previous studies on thermal groundwater in a thermal field in Russia showed that Archaea is dominated by a novel division in the phylum Euryarchaeota related to the order Thermoplasmatales (39 % of all archaea) and by another abundant group (33 % of all archaea) related to the phylum Crenarchaeota. Both groups are widely spread in hot springs all over the world [37]. Some Archaeal taxa such as Methanococcus, Thermoprotei and Thermococcus were also common within the samples under review by Brown et al. [30]. These are similar to the classes obtained in this study, indicating that Archaea are well adapted to extreme conditions and could be responsible for various functional processes within the ecosystem.
Fig. 3.
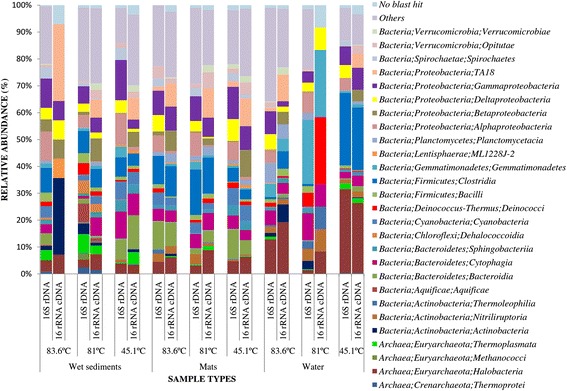
Relative abundance of the most abundant prokaryotic taxa at order level in samples from the hot springs of L. Magadi and Little Magadi. ‘Others’ represent all taxa that scored a relative abundance of below 1 % across all samples in both data sets
Active diversity taxonomic composition based 16S rRNA cDNA analysis
Bacterial taxonomic composition
The 1913 OTUs of 16S rRNA cDNA data set were dominated by bacterial phyla comprising Proteobacteria (8.3–50 %) and Actinobacteria (2.0–28.6 %) across all samples. The data confirm the predominance of previously defined groups belonging to Proteobacteria [38]. Other groups scoring high relative abundance in some samples include Bacteroidetes (26.4 % in wet sediment samples at 45.1 °C), Chloroflexi (5.3 % in water samples at 45.1 °C), Firmicutes (23.9 % in mat samples at 45.1 °C), Cyanobacteria (8 % in mat samples at 45.1 °C), Gemmatimonadetes (25 % in water samples at 81 °C), Lentisphaerae (7.1 % in wet sediment samples at 83.6 °C) and Planctomycetes (6.6 % in mat samples at 83.6 °C) (Fig. 2).
Unlike 16S rDNA dataset, the most abundant bacterial families found within cDNA dataset belonged to Methylophilaceae; scoring up to 8.3 % relative abundance in some samples, Corynebacteriaceae, Dermatophilaceae, Micrococcaceae, Nocardioidaceae, Bacteroidaceae; ML635J-40 aquatic group, Sphingomonadaceae, Burkholderiaceae, and Myxococcales; 0319-6G20, Alcanivoracaceae, Pseudomonadaceae (7.14 %), Desulfuromonadales; GR-WP33-58 (5.11 %), Cyanobacteria; Subsection III; Family I, Syntrophomonadaceae (4.7 %), Spirochaetaceae (3.7 %), Anaerolineaceae (3.44 %), Gemmatimonadetes; BD2-11 terrestrial group; uncultured bacterium 3.3 %, Rhodospirillaceae (3.3 %) and Lentisphaerae (ML1228J-2; uncultured bacterium) (2.6 %) (Additional file 7: Table S7).
Archaeal taxonomic composition
16S rRNA cDNA OTUs were distributed among three Archaeal phyla, similar to those obtained from the 16S rDNA dataset. These were Euryarchaeota (6.6–28.4 %) across all samples, Crenarchaeota (up to 1.38 %) in water samples at 81 °C and Thaumarchaeota (up to 1.15 %) in wet sediment samples at 45.1 °C (Fig. 3). The most abundant families belonged to Halobacteriaceae (21.31 %), Deep Sea Hydrothermal Vent Gp 6 (DHVEG-6) (8.59 %), and Marine Benthic Group D and DHVEG-1 (2.29 %). Crenarchaeota phyla members identified belonged to the families Desulfurococcaceae (1.11 %) in wet sediment samples at 81 °C and Sulfolobaceae (0.28 %) in wet water samples at 45.1 °C, while Thaumarchaeota were mainly assigned to uncultured archaeon with up to 1.15 % relative abundance in wet sediment samples at 45.1 °C (Additional file 7: Table S7). Previously, several 16S rRNA gene sequences related to novel Archaea (Euryarchaeota) were retrieved from the alkaline saltern at Lake Magadi [13]. Haloalkaliphilic Archaea related to Natronomonas, Natrialba, Natronolimnobius and Halorubrum spp. have also been isolated from Lake Magadi and Inner Mongolian soda lakes [39].
Microbial richness and diversity indices
Using rarefaction, the same number of sequences from each sample was used in comparison of community alpha and beta diversity measures. Paired t-tests at class taxonomic level of both 16S rDNA and 16 rRNA cDNA indicated that significant differences between samples based on alpha diversity indices whose values obtained were as follows: Shannon diversity index (H’); wet sediment 83.6 °C (7.9 vs 3.8), water 81 °C (9.1 vs 3.6) and Simpson (1/D); wet sediment 83.6 °C (34.54 vs 12.2); water 81 °C (7.76 vs 6) and water 45.1 °C (28.53 vs 10.3) respectively (Table 2).
Table 2.
Diversity indices computed on all OTU-based microbial taxonomic units within 16S rDNA and 16S rRNA cDNA datasets
| Sample ID | Dataset | No. of sequences | No. of OTUs | Shannon (H’) | Simpson (1/D) | Evenness |
|---|---|---|---|---|---|---|
| Wet sediment (83.6 °C) | 16S rDNA | 38753 | 238 | 5.5 | 34.54 | 0.658 |
| 16S rRNA cDNA | 1625 | 14 | 2.6 | 12.2 | 0.975 | |
| Mats (83.6 °C) | 16S rDNA | 7575 | 66 | 4.2 | 27.41 | 0.475 |
| 16S rRNA cDNA | 21451 | 212 | 5.3 | 32 | 0.632 | |
| Water (83.6 °C) | 16S rDNA | 19186 | 680 | 6.5 | 14.88 | 0.356 |
| 16S rRNA cDNA | 5124 | 151 | 5.0 | 18.1 | 0.594 | |
| Wet sediment (81 °C) | 16S rDNA | 36047 | 324 | 5.8 | 22.57 | 0.427 |
| 16S rRNA cDNA | 67235 | 361 | 5.9 | 53.6 | 0.636 | |
| Mats (81 °C) | 16S rDNA | 24252 | 282 | 5.6 | 34.55 | 0.61 |
| 16S rRNA cDNA | 33751 | 349 | 5.8 | 34.3 | 0.538 | |
| Water (81 °C) | 16S rDNA | 36126 | 568 | 6.3 | 7.76 | 0.278 |
| 16S rRNA cDNA | 638 | 12 | 2.5 | 6 | 0.866 | |
| Wet sediment (45.1 °C) | 16S rDNA | 52115 | 509 | 6.2 | 35.09 | 0.577 |
| 16S rRNA cDNA | 11471 | 87 | 4.4 | 27.7 | 0.784 | |
| Mats (45.1 °C) | 16S rDNA | 37446 | 382 | 6.0 | 22.45 | 0.785 |
| 16S rRNA cDNA | 48578 | 375 | 6.0 | 37.4 | 0.572 | |
| Water (45.1 °C) | 16S rDNA | 19789 | 377 | 6.0 | 28.53 | 0.461 |
| 16S rRNA cDNA | 24790 | 352 | 5.9 | 10.3 | 0.346 |
Total microbial diversity based on 16S rDNA and 16S rRNA cDNA ANOSIM at order level showed that there were significant differences in microbial community structure in the samples at 95 % level of confidence (P value, 0.009), and 0.383 R statistic value while active microbial diversity based on 16S rRNA cDNA had (P value, 0.01), and 0.333 R statistic value. This could be attributed to differences in temperature and pH of the specific sites during sampling. Samples from 45.1 °C harbored more closely related populations because it had less extreme conditions as compared to the two other hot springs.
Distance based redundancy analysis showed that the microbial community evenness significantly differed from each site for both 16S rDNA and 16S rRNA cDNA. 16S rDNA ordination of the three sample types showed a significance of 0.017 and while 16S rRNA cDNA dataset showed a significance of 0. 011. Samples from each site clustered close to each other in separate quadrants indicating high beta diversity between the three sampling sites.
NMDS analyses supported by OTU and taxonomic composition, divide the datasets into three ellipses: one for each hot spring. Some microbial taxa were shared between habitats in both 16S rDNA and 16S rRNA cDNA derived datasets. This scenario was more pronounced in 16S rDNA - derived dataset, indicating that DNA pool contained a “seed bank” of inactive and sporulating organisms [40], while fewer taxa were active within the ecosystem as shown in the 16S rRNA cDNA derived dataset (Fig. 4). Similar results were observed in a study on Ethiopian soda lakes where NMDS analyses supported both by OTU and taxonomic composition; divided the datasets into six well-separated habitats with relatively few OTUs that were shared between more than one or two habitats [41]. The taxa were also observed to cluster according to sample types (i.e. wet sediments, microbial mats and water samples). There was an overlap of taxonomic clusters between wet sediments and microbial mats from the three sample types in both 16S rDNA and 16S rRNA cDNA derived datasets. However, water samples formed separate clusters from the other two sample types in both 16S rDNA and 16S rRNA cDNA datasets (Figs. 4 and 5).
Fig. 4.
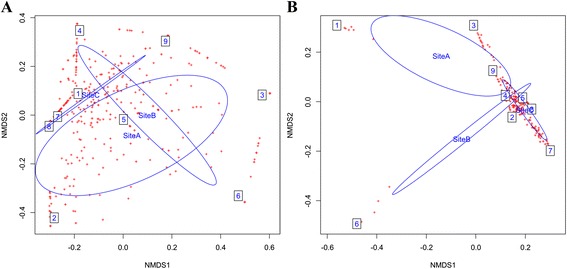
a Non-metric multidimensional scaling (NMDS) based on Bray-Curtis dissimilarities between microbial compositions of 16S rDNAdataset, grouped according to sampling sites. Site A, B and C represent hot springs 83.6 ºC, 81 ºC and 45.1 ºC respectively. The boxes 1 - 3 represent wet sediments, mats and water samples from 83.6 ºC, 4 - 6 represent wet sediments, mats and water samples from 81 ºC and 7 - 9 represent wet sediments, mats and water samples from 45.1 ºC. b Non-metric multidimensional scaling (NMDS) based on Bray-Curtis dissimilarities between microbial compositions of 16S rRNA cDNAdatasets grouped according to sampling sites. Site A, B and C represent hot springs 83.6 ºC, 81 ºC and 45.1 ºC respectively. The boxes 1 - 3 represent wet sediments, mats and water samples from 83.6 ºC, 4 - 6 represent wet sediments, mats and water samples from 81 ºC and 7 - 9 represent wet sediments, mats and water samples from 45.1 ºC
Fig. 5.
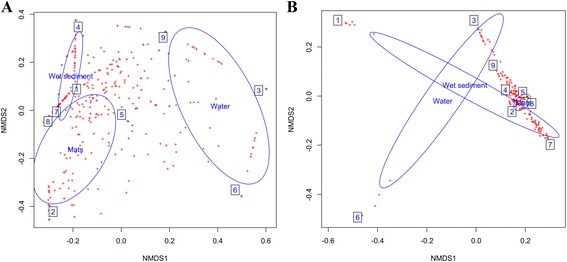
a Non-metric multidimensional scaling (NMDS) based on Bray-Curtis dissimilarities between microbial compositions of 16S rDNAdatasetgrouped according to sample types. Each ellipse represents a set of the three sample types collected from each hot spring. The boxes 1 - 3 represent wet sediments, mats and water samples from 83.6 ºC, 4 - 6 represent wet sediments, mats and water samples from 81 ºC and 7 - 9 represent wet sediments, mats and water samples from 45.1 ºC. b Non-metric multidimensional scaling (NMDS) based on Bray-Curtis dissimilarities between microbial compositions of 16S rRNA cDNA dataset grouped according to sample types. Each ellipse represents a set of the three sample types collected from each hot spring. The boxes 1 - 3 represent wet sediments, mats and water samples from 83.6 ºC, 4 - 6 represent wet sediments, mats and water samples from 81 ºC and 7 - 9 represent wet sediments, mats and water samples from 45.1 ºC
Hierarchical clustering between samples collected from lake Magadi and Little Magadi revealed samples from the two hot springs in Little Magadi “Nasikie eng’ida” were closer than samples from the hot spring in the main lake (Fig. 6 (a and b) and (Additional file 8: Figure S1 and Additional file 9: Figure S2).
Fig. 6.
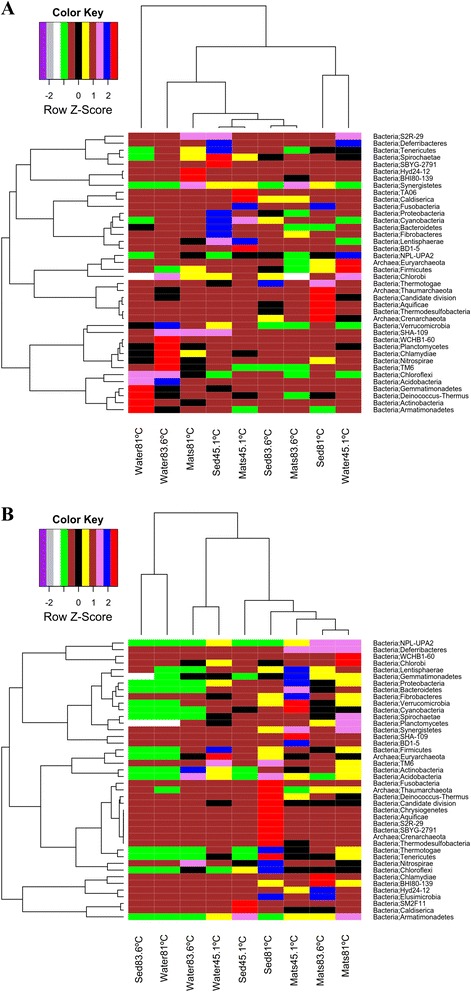
a Hierarchical clustering of 16S rDNA samples collected from the three hot springs of L. Magadi and Little Magadi. Phylum level was chosen to be used in hierarchical clustering to assess the relationships between samples and taxa. “Sed” represent wet sediment samples from respective temperature. b Hierarchical clustering of 16S rRNA cDNA samples collected from the three hot springs of L. Magadi and Little Magadi. Phylum level was chosen to be used in hierarchical clustering to assess the relationships between samples and taxa. “Sed” represent wet sediment samples from respective temperature
Conclusion
The combined findings of this study, show that estimated diversity and richness within the hot spring samples were found to be as high as those found in other environments such as soil and deep-sea hydrothermal environments [42]. The results confirm that different groups of microorganisms have the capacity to adapt and thrive even in the most hostile environments. Some of these groups (Acidobacteria; Blastocatella, Bryobacter and Telmatobacter genera, Bacteroidetes; Bacteroidales, Rhodothermaceae, Flavobacteriaceae, Sphingobacteriales and Chloroflexi; Dehalococcoidales and Thermomicrobia) also obtained from previous similar studies were reported to have a fermentative ability [41]. It was observed that from the cDNA dataset, photosynthetic taxa were represented by Cyanobacterial genera Leptolyngbya and Lyngbya, among other uncultured groups. Primary production within the hot springs is probably supported by some groups of non-sulfur purple bacteria from the family Rhodobacteraceae (specifically the genera Roseobacter that scored 1.1 % relative abundance), and purple sulfur bacteria from the family Ectothiorhodospiraceae present across samples at different relative abundance. The presence of Planctomycetes within samples could be an indicator that anaerobic ammonium oxidation may be another metabolic pathway supporting primary production in the low-oxygen, saline environment, since the dissolved oxygen concentration of the sampling sites ranged between 0.04 and 12.4 mg/l. Actinobacteria and Firmicutes are believed to have adaptive advantage under low-nutrient conditions of the highly alkaline, saline hot springs hence their high relative abundance levels. The presence of sulfate reducers in the family Desulfohalobiaceae (mainly Desulfonatronovibrio), suggested an internal sulfur cycle within the lake, as previously suggested for Khadin, Tuva, Russia and Natron soda lakes [42, 43]. Taxa typical for highly specialized metabolisms that were encountered in this study include Nitriliiruptor, known for their ability to catabolize nitriles or cyanides [44] and heterotrophic Oceanospirillaceae (Marinospirillum). Other functional taxa encountered include aerobic heterotrophs (e.g. Bacteroidetes, Marinicella) and fermentative anaerobes such as Thermoplasmatales among other uncultured groups. Euryarchaeota members were clustered into the classes, Halobacteria, Methanobacteria, Methanomicrobia, Methanococci, Thermococci and Thermoplasmata while Crenarchaeota phyla comprised Thermoprotei class. Previously, Methanobacteria and Methanomicrobia have been reported in oilfields, while Halobacteria and Thermoprotei have been reported in petroleum reservoirs [45]. Some Halobacteria members are important in organic fertilizer production industry as Lignin decomposers [46]. However, most of the genera identified in this study are known to be heterotrophs responsible for the primary degradation of organic matter [39]. The actual function of microbial taxa reported in this study could further be explored and established using culture dependent methods as well as mRNA transcripts.
In conclusion, this study presented microbial diversity analysis of samples collected from the hot springs of L. Magadi and Little Magadi based on both DNA and RNA, using Illumina Sequencing Technology. The results showed comparable profiles of microbial community using 16S rDNA and 16S rRNA cDNA derived datasets, hence indicating that the observed diversity is real. The findings showed a broad microbial distribution with water from the spring at 83.6 °C found to be the richest sample, constituting 680 observed species. Despite the fact that the sampling environment is multi-extreme due to high pH, temperature, and salinity, this study shows that there are stable and active microbial communities that have adapted to this environment. Culture dependent studies in future will help us unravel the survival mechanisms used by these polyextremophiles.
Acknowledgements
We thank National Commission for Science, Technology and Innovation (NACOSTI) for providing a Research Grant to this work and Higher Education Loans Board (HELB) for providing a partial scholarship for school fees. We also thank Kenya Wildlife Services (KWS) and the NACOSTI provided permits for sample collection in Lake Magadi.
Funding
The Research was funded by the National Commission for Science, Technology and Innovation.
Availability of data and material
The data is available on NCBI Sequence Read Archive with SRP# Study accessions: SRP061805. SRX1124606: RNA-Seq of Prokaryotes: Alkaline Hot springs and SRX1124607: DNA-Seq of Prokaryotes: Alkaline Hot springs.
Authors’ contributions
Conceived and designed the experiments: AKK EKN HIB RM. Performed the experiments: AKK EKN RWK RM. Analyzed the data: AKK EKN RM HMM. Contributed reagents/materials/sequencing costs/analysis tools: HIB EKN RM, RWK. Wrote the paper: AKK HIB RWK EKN HMM RM. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
All authors have consented to the publication of this manuscript.
Ethics approval and consent to participate
Not Applicable.
New clinical tools and procedures
Not Applicable.
Research Authorization
Research was authorized by National Commission for Science, Technology and Innovation and Kenya Wildlife Services.
Additional files
Total diversity environmental data sheet of the samples collected from the hot springs of L. Magadi and Little Magadi (XLS 25 kb)
Active diversity environmental data sheet of the samples collected from the hot springs of L. Magadi and Little Magadi (XLS 24 kb)
Total diversity Sequence Read Archive metadata sheet of the samples collected from the hot springs of L. Magadi and Little Magadi (XLSX 50 kb)
Active diversity Sequence Read Archive metadata sheet of the samples collected from the hot springs of L. Magadi and Little Magadi. (XLSX 50 kb)
Relative abundance of microbial taxa at phylum level in various samples collected from the three hot springs (83.6, 81 and 45.1 °C) of L. Magadi and Little Magadi. ‘No blast hit/Others’ represent unclassified sequences in both data sets. (XLSX 17 kb)
Total diversity relative abundance of microbial taxa at family level in various samples collected from the three hot springs (83.6, 81 and 45.1 °C) of L. Magadi and Little Magadi. ‘No blast hit/Others’ represent unclassified sequences in both data sets. (XLSX 45 kb)
Active diversity relative abundance of microbial taxa at family level in various samples collected from the three hot springs (83.6, 81 and 45.1 °C) of L. Magadi and Little Magadi. ‘No blast hit/Others’ represent unclassified sequences in both data sets. (XLSX 32 kb)
Comparative analysis (UPGMA similarity tree) of total microbial diversity of various sample types within hot springs of L. Magadi and Little Magadi. (DOCX 55 kb)
Comparative analysis (UPGMA similarity tree) of active microbial diversity of various sample types within hot springs of L. Magadi and Little Magadi. (DOCX 56 kb)
Contributor Information
Anne Kelly Kambura, Email: annnderitu@gmail.com.
Romano Kachiuru Mwirichia, Email: rkachiuru2000@yahoo.com.
Remmy Wekesa Kasili, Email: rkasili@gmail.com.
Edward Nderitu Karanja, Email: ekaranja@icipe.org.
Huxley Mae Makonde, Email: huxcly@yahoo.com.
Hamadi Iddi Boga, Email: hamadiboga@ttuc.ac.ke.
References
- 1.Thiel V. Extreme Environments. In J. Reitner and V. Thiel (eds), Encyclopedia of Geobiology, Encyclopedia of Earth Science Series, Springer, Heidelberg, 2011. p. 362-366.
- 2.Takai K, Nakamur K, Toki T, et al. Cell proliferation at 122 °C and isotopically heavy CH4 production by a hyperthermophillic methanogen under high-pressure cultivation. Proc Natl Acad Sci USA. 2008;105:10949–10954. doi: 10.1073/pnas.0712334105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rothschild LJ, Mancinelli RL. Life in extreme environments. Nature. 2001;409:1092–1101. doi: 10.1038/35059215. [DOI] [PubMed] [Google Scholar]
- 4.Melack JM, Kilham P. Photosynthetic rates of phytoplankton in East African Lakes. Limnol Oceanogr. 1974;19:743–755. doi: 10.4319/lo.1974.19.5.0743. [DOI] [Google Scholar]
- 5.Grant WD, Mwatha WE, Jones BE. Alkaliphiles: ecology, diversity and applications. FEMS Microbiol Rev. 1990;75:255–270. doi: 10.1111/j.1574-6968.1990.tb04099.x. [DOI] [Google Scholar]
- 6.Warren JK. Subaqueous salts: salinas and perennial lakes. Evaporites sediments, resources and hydrocarbons. Springer, Berlin, Germany; 2006.
- 7.Baumgarte S. Microbial diversity of soda lake habitats. PhD thesis. Braunschweig: Carolo-Wilhelmina University; 2003. [Google Scholar]
- 8.Tindall BJ, Ross HNM, Grant WD. An alkalophilic red halophilic bacterium with a low magnesium requirement from Kenyan Soda Lake. J Gen Microbiol. 1980;116:257–260. [Google Scholar]
- 9.Tindall BJ, Ross HNM, Grant WD. Natronobacterium gen.nov. and Natronococcus gen. nov., two genera of haloalkaliphilic archaebacteria. J Syst Appl Microbiol. 1984;5:41–57. doi: 10.1016/S0723-2020(84)80050-8. [DOI] [Google Scholar]
- 10.Mwatha WE, Grant WD. Natronobacterium vacuolata sp. nov., a haloalkaliphilic archaeon isolated from Lake Magadi, Kenya. Int J Syst Bacteriol. 1993;43:401–406. doi: 10.1099/00207713-43-3-401. [DOI] [Google Scholar]
- 11.Duckworth AW, Grant WD, Jones BE, van Steenbergen R. Phylogenetic diversity of soda Lake Alkaliphiles. FEMS Microbiol Ecol. 1996;19:181–191. doi: 10.1111/j.1574-6941.1996.tb00211.x. [DOI] [Google Scholar]
- 12.Jones BE, Grant WD, Duckworth AW, Owenson GG. Microbial diversity of soda lakes. Extremophiles. 1998;2:191–200. doi: 10.1007/s007920050060. [DOI] [PubMed] [Google Scholar]
- 13.Grant S, Grant D, Jones BE, Kato C, Li L. Novel Archaeal phenotypes from an East African alkaline environment. Extremophiles. 1999;3:139–145. doi: 10.1007/s007920050109. [DOI] [PubMed] [Google Scholar]
- 14.Zavarzin GA, Zhilina TN, Kevbrin VV. The alkaliphilic microbial community and its functional diversity. Microbiology. 1999;68:503–521. [Google Scholar]
- 15.Kambura AK, Boga HI, Mwirichia RK, Ngaira J. Isolation and characterization of bacterial isolates from Lake Magadi. J Trop Microbiol Biotechnol. 2012;8:20–28. [Google Scholar]
- 16.Pawlowski J, Lejzerowicz F, Esling P. Next-Generation Environmental Diversity Surveys of Foraminifera: Preparing the Future. Biol Bull. 2014;227(2);93–106. [DOI] [PubMed]
- 17.Behr HJ, Röhricht Record of seismotectonic events in siliceous cyanobacterial sediments (Magadi cherts), Lake Magadi, Kenya. Int J Earth Sci. 2000;89:268–283. doi: 10.1007/s005319900070. [DOI] [Google Scholar]
- 18.Sambrook KJ, Fritsch EF, Maniatis T. Molecular Cloning: a Laboratory Manual. 2. Cold Spring Harbor: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 19.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate – phenol - chloroform extraction. Anal Biochem. 1987;162:156. doi: 10.1016/0003-2697(87)90021-2. [DOI] [PubMed] [Google Scholar]
- 20.Urich T, Lanzén A, Qi J, Huson DH, Schleper C, et al. Simultaneous Assessment of Soil Microbial Community Structure and Function through Analysis of the Meta-Transcriptome. PLoS ONE. 2008;3(6):e2527. doi: 10.1371/journal.pone.0002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caporaso JG, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6(8):1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu K, Zhang T. Metagenomic and Metatranscriptomic Analysis of Microbial Community Structure and Gene Expression of Activated Sludge. PLoS ONE. 2012;7(5):e38183. doi: 10.1371/journal.pone.0038183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reeder J, Knight R. Rapidly denoising pyrosequencing amplicon reads exploiting rank-abundance distributions. Nat Methods. 2010;7:668–669. doi: 10.1038/nmeth0910-668b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gontcharova V, Youn E, Sun Y, et al. A comparison of bacterial composition in diabetic ulcers and contralateral intact skin. Open Microbiol J. 2010;4:8–19. doi: 10.2174/1874285801004010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41(D1):D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.R Development Core Team R . A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2012. [Google Scholar]
- 28.Oksanen J, Blanchet F, Kindt R, Legendre P, Minchin P, et al. vegan: Community Ecology Package. 2012.
- 29.DeLong EF, Preston CM, Mincer T, Rich V, Hallam SJ, et al. Community Genomics among Stratified Microbial Assemblages in the Ocean’s Interior. Science. 2006;311:496. doi: 10.1126/science.1120250. [DOI] [PubMed] [Google Scholar]
- 30.Brown MV, Philip GK, Bunge JA, Smith MC, Bissett A, et al. Microbial community structure in the North Pacific Ocean. ISME J. 2009;3:1374–1386. doi: 10.1038/ismej.2009.86. [DOI] [PubMed] [Google Scholar]
- 31.Liao L, Xu XW, Jiang XW, Wang CS, Zhang DS, et al. Microbial diversity in deep-sea sediment from the cobalt rich crust deposit region in the Pacific Ocean. FEMS Microbiol Ecol. 2011;78:565–585. doi: 10.1111/j.1574-6941.2011.01186.x. [DOI] [PubMed] [Google Scholar]
- 32.Nunoura T, Takaki Y, Kakuta J, Nishi S, Sugahara J, et al. Insights into the evolution of Archaea and eukaryotic protein modifier systems revealed by the genome of a novel archaeal group. Nucleic Acids Res. 2011;39:3204–3223. doi: 10.1093/nar/gkq1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nelson WC, Wollerman L, Bhaya D, Heidelberg JF. Analysis of insertion sequences in thermophilic cyanobacteria: exploring the mechanisms of establishing, maintaining, and withstanding high insertion sequence abundance. Appl Environ Microbiol. 2011;77:5458–5466. doi: 10.1128/AEM.05090-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakai R, Abe T, Takeyama H, Naganuma T. Metagenomic analysis of 0.2-mm-passable microorganisms in deep-sea hydrothermal fluid. Mar Biotechnol. 2011;13:900–908. doi: 10.1007/s10126-010-9351-6. [DOI] [PubMed] [Google Scholar]
- 35.Brazelton WJ, Baross JA. Abundant transposases encoded by the metagenome of a hydrothermal chimney biofilm. ISME J. 2009;3:1420–1424. doi: 10.1038/ismej.2009.79. [DOI] [PubMed] [Google Scholar]
- 36.Xie W, Wang FP, Guo L, Chen ZL, Sievert SM, et al. Comparative Metagenomics of microbial communities inhabiting deep-sea hydrothermal vent chimneys with contrasting chemistries. ISME J. 2011;5:414–426. doi: 10.1038/ismej.2010.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mardanov AV, Gumerov VM, Beletsky AV, Perevalova AA, Karpov GA, et al. Uncultured archaea dominate in the thermal groundwater of Uzon Caldera, Kamchatka. Extremophiles. 2011;15:365–372. doi: 10.1007/s00792-011-0368-1. [DOI] [PubMed] [Google Scholar]
- 38.Tekere M, Lötter A, Olivier J, Jonker N, Venter S. Metagenomic analysis of bacterial diversity of Siloam hot water spring, Limpopo, South Africa. Afr J Biotechnol. 2011;10:18005–18012. [Google Scholar]
- 39.Grant WD, Sorokin DY. Distribution and diversity of soda lake alkaliphiles. In: Horikoshi K, Antranikian G, Bull AT, Robb FT, Stetter KO, editors. Extremophiles Handbook. Tokyo: Springer; 2011. pp. 27–54. [Google Scholar]
- 40.Jones SE, Lennon JT. Dormancy contributes to the maintenance of microbial diversity. Proc Natl Acad Sci USA. 2010;107:5881–5886. doi: 10.1073/pnas.0912765107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lanze’n A, Simachew A, Gessesse A, Chmolowska D, Jonassen I, et al. Surprising Prokaryotic and Eukaryotic Diversity, Community Structure and Biogeography of Ethiopian Soda Lakes. PLoS ONE. 2013;8(8):e72577. doi:10.1371/journal.pone.0072577. [DOI] [PMC free article] [PubMed]
- 42.Zavarzin GA and Zhilina TN. Anaerobic chemotrophic alkaliphiles. In: Seckbach J, editor. Journey to Diverse Microbial Worlds: Adaptation to Exotic Environments: Kluwer Academic Publishers, London. 2000; pp. 191–108.
- 43.Sorokin DY, Kuenen JG, Muyzer G. The microbial sulfur cycle at extremely haloalkaline conditions of soda lakes. Front Microbiol. 2011;2:44. doi: 10.3389/fmicb.2011.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bengtsson MM, Sjøtun K, Øvreas L. Seasonal dynamics of bacterial biofilms on the kelp Laminaria hyperborea. Aquat Microb Ecol. 2010;60:71–83. doi: 10.3354/ame01409. [DOI] [Google Scholar]
- 45.Lenchi N, Inceoğlu O, Kebbouche-Gana S, Gana ML, Llirós M, et al. Diversity of Microbial Communities in Production and Injection Waters of Algerian Oilfields Revealed by 16S rRNA Gene Amplicon 454 Pyrosequencing. PLoS ONE. 2013;8(6):e66588. doi: 10.1371/journal.pone.0066588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nura A. Abboud and Amir Dakkak (2015). Halobacterium – A Model of Life in the Dead Sea. http://www.ecomena.org/halobacterium/. Accessed 25 June 2016.