

Abstract
Fatty acid amide hydrolase (FAAH) is a membrane anchored serine hydrolase that has a principle role in the metabolism of the endogenous cannabinoid anandamide. Docking studies using representative FAAH crystal structures revealed that compounds containing a novel piperidinyl thiazole isoxazoline core fit within the ligand binding domains. New potential FAAH inhibitors were designed and synthesized incorporating urea, carbamate, alkyldione and thiourea reactive centers as potential pharmacophores. A small library of candidate compounds (75) was then screened against human FAAH leading to the identification of new carbamate and urea based inhibitors (Ki = pM and nM respectively). Representative carbamate and urea based chemotypes displayed slow, time dependent inhibition kinetics leading to enzyme inactivation which was slowly reversible. However, evidence indicated that features of the mechanism of inactivation differ between the two pharmacophore types. Selected compounds were also evaluated for analgesic activity in the mouse-tail flick test.
Keywords: Fatty Acid Amide Hydrolase, Inhibition, Inactivation, Analgesia
Graphical abstract
Fatty acid amide hydrolase (FAAH) is a membrane anchored protein that has an important role in regulating the activity of the G-protein coupled receptors (GPCRs) CB1 and CB2 via regulation of metabolism of one of their endogenous ligands, anadamide1. CB1 receptors are expressed throughout the central nervous system, and act presynaptically to regulate neurotransmitter release. Fatty acid amides such as anandamide (AEA, arachidonyl ethanolamide), oleamide, and palmitoyl ethanolamide are endogenous agonists (endocannabinoids) of CB1 that have been implicated in a number of physiological effects including endogenous analgesia, feeding behavior and sleep induction2. FAAH catalyzes the deacylating hydrolysis of these lipid amide agonists thereby down regulating the levels of the cognate ligands. By blocking hydrolysis of these agonists, in particular AEA, through inhibition of FAAH, CB1-mediated effects can be increased and prolonged leading to physiological effects such as analgesia3. FAAH inhibition is an emerging target for analgesia and a variety of FAAH inhibitors have shown efficacy in pre-clinical chronic pain models through a mechanism of action that includes sites in the central and peripheral nervous systems3, 4. Despite this promising pre-clinical data, a recent trial in humans failed to show efficacy for osteoarthritis pain5.
FAAH is a member of the amidase enzyme family whose catalytic mechanism depends on a SER-SER-LYS catalytic triad for activity6, 7. The enzymatic mechanism involves nucleophilic attack of SER241 on the carbonyl of fatty acid amide substrates. With AEA, ethanolamine is eliminated leaving an acyl enzyme arachidonyl intermediate. The enzyme is then deacylated by hydrolysis of the covalent intermediate with water thereby releasing arachidonic acid. This completes the catalytic cycle restoring active enzyme. A variety of potent inhibitors of FAAH have been developed as potential drugs based on exploiting these features of the reaction mechanism that have led to both irreversible and reversible type inhibitors8. Electrophilic urea and carbamate pharmacophores have been used in a number of scaffolds. Compounds such as URB5979, and aryl ureas from Johnson and Johnson, Takeda, and Pfizer10–15 (Fig 1) display potent binding properties leading to enzyme inactivation through covalent modification of the active site SER241. Co-crystallographic studies with inhibitors and mammalian FAAH have helped map critical structural features of the enzyme16, 17. The cytoplasmic port (CP) domain, a hydrophilic region adjacent to the catalytic site, interacts with the polar head groups of substrates and inhibitors. Also, adjacent to the catalytic domain are a hydrophobic acyl binding pocket (ABP), and membrane access channel (MAC) that bind the extended lipid chains of substrates and hydrophobic scaffolds of inhibitors. We now report on new acyl urea and carbamate inhibitors of FAAH with high in vitro potency, and their evaluation for analgesia in mice.
Figure 1.

Oxathiapiprolin fungicide; Piperidine and piperazine aryl urea FAAH inhibitors (PF-750, JNJ-1661010); α-ketooxazole and carbamate FAAH inhibitors (OL-135, URB597).
New bioactive chemotypes can be derived from primary scaffolds by addition or linking of secondary heterocycles guided by compatibility with structural or mechanistic features of target proteins. The aryl urea FAAH inhibitors reported by both Takeda and Johnson and Johnson contain piperazine thiazole and thiadiazole scaffolds which are similar to the piperidinyl thiazole central core of Oxathiapiprolin (Fig 1), an antifungal agent discovered by DuPont that is used for control of plant diseases caused by oomycetes.18 Although there are structural similarities to FAAH inhibitors, the mechanism of action of Oxathiapiprolin is not thought to involve a homologue of FAAH.19 This is due in part to the presence of amidase signature domains in Phytophthora infestans (a target organism) which appear to be restricted to the glutamyl t-RNA amidotransferase protein family. In addition, Oxathiapiprolin differs significantly from FAAH chemotypes since it has an atypical substitution off the piperidine ring and extended aryl isoxazoline substitution from the thiazole ring. We hypothesized that because of the extended ABP and adjacent MAC regions in FAAH that further extension from the thiazole might be well tolerated, and that appropriate substitution of piperidinyl ring with electrophilic substituents might yield efficacious FAAH inhibitors.
Crystallographic results with FAAH and inhibitory ligands have revealed a dynamic relation between the ABP and CP domains, and alterations in the flexible ABP and MAC regions depending on chemotype and mechanism of inhibition.17, 20 For example, in the crystallographic structure 2WJ2 (PDB code) containing α-ketoheterocycle inhibitor OL-135, the conformation of the ABP is associated with the MAC in the “open” state.17 OL-135 forms a covalent tetrahedral transition state mimic with SER241 that is also fully reversible. Alternatively, crystallographic structure 2WAP (PDB code) was determined from FAAH covalently inactivated with an aryl urea (PF-3845) that yields a carbamoylated SER241.4 2WAP depicts an endpoint inactivation with reorganized ABP and MAC domains viewed as a “closed” state. Docking experiments with the aryl carbamate 1a and aryl urea 2c (Table 1) containing the piperidinyl thiazole isoxazoline core yielded estimated binding constants in the low nM range when docked to either 2WAP or 2WJ2. Fig 2 depicts a view of compound 1a docked to 2WAP. In this view compound 1a extends deeply into the ABP domain in an extended linear conformation. In addition to hydrophobic accommodation in the ABP domain additional interactions appear to contribute to the binding affinity: The terminal aryl ring is positioned close to PHE381 showing an edge facing π-π interaction. The thiazole ring is near MET495 showing possible H-bonding with the ring nitrogen. PHE192 is positioned close to the piperidine ring with a C-H Ar interaction from the 3-position methylene. The carbamate aryl ring is positioned close to MET191 with apparent stabilization through a C-H Ar interaction. Docking of the aryl urea 2c to structure 2WAP was nearly identical to compound 1a. A key feature for both compound 1a and 2c is the nearly covalent bond distance (2.0Å) between SER241 and the carbonyl thereby supporting the likelihood of covalent enzyme modification by these inhibitors.
Table 1.
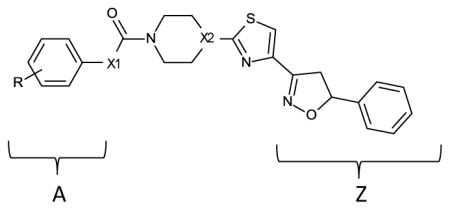
| |||||
|---|---|---|---|---|---|
| Compound | X1 | X2 | R | Apparent IC50 (nM) | Ki (pM) estimate |
| 1a | O | CH | H | 0.07 | 6.3 |
| 1b | O | CH | 4-CN | 0.14 | 12.8 |
| 1c | O | CH | 3-CN | 0.03 | 2.5 |
| 1d | O | CH | 4-Cl | 0.08 | 7.2 |
| 1e | O | CH | 4-CF3 | 0.36 | 33.6 |
| 1f | O | CH | 2-CL | 0.06 | 5.3 |
| 1g | O | CH | 3,5-CH3 | 42 | 3962 |
| 1h | O | CH | 2-CH3 | 0.04 | 3.4 |
| 1i | O | CH | 2,5-CH3 | 35 | 3302 |
| 1j | O | CH | 3-CL | 0.41 | 38.3 |
| 1k | O | CH | 2,6-CH3 | 2000 | 188,680 |
| 1l | O | CH | 3-CH3 | 0.6 | 56.3 |
| 1m | O | CH | 4-CH3 | 0.2 | 18.5 |
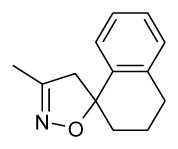
| 1n (notea) | O | CH | H | 29 | 2735 |
| 1o (noteb) | O | CH | H | 0.02 | INDh |
| 1p | O | CH | 4-OCH3 | 0.01 | INDh |
| 2a | NH | CH | 2-CH3 | 600 | 56,603 |
| 2b (notec) | NH | CH | H | 222 | 20,943 |
| 2c | NH | CH | H | 190 | 17,924 |
| 2d (noted) | NH | CH | NAg | 382 | 36,037 |
| 2e (notee) | NH | CH | H | 9.8 | 924 |
| 2f (notef) | NH | CH | H | 25 | 2358 |
| 3a | O | N | H | 1.56 | 147 |
| 3b | O | N | 4-CN | 0.09 | 8.1 |
| 3c | O | N | 3-CN | 0.2 | 18.5 |
| 3d | O | N | 4-CL | 0.01 | INDh |
| 3e | O | N | 4-CF3 | 0.16 | 14.7 |
| 3f | O | N | 4-NO2 | 0.2 | 18.5 |
fragment Z =
fragment A =
fragment A =
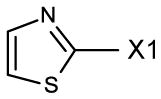
fragment A =
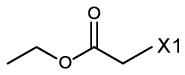
fragment A =
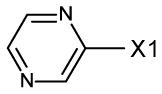
fragment A =
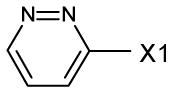
NA(not applicable)
IND(indeterminable)
IC50 values were determined as described in Experimental Procedures. Reactions were measured over 40 min, but because inhibition was time dependent, only initial linear rates were used (0 to about 25min).
Figure 2.

Docking of compound 1a (green carbon backbone) to FAAH structure 2WAP.
Docking of 1a and 2c to 2WJ2, representative of the “open” conformation of the MAC and associated ABP domains repositions the aryl carbamate and urea portions of the molecules extending further into the CP domain. The extended aryl isoxazoline portions of the compounds were accommodated in the ABP and MAC domain similarly to bound OL-135. In these experiments the aryl ring of the carbamate (1a) was positioned very close to the S of CYS269 (3.6Å) with the ring hydrogens capable of interacting favorably with the sulfur lone pairs. An S-H Ar interaction appeared also be possible, but would be highly dependent on specific orientation.21 In contrast, the urea aryl ring of 2c was positioned close to the backbone amide of CYS269 forming a potential N-H Ar interaction, however it was farther away from the CYS269 thiol (6.5Å) relative to the carbamate aryl ring of 1a which can perhaps be attributed to the non-rotation of the urea amide bond in 2c.
As a result of the favorable modeling results with respect to estimated binding affinity and active site orientation, targeted synthesis then yielded a variety of analogs containing aryl carbamate and ureas, as well as alkyl carbamates, aryl thioureas, isothioureas, and hydrazone electrophilic substituents.
A small library of about 75 compounds was then screened in vitro for inhibition of FAAH. Out of this initial pool, active compounds with the highest level of activity resided in either aryl urea or aryl carbamate substituted compounds (region A, figure in Table 1). Interestingly, in the urea series when region A was a thiazole or an acyl ester, IC50 values were still observed below 1 μM (Table 1). Within the urea series when region A was substituted with the weakly basic diazine heterocycles (pyrimidine, pyridazine, 2e and 2f respectively) a large increase in binding affinity was observed relative to simple phenyl substitution (2c). This trend is qualitatively similar to the effects of nitrogen addition within the central activating oxazole of the α-ketoheterocycle FAAH inhibitors.17 As listed in Table 1, better binding affinity was observed with aryl carbamates relative to acyl ureas.
Additional analogs were also prepared around piperazine ring substitution as used in other FAAH inhibitor chemotypes. Attention was given to the aryl carbamates because of better intrinsic binding affinity and enzyme inactivation potency relative to the aryl urea series. As listed in Table 1, many of the piperidine and piperazine aryl carbamates have very high binding affinity with observed IC50 values and Ki estimates reaching the pM range. The IC50 values determined for several standard inhibitors under our assay conditions (0.24 nM URB597 and 0.4 nM OL-135, Fig 1) were generally lower than those reported in the literature (3 nM URB597 and 0.3, 4.7 nM OL-135).9, 22, 23 This can likely be attributed to the low concentration of active enzyme used in the fluorometric assay (8 pM). For several compounds the IC50 values (within experimental error) reached a limiting value equivalent to [Et]/2 indicating that a Ki value could not be estimated under the conditions of the experiments. Nevertheless, the relative values for the majority of compounds in the analog sets are comparable for structure activity purposes. Among the piperidinyl analogs listed in Table 1, di-substitution of the phenyl carbamate ring significantly decreased relative binding affinity. In particular, the 2, 6-dimethyl substitution (1k) decreased binding affinity greater than 4 orders of magnitude relative to the unsubstituted analog (1a). Mono-substitution in the 2, 3, or 4 positions on the phenyl carbamate ring in the piperidine series was relatively well tolerated without much shift in binding affinity observed among analogs with the exception being the 3 substituted CL and CH3 analogs. Within the piperazine series substitution in the 4 position of the phenyl ring (3b, 3d, 3e) showed similar inhibition values compared to identically substituted piperidine analogs (1b, 1d, 1e). Interestingly, when the phenyl ring is unsubstituted (3a), or substituted in the 3 position (3c) binding affinity decreased relative to the corresponding piperidine analog.
Representative aryl carbamates and ureas were examined in more detail to explore the mechanism of inhibition. Both the aryl carbamates and ureas examined showed time dependent loss of enzyme activity consistent with inactivation of the enzyme as observed with other carbamate and urea chemotypes. This response is consistent with several possible mechanisms of inhibition. Reversible, slow-tight binding inhibition, reversible or irreversible mechanism-based inhibition, or irreversible modification of active site residues due to reactive propinquity can yield time dependent inhibition kinetics. A kinact for representative compounds was determined using a 2 step model; an initial binding equilibrium followed by a first order inactivation event, and competitive with substrate binding and catalysis. The individual ki’ values determined at different concentrations of inhibitor were used to estimate a kinact. Determination of ki’ values for compound 1l are shown in Fig 3A. A representative fit of the data to determine kinact is depicted in Fig 3B for the aryl urea 2b (Table 1). Table 2 summarizes the inactivation kinetics for representative ureas and carbamates. Based on comparison of the second order inactivation constants the ureas were less effective than the carbamates, and directly related to the lower binding affinity for the enzyme. The kinact values generally correlated with the electrophilicity of the activating group on the carbamate or urea. For example, in the urea series, benzyl (2c) is > thiazole (2b) is > acyl ester (2d). Taken together with the docking experiments, the results for the urea and carbamates are consistent with inactivation of the enzyme through covalent enzyme modification of SER241 as observed with other FAAH chemotypes.13, 14
Figure 3.

A, Time dependent loss of FAAH activity in the presence of increasing concentrations of compound 1l. Baseline (open circles), 10nM (dark circles), 1 nM (open squares), 0.1 nM (dark squares), and the uninhibited reaction (open triangles). The solid lines represent the computer fit of the data using the apparent inactivation constant (ki’) determined at each concentration of 1l. B, computer fit of the apparent ki’ values for compound 2b to determine kinact as described in Experimental procedures.
Table 2.
| Compound | kinact (sec−1) | Ki (nM) | kinact/Ki (M−1sec−1) |
|---|---|---|---|
| 1a | .0004 | .007 | 6.1 × 107 |
| 1b | .0012 | .013 | 9.2 × 107 |
| 1l | .0014 | .056 | 2.5 × 107 |
| 2c | .0043 | 18 | 2.4 × 105 |
| 2b | .0025 | 21 | 1.2 × 105 |
| 2d | .0015 | 36 | 4.2 × 105 |
The Kinact and in vitro efficacies of the selected acyl ureas and carbamates were determined as described in Experimental procedures.
To examine the reversibility of inactivation by the aryl carbamates and ureas, FAAH samples that had been completely inactivated by compounds 1a, 2c, 2d, and JNJ-1661010 were extensively dialyzed at 4°C to determine if enzymatic activity could be regained. Activity was sampled at 24, 48, and 144 hrs. The results are shown in Fig 4. At 24 hrs the ureas, 2c and 2d showed some recovery of activity relative to the others. Progressive recovery of activity for 2c and 2d treated enzyme was observed over the time course reaching about 60% recovery of activity. FAAH inactivation by the ureas appears to be predominately reversible. Interestingly, the aryl carbamate (1a) inactivated enzyme also showed some recover of activity (30%) at 144hrs, and was similar to recovery observed for JNJ-1661010. The extent of recovered activity for JNJ-1661010 inactivated enzyme was similar to that previously reported at 4°C.13 Control, untreated enzyme maintained about 80% activity over the duration of the experiment. These results support an inactivation mechanism through reversible intermediates. Covalent modification of the enzyme by 1a would be consistent with the very slow partial recovery of activity as observed with the acylating inhibitor, JNJ-1661010.
Figure 4.
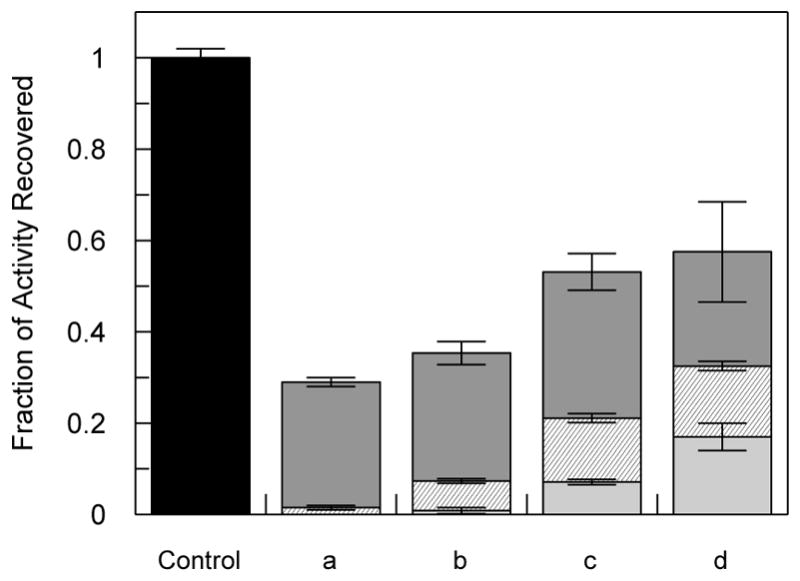
Recovery of FAAH activity (relative to controls) through exhaustive dialysis after inactivation by various compounds (a) compound 1a; (b) JNJ-1661010; (c) compound 2c; (d) compound 2d as described in Experimental procedures. Bars: light gray, 24 hrs; hash marks, 48 hrs; dark gray, 144 hrs; and black, range of control activity over the time course. Structures are given in Table 1 and Fig 1.
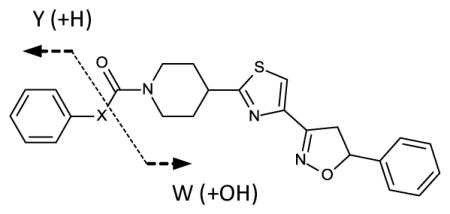
Both the representative aryl carbamates and ureas displayed slow, time dependent inactivation, and slow reversibility. This is characteristic of either a mechanism-based inactivating substrate, or formation of an inhibitory transition-state type mimic. The former type would suggest hydrolysis of the inhibitor forming a very slowly released acylated enzyme intermediate with initial release of either phenol or aniline. To investigate this, single turnover (inactivation) experiments with enzyme and added inhibitor were analyzed by LC/APCI MS for the presence of hydrolytic products in the incubation mixtures using compounds 2c and 1a respectively. Mixtures were analyzed for parent inhibitor, aniline (2c) or phenol (1a), and evidence of the carbamic acid piperidinyl thiazole isoxazoline core. For compound 1a, phenol and parent inhibitor were detectable in the reaction mix. However for compound 2c, parent inhibitor, aniline, and evidence of the carbamic acid core were observed (Table 3). The product ion corresponding to m/z = 359.4 is consistent with the sodium adduct of the free piperidinyl scaffold resulting from instability of the carbamic acid. Only parent inhibitor was detected in control incubations containing all components except enzyme for both 2c and 1a.
Table 3.
| |||
|---|---|---|---|
| Compound | Parent | Y | W(−CHO2) |
| 1a | 434.5 [M+H]+ | 113.1 [M+H3O]+ | Not Found |
| 2c | 433.5 [M+H]+ | 94.1 [M+H]+ | 359.4 [M+2Na]+ |
m/z values for the parent and product ions of the aryl carbamate (1a) and urea (2c). HPLC/MS conditions are described in Experimental procedures. Under the conditions of the experiment phenol was detected primarily as an adduct (water), although the parent [M+H] + = 95.1 was present in trace amounts. The product ion m/z=359.4 was also detected in control extraction of incubation buffers containing only the piperidinyl scaffold. For 1a X is O; for 2c X is N.
FAAH is a serine hydrolase, but as a member of the amidase enzyme family contains a catalytic triad that is unusual, and not found in other typical serine hydrolases such as proteases and esterases16. However, some features of the reaction mechanism appear similar, such as the use of an activated serine nucleophile, with some potent FAAH inhibitor chemotypes showing some degree of non-selectivity inhibiting unrelated serine hydrolases24. In preliminary tests, to probe FAAH specificity, we examined the ability of selected compounds to inhibit mammalian pancreatic esterase and elastase activities. All compounds tested showed at most, only slight inhibition of pancreatic elastase at the highest concentration tested (10 μM). IC50 values for 2b, 2c, 1f, 1d, 1h, and 1a were >10, 5.6, 0.87, 0.47, 0.84, and 2.4 uM respectively for inhibition of liver esterase. These values were orders of magnitude less potent compared to FAAH inhibition. These initial results indicated promising specificity for FAAH inhibition by these compounds.
We then sought to determine in vivo efficacy of these compounds using the mouse tail-flick test. Previous studies have shown that FAAH blockade produces a short-lasting analgesia in this assay as reflected by an increase latency to respond to noxious heat stimulation of the tail24. Since the bioavailablity properties for our aryl urea, and aryl carbamate inhibitor types were unknown, compounds were selected from both classes for in vivo evaluation. The 2 most potent aryl ureas (2e, 2f), and 4 of the more potent aryl carbamates (1a, 1b, 1o, and 1p) were screened at 50 mg/kg intraperitoneal (IP) dosing (Fig 5). Compounds 1a, 1b, 2e and 2f displayed significant analgesic effects at this dose and were tested at further doses to establish a dose-response relationship. Compounds 1o and 1p did not produce analgesia (Fig 5C, D). Lower doses of compound 1a, 2e and 2f failed to produce significant analgesia, however, 1b produced clear dose-dependent effects down to 5 mg/kg. Summary data for all tested compounds over the entire time course of testing is shown in Fig 5G.
Figure 5.

in vivo efficacies of novel FAAH inhibitors (Table1) in the mouse tail flick test. A–F shows tail flick latencies in the 52° C tail flick test following intraperitoneal injection of doses of novel FAAH inhibitors G shows summary data for all compounds where compound 1b shows the best in vivo potency. * p < 0.05, ** p < 0.01, *** p < 0.001 two way ANOVA with Bonferroni posthoc test.
The aryl isoxazoline carbamate and urea FAAH inhibitors reported here are among the most potent in vitro FAAH inhibitors identified. Contributions to the high binding affinities implicated through docking experiments are the extended interactions of the aryl isoxazoline moiety within the ABP domain, and interactions of both the aryl carbamate and urea portions of the chemotypes with key residues in the CP domain of FAAH. The SAR relationships are supported by the modeling results, for example, the bulky spiro-tetralin substitution on the isoxazoline (1n) ring showed a significant decrease in binding affinity relative to other extended aryl isoxazoline analogs. This is consistent with steric constraints imposed by the tetralin relative to the extended aryl isoxazoline within the ABP, and interaction with residues such as PHE192 and MET495. The aryl carbamate series showed higher binding affinity to FAAH than the aryl urea series where the common piperidinyl backbone is shared suggesting differences in interaction with the CP domain likely also occur between the two chemotypes. This was supported through experimental modeling (2WJ2) where favorable interactions with CYS269 appeared diminished with aryl urea 2c relative to the aryl carbamate 1a. Additionally, the greater electrophilicity of the aryl carbamates relative to the aryl ureas would lead to a greater interaction potential with SER241, and is likely reflected in the higher binding affinity of the aryl carbamates.
Previous structural observations have identified chemotype dependent conformational differences in critical active site residues that are coupled to corresponding structural changes in the ABP region20. This indicates a dynamic relationship between the ABP and CP domains. This is observed in crystallographic structures that show dynamic progress through the reaction states of the enzyme with reorientation of critical binding domains. The docked view of 1a (Fig 2) depicts the covalent approach of SER241 (Oγ) to the carbonyl in the “closed” conformation of the MAC and associated ABP domains, and is consistent with an expected inactivation mechanism leading to covalent modification of SER241, likely through carbamoylation. This was supported experimentally by detection of phenol as the resulting elimination product, and the absence of an extractable carbamic piperidinyl core in the reaction mixture. The absence of the latter indicates the formation of a non-dissociable carbamoylated intermediate supported by the view in Fig 2. Compound 1a displayed time-dependent inactivation, and very slow partial reversibility. The data suggests 1a is a mechanism based inhibitory substrate similar to JNJ-1661010.
The data also indicates that the distribution and nature of inactivating intermediates formed during inactivation of FAAH by 2c is distinct from that of 1a. This is based on the recovery of both the aniline leaving group, and the piperidinyl thiazole isoxazoline core as hydrolysis products. While this does not eliminate the presence of a stable carbamoylated intermediate (SER241), it nevertheless indicates that a significant proportion of the inactivating intermediate is dissociable. This is also supported by significant reversibility of inactivated enzyme by 2c upon dialysis. The nature of FAAH inhibition by 2c may therefore be through the reversible formation of a quaternary transition state mimic resulting from attack of SER241 Oγ on the urea carbonyl, similar to OL-135 inactivation. The ability to recover both hydrolysis products of 2c is consistent with this, and would likely result from partitioning of the quaternary intermediate; collapsing to either parent, carbamoylated intermediate, or hydrolysis products during aqueous denaturation of the complex. The observed differences in inactivation of FAAH by 1a or 2c can likely be attributed to the greater intrinsic susceptibility of aryl carbamates to nucleophilic attack relative to aryl ureas.
The inactivation rates (kinact) determined for the three carbamate and urea analogs are slower than those reported for other chemotypes such as carbamate URB597, and the biarylether urea inhibitor PF-3845 (0.0033 sec−1 each)4. This may be due to the relative intrinsic susceptibility differences among the individual electrophilic substituents. Among the aryl carbamates tested the greater kinact for 1b over 1a is consistent with greater susceptibility of the carbonyl toward nucleophilic attack because of the greater electron withdrawing character of the 4-CN (σ = +.66) on the phenyl ring of 1b. However, if ring substitution were solely dominant, then the kinact for 1l would be expected to be similar to that for 1a since only modest effects from the 3-CH3 (σ= −.069) on the aryl leaving group would be expected, however the kinact for 1l is about 2 fold higher than 1a. From the docked example of 1a (Fig 2) it is evident that the terminal aryl ring of the carbamate potentially interacts significantly with MET191 in the oxyanion pocket. The greater kinact for 1l may therefore result from greater delocalization through a C-H Ar type interaction due to the electron donating effect of the 3-CH3 substitution.
We chose a subset of compounds for in vivo testing in mice. Inhibition of FAAH results in increased AEA levels in the periphery and in the central nervous system2, 6. One result of this increased AEA concentration is analgesia via the activation of CB1 receptors. Recent evidence suggests that at least part of this effect is mediated by a peripheral mechanism25–27. We observed strong, dose-dependent analgesia in the tail flick test with 1b whereas several other active compounds only produced significant effects at 50 mg/kg. While the disposition of 1b in vivo is not currently known, the strong in vitro and in vivo potency for this compound make it an excellent starting point for further optimization based on this scaffold.
Experimental Procedures
Materials – JNJ-1661010, N-succinyl-ala-ala-ala-ρ-nitroanilide, 4-nitrophenyl butyrate, amino-4-methyl coumarin, porcine liver esterase, porcine pancreatic elastase, and the computer program GraFit (ver 3, Erithacus software) were from Sigma Aldrich. Sephacryl S100 was obtained from GE Healthcare. Expression vector pMAL-c4x, E. coli T7 Express cells, and restriction enzymes were from New England BioLabs. EDTA-free protease inhibitor cocktail was from Roche Life sciences. The human FAAH gene was purchased from Open BioSystems, GE Healthcare. Microplates were from Costar. Enzyme assays using chromogenic or fluorogenic substrates were measured on either a SpectraMax Plus or SpectraMax Gemini plate reader (Molecular Devices, Inc.), respectively. URB597 was from Asinex, Inc. NMR measurements were made on either a Varian INOVA 400 MHz system equipped with Nalorac indirect detection probe or a Brucker 500 MHz Avance III spectrometer using a 5mm 1H, X BBO probe. Mass analysis supporting chemical synthesis was performed using a Waters Alliance e2695 liquid chromatography system coupled to a SQ detector 2 (single quad) mass detector. Enzymatic reaction products were analyzed using an Agilent 1100 liquid chromatography system coupled to a Thermo LTQ mass spectrometer. Slide-A-Lyzer® mini dialysis units were from Thermo Scientific. Melting points were determined using a Thomas Hoover apparatus. All IC50 and enzyme kinetic data were fit with the computer program GraFit. All other chemicals, reagents, and materials were of reagent grade or better.
Compound Preparation
The synthetic chemistry used to prepare the compounds described in this article has been previously described in patent WO201107220728. As an example, phenyl 4-[4-(4, 5-dihydro-5-phenyl-3-isoxazolyl)-2-thiazolyl]-1-piperidinecarboxylate (Compound 1a, Table A) can be prepared in the following series of steps:
Preparation of 1,1-dimethylethyl 4-[4-(4,5-dihydro-5-phenyl-3-isoxazolyl)-2-thiazolyl]-1-piperidinecarboxylate
To a mixture of 1,1-dimethylethyl 4-(4-formyl-2-thiazolyl)-1-piperidinecarboxylate (1.0 g, 3.4 mmol) in ethanol (5 mL) was added an aqueous solution of hydroxylamine (50 wt. %, 0.25 mL, 4.0 mmol). The reaction mixture was heated at 60 °C for 1 h, during which time the reaction mixture became homogeneous. The resulting reaction solution was cooled to room temperature and diluted with tetrahydrofuran (10 mL). Styrene (0.57 mL, 5 mmol) was added to the reaction mixture, followed by a portion wise addition of Clorox® (aqueous sodium hypochlorite solution) (10.5 mL) over 3 h. The reaction mixture was stirred overnight at room temperature and then filtered. The solid collected by filtration was washed with water and diethyl ether and then air dried to give the title compound as a white powder (610 mg). The filtrate was diluted with saturated aqueous sodium bicarbonate solution and extracted with diethyl ether. The extract was dried (MgSO4) and concentrated under reduced pressure to give more of the title compound as yellow oil (850 mg). The oil was diluted with diethyl ether (4 mL) and upon standing provided the title compound as a white solid (233 mg). 1H NMR (CDCl3) δ 1.47 (s, 9H), 1.7 (m, 2H), 2.1 (m, 2H), 2.85 (m, 2H), 3.2 (m, 1H), 3.45 (m, 1H), 3.84 (m, 1H) 4.2 (br s, 2H), 5.75 (m, 1H), 7.25–7.40 (m, 5H), 7.61 (s, 1H).
Preparation of 4-[4-(4,5-dihydro-5-phenyl-3-isoxazolyl)-2-thiazolyl]piperidine
To a solution of 1,1-dimethylethyl 4-[4-(4,5-dihydro-5-phenyl-3-isoxazolyl)-2-thiazolyl]-1-piperidinecarboxylate (0.815 g, 1.97 mmol) in dichloromethane (50 mL) was added a solution of hydrogen chloride in diethyl ether (2 M, 10 mL, 20 mmol). The reaction mixture was stirred at room temperature for 1 h to give a gummy precipitate. Methanol was added to dissolve the precipitate, and the reaction mixture was stirred for an additional 1 h. The reaction mixture was concentrated under reduced pressure and partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The organic layer was dried (MgSO4) and concentrated to give the title compound as a clear oil (0.31 g), which solidified on standing. 1H NMR (CDCl3) δ 1.65 (br s, 1 H), 1.7 (m, 2H), 2.1 (m, 2H), 2.75 (m, 2H), 3.1–3.25 (m, 3H), 3.41 (m, 1H), 3.83 (m, 1H), 5.75 (m, 1H), 7.25–7.40 (m, 5H), 7.60 (s, 1H).
Preparation of phenyl 4-[4-(4,5-dihydro-5-phenyl-3-isoxazolyl)-2-thiazolyl]-1- piperidinecarboxylate
To a solution of 4-[4-(4,5-dihydro-5-phenyl-3-isoxazolyl)-2-thiazolyl]piperidine (3.3 g, 10 mmol) and triethylamine (2 mL, 14 mmol) in dichloromethane (40 mL) cooled to −5° C, was added a solution of phenyl chloroformate (1.6 g, 10 mmol) in dichloromethane (10 mL) dropwise over 5 minutes. The reaction mixture was stirred at −5° C for 30 minutes and then allowed to warm to room temperature. After 2 h, the mixture was washed with 1 N hydrochloric acid and brine, dried (MgSO4) and concentrated under reduced pressure to give the title compound as white foam (4.3 g). A 1 g sample was crystallized from ethanol (20 mL) to give a white powder (0.81 g) melting at 123–125° C. 1H NMR (CDCl3) δ 1.85 (m, 2H), 2.20 (m, 2H), 2.95–3.22 (m, 2H), 3.30 (m, 1H), 3.45 (m, 1H), 3.85 (m, 1H), 4.30–4.50 (m, 2H), 5.75 (m, 1H), 7.15 (m, 2H), 7.22 (m, 1H), 7.25–7.42 (m, 7H), 7.63 (s, 1H); m/z AP+ (M+1) = 434.
Ligand Docking
Ligand docking to the crystal structures FAAH was explored using AutoDock 4.2.6 and AutoDock Tools 1.5.6.29 Python Molecular viewer was used for graphics.30 Ligand and protein files were prepared using standard AutoDock protocols. Default values for number of ligand rotatable bonds and torsion angles were used which included assignment of urea amide bonds as non-rotatable. Medium level exhaustiveness was used in docking conformation searches.
FAAH Expression and Purification – recombinant human FAAH was expressed in truncated form, in which the transmembrane (TM) portion of the enzyme was removed from the N-terminal (amino acids 1–33), and then heterologously expressed as a MBP (maltose-binding protein) fusion protein in E. coli (MBP-ΔTM-FAAH) similar to that previously described31. The region of the gene corresponding to amino acids 34 to 579 was cloned into pMAL-c4x (New England BioLabs, Inc.) using EcoR1 and Sal1 restriction sites. E. coli T7 Express cells, containing the FAAH constructs, were used for expression of protein by induction with IPTG (isopropyl-β-D-thiogalactopyranoside) (100 μM) overnight at room temperature in Lennox Broth with 0.2% glucose. After harvest, the cells were resuspended in 20 mM Hepes buffer (pH 7.4) containing 200 mM NaCl, 2mM DTT (dithiothreitol), and protease inhibitor cocktail. The cell suspension was lysed by sonication, and the cell debris removed by centrifugation. The soluble extract was adjusted to 2.5 mg/mL protein, and the FAAH fusion protein (~105 kDa) loaded onto a 5 mL column of amylose affinity resin. The enzyme was eluted using 15 mM maltose as per manufacturer's (New England BioLabs, Inc.) instruction. Fractions containing FAAH were concentrated and further purified using Sephacryl S100 (HIPrep 26/60, GE Healthcare, Inc.) chromatography. Fractions enriched in FAAH were pooled, concentrated, and made 10% in glycerol then stored at −80°C until use. Stock samples used for single turnover (inactivation) experiments were stored without glycerol. All column chromatography steps used the Hepes buffer described above. Protein purity was about 90% determined by SDS-PAGE.
FAAH Assay and Compound Evaluation
Enzyme activity was measured using the fluorogenic substrate, decanoyl 7-amino-4-methyl coumarin (D-AMC) as described in the literature32. Briefly, the assay buffer consisted of 125 mM Tris-CL, 1 mM EDTA, and 0.1% BSA (pH 8.0). D-AMC was used at final concentration of 5 μM in all assays. Under these conditions specific activity of usual enzyme preparations were about 4000 to 6000 RFU min−1mg−1, although some preparations reached 15,000 RFU/mg−1/min−1. Reactions were carried out in black 96-well microplates using a SpectraMax Gemini (Molecular Devices, Inc.) fluorescence plate reader in a reaction volume of 200 μL per well at 37 °C. Reaction rates were monitored at an emission wavelength of 430 nm using an excitation wavelength of 351 nm over 30 to 40 minutes. Stock experimental compounds were dissolved in DMSO, and initially evaluated at a single diluted test concentration of 2 μM. Reactions were initiated by the addition of enzyme using 3 ug protein per well. Compounds inhibiting the reaction ≥ 90% were subsequently retested in microplates using serial dilution of compounds to determine IC50 values. The final concentrations of DMSO in the assay were 1% or lower.
Enzyme Inhibition and Kinetic Analysis
IC50 values were determined by computer fit of the dose response data using initial rates over a 9 dose range. Approximate inhibition constants (Ki values) were determined for experimental compounds using the relationship33, IC50 = Ki (1+[S]/Km) + [Et]/2. A Km value of 0.52 μM was used for D-AMC as reported in the literature32. To determine inactivation constants (kinact) for specific compounds the apparent first order rates of inactivation (ki′) of the enzyme were determined at several different concentrations of inhibitor by computer fit to the relationship, Fo=F∞(1-e−ki′t) where Fo is initial observed fluorescence and F∞ is the maximum final observed fluorescence. The apparent inactivation constants at each corresponding inhibitor concentration were then used to determine the kinact and the inhibition constant Ki from the relationship; ki′ = ([I]/Ki) kinact/(1 + [I]/Ki + [S]/Km) through solution (fit) of the resulting simultaneous equations. To assess the reversibility of aryl carbamate and urea enzyme inactivation, 50 μL (158 μg protein) of stock enzyme was mixed with 50 μL of assay buffer (described above), and incubated in the presence of excess inhibitor for 2 hr at 25°C. Control enzyme sample was treated identically, but without inhibitor. The samples were then assayed to insure complete inactivation of treated enzyme versus the control. Following inactivation, control and treated enzyme samples were dialyzed against multiple 1L changes (3) of 20mM Hepes buffer (pH7.4) containing 200mM NaCL, and 0.5 mM DTT (dithiothreitol) over a 144 hr period at 4°C. At various intervals during dialysis, samples were assayed as described above. To assess product release from aryl carbamate and urea inhibitors as a result of turnover during enzyme inactivation, the enzyme (50uL; 180 ug protein, high specific activity preparation, 15,730 RFU min−1mg−1) was first inactivated with inhibitor at 35°C as described above. The final concentrations of compound 1a and 2c (Table 1) were 200 nM and 10 uM respectively. The enzyme solutions (100uL) were then extracted with 100 uL of Hexane: Ethyl acetate (60:40) by vigorously vortexing the mixture (2×20 sec). Samples were centrifuged to separate the phases, the organic layer removed, and evaporated under reduced pressure at room temperature. 100uL of ACN: H20 (1:1) was added to resuspend the sample. Analysis was by LC MS. Sample separation was on an ACE 3 (50×4.6mm) C18 column equilibrated in H20 (0.1% FA): ACN (0.1% FA) [90:10]. A linear gradient (0.8 mL/min) from 1 to 10 min reaching H20 (0.1% FA): ACN (0.1% FA) [5:95] was used to elute analytes. MS analysis was made using an APCI source (vaporizer temperature 450°C) on a Thermo LTQ mass spectrometer. Detection was in positive polarity. The concentration of active enzyme used in assays was determined by stoichiometric titration of the enzyme with compound 1a (table 1). Stock enzyme (10 uL) was mixed with varying concentrations of inhibitor in assay buffer (10uL), and incubated at 25°C for 20 min to reach an endpoint. The enzyme samples were then assayed as above. A plot of the fraction of active enzyme remaining versus [I] was used to estimate active enzyme concentration.
Evaluation of FAAH Inhibitor Selectivity
The specificity of FAAH inhibition relative to other mechanistically similar enzymes, such as porcine liver esterase and porcine pancreatic elastase, was also explored for selected compounds. Both enzymes and substrates were obtained from commercial sources, and assayed in microplate format. N-succinyl-ala-ala-ala-ρ-nitroanilide was used as a substrate for pancreatic elastase, and 4-nitrophenyl butyrate was used as a substrate for measuring liver esterase activity. Enzyme activity was measured by following the release of ρ-nitroaniline and ρ-nitrophenol at 400 nm from the respective chromogenic substrates. The assay reaction mixture contained enzyme, 100uM substrate, 0.125 M TrisCl, and 0.2 mM EDTA, pH 8.0 in a total volume of 200 uL. Reactions were started by the addition of substrate. Control reactions give linear reaction rates (20 to 50 mOD/min) over at least 5 min.
Animal experiments
Male ICR mice (18–22g) were purchased from Harlan. Animals were housed in a climate-controlled room on a 12–12hr light/dark cycle where food and water were available ad libitum. All experiments were performed under an approved protocol in accordance with the policies and recommendations from IASP, NIH and IACUC of the University of Arizona. Stock solutions of compounds were made in 100% dimethyl sulfoxide (DMSO). All compounds were diluted to final doses in Ringer's solution for injection. Final concentrations of DMSO in solutions used for injection in all tests were less than 10%. For intraperitoneal injections in mice the mouse was restrained and its head was held down at an angle to allow the abdominal contents to move away from the injection site. The needle was placed parallel to the linea alba and inserted at a 30–45 degree angle in one of the two lower quadrants of the abdomen. Proper placement of the needle was verified prior to injection by withdrawing the syringe plunger. A lack of intestinal or bladder content in the syringe confirmed a good placement of the needle. The injection was then administered. A volume of 0.1 mL was used. In all experiments observers and lab personnel conducting the injections were blinded to the experimental conditions.
Tail Flick
Analgesia was measured using the tail immersion method34. The mice were restrained with the tail extending out. The distal portion of the tail (2–3 cm) was immersed in a water bath thermostatically controlled at 52°C ± .5. The tail withdrawal reaction time (in seconds) was initially recorded as the tail flick latency before drug administration and then recorded at 10, 20, 30, 45, and 60 minutes after the administration of the test compound. A cutoff latency of 10 seconds was maintained to prevent tissue damage. Percent maximum possible effect (%MPE) was calculated as %MPE = [(Test latency-Baseline latency)/(Cutoff latency-Baseline latency)]*100. All statistical analysis was done using Graph Pad Prism for Mac OS X Version 6.0c. Comparisons between groups were made by two-way anova with Bonferroni’s multiple comparison tests. At least 6 mice were used for each compound at each dose. Data are presented as mean ± SEM in all cases.
Acknowledgments
The authors would like to thank Drs. Pat Confalone, William Barnett, Stephen Gutteridge, and Scott Dax, for their interest and support of the project, and DuPont crop protection management for allowing exploration of the research area. We also thank Dr. John Andreassi for helpful discussion. We also acknowledge the skilled technical assistance of Daniel Rhoades and Boris Klyashchitsky. Special thanks to Dr. Seetha Krishnan (Syngene International Ltd, Bangalore, India) for molecular biology support and Dr. Satheesh Anand (DuPont) for preliminary assessment of analgesic activity. Work in the Price lab was supported by NIH grant GM102575.
Abbreviations
- FAAH
fatty acid amide hydrolase
- AEA
arachidonyl ethanolamide
- CB1
cannabinoid receptor type 1
- CB2
cannabinoid receptor type 2
- CP
cytoplasmic port
- ABP
acyl binding pocket
- MAC
membrane access channel
- D-AMC
decanoyl 7-amino-4-methyl coumarin
- Ar
aromatic
- SE
standard error
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Stephen O. Pember, Email: steve.o.pember@dupont.com.
Galo L. Mejia, Email: glm140230@utdallas.edu.
Theodore J. Price, Email: theodore.price@utdallas.edu.
Robert J. Pasteris, Email: robert.j.pasteris@dupont.com.
References
- 1.Lambert DM, Fowler CJ. J Med Chem. 2005;48:5059. doi: 10.1021/jm058183t. [DOI] [PubMed] [Google Scholar]
- 2.Piomelli D. Nat Rev Neurosci. 2003;4:873. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- 3.Blankman JL, Cravatt BF. Pharmacol Rev. 2013;65:849. doi: 10.1124/pr.112.006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, Weerapana E, Sadagopan N, Liimatta M, Smith SE, Lazerwith S, Stiff C, Kamtekar S, Bhattacharya K, Zhang Y, Swaney S, Van Becelaere K, Stevens RC, Cravatt BF. Chem Biol. 2009;16:411. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huggins JP, Smart TS, Langman S, Taylor L, Young T. Pain. 2012;153:1837. doi: 10.1016/j.pain.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 6.McKinney MK, Cravatt BF. Annu Rev Biochem. 2005;74:411. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]
- 7.Ahn K, Johnson DS, Cravatt BF. Expert Opin Drug Discov. 2009;4:763. doi: 10.1517/17460440903018857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Otrubova K, Ezzili C, Boger DL. Bioorg Med Chem Lett. 2011;21:4674. doi: 10.1016/j.bmcl.2011.06.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mor M, Rivara S, Lodola A, Plazzi PV, Tarzia G, Duranti A, Tontini A, Piersanti G, Kathuria S, Piomelli D. J Med Chem. 2004;47:4998. doi: 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]
- 10.Apodaca R, Breitenbucher JG, Pattabiraman K, Seierstad M, Xiao W. 2007005510. WO. 2007
- 11.Kiyota Y, Kori M, Matsumoto T, Miyazaki J. 2006054652. WO. 2006
- 12.Kono M, Matsumoto T, Kawamura T, Nishimura A, Kiyota Y, Oki H, Miyazaki J, Igaki S, Behnke CA, Shimojo M, Kori M. Bioorg Med Chem. 2013;21:28. doi: 10.1016/j.bmc.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 13.Keith JM, Apodaca R, Xiao W, Seierstad M, Pattabiraman K, Wu J, Webb M, Karbarz MJ, Brown S, Wilson S, Scott B, Tham CS, Luo L, Palmer J, Wennerholm M, Chaplan S, Breitenbucher JG. Bioorg Med Chem Lett. 2008;18:4838. doi: 10.1016/j.bmcl.2008.07.081. [DOI] [PubMed] [Google Scholar]
- 14.Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent RA, Nomanbhoy TK, Alexander JP, Cravatt BF. Biochemistry. 2007;46:13019. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]
- 15.Johnson DS, Ahn K, Kesten S, Lazerwith SE, Song Y, Morris M, Fay L, Gregory T, Stiff C, Dunbar JB, Jr, Liimatta M, Beidler D, Smith S, Nomanbhoy TK, Cravatt BF. Bioorg Med Chem Lett. 2009;19:2865. doi: 10.1016/j.bmcl.2009.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Science. 2002;298:1793. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]
- 17.Mileni M, Garfunkle J, DeMartino JK, Cravatt BF, Boger DL, Stevens RC. J Am Chem Soc. 2009;131:10497. doi: 10.1021/ja902694n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pasteris RJ, Hanagan MA, Bisaha JJ, Finkelstein BL, Hoffman LE, Gregory V, Andreassi JL, Sweigard JA, Klyashchitsky BA, Henry YT, Berger RA. Bioorg Med Chem. 2015 doi: 10.1016/j.bmc.2015.07.064. [DOI] [PubMed] [Google Scholar]
- 19.Andreassi JL, Gutteridge S, Pember SO, Sweigard JA. 2013009971 A1, . United States Patent WO. 2013
- 20.Mileni M, Kamtekar S, Wood DC, Benson TE, Cravatt BF, Stevens RC. J Mol Biol. 2010;400:743. doi: 10.1016/j.jmb.2010.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duan G, Smith VH, Weaver DF. Molecular Physics. 2001;99:1689. [Google Scholar]
- 22.Boger DL, Miyauchi H, Du W, Hardouin C, Fecik RA, Cheng H, Hwang I, Hedrick MP, Leung D, Acevedo O, Guimaraes CR, Jorgensen WL, Cravatt BF. J Med Chem. 2005;48:1849. doi: 10.1021/jm049614v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garfunkle J, Ezzili C, Rayl TJ, Hochstatter DG, Hwang I, Boger DL. J Med Chem. 2008;51:4392. doi: 10.1021/jm800136b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seierstad M, Breitenbucher JG. J Med Chem. 2008;51:7327. doi: 10.1021/jm800311k. [DOI] [PubMed] [Google Scholar]
- 25.Clapper JR, Moreno-Sanz G, Russo R, Guijarro A, Vacondio F, Duranti A, Tontini A, Sanchini S, Sciolino NR, Spradley JM, Hohmann AG, Calignano A, Mor M, Tarzia G, Piomelli D. Nat Neurosci. 2010;13:1265. doi: 10.1038/nn.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moreno-Sanz G, Sasso O, Guijarro A, Oluyemi O, Bertorelli R, Reggiani A, Piomelli D. Br J Pharmacol. 2012;167:1620. doi: 10.1111/j.1476-5381.2012.02098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sasso O, Bertorelli R, Bandiera T, Scarpelli R, Colombano G, Armirotti A, Moreno-Sanz G, Reggiani A, Piomelli D. Pharmacol Res. 2012;65:553. doi: 10.1016/j.phrs.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dung MH, Pasteris RJ. 2011072207. WO. 2011
- 29.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. J Comput Chem. 2009;30:2785. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanner MF. J Mol Graph Model. 1999;17:57. [PubMed] [Google Scholar]
- 31.Labar G, Vliet FV, Wouters J, Lambert DM. Amino Acids. 2008;34:127. doi: 10.1007/s00726-007-0540-1. [DOI] [PubMed] [Google Scholar]
- 32.Kage KL, Richardson PL, Traphagen L, Severin J, Pereda-Lopez A, Lubben T, Davis-Taber R, Vos MH, Bartley D, Walter K, Harlan J, Solomon L, Warrior U, Holzman TF, Faltynek C, Surowy CS, Scott VE. J Neurosci Methods. 2007;161:47. doi: 10.1016/j.jneumeth.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 33.Robert A. Copeland Enzymes: A Practical Introduction to Structure, Mechanism and Data Analysis. Wiley-VCH Inc; 2000. pp. 305–317. [Google Scholar]
- 34.Janssen PA, Niemegeers CJ, Dony JG. Arzneimittelforschung. 1963;13:502. [PubMed] [Google Scholar]