

Abstract
Objective
To use linkage analysis and whole exome sequencing to identify the genetic mutation in a multigenerational Australian family with Charcot–Marie–Tooth disease type 2 (CMT2) and pyramidal signs.
Methods
Genome-wide linkage analysis was performed to map the locus. Whole exome sequencing was undertaken on selected individuals (3 affected, 1 normal), and segregation analysis and mutation screening were carried out using high-resolution melt analysis. The GEM.app database was queried to identify additional families with mutations.
Results
Significant linkage (2-point LOD score ≥ +3) and haplotype analysis mapped a new locus for CMT2 and pyramidal signs to a 6.6Mb interval on chromosome 22q12.1–q12.3. Whole exome sequencing identified a novel mutation (p.R252W) in the microrchidia CW-type zinc finger 2 (MORC2) gene mapping within the linkage region. The mutation fully segregated with the disease phenotype in the family. Screening additional families and querying unsolved CMT2 exomes, we identified the p.R252W mutation in 2 unrelated early onset CMT2 families and a second mutation p.E236G in 2 unrelated CMT2 families. Both the mutations occurred at highly conserved amino acid residues and were absent in the normal population.
Interpretation
We have identified a new locus in which MORC2 mutations are the likely pathogenic cause of CMT2 and pyramidal signs in these families. MORC2 encodes the human CW-type zinc finger 2 protein, which is a chromatin modifier involved in the regulation of DNA repair as well as gene transcription.
Charcot–Marie–Tooth disease (CMT) is a clinically and genetically heterogeneous group of disorders affecting the motor and sensory neurons in the peripheral nervous system. The CMT phenotype is characterized by progressive weakness and atrophy of distal muscles, high arched feet (pes cavus), and loss of deep tendon reflexes. CMT has traditionally been divided into 2 groups, demyelinating (CMT1) and axonal (CMT2), based on median motor nerve conduction velocities (NCV). CMT1 patients have NCV < 38m/s in the median nerve. Patients with CMT2 usually show normal or a slight reduction of NCV with reduced compound amplitudes of motor and sensory nerve action potentials indicating degeneration of axons.1
We previously reported genetic studies in an Australian family (CMT105) diagnosed with CMT2 and pyramidal signs.2–4 This family has also been the subject of a clinical report.5 As mutations in all the known CMT2 genes had been excluded, the family was suitable for a combined genetic linkage and whole exome sequencing (WES) approach to identify the pathogenic gene. Here we report a new CMT2 locus and gene mutations likely to be causing CMT2 with pyramidal signs.
Patients and Methods
Family Ascertainment
Thirty-two family members were examined. Participants provided written consent according to protocols approved by the Sydney Local Health District Human Ethics Committee (Concord Hospital, Sydney). Genomic DNA was extracted from peripheral blood using standard methods.
Linkage and Haplotype Analysis
A genome scan was initially performed on 25 family members at deCODE Genetics (Reykjavik, Iceland) using an 8cM linkage panel. Two-point and multipoint linkage analyses were performed using the SuperLink (V1.5) and SimWalk (V2.91) modules from the easyLINKAGE Plus package (V5.02), respectively.6 Several suggestive LOD scores (≥1.5) from the initial genome scan were further analyzed with additional markers as previously described,7 on an extended pedigree of the family (32 members). Four in-house microsatellite markers (O22AX21TG, O22A21XTG, O22A25XAC, and O22A22XAC) were designed using the dinucleotide repeat sequences annotated in the microsatellite option of the Repeats Track in the UCSC Genome Browser (hg19 assembly). Primer sequences for the in-house markers are available on request. Extended haplotypes of the family members were constructed according to the physical location of the markers and based on the minimal number of intermarker recombination events.
WES and Bioinformatics
Genomic DNA (2.5 μg) from 3 affected members (III-2, III-13, and IV-2) and a married-in healthy control (III-6; Fig 1) were sent for WES at Axeq Technologies (Seoul, South Korea) as previously described.8 Candidate variants were screened through 1,000 neurologically normal chromosomes as previously described and also queried against 30 in-house exomes from neurologically normal controls.8 Variants were annotated using ANNOVAR software.9
FIGURE 1.
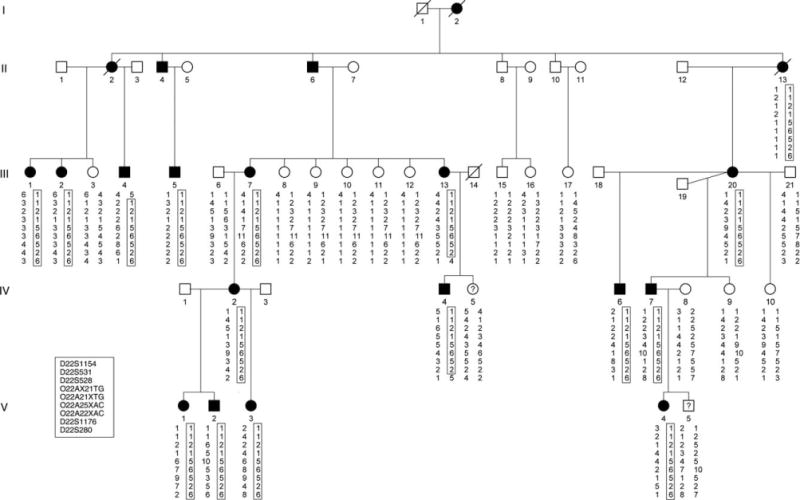
Haplotype analysis of markers from chromosome 22q12.1–q12.3 in family (CMT105) with autosomal dominant Charcot–Marie–Tooth disease type 2 with pyramidal signs. The haplotype segregating with the disease is boxed. The markers are ordered from centromere (top) to telomere (bottom). Solid symbols denote affected individuals; open symbols denote unaffected individuals. Symbols with a question mark denote unknown phenotype. A diagonal line through a symbol denotes deceased individual. For the 6.6Mb linkage interval, individual III-4 defines the proximal boundary at D22S1154 and individuals III-13 and IV-4 define the distal boundary at D22S280.
Mutation Screening
Mutation analysis was performed using high-resolution melt (HRM) protocols as previously described.8 Primers were designed to amplify the coding exons and adjacent exon/intron splice sites and are available on request. Index individuals from 45 unrelated CMT2 families underwent mutation analysis of the MORC2 gene. In addition, the GEM.app database was queried for MORC2 variants in exome data submitted from CMT2 index cases.10 Annotation of the MORC2 mutations, are based on the human MORC2 isoform 1, which encodes the longest isoform of 1,032 amino acids (accession number NM_001303256.1). Annotations of the MORC2 mutations reported by Sevilla et al are based on the human MORC2 isoform 3, which encodes a shorter isoform of 970 amino acids (NM_014941.2).11
Results
Mapping a New Locus for Axonal CMT with Pyramidal Signs to Chromosome 22q12.1–q12.3
Initial genome-wide linkage data based on genotyping 25 individuals gave a maximum 2-point LOD score of 1.9 at zero recombination for the marker D22S531 (data not shown). After expanding the family and regenotyping 32 family members, the marker D22S531 gave a LOD score of 3.15 at zero recombination (Supplementary Table S1). Fine mapping was performed and significant linkage was obtained for several markers, with the maximum LOD score (Zmax = 6.33 at θ = 0) being at marker O22A25XAC. A disease haplotype segregating with affected individuals was observed between the markers D22S1154 and D22S820 (see Fig 1). One affected male (III:4) showed a recombination event between the markers D22S1154 and D22S531. Two affected individuals (III:13 and IV:4) showed a recombination event between the markers D22S1176 and D22S280. The combined recombination information refined the linkage region to a 6.6Mb interval flanked by the markers D22S1154 and D22S280 (Fig 2A).
FIGURE 2.
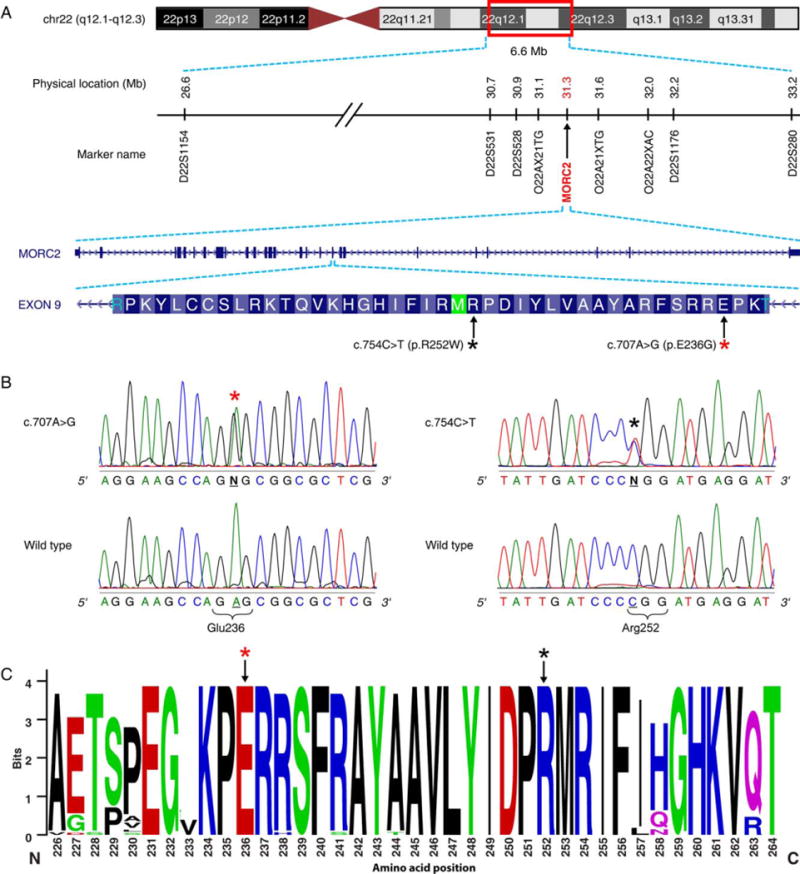
Genetic basis of axonal Charcot–Marie–Tooth disease (CMT) with pyramidal signs. (A) Physical map of markers defining the 6.6Mb linkage region harboring the MORC2 gene on chromosome 22q12.1–q12.3. The mutations occur in exon 9, and the asterisks denote the amino acid residue substituted for the p.R252W (black) and p.E236G (red) mutations. (B) Sequence traces of normal and affected individuals with the p.E236G (left panel) and p.R252W (right panel) mutation. The asterisks designate the base change resulting in the p.R252W (black) and p.E236G (red) missense mutations that segregate with CMT with pyramidal signs in the respective families. (C) Sequence logo showing the conservation of the amino acid residue E236 (red asterisk) and R252 (black asterisk). The region of the protein in which the E236 and E252 residues are located is also highly conserved, as reflected in the height of the stack at each amino acid position.
WES Identifies a Mutation in the MORC2 Gene
The average coverage of targeted regions in the 4 individuals sequenced was 93.9% and 80.5% reads at 1× and 10×, respectively. The average coverage of the genes in the 6.6Mb linkage interval was 89.9% at 1× to 57.70% at 203. Greater than 62,000 sequence variants were identified in each individual. No mutations were identified in the known peripheral neuropathy genes. Variant filtering identified a missense mutation (c.754C>T) in the MORC2 gene, which localized within the linkage interval. The mutation occurred in exon 9 and corresponded to a p.R252W missense mutation. Sanger sequencing confirmed the c.754C>T mutation in the affected individuals who initially underwent WES, and the mutation fully segregated with the disease phenotype in the entire family. The mutated amino acid residue localized in a highly conserved region of the protein that is associated with the GHL (Gyrase B, Hsp90, and MutL)-ATPase domain of the MORC2 protein (Fig 2B, C).12
Identification of MORC2 Mutations in Additional Unrelated Families Supports the Role for MORC2 in CMT2 Neuropathy
A combination of mutation screening CMT2 families and querying CMT2 WES data in the GEM.app database identified an additional 4 families with MORC2 mutations. The p.R252W was identified in 2 unrelated CMT2 families (Fig 3). The mutation in CMT895 was in an affected female from a consanguineous marriage. We validated paternity (data not shown), and as the parents do not carry the mutation we can confirm this is a de novo mutation. A second MORC2 missense mutation c.707A>G (p.E236G) was identified in 2 unrelated CMT2 families. The second p.E236G mutation also occurred in exon 9 and localized to the GHL-ATPase domain of the MORC2 protein (see Fig 2B, C). For both p.R252W and p.E236G, HRM genotyping excluded the mutation in 500 neurologically normal control samples (1,000 chromosomes) and they were also absent in NHLB1 EVS 6500. Both the mutations were processed through several prediction programs for pathogenicity. The missense changes were highly conserved with a PhastCons score of 1. The GERP (genomic evolutionary rate profiling) scores for c.707A>G (p.E236G) and c.754C>T (p.R252W) were 5.56 and 2.13, respectively. Both missense changes were predicted to be damaging in 4 of the 5 (Polyphen 2, Mutation Taster, Mutation Assessor, LRT, and SIFT) protein prediction programs (Supplementary Table S2).
FIGURE 3.
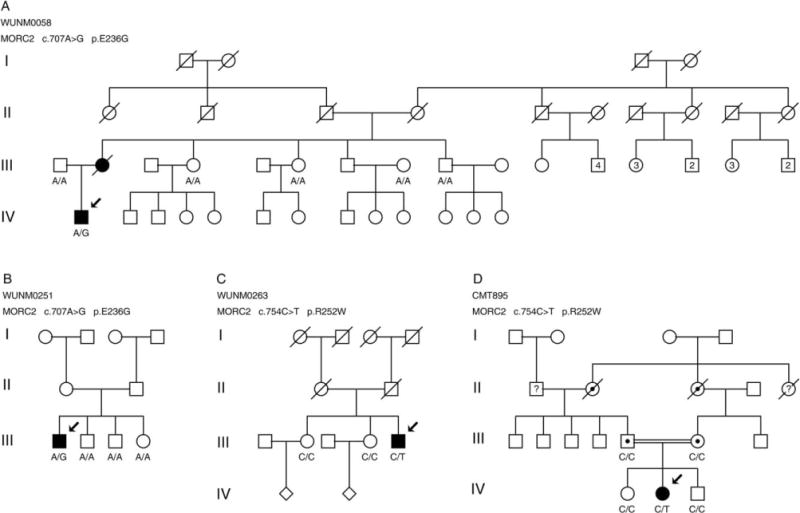
Segregation of the MORC2 mutations c.707A>G p.E236G (A, B) and c.754C>T p.R252W (C, D) in additional unrelated Charcot–Marie–Tooth disease type 2 families. The genotypes are shown below individuals who were available for genotyping. Solid symbols denote clinically affected individuals. Arrows denote the individuals in whom the MORC2 mutation was initially identified. Individuals in each generation are counted consecutively from left to right starting with 1.
In addition to the 2 mutations, several families with other MORC2 gene variants were identified (Table 1). Five of the variants did not cosegregate with the disease in the respective families and were deemed nonpathogenic. Two of the variants (p.G444R and p.Q96E) were absent in control exomes and published databases. Pathogenic predictions for both variants were equivalent to the predictions for p.R252W and p.E236G (see Supplementary Table S2) and are likely to be pathogenic; however, this should be further supported by additional families with the same mutations or functional evidence.
TABLE 1.
Non-segregating and unconfirmed MORC2 variants found in additional CMT2 families
| Family | Variant | AA change | |
|---|---|---|---|
| Non-Segregating | CMT102 | c.848G>A | p.R283H |
| CMT131 | c.1753C>T | p.R585C | |
| CMT739 | c.1396G>C | p.D456H | |
| CMT775 | c.2270A>G | p.E757G | |
| CMT791 | c.743A>G | p.Y248C | |
| Uncertain/unconfirmed | Austrian 1 | c.1330C>T | p.G444Ra |
| Sporadic | c.286C>G | p.Q96Ea |
Excluded in > 1000 control chromosomes
Excluded in > 2000 control exomes
Clinical Characterization of Families with the p.R252W Mutation
FAMILY CMT105
This family has been previously reported.3,5 Sixteen individuals assessed were clinically affected, and 2 individuals (IV-5 and V-5) were at risk with an unknown phenotype (see Fig 1). This is a large family with an early onset (children to young adults) and a length-dependent axonal motor and sensory neuropathy with pyramidal signs. Clinically, there is pes cavus, distal muscular atrophy, and weakness of the intrinsic feet muscles, later progressing to hand muscles and finger contractures. Over decades, the weakness spreads proximally, resulting in severe disability requiring walking aids by middle age and wheeled transport for some individuals later in life.
There were some additional unusual clinical features that are summarized and compared to the additional families reported in this study (Table 2). The disorder in children could present with toe walking and developmental delay. Learning difficulties were present in many affected individuals. The pyramidal features were usually increased muscle tone in the legs without frank spasticity, and 5 had extensor plantar reflexes. Three individuals had a distinctive high-pitched speech, and some had deafness. Two affected individuals had mild pigmentary retinal changes and color perception abnormalities. Median sensory action potentials were present in childhood but were absent in adults. Sural sensory responses were unobtainable even in early disease. Nerve biopsy showed a reduction of myelinated axons of all diameters, but teased fibers showed no demyelination. Brain magnetic resonance imaging (MRI) showed mild periventricular leukomalacia in 1 affected child. This may be further evidence of central involvement; however, brain MRIs were not systematically studied. Karyotype analysis showed no chromosomal abnormalities in the analyzed affected individual with a G banding resolution of 400 to 550 bands per haploid set. Similarly, no microscopic abnormalities were detected by the comparative genomic hybridization microarray at an effective resolution of 0.25Mb (data not shown).
TABLE 2.
Clinical Summary of MORC2 Families
| Phenotype of MORC2 Families | Family | ||||
|---|---|---|---|---|---|
| CMT105a | WUNM0263 | CMT895 | WUNM0058 | WUNM0251 | |
| Age of onset | Childhood, early adult | Onset age 8 years, asymmetrical foot drop | Late walking | Ankle weakness age 7 years, foot drop | 2 years distal leg weakness |
| Progression & disability | Walking aids in middle age, WC in later life | WC age 40 years | — | WC age 35 years | WC age 25 years |
| Weakness | Early distal, later proximal | Distal & asymmetrical, proximal later | Distal & proximal upper & lower limbs | Distal in childhood, proximal in teens | Distal then proximal |
| UMN signsb | Yes | No | No | No | No |
| Reflexes | Absent | Absent | Absent | Absent | Absent |
| Sensory nerve conduction study | Sural SNAP absent in childhood median sensory nerve action potential absent in adults | Median & sural SNAPs absent age 43 years | Median SNAP reduced & delayed | Sural SNAP absent, median SNAP = 5 μV (age 13 years) | Median SNAP = 8 μV & sural SNAP = 2 μV |
| Motor nerve conduction study | MCV = 42–60, PCV = 0–60 | MCV = 44 (CMAP = 0.5mV), peroneal CMAPs absent | Median CMAPs & peroneal CMAPs absent | MCV = 57 (CMAP = 8mV | MCV = 43 (1.0mV) & peroneal CMAP = 0.9mV |
| MRI | MRI muscle: fatty atrophy of gastrocnemius & soleus & TA; MRI brain: mild periventricular leukomalacia, age 9 years | — | MRI calves: fatty involution, all compartments | — | — |
| Other features | Deafness, high-pitched speech, pigmentary, retinopathy (in 1 branch of family) | Seizures | Dysmorphic face with prognathism | Childhood seizures | Nocturnal hypoventilation |
| Learning difficulties | Developmental delay in 2 branches of the family | — | Developmental delay | Mild learning difficulties | — |
= no data; CMAP = compound muscle action potential amplitude; MCV = median motor conduction velocity (m/s); MRI = magnetic resonance imaging; PCV = peroneal motor conduction velocity (m/s); SNAP = sensory nerve action potential; TA = tibialis anterior; UMN = upper motor neuron; WC = wheelchair.
The phenotype of this family has been previously published.
UMN signs = increased bone ± Babinski reflex.
FAMILY WUNM0263
The index individual (III-5) is a sporadic case with a severe motor and sensory neuropathy (see Fig 3C) that presented with asymmetric foot drop at the age of 8 years (left more than right). He required leg braces throughout adolescence, and began using a wheelchair at age 20 years, when proximal leg and hand weakness developed. He was wheelchair dependent by 40 years, and completely bedbound by age 65 years. Rare seizures of unknown cause developed at the age of 45 years. Neurophysiology at age 42 years showed a severe motor and sensory axonal neuropathy.
FAMILY CMT895
The index patient (IV-2) is a 27-year-old female who had developmental delay with late walking and a tendency to fall (see Fig 3D). As an adult she worked in a sheltered workshop. She was thin and had a dysmorphic face with prognathism. She had a recurrent throat-clearing cough. Her parents were first cousins. Her maternal grandmother’s sister had similar problems with gait and development. She had wasting of the hands, arms, upper and lower legs, and feet. There were no hamstring or Achilles tendon contractures. There was mild weakness proximally of the deltoid and hip flexors. There was moderate weakness of the intrinsic hand muscles with finger contractures. Tendon reflexes were absent. There was a level to sharp appreciation above the ankles. Vibration sense was reduced at the ankles. WES excluded known neuropathy gene mutations.
Clinical Characterization of Families with the p.E236G Mutation
FAMILY WUNM0058
The index individual (III-2; see Fig 3A) had nocturnal seizures as a child, and presented with asymmetric ankle weakness at age 7 years. Proximal weakness developed in her teens, and she became wheelchair dependent after a leg fracture in her 30s. Her son (IV-1) developed foot drop at the age of 6 years, used a wheelchair at age 27 years, and was dependent by 35 years. He also had seizures, moderate learning difficulties, and childhood onset diabetes mellitus. Nerve conduction studies were normal at age 3 years, but by age 13 years showed a motor and sensory axonal neuropathy.
FAMILY WUNM0251
The index case (III-1) is a sporadic case who presented at age 2 years with asymmetric distal leg weakness that progressed to include the proximal legs such that a modified Gowers maneuver was required for standing (see Fig 3B). He began using a wheelchair at age 18 years, became wheelchair dependent at 25 years, and began using BiPAP at age 28 years for nocturnal hypoventilation. Weakness was accompanied by progressive distal sensory loss. Nerve conduction study at age 12 years showed a severe motor and sensory axonal neuropathy.
Discussion
We have established linkage to a 6.6Mb region on chromosome 22q12.1–12.3 for this axonal form of CMT with pyramidal signs in family CMT105 and utilized WES to identify a mutation (p.R252W) in the MORC2 gene that lies within the linkage region. The identification of 2 additional unrelated CMT2 families with the same p.R252W mutation as well as identification of a second mutation (p.E263G) in 2 unrelated CMT2 families provides compelling evidence for a pathogenic role of MORC2 in CMT2 neuropathy. Known CMT2 loci were previously excluded in CMT105, and the results from WES on this family confirmed this initial finding.2–4
Human microrchidia CW-type zinc finger 2 (MORC2), also known as KIAA0852, ZCW3, AC004542.C22.1, and ZCWCC1, is ubiquitously expressed and is 1 of 5 members of the highly conserved MORC protein family.12,13 The MORC proteins share conserved protein domains including a CW-type zinc finger domain, a GHL-ATPase domain at the amino terminus, and varied coiled-coil domains.12
The MORC2 protein is predominantly expressed in the nucleus, where it promotes chromatin remodeling during DNA repair and transcription regulation.14–16 MORC2 target genes have a wide range of biological functions including cell apoptosis, cell cycle, cell adhesion, cell mobility, material transport, and metabolism. The majority of these target genes are downregulated following MORC2 expression.16 MORC2 represses gene activity by interacting and recruiting histone deacetylases to promoters. Deregulation of gene transcription has been implicated in several neurodegenerative disorders including ataxia telangiectasia.17
Following DNA damage, MORC2 is recruited to the chromatin, where it uses energy from adenosine triphosphate (ATP) hydrolysis to replace canonical H2A-H2B dimers with a mixture of H2AZ-H2B dimers. This facilitates chromatin relaxation to enable access of DNA repair machinery to the damaged site.14,18 Both the mutations reported here are located in the GHL-ATPase domain and could therefore affect DNA repair. Interestingly, mutations in the Hint1 gene, which also play a role in the DNA damage response,19 cause axonal peripheral neuropathy with neuromyotonia and distal motor neuropathy.20,21
Recently, a novel cytoplasmic function of MORC2 was described in which it regulates activity of the enzyme ATP-citrate lyase (ACLY).22 ACLY catalyzes the formation of acetyl–coenzyme A (CoA) and oxaloacetate from mitochondrial-derived citrate and CoA. Acetyl-CoA is an important substrate for many biological reactions including lipogenesis,22 acetylation reactions such as histone acetylation,23 and the production of the neurotransmitter acetylcholine.24
While this work was under revision, the article by Sevilla et al was published online.11 They also report the MORC2 (p.R190W) mutation, which is the same as the p.R252W described in our largest family CMT105. No pyramidal signs were described in their families. Family CMT105 has prominent sensory signs as described by Sevilla et al, but in addition has pyramidal signs with increased reflexes, tone, and Babinski signs5 previously reported. Affected individuals in CMT105 presented with distal weakness progressing to proximal weakness typical of a length-dependent neuropathy, which we would not describe as spinal muscular atrophy. Further studies of larger patient numbers with this genotype are needed to fully characterize the phenotype and to understand the functional consequences of this common MORC2 mutation.
In summary, we report pathogenic mutations in the gene encoding MORC2 in families with CMT2 with pyramidal signs. Based on the current known roles of MORC2, the mutations may affect functions including DNA repair and gene regulation in both the central and peripheral nervous systems. Our findings expand the spectrum of genetic causes in axonal neuropathies.
Supplementary Material
Acknowledgments
This work was supported by National Health and Medical Research Council project grants (APP1046680, APP1007705, M.L.K., G.A.N.) and the Austrian Science Fund (P27634-B19, M.A.-G.). Research on families WUNM0251, WUNM0058, and WUNM0263 was funded by an NIH National Institute of Neurological Disorders and Stroke grant (K08NS075094, M.B.H.) and the Children’s Discovery Institute (R.H.B.). A PhD education travel scholarship from the Kingdom of Saudi Arabia supported O.M.A. A postgraduate scholarship from the Family Trust of Lord Selborne supported A.H.S.
We thank the CMT Association of Australia; the CMT families and their friends; P. Allred for input; Cedars Sinai Medical Center, Los Angeles, for help in sample coordination and collection; and Drs M. Brewer and G. Perez-Siles for helpful comments during manuscript preparation.
Footnotes
Potential Conflicts of Interest
Nothing to report.
References
- 1.Harding AE, Thomas PK. Genetic aspects of hereditary motor and sensory neuropathy (types I and II) J Med Genet. 1980;17:329–336. doi: 10.1136/jmg.17.5.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albulym OM, Zhu D, Reddel S, et al. The MFN2V705I variant is not a disease-causing mutation: a segregation analysis in a CMT2 family. J Neurodegener Dis. 2013;2013:1–5. doi: 10.1155/2013/495873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vucic S, Kennerson M, Zhu D, et al. CMT with pyramidal features. Charcot-Marie-Tooth Neurology. 2003;60:696–699. doi: 10.1212/01.wnl.0000048561.61921.71. [DOI] [PubMed] [Google Scholar]
- 4.Zhu D, Kennerson ML, Walizada G, et al. Charcot-Marie-Tooth with pyramidal signs is genetically heterogeneous: families with and without MFN2 mutations. Neurology. 2005;65:496–497. doi: 10.1212/01.wnl.0000171345.62270.29. [DOI] [PubMed] [Google Scholar]
- 5.Frith JA, McLeod JG, Nicholson GA, et al. Peroneal muscular atrophy with pyramidal tract features (hereditary motor and sensory neuropathy type V): a clinical, neurophysiological, and pathological study of a large kindred. J Neurol Neurosurg Psychiatry. 1994;57:1343–1346. doi: 10.1136/jnnp.57.11.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lindner TH, Hoffmann K. easyLINKAGE: a PERL script for easy and automated two-/multi-point linkage analyses. Bioinformatics. 2005;21:405–407. doi: 10.1093/bioinformatics/bti009. [DOI] [PubMed] [Google Scholar]
- 7.Kennerson M, Nicholson G, Kowalski B, et al. X-linked distal hereditary motor neuropathy maps to the DSMAX locus on chromosome Xq13.1-q21. Neurology. 2009;72:246–252. doi: 10.1212/01.wnl.0000339483.86094.a5. [DOI] [PubMed] [Google Scholar]
- 8.Kennerson ML, Yiu EM, Chuang DT, et al. A new locus for X-linked dominant Charcot-Marie-Tooth disease (CMTX6) is caused by mutations in the pyruvate dehydrogenase kinase isoenzyme 3 (PDK3) gene. Hum Mol Genet. 2013;22:1404–1416. doi: 10.1093/hmg/dds557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonzalez MA, Lebrigio RF, Van Booven D, et al. GEnomes Management Application (GEM.app): a new software tool for large-scale collaborative genome analysis. Hum Mutat. 2013;34:842–846. doi: 10.1002/humu.22305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sevilla T, Lupo V, Martinez-Rubio D, et al. Mutations in the MORC2 gene cause axonal Charcot-Marie-Tooth disease. Brain. 2015 Oct 24; doi: 10.1093/brain/awv311. pii: awv311 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 12.Wang GL, Wang CY, Cai XZ, et al. Identification and expression analysis of a novel CW-type zinc finger protein MORC2 in cancer cells. Anat Rec (Hoboken) 2010;293:1002–1009. doi: 10.1002/ar.21119. [DOI] [PubMed] [Google Scholar]
- 13.Li DQ, Nair SS, Kumar R. The MORC family: new epigenetic regulators of transcription and DNA damage response. Epigenetics. 2013;8:685–693. doi: 10.4161/epi.24976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li DQ, Nair SS, Ohshiro K, et al. MORC2 signaling integrates phosphorylation-dependent, ATPase-coupled chromatin remodeling during the DNA damage response. Cell Rep. 2012;2:1657–1659. doi: 10.1016/j.celrep.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shao Y, Li Y, Zhang J, et al. Involvement of histone deacetylation in MORC2-mediated down-regulation of carbonic anhydrase IX. Nucleic Acids Res. 2010;38:2813–2824. doi: 10.1093/nar/gkq006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Q, Song Y, Chen W, et al. By recruiting HDAC1, MORC2 suppresses p21 Waf1/Cip1 in gastric cancer. Oncotarget. 2015;6:16461–16470. doi: 10.18632/oncotarget.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, Chen J, Ricupero CL, et al. Nuclear accumulation of HDAC4 in ATM deficiency promotes neurodegeneration in ataxia telangiectasia. Nat Med. 2012;18:783–790. doi: 10.1038/nm.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu Y, Ayrapetov MK, Xu C, et al. Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol Cell. 2012;48:723–733. doi: 10.1016/j.molcel.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Balajee AS, Su T, et al. The HINT1 tumor suppressor regulates both gamma-H2AX and ATM in response to DNA damage. J Cell Biol. 2008;183:253–265. doi: 10.1083/jcb.200711150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zimon M, Baets J, Almeida-Souza L, et al. Loss-of-function mutations in HINT1 cause axonal neuropathy with neuromyotonia. Nat Genet. 2012;44:1080–1083. doi: 10.1038/ng.2406. [DOI] [PubMed] [Google Scholar]
- 21.Zhao H, Race V, Matthijs G, et al. Exome sequencing reveals HINT1 mutations as a cause of distal hereditary motor neuropathy. Eur J Hum Genet. 2014;22:847–850. doi: 10.1038/ejhg.2013.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanchez-Solana B, Li DQ, Kumar R. Cytosolic functions of MORC2 in lipogenesis and adipogenesis. Biochim Biophys Acta. 2014;1843:316–326. doi: 10.1016/j.bbamcr.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi H, McCaffery JM, Irizarry RA, et al. Nucleocytosolic acetyl-coenzyme A synthetase is required for histone acetylation and global transcription. Mol Cell. 2006;23:207–217. doi: 10.1016/j.molcel.2006.05.040. [DOI] [PubMed] [Google Scholar]
- 24.Jope RS, Jenden DJ. The utilization of choline and acetyl coenzyme A for the synthesis of acetylcholine. J Neurochem. 1980;35:318–325. doi: 10.1111/j.1471-4159.1980.tb06267.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.