

Abstract
Streptococcus oralis, an oral commensal, belongs to the mitis group of streptococci and occasionally causes opportunistic infections, such as bacterial endocarditis and bacteremia. Recently, we found that the hydrogen peroxide (H2O2) produced by S. oralis is sufficient to kill human monocytes and epithelial cells, implying that streptococcal H2O2 is a cytotoxin. In the present study, we investigated whether streptococcal H2O2 impacts lysosomes, organelles of the intracellular digestive system, in relation to cell death. S. oralis infection induced the death of RAW 264 macrophages in an H2O2-dependent manner, which was exemplified by the fact that exogenous H2O2 also induced cell death. Infection with either a mutant lacking spxB, which encodes pyruvate oxidase responsible for H2O2 production, or Streptococcus mutans, which does not produce H2O2, showed less cytotoxicity. Visualization of lysosomes with LysoTracker revealed lysosome deacidification after infection with S. oralis or exposure to H2O2, which was corroborated by acridine orange staining. Similarly, fluorescent labeling of lysosome-associated membrane protein-1 gradually disappeared during infection with S. oralis or exposure to H2O2. The deacidification and the following induction of cell death were inhibited by chelating iron in lysosomes. Moreover, fluorescent staining of cathepsin B indicated lysosomal destruction. However, treatment of infected cells with a specific inhibitor of cathepsin B had negligible effects on cell death; instead, it suppressed the detachment of dead cells from the culture plates. These results suggest that streptococcal H2O2 induces cell death with lysosomal destruction and then the released lysosomal cathepsins contribute to the detachment of the dead cells.
INTRODUCTION
Streptococcus oralis, a commensal bacterium in the oral cavity, belongs to the group of oral inhabitants of the mitis group of streptococci (1–4). Members of this group, including Streptococcus sanguinis and Streptococcus gordonii, are dominant colonizers of human teeth and are involved in dental plaque formation (1–3). Pathogenic streptococcal species, such as Streptococcus pneumoniae and Streptococcus mitis, are also members of the mitis group (2, 4–6). This group has been implicated as a possible cause of human cardiovascular diseases, including infective endocarditis and atherosclerosis. S. oralis is frequently isolated from cases of infective endocarditis (7–10), and ribosomal DNAs of the mitis group were detected in atheromatous plaque (11). The rate of bacteremia caused by members of the mitis group of streptococci is comparable to that caused by group A or B streptococci (12).
The members of the mitis group of streptococci produce hydrogen peroxide (H2O2) (2, 13, 14), which is important in bacterial competition in microbial communities, such as oral biofilms (13, 14). S. sanguinis and S. gordonii produce H2O2 in quantities sufficient to reduce the growth of many oral bacteria, including the cariogenic bacterium Streptococcus mutans and several periodontal pathogens (13, 14).
Recently, we found that S. oralis induces the death of THP-1 macrophages in vitro by H2O2-mediated cytotoxicity (15). The cytotoxic effects of streptococcal H2O2 on THP-1 macrophages were also observed with S. sanguinis (15, 16), suggesting that H2O2 contributes to the pathogenicity of the mitis group of streptococci. In fact, the H2O2 produced by S. pneumoniae is a cytotoxic virulence factor (17–21). A recent study showed that pneumococcal H2O2 induces the activation of stress-related cellular signaling pathways (21). We also found that streptococcal H2O2 stimulates the production of cytokines, such as tumor necrosis factor alpha and interleukin-6 (15, 22). H2O2 is the simplest peroxide, a strong oxidizer, and a cytotoxic agent (23–25). Therefore, the H2O2 produced by oral streptococci has the potential to disturb host defense systems in multiple ways.
Since the cytotoxic mechanism of streptococcal H2O2 is not well understood, we investigated the morphological changes and functional impairment of intracellular organelles that occur during streptococcus H2O2-induced or exogenously added H2O2-induced macrophage death using the RAW 264 cell line. We found that exposure to S. oralis and H2O2 mediated damage to the lysosome with a reduction of the acidic environment in lysosomes. Lysosomes are involved in the intracellular degradation of various cellular components, such as proteins, nucleic acids, carbohydrates, and lipids (26, 27). Our findings suggest that the lysosomal dysfunction induced by streptococcal H2O2 is associated with macrophage death.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
S. oralis ATCC 35037, a type strain originally isolated from the human mouth (28), was obtained from the Japan Collection of Microorganisms at the RIKEN BioResource Center (Tsukuba, Japan). The pyruvate oxidase gene (spxB) deletion mutant (the spxB knockout [KO] mutant) was generated from wild-type (WT) strain S. oralis ATCC 35037 as described previously (15). The S. oralis WT strain produces 1 to 2 mM H2O2, whereas the spxB KO mutant produces less than 0.2 mM H2O2 (15). A mutant with an spxB revertant mutation (the spxB Rev mutant) (15), which produced H2O2 at a level similar to that produced by the WT, was also used.
S. mutans MT8148, a representative cariogenic strain of S. mutans serotype c (1, 29), was selected from the stock culture collection in the Department of Oral and Molecular Microbiology, Osaka University Graduate School of Dentistry (Suita-Osaka, Japan). S. mutans does not produce detectable H2O2 (1, 2) and was used as an H2O2-nonproducing streptococcus. These bacteria were cultured in brain heart infusion (BHI) broth (Becton Dickinson, Sparks, MD, USA).
Cell culture.
Mouse macrophage line RAW 264 (RBC0535) was obtained from the RIKEN BioResource Center. This cell line originates from ECA8510291 of the culture collections of Public Health England (London, United Kingdom; https://www.phe-culturecollections.org.uk/products/celllines/generalcell/detail.jsp?refId=85062803&collection=ecacc_gcIt). The cells were cultured in RPMI 1640 medium (Nacalai Tesque, Kyoto, Japan) supplemented with 5% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA), penicillin (100 U ml−1), and streptomycin (100 μg ml−1) at 37°C in a 5% CO2 atmosphere.
Macrophage death.
Streptococcal strains were grown to the exponential phase. Prior to infection, the culture medium with the RAW 264 cells was changed to fresh medium containing no antibiotics. RAW 264 cells (2 × 105 cells) in 24-well culture plates (Asahi Glass, Tokyo, Japan) were then infected with 2 × 107 or 4 × 107 CFU of viable streptococcal strains (multiplicity of infection [MOI] = 100 or 200) for 3 h. The cells were washed with phosphate-buffered saline (PBS; pH 7.2) to remove extracellular nonadherent bacteria and cultured for 21 h in fresh medium containing antibiotics (see Fig. S1 in the supplemental material). The cells were then stained with 0.2% trypan blue (Sigma-Aldrich, St. Louis, MO, USA) in PBS, and the numbers of viable and dead cells were counted using light microscopy (Nikon TMS-F; Nikon, Tokyo, Japan), as described previously (15, 16). Cell death induced by 1 mM H2O2 (Nacalai Tesque) was similarly examined.
Hydrogen peroxide measurement.
The H2O2 concentration in culture supernatants of RAW 264 cells infected with S. oralis was quantitatively determined using a hydrogen peroxide colorimetric detection kit (Enzo Life Science, Plymouth Meeting, PA, USA). RAW 264 cells were cultured in 24-well culture plates and were exposed to the S. oralis WT (MOI = 100 or 200) for 3 h. After washing steps with PBS, the cells were cultured for an additional 21 h (total, 24 h) in fresh medium containing antibiotics. The culture supernatants were collected at 1, 3, 6, and 24 h after infection and diluted 50-fold in PBS. The H2O2 concentration was determined according to the manufacturer's instructions. As a control, the H2O2 concentration in the culture supernatants of S. oralis cells grown in the same medium was also measured.
Acidic lysosome staining.
RAW 264 cells were cultured on cell desks (Cell Desk LF; Sumitomo Bakelite, Tokyo, Japan) in 24-well culture plates, exposed to the S. oralis WT (MOI = 200) or H2O2 (1 mM) for 3 h, washed with PBS, and cultured for an additional 3 h (total, 6 h) or 21 h (total, 24 h) in fresh medium containing antibiotics. The cells were stained with 50 nM LysoTracker red probe (Molecular Probes, Carlsbad, CA, USA) and SYBR green II (1:2,000 dilution; TaKaRa Bio, Otsu, Japan) in culture medium for 15 min, washed with PBS, and observed using a Carl Zeiss Axioplan 2 fluorescence microscope system (Carl Zeiss, Oberkochen, Germany). LysoTracker red is an acidotropic red fluorescent probe which accumulates on acidic lysosomes. SYBR green (green fluorescence) is a DNA-binding dye which stains the nuclei. LysoTracker-fluorescent areas of cells from four fluorescent images were analyzed using ImageJ software (http://rsbweb.nih.gov/ij/), followed by normalization to the cell number.
The acidic pH in lysosomes was also monitored with a lysosomotropic fluorochrome, acridine orange. The cells were exposed to the S. oralis WT or spxB KO mutant, S. mutans MT8148, or H2O2 and stained with 5 μM acridine orange (Wako Pure Chemicals, Osaka, Japan) for 15 min. The fluorescence of acridine orange was observed using fluorescence microscopy. When acridine orange accumulates in lysosomes, it emits a red fluorescence by excitation with blue light. In contrast, it emits green fluorescence in the cytosol and nucleus. The disappearance of the red fluorescence indicates a reduction of lysosome acidification (deacidification).
Immunofluorescent staining of LAMP-1 and cathepsin B.
RAW 264 cells were treated as described above, and the cells were fixed overnight with 10% formaldehyde at 4°C, followed by permeabilization with PBS containing 0.2% Triton X-100 for 1 h at room temperature. The cells were then blocked with StartingBlock buffer (Thermo Scientific, Rockford, IL, USA) for 1 h and reacted with an Alexa Fluor 488-conjugated rat anti-mouse lysosome-associated membrane protein-1 (LAMP-1) monoclonal antibody (1 μg ml−1; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and 4′,6-diamidino-2-phenylindole (DAPI; 1 μg ml−1; Dojindo Molecular Technologies, Kumamoto, Japan) in PBS containing 0.1% bovine serum albumin (BSA; Sigma-Aldrich) for 1 h. After washing with PBS, cell fluorescence was observed. Green fluorescence indicated the accumulation of LAMP-1. The fluorescent areas of the cells on the images indicating the accumulation of LAMP-1 were analyzed using ImageJ software and normalized to the cell number.
Lysosomal cathepsin B was also stained. RAW 264 cells exposed to the S. oralis WT or spxB KO mutant, S. mutans MT8148, or H2O2 were reacted with anti-cathepsin B antibodies (1 μg ml−1; R&D Systems, Minneapolis, MN, USA) in PBS containing 0.1% BSA and then stained with Alexa Fluor 594-conjugated anti-goat IgG antibodies (0.5 μg ml−1; Jackson ImmunoResearch, West Grove, PA, USA).
Effect of catalase and deferoxamine on viability and lysosomes of RAW 264 cells.
Prior to infection, RAW 264 cells were treated with 50 or 200 U ml−1 of catalase (an H2O2-decomposing enzyme; Sigma-Aldrich) or 0.5 or 2 mM deferoxamine mesylate (a bacterial siderophore that chelates iron [Fe]; Sigma-Aldrich) (30, 31). The cells were then infected with viable S. oralis strains (MOI = 200) for 3 h. The cells were washed with PBS and cultured in fresh medium containing catalase and antibiotics for 21 h. Viability was determined by trypan blue staining. For acidic lysosomal staining, the cells were stained with LysoTracker after 3 h of infection. The cells were also immunostained using LAMP-1 antibodies.
Effect of cathepsin B inhibitor on detachment of dead cells.
Prior to treatment, RAW 264 cells were incubated with 20 μM CA-074 Me (a membrane-permeant methyl ester of CA-074; Peptide Institute, Osaka, Japan), a specific inhibitor of cathepsin B (32), for 1 h. Then, the cells were exposed to viable S. oralis WT cells (MOI = 100 or 200) or 1 mM H2O2 for 3 h in the presence of CA-074 Me. The cells were washed with PBS and cultured in fresh medium containing antibiotics and CA-074 Me (10 μM) for 21 h. The cells were stained with trypan blue, and the number of adherent, viable, and dead cells was counted (see Fig. S2 in the supplemental material).
For fluorescence microscopy observation, RAW 264 cells cultured on the cell desk were fixed with 10% formaldehyde, followed by permeabilization with 0.2% Triton X-100. Then, DNA and actin filaments were labeled with SYBR green II (1:2,000 dilution) and Alexa Fluor 594-conjugated phalloidin (1:200 dilution; Molecular Probes, Eugene, OR, USA) in PBS for 15 min. After washing with PBS, the cell fluorescence was observed.
Statistical analysis.
Statistical analyses were performed using QuickCalcs software (GraphPad Software, La Jolla, CA, USA). Differences were examined for statistical significance using an independent Student's t test, with a P value of <0.05 indicating statistical significance.
RESULTS
S. oralis-induced death of RAW 264 macrophages.
We previously reported that infection with members of the mitis group of streptococci that inhabit the oral cavity, such as S. oralis and S. sanguinis, induces the death of THP-1 macrophages and several epithelial cell lines, with streptococcal H2O2 contributing to this process (15, 16, 22). In the present study, the RAW 264 macrophage line was infected with viable S. oralis ATCC 35037 at an MOI of 100 or 200 for 3 h in antibiotic-free medium. The cells were then washed with PBS to remove extracellular nonadherent bacteria (see Fig. S1 in the supplemental material). At this point, no change in cellular morphology was observed and most cells appeared to be viable. However, at 24 h after infection, macrophage death was apparent (Fig. 1; also see Fig. S3 in the supplemental material). To elucidate the contribution of H2O2, the S. oralis spxB KO strain, which is deficient in H2O2 production, was subjected to the experiments. The spxB KO mutant showed reduced cytotoxicity, even at an MOI of 200. The ability of a revertant mutant, the spxB Rev mutant, to induce the death of RAW 264 macrophages was comparable to that of the WT (data not shown). S. mutans, a cariogenic oral streptococcus that does not produce H2O2, showed no cytotoxicity to RAW 264 cells. Furthermore, treatment with H2O2 (1 mM) alone was sufficient to induce cell death (Fig. 1).
FIG 1.
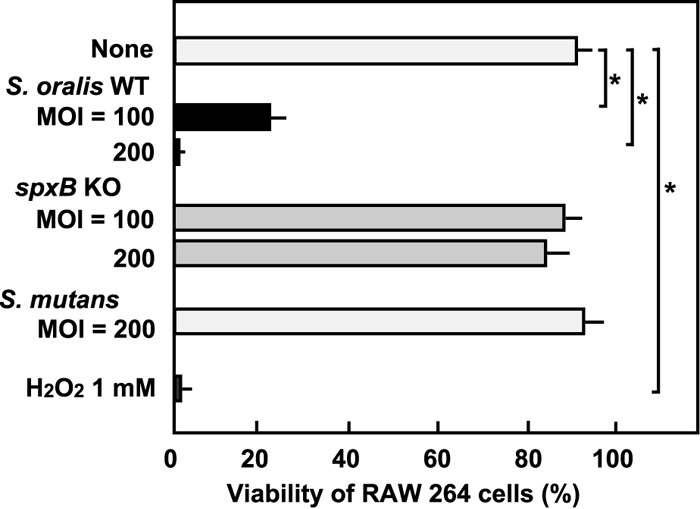
Death of RAW 264 macrophages. RAW 264 cells were exposed to the S. oralis WT or spxB KO mutant, S. mutans MT8148, or H2O2 for 3 h. Following washing steps, the cells were cultured in fresh medium containing antibiotics for 21 h (see also Fig. S1 in the supplemental material). Viable cells were counted after trypan blue staining. Data are shown as the mean ± SD for triplicate samples. *, P < 0.05 compared with the untreated control (None). The results of live/dead fluorescent staining are shown in Fig. S3 in the supplemental material.
Since mammalian cells produce H2O2-decomposing enzymes such as catalase, we determined the H2O2 concentration in the culture supernatants of RAW 264 cells infected with the S. oralis WT. In the infected culture, the H2O2 concentration gradually increased. At 3 h after infection at an MOI of 200, the concentration reached approximately 0.2 mM. The concentration was sufficient to induce the death of RAW 264 cells (Fig. 2A). S. oralis alone cultured in the same medium produced approximately 0.4 mM H2O2 at 3 h (Fig. 2B).
FIG 2.
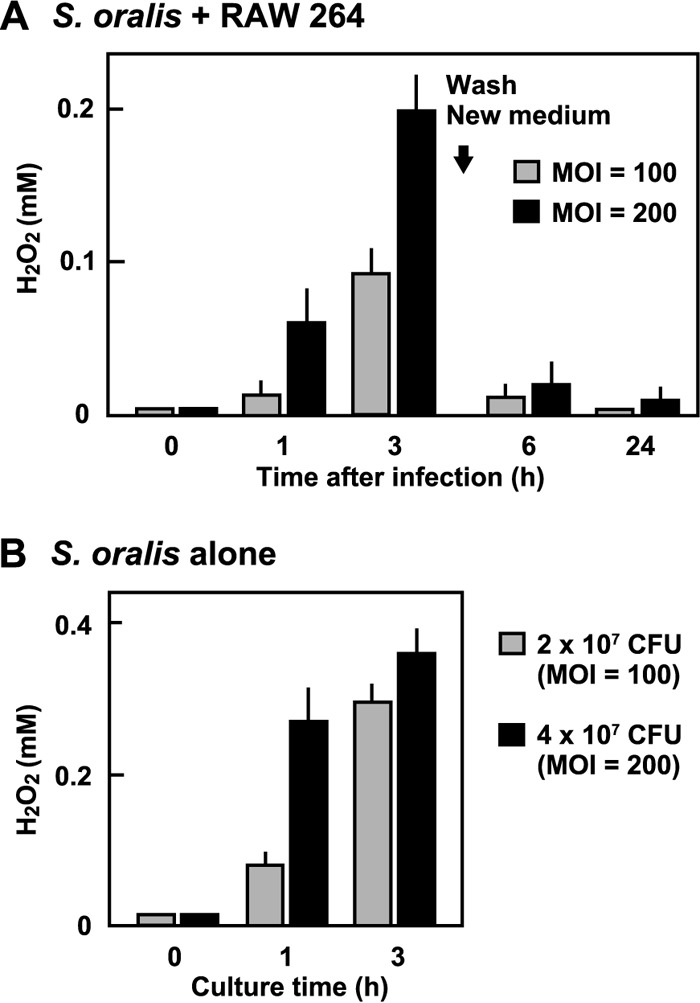
H2O2 concentration in culture medium of RAW 264 cells infected with S. oralis. (A) RAW 264 cells were infected with the S. oralis WT for 3 h. The cells were washed with PBS and cultured for an additional 21 h (total, 24 h) in fresh medium containing antibiotics. The H2O2 concentration of the culture supernatants was determined using a hydrogen peroxide colorimetric detection kit. (B) The H2O2 concentration of the culture supernatants of S. oralis cultured in the same medium was also determined.
Fluorescent study of acidic pH of lysosomes.
During the present study, we found that H2O2 damaged some intracellular organelles, including lysosomes. To visualize changes in lysosomes during cell death, the cells were stained with LysoTracker, an acidotropic fluorescent probe (Fig. 3). The staining revealed that the acidic pH of lysosomes was gradually reduced by the infection with the S. oralis WT by 3 h (Fig. 3, left), suggesting disappearance of the proton gradient over the lysosomal membrane. Exposure to H2O2 also induced similar pH increases, i.e., deacidification in the lysosomes (Fig. 3, right). The deacidification was detectable at 1 h, and the process seemed to occur rapidly preceding cell death.
FIG 3.
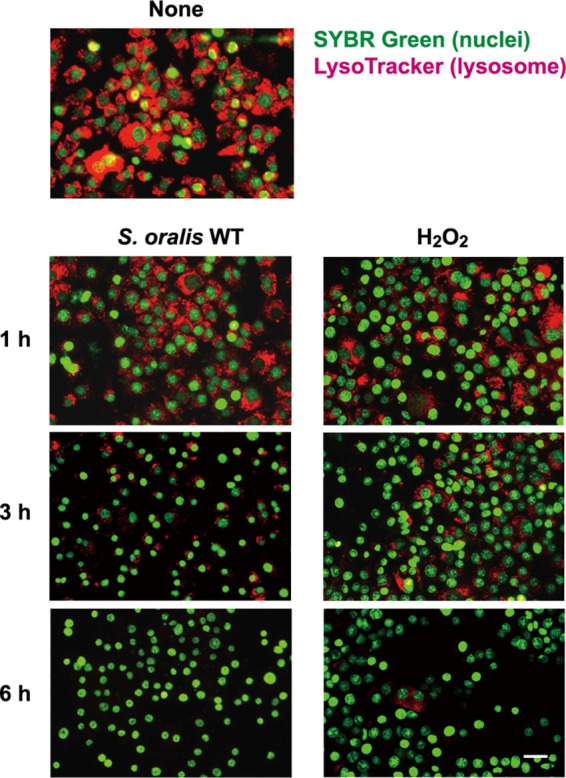
S. oralis and H2O2 mediate lysosomal damage. RAW 264 cells were exposed to the S. oralis WT or H2O2 for 3 h. Then, the cells were cultured for an additional 3 h (total, 6 h) in fresh medium containing antibiotics. At 1, 3, and 6 h after exposure, the cells were stained with LysoTracker red and SYBR green II. Lysosomal deacidification was monitored using LysoTracker red (red), which accumulates in acidic organelles. Bar = 20 μm.
To estimate the contribution of streptococcal H2O2 to the deacidification, the S. oralis spxB KO mutant and S. mutans were employed in the infection assay. Infection with the spxB KO mutant or S. mutans had little or no effect on LysoTracker fluorescence (Fig. 4A, top), suggesting that the deacidification in the lysosomes was related to the H2O2 produced by the streptococcus. Quantitative analyses of fluorescent images confirmed the deacidification in lysosomes of macrophages exposed to the S. oralis WT or H2O2 (Fig. 4B). The reduction in the acidic pH of the lysosomes was further examined by acridine orange staining. The dye emits red fluorescence in the acidic environment of intact lysosomes, and disappearance of the red fluorescence indicates an increase in the lysosomal pH. The staining revealed the deacidification in the lysosomes of the RAW 264 cells exposed to the S. oralis WT or H2O2 (Fig. 4A, bottom). In addition, infection with the spxB Rev strain also induced deacidification (data not shown). Thus, these results demonstrate that exposure to the S. oralis WT or H2O2 mediates a reduction of the acidic environment in lysosomes.
FIG 4.

The H2O2 produced by S. oralis induces lysosomal deacidification. (A) RAW 264 cells were exposed to the S. oralis WT or spxB KO mutant, S. mutans MT8148, or H2O2 for 3 h. (Top) The cells were stained with LysoTracker red and SYBR green II or acridine orange. LysoTracker red accumulates in acidic organelles. (Bottom) The acidic environment in lysosomes was also monitored by acridine orange staining. Acridine orange emits red fluorescence in acidic environments, and thus, the disappearance of red fluorescence reflects lysosome deacidification. Bar = 20 μm. (B) Measurements of the LysoTracker-stained fluorescent areas were conducted using ImageJ software. The average fluorescence area of control cells (None) was set to 100%. The results are shown as the mean ± SD for four samples. *, P < 0.05 compared with the untreated control (None).
Immunofluorescent staining of LAMP-1 and cathepsin B.
The deacidification described above was suspected to be associated with lysosome impairment. To monitor the lysosomal integrity in RAW 264 cells exposed to the S. oralis WT or H2O2, lysosome-specific protein LAMP-1 was visualized by immunofluorescent staining (Fig. 5). The number of LAMP-1-positive lysosomes was reduced in a time-dependent manner. Most cells were dead at 24 h; however, a small and weak LAMP-1 fluorescence was still visible, suggesting that some lysosomal debris was associated with the dead cells. In addition, no or little change in LAMP-1 fluorescence was observed in cells infected with the S. oralis spxB KO mutant or S. mutans (Fig. 6A, top). Further, quantitative analyses of fluorescent images confirmed a significant decrease in the amount of LAMP-1 in macrophages exposed to the S. oralis WT or H2O2 (Fig. 6B).
FIG 5.
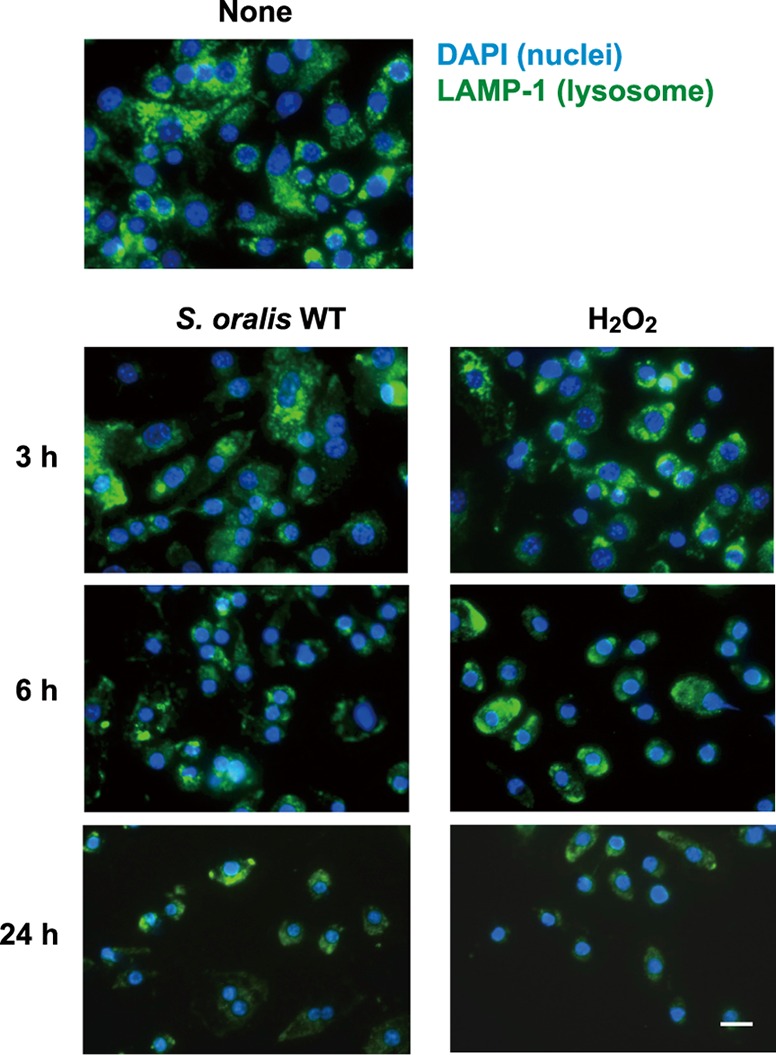
S. oralis and H2O2 mediate lysosomal destruction. RAW 264 cells were exposed to the S. oralis WT or H2O2 for 3 h and cultured for an additional 3 and 21 h (total, 6 and 24 h, respectively) in fresh medium containing antibiotics. At the indicated times after exposure, nuclei and LAMP-1 were fluorescently labeled with DAPI and Alexa Fluor 488-conjugated anti-LAMP-1 monoclonal antibody, respectively. Bar = 10 μm.
FIG 6.

The H2O2 produced by S. oralis induces lysosomal destruction. (A) RAW 264 cells were exposed to the S. oralis WT or spxB KO mutant, S. mutans MT8148, or H2O2 for 3 h. The cells were then washed and cultured for an additional 3 h in fresh medium containing antibiotics. (Top) LAMP-1 and DNA were labeled with Alexa Fluor 488-conjugated anti-LAMP-1 monoclonal antibody and DAPI, respectively. (Bottom) Cathepsin B was also labeled with Alexa Fluor 594-conjugated anti-goat IgG. Bar = 10 μm. (B) Measurements of the LAMP-1-positive areas were conducted using ImageJ software. The average fluorescence area for untreated control cells (None) was set to 100%. The results are shown as the mean ± SD for four samples. *, P < 0.05 compared with the control (None).
Lysosome destruction was also demonstrated by immunofluorescent staining of cathepsin B, a lysosomal cysteine protease (Fig. 6A, bottom). Decreased cathepsin B fluorescence was observed in cells exposed to the S. oralis WT or H2O2, confirming the impairment of lysosomes. Again, no significant change in cathepsin B fluorescence was observed in cells infected with the S. oralis spxB KO mutant or S. mutans. The destruction of lysosomes was observed in spxB Rev mutant-infected RAW 264 cells (data not shown). These results indicate that streptococcal H2O2 impairs lysosomes.
Effect of catalase and deferoxamine on cell death and lysosomes in S. oralis-infected RAW 264 cells.
To confirm the contribution of streptococcal H2O2 to cell death and lysosomal impairment in RAW 264 cells infected with S. oralis, we investigated the effect of catalase, an H2O2-decomposing enzyme, on S. oralis-induced cell death. Exogenously added catalase was shown to reduce cell death in macrophages infected with the S. oralis WT (Fig. 7A). In the presence of catalase, the deacidification and degradation of lysosomes were also reduced (Fig. 7B).
FIG 7.
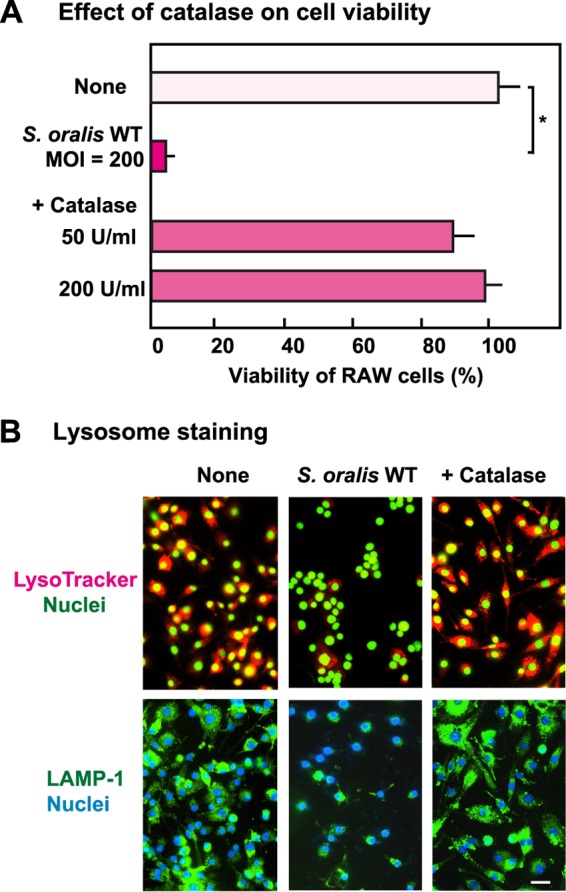
Effect of catalase on lysosomal damage in S. oralis-infected macrophages. RAW 264 cells pretreated with 50 or 200 U ml−1 of catalase were infected with viable S. oralis WT for 3 h. The cells were washed with PBS and cultured in fresh medium containing catalase and antibiotics for 21 h. (A) Viability was determined by trypan blue staining. *, P < 0.05 compared with the untreated control (None). (B) The cells were stained with LysoTracker after 3 h of infection (top) and were also immunostained using LAMP-1 antibodies after 6 h of infection (bottom). Bar = 10 μm.
Deferoxamine has been reported to inhibit the generation of hydroxyl radicals from H2O2 within lysosomes (30, 31). As shown in Fig. 8A, deferoxamine reduced the proportion of RAW 264 cells infected with the S. oralis WT that died. Moreover, LysoTracker and LAMP-1 staining showed reduced lysosomal damage in the presence of deferoxamine (Fig. 8B).
FIG 8.
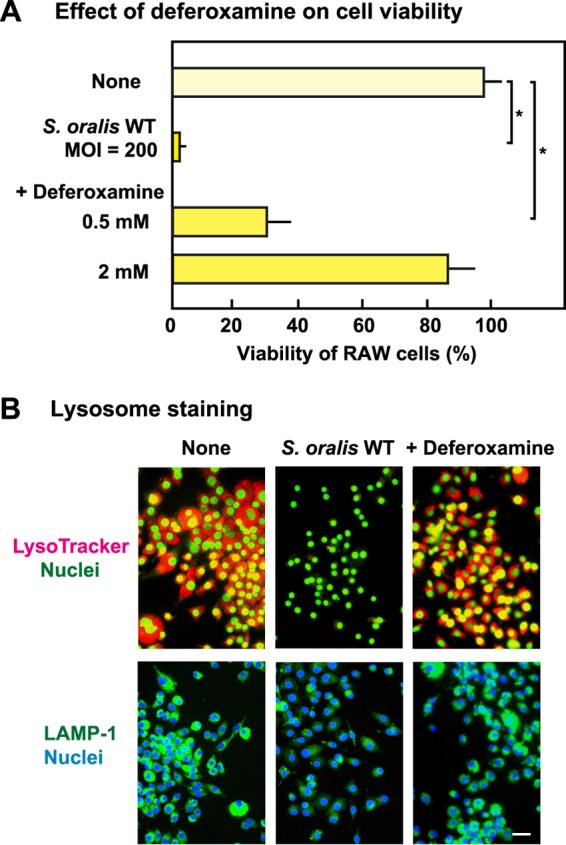
Effect of deferoxamine on lysosomal damage in S. oralis-infected macrophages. RAW 264 cells were treated with 0.5 or 2 mM deferoxamine and then infected with viable S. oralis strains for 3 h. The cells were washed with PBS and cultured in fresh medium containing antibiotics for 21 h. (A) Viability was determined by trypan blue staining. *, P < 0.05 compared with the untreated control (None). (B) The cells were stained with LysoTracker after 3 h of infection (top) and were also immunostained using LAMP-1 antibodies after 6 h of infection (bottom). Bar = 10 μm.
Effect of a cathepsin B inhibitor on cell death and detachment of dead cells.
The results presented above suggest that streptococcal H2O2 triggers the destruction of lysosomes. Proteases released from damaged lysosomes are involved in cell death (26, 27, 33, 34). Therefore, we investigated the effect of a cathepsin B inhibitor, CA-074 Me, on cell death (see Fig. S2 in the supplemental material). Even in the presence of CA-074 Me, both the S. oralis WT and H2O2 induced the death of RAW 264 cells, whereas only a modest yet statistically significant reduction in the level of death was observed in cells exposed to either S. oralis at an MOI of 200 or 1 mM H2O2. (Fig. 9A). However, when dead but adherent cells (see Fig. S2 in the supplemental material) were counted, the number of adherent cells in the presence of CA-074 Me was significantly higher than that for the control (Fig. 9B).
FIG 9.

Effect of a cathepsin B inhibitor on cell death and detachment of dead cells. RAW 264 cells pretreated with the cathepsin B inhibitor CA-074 Me (20 μM) were exposed to the S. oralis WT (MOI = 100 or 200) or H2O2 (1 mM) for 3 h. The cells were washed and cultured for an additional 21 h (total, 24 h) in fresh medium containing CA-074 Me (10 μM) and antibiotics (see also Fig. S2 in the supplemental material). (A) The viable cells were counted after trypan blue staining. (B) The adherent cells (both viable and dead) were also counted. Data are shown as the mean ± SD for triplicate samples. *, P < 0.05 compared with the untreated control (None, No inhibitor). (C) The cells were stained with SYBR green II (green; nuclei) together with Alexa Fluor 594-conjugated phalloidin (red; actin filaments). Bar = 10 μm.
The inhibitory effect of CA-074 Me on the detachment of dead RAW 264 cells was confirmed by fluorescence microscopy (Fig. 9C). Without the inhibitor, more than half of the cells detached from the culture plates. Fluorescent staining using Alexa Fluor 594-phalloidin showed that most of the actin filaments of the cells disappeared (Fig. 9C, top). In the presence of CA-074 Me, larger numbers of cells were still attached to the culture plates, and certain actin filaments were still visible (Fig. 9C, bottom). Thus, the cathepsin B released from the injured lysosomes was involved in the detachment of dead macrophages (Fig. 10); however, the cell death process itself seemed to be independent of cathepsin B.
FIG 10.
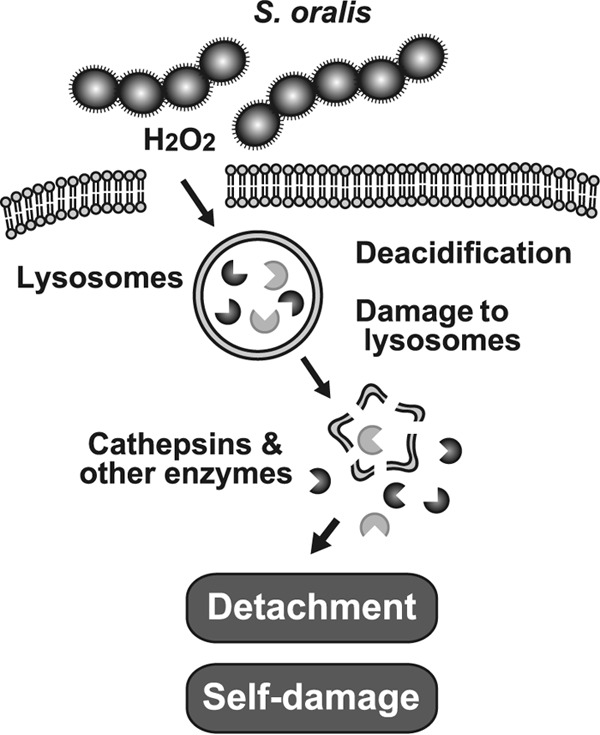
Proposed model illustrating the role of H2O2 in S. oralis-induced macrophage death. H2O2 causes lysosomal deacidification and the destruction of lysosomal integrity via Fenton's reaction, followed by leakage of cathepsins and other lysosomal hydrolytic enzymes. These cytotoxic enzymes degrade cellular components and induce self-damage in S. oralis-infected cells. Cathepsins are implicated in the detachment of the dead cells from the culture plates.
DISCUSSION
Macrophages and monocytes are major contributors to host immune responses against bacterial infections (35, 36). We previously reported that members of the mitis group of streptococci that inhabit the oral cavity induce the death of THP-1 macrophages (15, 16), and their cytotoxicity is mediated by streptococcal H2O2 (15, 22). The cytotoxicity and tissue-damaging effects of streptococcal H2O2 are factors of streptococcal pathogenicity. The H2O2 enables bacteria to escape from macrophage phagocytosis and thus contributes to the onset of bacteremia and infectious endocarditis. Regarding S. pneumoniae, several investigations revealed that the H2O2 produced by S. pneumoniae is one of the virulence factors of this species (17–21). H2O2 contributes to pneumococcal lung and blood infections in experimental animals (17, 19). Another study showed that pneumococcal H2O2 induces microglial and neuronal apoptosis and enhances the severity of experimental meningitis (18). Bioluminescent imaging in infected mice showed that H2O2 contributes to prolonged nasopharyngeal colonization by S. pneumoniae (19). It is therefore conceivable that the H2O2 produced by oral streptococci contributes to their virulence.
In the present study, the effect of streptococcal H2O2 on lysosomal integrity was investigated using the RAW 264 macrophage line. We found that the S. oralis WT and H2O2 mediated the damage to the lysosomes with a reduction of the acidic environment of the lysosomes. Under normal conditions, the pH within lysosomes is maintained at about 5 (26, 27). Deacidification occurred relatively quickly, within 3 h after exposure to the S. oralis WT or H2O2. At this point, most cells were still viable (see Fig. S3 in the supplemental material), and lysosomal deacidification was possibly related to the macrophage death process.
Reactive oxygen species (ROS), including H2O2, are generated from mammalian cells as by-products of mitochondrial respiration and metabolite reactions in response to various stimuli, such as heat shock, cytokine stimulation, and infection by pathogens (23–25, 37–39). Macrophages produce large amounts of ROS when activated by pathogens and proinflammatory cytokines (23, 24, 35–37). The chemical properties of H2O2 have been widely studied, and it is accepted that the cytotoxicity of H2O2 is due to oxygen formation, lipid peroxidation, and damage to proteins and nucleic acids (24, 25). In general, a high concentration of H2O2 (more than 100 μM) induces cell death, whereas a low concentration of H2O2 promotes cell proliferation (23, 37, 38, 40–42). We also found an increased production of proinflammatory cytokines from cells exposed to the H2O2-producing oral streptococci (15, 16, 22).
Early studies reported that the ROS released from mitochondria mediates lysosomal impairment and induces cell death (30, 31, 41, 42). Lysosomes are organelles filled with cytotoxic hydrolytic enzymes, including proteases, and their dysfunction is considered to induce cell death (26, 27). Therefore, we examined the effect of streptococcal H2O2 on lysosomal integrity. LysoTracker and acridine orange fluorescent staining demonstrated that the streptococcal H2O2 elicited a reduction of the acidic lysosomal environment within 3 h (Fig. 3 and 4). Immunofluorescent studies of LAMP-1 and cathepsin B revealed the destruction of lysosomes (Fig. 5 and 6). The protective effect of catalase on S. oralis-induced cell death also demonstrated a direct contribution of streptococcal H2O2 to the damage to the lysosome and cell death (Fig. 7). It is noted that this deacidification occurred relatively quickly and preceded cell death (Fig. 3; see also Fig. S3 in the supplemental material). Since oral streptococci produce carboxylic acids, such as formic, acetic, and lactic acids, as metabolic products (1, 2), it can be assumed that these acids may participate in lysosomal impairment. However, both the S. oralis spxB KO mutant and S. mutans showed no or little effect on lysosomes (Fig. 4 and 6); thus, the contribution of these acids is negligible.
Lysosomes are linked with major pathways of cell death, i.e., apoptosis, necrosis, and pyroptosis (26, 27, 33). Impairment of lysosomes induces lysosomal membrane permeability, followed by leakage of the cytotoxic content, including hydrolytic enzymes, such as cathepsins (cathepsins B, C, and D and other cathepsins), lipases, nucleases, and glycosidases (26, 27, 33, 41, 43, 44). Several studies demonstrated that the cathepsins released from the damaged lysosomes trigger lysosome-associated cell death (26, 33, 34, 41, 43–45). It should be noted that osmotic lysis and various agents which induce lysosomal membrane permeabilization are reported to cause cell death (33, 44, 46–48). Selective lysosome disruption with l-leucyl-l-leucine methyl ester (LeuLeuOMe) induces apoptosis (33, 47, 48). LeuLeuOMe accumulates in lysosomes, followed by conversion to a membranolytic form. It finally destroys lysosomal membrane integrity, resulting in cell death (33, 47, 48).
Regarding the lysosome-associated cell death induced by oxidative stress, previous studies revealed that Fe ions within lysosomes catalyze the peroxidation of H2O2 through Fenton's reaction and produce highly reactive oxygen radicals (30, 31, 42, 49, 50). The resultant oxygen radicals damage and destabilize lysosomal membranes (30, 31, 42, 49, 50). Thus, Fe ions within lysosomes play an important role in H2O2-induced cell death (30, 31, 42, 49, 50). It was reported that deferoxamine inhibits the production of oxygen radicals within lysosomes by chelating Fe ions and thus reduces the level of H2O2-induced cell death (30, 31). In the present study, deferoxamine was shown to reduce S. oralis-induced macrophage death (Fig. 8), suggesting that Fe ions within lysosomes contribute to cell death. Taken together, lysosomal impairment is considered to be involved in macrophage death. At present, it is uncertain whether the lysosome-associated cell death results from apoptosis, necrosis, or pyroptosis (26, 33, 34, 41, 48, 50). It is possible that the streptococcal H2O2-induced macrophage death is mediated by multiple cell death pathways.
Hydrolyzing enzymes, including cathepsins released from lysosomes, could trigger a forward loop promoting lysosome rupture and cell death (26, 27, 34). It was difficult to investigate all the contributions of the many lysosome-hydrolyzing enzymes regarding streptococcal H2O2-mediated cell death; however, we investigated the possible contribution of cathepsin B using CA-074 Me, a specific cathepsin B inhibitor (32). Contrary to our expectations, the inhibitor did not inhibit cell death (Fig. 9). At present, we can only speculate that multiple lysosomal cytotoxic hydrolyzing enzymes, including other proteases, could be involved in streptococcal H2O2-induced cell death.
However, CA-074 Me reduced the detachment of dead cells (Fig. 9). Similar results were obtained with E-64, another cysteine protease inhibitor (32). These findings suggest that cathepsin B and other cathepsins contribute to the detachment of dead cells. In this regard, early studies reported that cathepsins are major proteases involved in extracellular matrix degradation (51). Thus, cathepsin-mediated degradation of matrix proteins is possibly associated with the detachment of dead cells.
In summary, this study revealed a novel finding that the H2O2 produced by oral streptococci induces the rapid deacidification and destruction of lysosomes (Fig. 10). Subsequently, the released lysosomal cathepsins participate in the detachment of dead macrophages (Fig. 10). Lysosomal impairment enables bacteria to escape from macrophage phagocytosis. Since our preliminary study suggested that H2O2 damages other cellular organelles, including mitochondria, impairment of these organelles should be further investigated.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by JSPS KAKENHI grants (grants 26463112, 26462779, and 15H05012) from the Japan Society for the Promotion of Science.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00134-16.
REFERENCES
- 1.Hamada S, Slade HD. 1980. Biology, immunology, and cariogenicity of Streptococcus mutans. Microbiol Rev 44:331–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coykendall AL. 1989. Classification and identification of the viridans streptococci. Clin Microbiol Rev 2:315–328. doi: 10.1128/ClinMicrobiolRev2.3.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nobbs AH, Lamont RJ, Jenkinson HF. 2009. Streptococcus adherence and colonization. Microbiol Mol Biol Rev 73:407–450. doi: 10.1128/MMBR.00014-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kilian M, Mikkeksen L, Henrichsen J. 1989. Taxonomic study of viridans streptococci: description of Streptococcus gordonii sp. nov. and emended descriptions of Streptococcus sanguis (White and Niven 1946), Streptococcus oralis (Bridge and Sneath 1982), and Streptococcus mitis (Andrewes and Horder 1906). Int J Syst Evol Microbiol 39:471–484. [Google Scholar]
- 5.Kawamura Y, Hou X-G, Sultana F, Miura H, Ezaki T. 1995. Determination of 16S rRNA sequences of Streptococcus mitis and Streptococcus gordonii and phylogenetic relationships among members of the genus streptococcus. Int J Syst Bacteriol 45:406–408. doi: 10.1099/00207713-45-2-406. [DOI] [PubMed] [Google Scholar]
- 6.Mitchell J. 2011. Streptococcus mitis: walking the line between commensalism and pathogenesis. Mol Oral Microbiol 26:89–98. doi: 10.1111/j.2041-1014.2010.00601.x. [DOI] [PubMed] [Google Scholar]
- 7.Douglas CW, Heath J, Hampton KK, Preston FE. 1993. Identity of viridans streptococci isolated from cases of infective endocarditis. J Med Microbiol 39:179–182. doi: 10.1099/00222615-39-3-179. [DOI] [PubMed] [Google Scholar]
- 8.Dyson C, Barnes RA, Harrison GAJ. 1999. Infective endocarditis: an epidemiological review of 128 episodes. J Infect 38:87–93. doi: 10.1016/S0163-4453(99)90074-9. [DOI] [PubMed] [Google Scholar]
- 9.Moreillon P, Que YA. 2004. Infective endocarditis. Lancet 363:139–149. doi: 10.1016/S0140-6736(03)15266-X. [DOI] [PubMed] [Google Scholar]
- 10.Parahitiyawa NB, Jin LJ, Leung WK, Yam WC, Samaranayake LP. 2009. Microbiology of odontogenic bacteremia: beyond endocarditis. Clin Microbiol Rev 22:46–64. doi: 10.1128/CMR.00028-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiu B. 1999. Multiple infections in carotid atherosclerotic plaques. Am Heart J 138:S534–S536. doi: 10.1016/S0002-8703(99)70294-2. [DOI] [PubMed] [Google Scholar]
- 12.Public Health England. 2014. Pyogenic and non-pyogenic streptococcal bacteraemia in England, Wales and Northern Ireland: 2013. Health Protection Reports, vol. 8, no. 44. Public Health England, London, United Kingdom. [Google Scholar]
- 13.Kreth J, Zhang Y, Herzberg MC. 2008. Streptococcal antagonism in oral biofilms: Streptococcus sanguinis and Streptococcus gordonii interference with Streptococcus mutans. J Bacteriol 190:4632–4640. doi: 10.1128/JB.00276-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu L, Kreth J. 2012. The role of hydrogen peroxide in environmental adaptation of oral microbial communities. Oxid Med Cell Longev Article 2012:717843. doi: 10.1155/2012/717843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okahashi N, Nakata M, Sumitomo T, Terao Y, Kawabata S. 2013. Hydrogen peroxide produced by oral streptococci induces macrophage cell death. PLoS One 8:e62563. doi: 10.1371/journal.pone.0062563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okahashi N, Okinaga T, Sakurai A, Terao Y, Nakata M, Nakashima K, Shintani S, Kawabata S, Ooshima T, Nishihara T. 2011. Streptococcus sanguinis induces foam cell formation and cell death of macrophages in association with production of reactive oxygen species. FEMS Microbiol Lett 323:164–170. doi: 10.1111/j.1574-6968.2011.02375.x. [DOI] [PubMed] [Google Scholar]
- 17.Spellerberg B, Cundell DR, Sandros J, Pearce BJ, Idanpaan-Heikkila I, Rosenow C, Masure HR. 1996. Pyruvate oxidase, as a determinant of virulence in Streptococcus pneumoniae. Mol Microbiol 4:803–813. doi: 10.1046/j.1365-2958.1996.425954.x. [DOI] [PubMed] [Google Scholar]
- 18.Braun JS, Sublett JE, Freyer D, Mitchell TJ, Cleveland JL, Tuomanen EI, Weber JR. 2002. Pneumococcal pneumolysin and H2O2 mediate brain cell apoptosis during meningitis. J Clin Invest 109:19–27. doi: 10.1172/JCI12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orihuela CJ, Gao G, Francis KP, Yu J, Tuomanen EI. 2004. Tissue-specific contribution of pneumococcal virulence factors to pathogenesis. J Infect Dis 190:1661–1669. doi: 10.1086/424596. [DOI] [PubMed] [Google Scholar]
- 20.Rai P, Parrish M, Tay IJJ, Li N, Ackerman S, He F, Kwang J, Chow V, Engelward BP. 2015. Streptococcus pneumoniae secretes hydrogen peroxide leading to DNA damage and apoptosis in lung cells. Proc Natl Acad Sci U S A 112:E3421–30. doi: 10.1073/pnas.1424144112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loose M, Hudel M, Zimmer K-P, Garcia E, Hammerschmidt S, Lucas R, Chakraborty T, Pillich H. 2015. Pneumococcal hydrogen peroxide-induced stress signaling regulates inflammatory genes. J Infect Dis 211:306–316. doi: 10.1093/infdis/jiu428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okahashi N, Sumitomo T, Nakata M, Sakurai A, Kuwata H, Kawabata S. 2014. Hydrogen peroxide contributes to the epithelial cell death induced by the oral mitis group of streptococci. PLoS One 9:e88136. doi: 10.1371/journal.pone.0088136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finkel T, Holbrook NJ. 2000. Oxidants, oxidative stress and the biology of aging. Nature 408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 24.Watt BE, Proudfoot AT, Vale JA. 2004. Hydrogen peroxide poisoning. Toxicol Rev 23:51–57. doi: 10.2165/00139709-200423010-00006. [DOI] [PubMed] [Google Scholar]
- 25.Bergamini CM, Gambetti S, Dondi A, Cervellati C. 2004. Oxygen, reactive oxygen species and tissue damage. Curr Pharm Des 10:1611–1626. doi: 10.2174/1381612043384664. [DOI] [PubMed] [Google Scholar]
- 26.Turk B, Turk V. 2009. Lysosomes as “suicide bags” in cell death: myth or reality? J Biol Chem 284:21783–21787. doi: 10.1074/jbc.R109.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mrschtik M, Ryan KM. 2015. Lysosomal proteins in cell death and autophagy. FEBS J 282:1858–1870. doi: 10.1111/febs.13253. [DOI] [PubMed] [Google Scholar]
- 28.Bridge PD, Sneath PH. 1982. Streptococcus gallinarum sp. nov. and Streptococcus oralis sp. nov. Int J Syst Bacteriol 32:410–415. doi: 10.1099/00207713-32-4-410. [DOI] [Google Scholar]
- 29.Okahashi N, Asakawa H, Koga T, Masuda N, Hamada S. 1984. Clinical isolates of Streptococcus mutans serotype c with altered colony morphology due to fructan synthesis. Infect Immun 44:617–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zdolsek J, Zhang H, Roberg K, Brunk U. 1993. H2O2-mediated damage to lysosomal membranes of J-774 cells. Free Radic Res Commun 18:71–85. doi: 10.3109/10715769309147344. [DOI] [PubMed] [Google Scholar]
- 31.Yu Z, Persson HL, Eaton JW, Brunk UT. 2003. Intralysosomal iron: a major determinant of oxidant-induced cell death. Free Radic Biol Med 34:1243–1252. doi: 10.1016/S0891-5849(03)00109-6. [DOI] [PubMed] [Google Scholar]
- 32.Katunuma N. 2011. Structure-based development of specific inhibitors for individual cathepsins and their medical applications. Proc Jpn Acad Ser B Phys Biol Sci 87:29–39. doi: 10.2183/pjab.87.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cirman T, Oresic K, Mazovec GD, Turk V, Reed JC, Myers RM, Salvesen GS, Turk B. 2004. Selective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of Bid by multiple papain-like lysosomal cathepsins. J Biol Chem 279:3578–3587. doi: 10.1074/jbc.M308347200. [DOI] [PubMed] [Google Scholar]
- 34.Jacobson LS, Lima H Jr, Goldberg MF, Gocheva V, Tsiperson V, Sutterwala FS, Joyce JA, Gapp BV, Blomen VA, Chandran K, Brummelkamp TR, Diaz-Griffero F, Brojatsch J. 2013. Cathepsin-mediated necrosis controls the adaptive immune response by Th2 (T helper type 2)-associated adjuvants. J Biol Chem 288:7481–7491. doi: 10.1074/jbc.M112.400655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moore KJ, Tabas I. 2011. Macrophages in the pathogenesis of atherosclerosis. Cell 145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wynn TA, Chawla A, Pollard JW. 2013. Macrophage biology in development, homeostasis and disease. Nature 496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pelletier M, Lepow TS, Billingham LK, Murphy MP, Siegel RM. 2012. New tricks from an old dog: mitochondrial redox signaling in cellular inflammation. Semin Immunol 24:384–392. doi: 10.1016/j.smim.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giorgio M, Trinei M, Migliaccio E, Pelicci PG. 2007. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol 8:722–728. doi: 10.1038/nrm2240. [DOI] [PubMed] [Google Scholar]
- 39.Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. 2007. Mitochondria, oxidative stress and cell death. Apoptosis 12:913–922. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- 40.Ghibelli L, Nosseri C, Coppola S, Maresca V, Dini L. 1995. The increase in H2O2-induced apoptosis by ADP-ribosylation inhibitors is related to cell blebbing. Exp Cell Res 221:470–477. doi: 10.1006/excr.1995.1398. [DOI] [PubMed] [Google Scholar]
- 41.Brunk UT, Svensson I. 1999. Oxidative stress, growth factor starvation and Fas activation may all cause apoptosis through lysosomal leak. Redox Rep 4:3–11. doi: 10.1179/135100099101534675. [DOI] [PubMed] [Google Scholar]
- 42.Chandra J, Samali A, Orrenius S. 2000. Triggering and modulation of apoptosis by oxidative stress. Free Radic Biol Med 29:323–333. doi: 10.1016/S0891-5849(00)00302-6. [DOI] [PubMed] [Google Scholar]
- 43.Lockshin RA, Zakeri Z. 2002. Caspase-independent cell deaths. Curr Opin Cell Biol 14:727–733. doi: 10.1016/S0955-0674(02)00383-6. [DOI] [PubMed] [Google Scholar]
- 44.Tardy C, Codogno P, Autefage H, Levade T, Andrieu-Abadie N. 2006. Lysosomes and lysosomal proteins in cancer cell death (new players of an old struggle). Biochim Biophys Acta 1765:101–125. doi: 10.1016/j.bbcan.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 45.Hoegen T, Tremel N, Klein M, Angele B, Wagner H, Kirschning C, Pfister H-W, Fontana A, Hammerschmidt S, Koedel U. 2011. The NLRP3 inflammasome contributes to brain injury in pneumococcal meningitis and is activated through ATP-dependent lysosomal cathepsin B release. J Immunol 187:5440–5451. doi: 10.4049/jimmunol.1100790. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Y, Yang N-D, Zhou F, Shen T, Duan T, Zhou J, Shi Y, Zhu X-Q, Shen H-M. 2012. (−)-Epigallocatechin-3-gallate induces non-apoptotic cell death in human cancer cells via ROS-mediated lysosomal membrane permeabilization. PLoS One 7:e46749. doi: 10.1371/journal.pone.0046749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Repnik U, Česen MH, Turk B. 2014. Lysosomal membrane permeabilization in cell death: concepts and challenges. Mitochondrion 19:49–57. doi: 10.1016/j.mito.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 48.Uchimoto T, Nohara H, Kamehara R, Iwamura M, Watanabe N, Kobayashi Y. 1999. Mechanism of apoptosis induced by a lysosomotrpic agent, l-leucyl-l-leucine methyl ester. Apoptosis 4:357–362. doi: 10.1023/A:1009695221038. [DOI] [PubMed] [Google Scholar]
- 49.Yoshioka Y, Kitano T, Kishino T, Yamamuro A, Maeda S. 2006. Nitric oxide protects macrophages from hydrogen peroxide-induced apoptosis by inducing the formation of catalase. J Immunol 176:4675–4681. doi: 10.4049/jimmunol.176.8.4675. [DOI] [PubMed] [Google Scholar]
- 50.Tenopoulou M, Doulias P-T, Barbouti A, Brunk U, Galaris D. 2005. Role of compartmentalized redox-active iron in hydrogen peroxide-induced DNA damage and apoptosis. Biochem J 387:703–710. doi: 10.1042/BJ20041650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fonovic M, Turk B. 2014. Cysteine cathepsins and extracellular matrix degradation. Biochim Biophys Acta 1840:2560–2570. doi: 10.1016/j.bbagen.2014.03.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.