

Abstract
Pseudomonas aeruginosa causes hospital-acquired pneumonia and is associated with high mortality. An effective response to such an infection includes efficient clearance of pathogenic organisms while limiting collateral damage from the host inflammatory response, known as host resistance and host tolerance, respectively. P. aeruginosa expresses a type III secretion system (T3SS) needle complex that induces NLRC4 (NOD-like receptor C4) activation, interleukin-1β (IL-1β) production, and host tissue damage. Chitinase 3-like-1 (Chil1) is expressed during infection and binds to its receptor, IL-13 receptor α2 (IL-13Rα2), to regulate the pathogen-host response during Streptococcus pneumoniae infection, but the role Chil1 plays in balancing the host resistance and host tolerance during P. aeruginosa pneumonia is not known. We conducted experiments using C57BL/6 mice with or without a genetic deficiency of Chil1 and demonstrated that Chil1-deficient mice succumb to P. aeruginosa infection more rapidly than the wild type (WT). The decreased survival time in infected Chil1-deficient mice is associated with more neutrophils recruited to the airways, more lung parenchymal damage, and increased pulmonary consolidation while maintaining equivalent bacterial killing compared to WT mice. Infected Chil1-deficient mice and bone marrow-derived macrophages (BMDMs) from Chil1-deficient mice have increased production of tumor necrosis factor alpha (TNF-α) and IL-1β compared to infected WT mice and macrophages. Infection of Chil1-deficient BMDMs with non-NLRC4-triggering P. aeruginosa, which is deficient in the T3SS needle complex, did not alter the excessive IL-1β production compared to BMDMs from WT mice. The addition of recombinant Chil1 decreases the excessive IL-1β production but only partially rescues stimulated BMDMs from IL-13Rα2-deficient mice. Our data provide mechanistic insights into how Chil1 regulates P. aeruginosa-induced host responses.
INTRODUCTION
Hospital-acquired pneumonia is a common cause of hospital-associated death and morbidity (1, 2). Pseudomonas aeruginosa causes both acute and chronic respiratory infections. P. aeruginosa is a common cause of opportunistic infection in hospitalized patients and is known to chronically colonize patients with structural lung diseases, such as cystic fibrosis, bronchiectasis, and chronic obstructive pulmonary disease (COPD), as opposed to nonstructural lung diseases, such as pulmonary edema, chronic thrombotic disease, or atelectasis (3, 4). Acute lower respiratory tract infections caused by P. aeruginosa can lead to severe complications, such as acute respiratory distress syndrome (ARDS) and sepsis (5, 6). The clinical outcomes associated with P. aeruginosa pneumonia are the product of the immune system's ability to recognize and clear pathogenic organisms, known as host resistance (7–9). Recent investigations have led to a better appreciation that the pathogen-mediated immune response also causes collateral damage to host tissues independently of the bacterial burden, which also can contribute to the overall clinical outcome during infection (10–12). The concept of preventing unnecessary damage and promoting repair to host tissues during infection is known as host tolerance, disease tolerance for infection, or tissue resilience (13, 14). Some of the means by which the host induces tolerance are mechanisms that prevent damage to host tissues, promote the repair of host tissue when damage occurs, and limit excessive energy utilization through mechanisms that prevent overexuberant microbially induced immune responses (10, 11). An imbalance between host resistance and host tolerance can lead to poor patient outcomes, such as the development of the above-mentioned ARDS or sepsis.
Pseudomonas expresses various pathogen-associated molecular patterns that are recognized by the germ line-encoded pattern recognition receptors, such as Toll-like receptors (TLRs) and NOD-like receptors (NLRs). Two well-characterized bacterial surface virulence factors are the TLR4 agonist lipopolysaccharide (LPS) and the TLR5 agonist polar flagellin. P. aeruginosa also possesses a functional type III secretion system (T3SS) that injects bacterium-derived proteins into host cells to promote bacterial survival and evasion of the immune system (9). The P. aeruginosa T3SS apparatus is composed of a needle-like complex that penetrates the host tissue cell membrane to insert T3SS-derived proteins. Additionally, the needle complex induces NOD-like receptor C4 (NLRC4) inflammasome activation (15, 16). Two of the T3SS-derived proteins are ExoU and ExoT (17). ExoT is an ADP ribosyltransferase that alters the cellular cytoskeletal arrangement and membrane disruption (18). The T3SS-derived protein ExoU is a phospholipase A2 that inhibits NLRC4 activation (19). Together, TLR activation leads to tumor necrosis factor alpha (TNF-α), pro-interleukin-1β (IL-1β), and pro-IL-18 production, while the type III secretion system induces inflammasome activation and subsequent cleavage of pro-IL-1β to active IL-1β. These stimuli culminate in neutrophil recruitment to the lungs and airways, which promotes bacterial clearance (8).
Chitinase 3-like-1 (Chil1 [formerly Chi3l1]) is a member of the 18-glycosyl hydrolase protein family that possesses the ability to bind its natural substrate, chitin, but does not possess catalytic activity (20). Chil1 is also known as BRP-39 in mouse and YKL-40 in humans. Chil1 is a secreted protein and is known to be expressed by both bone marrow-derived macrophages (BMDMs) and stromal cells (20, 21). The protein is also expressed in healthy volunteers into whom Escherichia coli endotoxin is injected; in the sera of patients with pneumococcal pneumonia; and in patients with COPD, asthma, cancer, arthritis, and lung fibrosis (20, 22–25). Chil1 was recently reported to signal through the cell surface receptor IL-13 receptor α2 (IL-13Rα2) (26). Chil1 binding to IL-13Rα2 induces the formation of a multiprotein complex that supports downstream cell survival pathways (26). Our laboratory has demonstrated that Chil1 promotes bacterial resistance and host tolerance during pneumococcal pneumonia; however, the roles that Chil1 plays in pneumonia caused by Gram-negative bacteria, such as P. aeruginosa pneumonia, are not known (26, 27). We asked whether Chil1 plays a role in bacterial resistance and host tolerance during P. aeruginosa pneumonia. We hypothesized that P. aeruginosa infection in the genetic absence of Chil1 would lead to lower host resistance (as defined by a decreased bacterial burden) and host tolerance (as defined by less host tissue damage and fewer inflammatory cells recruited to the site of infection) than infection in wild-type (WT) mice through the ability of Chil1 to promote macrophage survival and cytokine modulation.
MATERIALS AND METHODS
Mice.
The Yale Animal Use and Care Committee approved all the animal protocols. Chil1-deficient mice on a C57BL/6 background were previously generated (21, 28). IL-13Rα2-deficient mice were purchased from Jackson Laboratory and backcrossed onto a C57BL/6 background (26, 27).
Bacteria.
PA103 (a WT P. aeruginosa strain that expresses a functional type III secretion system but does not possess a polar flagellum), PA103 DUT (PA103 with genetic absence of ExoU and ExoT), and PA103-PSCI (PA103 with genetic absence of the T3SS needle complex) stocks were stored at −80°C in 15% glycerol. The bacteria were cultured overnight in Luria-Bertani medium at 37°C in a rotator at 250 rpm. The bacteria were subcultured in Luria-Bertani medium at 37°C in a rotator at 250 rpm the next morning for approximately 1.5 h at 1:5, 1:10, and 1:20 dilutions. The bacterial count was estimated from the optical density at 600 nm (OD600), and the bacteria were diluted to final CFU concentrations as needed for each experiment. Bacteria from the inoculum were serially diluted and cultured on Vogel-Bonner medium agar to determine the final infectious inoculum concentration.
Pneumonia model.
Eight- to 12-week-old age- and sex-matched mice were lightly sedated with ketamine-xylocaine via intraperitoneal injection. For each mouse, after adequate sedation, an incision was made over the trachea, and the soft tissue was cleared. Fifty microliters of phosphate-buffered saline (PBS) (control) or PA103 in PBS was injected into the trachea. Mice were euthanized at 4, 8, 12, 16, and 24 h after infection. Bronchoalveolar lavage (BAL) was performed using 0.8 ml PBS in two aliquots. A 10-μl aliquot of BAL fluid was serially diluted and plated on Vogel-Bonner medium agar for determination of the BAL fluid bacterial load. The BAL fluid was centrifuged at 10,000 × g for 10 min, and the supernatant was collected and stored at −80°C until it was assayed. The cell pellet was resuspended in 100 μl of PBS, and red blood cell (RBC) counts were determined using a Beckman Coulter AcT 10. The RBCs were lysed with RBC lysis buffer prior to white blood cell (WBC) counting by hemocytometer. The BAL fluid WBCs were stained with Diff-Quik to differentiate cell types. A minimum of 100 cells were counted for differentials.
Bronchoalveolar lavage fluid Chil1 and cytokine analysis.
The concentrations of Chil1, TNF-α, and IL-1β were determined using commercially available enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems) following the manufacturer's protocols.
Real-time quantitative RT-PCR.
After performing BAL, the whole lung was collected and flash-frozen using liquid nitrogen. The whole lung was stored at −80°C until RNA purification. The whole lung was homogenized in TRIzol (Life Technologies), and RNA was purified using RNeasy (Qiagen) following the manufacturer's protocol. The RNA concentration was determined using Nanodrop. An iScript cDNA kit (Bio-Rad) was used to make a cDNA library according to the manufacturer's protocol. Sequences for quantitative reverse transcription-PCR (RT-PCR) were obtained using GenBank (http://pga.mgh.harvard.edu/primerbank).
Histopathology and immunofluorescence.
After performing BAL, the mouse lung was instilled with 0.5% low-melting-point agarose, fixed with 10% paraformaldehyde, and then stored at 4°C overnight. The lung was then cleared of any mediastinal tissue and stored in 70% ethanol. Lung tissue was embedded, cut, and hematoxylin and eosin (H&E) stained by the Yale Histopathology Department. Double immunofluorescence was performed using Gr-1 (BD Pharmingen), CD68 (eBioscience), and terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) (Roche) to identify neutrophils, macrophages, and cells undergoing cell death, respectively. Fluorescence was detected using a Nikon Eclipse Ti microscope with a Lumencor filter.
In vitro experiments with bone marrow-derived macrophages.
Bone morrow cells were removed from the tibias and fibulas of WT mice or Chil1-deficient mice. The bone marrow cells were resuspended in 30 ml of conditioning medium (RPMI with 20% L-929 cell [Sigma] conditioning medium and 1% penicillin-streptomycin [pen-strep]) and plated in 5 tissue culture dishes (Falcon). The cells were cultured for 7 days, with the conditioning medium changed 3 days after the cells were harvested from the bone marrow. After a total of 7 days of differentiating in conditioning medium, the adherent cells were removed with a cell scraper (Falcon) and washed three times with sterile PBS. The cells were counted using a Beckman Coulter AcT 10 and plated on a 24-well plate at 105 cells per well in conditioning medium without antibiotics and rested overnight (∼15 h). For experiments requiring Pseudomonas infection, cells were primed with LPS derived from Pseudomonas (Sigma L8643) for 2.5 h and then stimulated with various strains of PA103 or PBS control, as indicated. For experiments where recombinant mouse Chil1 (rmChil1) was used, cells were primed with rmChil1 and LPS for 2.5 h prior to exposure to P. aeruginosa or PBS control.
Statistical analysis.
Statistical analysis was performed using GraphPad Prism software and Microsoft Excel. Normally distributed data were expressed as means and standard errors of the mean (SEM) and were assessed for significance using Student's t test or analysis of variance (ANOVA), as appropriate. Data that were not normally distributed were analyzed for significance using a Kruskal-Wallis test, followed by the Mann-Whitney U test to compare individual groups. Statistical significance was defined as a P value of <0.05.
RESULTS
Chil1 expression prolongs survival during P. aeruginosa pneumonia.
Chil1 expression is induced in the presence of various infectious and noninfectious stimuli, including endotoxin and hyperoxia (22, 27, 29). We first sought to determine if the expression of Chil1 would impact survival during P. aeruginosa pneumonia. WT and Chil1-deficient mice were infected with a lethal dose (107 CFU) of PA103, and survival was monitored. The survival times of infected WT mice were doubled compared to those of infected Chil1-deficient mice (Fig. 1). This suggests that Chil1 plays a role in regulating host antibacterial responses affecting survival during P. aeruginosa pneumonia.
FIG 1.
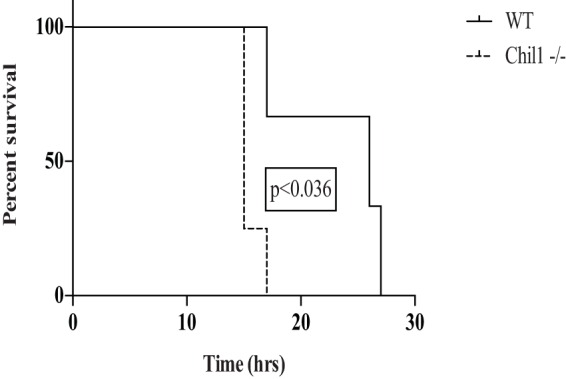
Infected mice expressing Chil1 have prolonged survival compared to Chil1-deficient mice. WT and Chil1-deficient mice were infected with 107 CFU of PA103, and survival was monitored. The survival time of WT mice was 25 h postinfection, while the survival time of Chil1-deficient mice was 14 h postinfection. The data represent three independent experiments with 8 to 10 mice per group.
P. aeruginosa-induced production of Chil1 limits inflammatory cell recruitment to the lung.
After determining that Chil1 prolongs survival during P. aeruginosa pneumonia, we investigated if Chil1 regulates host tolerance (as measured by inflammatory response and host tissue injury) or host resistance (as measured by bacterial clearance). We determined that 106 CFU would induce mortality in a significant fraction of the mice at 24 h postinfection (data not shown). Therefore, 105 CFU was chosen for the inoculum to investigate mechanisms of acute lung injury using a nonlethal model.
First, we investigated the role that Chil1 plays in regulating P. aeruginosa-induced inflammation. WT and Chil1-deficient mice were infected with 105 CFU of PA103, and BAL was performed at various time points after infection. Infected Chil1-deficient mice had more total WBCs recovered from BAL fluid than infected WT mice at 8 h, 12 h, and 16 h postinfection (Fig. 2A). Pseudomonas infection is known to induce robust recruitment of neutrophils to the lung during infection, which is essential for host survival; however, excessive neutrophil recruitment may play a role in decreasing host tolerance through excessive inflammatory-mediator release (8, 30). Infection of both WT and Chil1-deficient mice induced rapid and sustained neutrophil recruitment; however, Chil1-deficient mice recruited significantly more neutrophils than WT mice at 8 h, 12 h, and 16 h postinfection (Fig. 2B). Infected WT and Chil1-deficient mice had similar numbers of macrophages and lymphocytes recruited in BAL fluid (Fig. 2C and D, respectively). The excessive neutrophil recruitment seen in Chil1-deficient mice was associated with increased pulmonary consolidation and tissue edema as early as 8 h postinfection (Fig. 2F).
FIG 2.

Chil1 expression is important in regulating neutrophil recruitment to the lung. WT and Chil1-deficient mice were infected with 105 CFU of PA103, with BAL fluid recovered at the times indicated. (A to D) Infected Chil1-deficient mice had more total WBCs (A) and neutrophils (B) but statistically similar numbers of macrophages (C) and lymphocytes (D) recruited in BAL fluid compared to infected WT mice. (E) BAL fluid was recovered, serially diluted, and then placed on Vogel-Bonner minimal medium (VBM) plates to determine the bacterial load. BAL fluid bacterial loads were similar in WT and Chil1-deficient mice throughout the infection course. (F) Infected Chil1-deficient mice had more pulmonary consolidation than infected WT mice. *, P < 0.05; **, P < 0.001; NS, not significant. The data represent three independent experiments with 8 to 10 mice per group. The error bars indicate SEM.
Chil1-deficient mice maintain bacterial clearance during Pseudomonas pneumonia.
Neutrophils are prominent phagocytes during P. aeruginosa pneumonia. This is demonstrated by increased mortality due to bacterial overgrowth in P. aeruginosa-infected mice when neutrophils are depleted (8, 30, 31). In our model, Chil1-deficient mice recruited more neutrophils during P. aeruginosa infection than WT mice (Fig. 2B). We determined if host resistance is affected in the setting of recruitment of increased numbers of phagocytic cells to the lung observed in infected Chil1-deficient mice compared to infected WT mice. CFU recovered by BAL were measured at various time points after infection. Interestingly, infected WT and Chil1-deficient mice had similar bacterial loads throughout the infection course, suggesting no differences in overall bacterial resistance (Fig. 2E). Together, these data show that Chil1 expression promotes survival by promoting host tolerance by limiting neutrophil recruitment while maintaining adequate bacterial control despite lower phagocyte recruitment in the WT animals.
Chil1 limits pulmonary damage and promotes host health.
Mechanisms of host tolerance are beginning to be explored. One mechanism by which the host induces tolerance for an infectious organism is by preventing overexuberant inflammation and thus preventing excessive tissue damage (10). We sought to determine if Chil1 promotes survival by limiting host tissue damage. Two markers of host tissue damage during pneumonia are RBC and protein leakage recovered in BAL fluid. We measured RBCs recovered in BAL fluid from infected WT and Chil1-deficient mice, and as expected, both the WT and Chil1-deficient mice had evidence of lung hemorrhage; however, the Chil1-deficient mice had more RBCs recovered than the WT mice (Fig. 3A). Similar results were noted in protein measured in recovered BAL fluid early after infection, but this trend was lost by 12 h after infection (Fig. 3B). These results suggest that Chil1 limits host tissue damage during P. aeruginosa pneumonia.
FIG 3.

Infected Chil1-expressing mice have less pulmonary injury and improved host health during PA103 infection. WT and Chil1-deficient mice were infected with 105 CFU of PA103. BAL fluid was collected, and temperature and weight loss were measured at the times indicated. (A and B) Infected Chil1-deficient mice had increased pulmonary injury, as assessed by BAL hemorrhage (A) and BAL fluid protein recovery (B). PA, P. aeruginosa. (C and D) Infected Chil1-deficient mice had more weight loss compared to their preinfection weight than infected WT mice (C), while temperature changes from baseline were similar in the two groups (D). Cont, control. *, P < 0.05; NS, not significant. The data represent three independent experiments with 8 to 10 mice per group. The error bars indicate SEM.
We wanted to determine if Chil1 promotes host health during P. aeruginosa infection. Clinically, markers of general host health during infection include fever curves and weight changes. We measured these markers of general host health throughout the infection course. As expected, both WT and Chil1-deficient mice lost weight during the infection time course, but Chil1-deficient mice were less able to maintain their weight close to baseline than WT mice (Fig. 3C). Interestingly, WT and Chil1-deficient mice had similar changes in temperature during the infection course (Fig. 3D).
Pseudomonas infection induces robust Chil1 expression, and Chil1 regulates TNF-α production in vivo.
First, we sought to determine if PA103 induces Chil1 expression. We infected WT mice with 105 CFU of PA103 and determined the time course of Chil1 expression. Chil1 can be detected under basal conditions in BAL fluid (85.12 ± 10.00 pg/ml). Chil1 is rapidly induced in the BAL fluid with PA103 infection, with peak induction at 8 h postinfection, and is continuously expressed throughout the time course investigated. The peak concentration of Chil1 at 8 h postinfection is 310 ± 35.19 ng/ml, which is a 3-log-fold-higher concentration than baseline (Fig. 4A).
FIG 4.

P. aeruginosa induces robust Chil1 expression and differentially regulates TNF-α and IL-1β production. WT and Chil1-deficient mice were infected with 105 CFU of PA103, and BAL fluid was recovered for cytokine analysis. (A) PA103 induces robust and sustained Chil1 production throughout the infection course. (B) BAL fluid was recovered at 8 h postinfection. Infected Chil1-deficient mice express more TNF-α. (C and D) Lung tissue expression of pro-IL-1β did not differ between infected WT and infected Chil1-deficient mice at 8 h postinfection, while more IL-1β was recovered from infected Chil1-deficient mice than from infected WT mice. (E) Lung tissue from infected WT and Chil1-deficient mice was collected 8 h postinfection. Sections were labeled with CD68 (a macrophage marker), a cell death marker (TUNEL stain), and a nucleus marker (DAPI [4′,6-diamidino-2-phenylindole]). Infected Chil1-deficient mice had increased macrophage death compared to WT mice. Magnification, ×10. Inset magnification, ×40. Bar, 50 μm. (F) Infected Chil1-deficient mice had more TUNEL stain-CD68-DAPI-positive cells than infected WT mice. *, P < 0.05; **, P < 0.001; NS, not significant. The data represent three independent experiments with 8 to 10 mice per group. The immunohistochemistry represents at least 40 CD68-DAPI-positive cells counted at ×20 magnification from 3 independent mice per condition. The error bars indicate SEM.
TLR activation is a well-described feature of P. aeruginosa infection that leads to the production of cytokines known to induce host resistance (bacterial clearance) and inflammatory-cell recruitment to the site of infection (32–34). PA103 expresses the TLR4 agonist lipopolysaccharide; therefore, we measured TLR-associated cytokines to investigate if Chil1 modulates TLR activation (35, 36). We chose to measure cytokine production at 8 h postinfection, as that was the time at which Chil1 was most robustly produced. First, we measured TNF-α, a well-described cytokine produced during P. aeruginosa infection that promotes host resistance and clearance of bacteria (8, 37). Surprisingly, TNF-α production was exaggerated in Chil1-deficient mice compared to WT mice, although the bacterial loads remained similar in the two groups (Fig. 4B). This suggests that in the setting of equivalent bacterial loads in both WT and Chil1-deficient mice, Chil1-deficient mice exhibit increased TNF-α compared to WT mice and that the excessive TNF-α production may contribute to increased tissue damage.
Chil1 limits IL-1β production and is associated with macrophage death.
Next, we turned our attention to the role that Chil1 plays in regulating IL-1β production, given the known role of Chil1 in promoting cell survival in infectious and noninfectious states (20). First, we sought to determine if Chil1 plays a role in regulating TLR-mediated pro-IL-1β production. We measured pro-IL-1β expression in lung tissue from infected WT and Chil1-deficient mice at 8 h postinfection. We determined that infection of WT and Chil1-deficient mice induces similar levels of pro-IL-1β expression (Fig. 4C). We next measured the active form of IL-1β cytokine production in BAL fluid and determined that infected Chil1-deficient mice produce more IL-1β than infected WT mice (Fig. 4D). This suggests that Chil1 plays a role in regulating IL-1β production through inflammasome regulation rather than TLR-mediated pro-IL-1β expression.
Inflammasomes cleave pro-IL-1β to active IL-1β and induce caspase-dependent cell death through pyroptosis (17). As noted above, Chil1 limits IL-1β production via a TLR-independent mechanism, suggesting that Chil1 may regulate inflammasome activation. Next, we asked if Chil1 could regulate P. aeruginosa-induced macrophage death. We stained lung tissue from infected WT and Chil1-deficient mice with TUNEL stain (a cell death marker) and CD68 (a macrophage marker). Chil1-deficient mice infected with P. aeruginosa had more double-positive staining cells, where CD68 and TUNEL stain colocalized, than WT mice infected with P. aeruginosa (Fig. 4E and F).
Pseudomonas stimulation of macrophages induces early Chil1 expression.
Since macrophages are well-described early responders during pneumonia to promote neutrophil recruitment and macrophages produce Chil1 in noninfectious states (21), we next sought to determine if macrophages express Chil1 and its receptor, IL-13Rα2. We primed WT BMDMs with LPS for 2.5 h and then stimulated them with PA103 at a multiplicity of infection (MOI) of 20:1 for 15 min, 45 min, and 75 min, and then measured Chil1 release. We determined that Chil1 is released early after stimulation with P. aeruginosa, suggesting that Chil1 may be stored in a preformed state for immediate release (Fig. 5A). Next, we sought to determine if BMDMs could produce Chil1 with prolonged LPS exposure. We did not use P. aeruginosa stimulation for these experiments due to bacterial overgrowth at these late time points. We found that prolonged LPS stimulation does not induce additional Chil1 expression (Fig. 5B). Next, we sought to determine if BMDMs express IL-13Rα2 and if stimulation of the macrophages alters IL-13Rα2 expression. We again stimulated LPS-primed BMDMs with PA103 for 45 min and measured IL-13Rα2 expression. We found that BMDMs express IL-13Rα2, but that stimulation with bacteria did not alter the IL-13Rα2 expression level (Fig. 5C).
FIG 5.

Macrophages express Chil1 and its receptor, IL-13Rα2. Bone marrow-derived macrophages were primed with LPS for 2.5 h and then stimulated with PA103 or PBS control for the times indicated. Chil1 (A and B) and IL-13Rα2 (C) expression was determined by ELISA and quantitative PCR, respectively. (A) LPS-primed and PA103-stimulated BMDMs expressed Chil1 early after stimulation, but this response was not sustained. (B) Prolonged LPS stimulation alone of BMDMs for 6 h and 24 h did not produce more Chil1. (C) BMDMs were collected, and RNA was isolated. IL-13Rα2 expression was determined relative to RS18 expression. Neither LPS priming nor LPS priming with PA103 stimulation altered the expression of IL-13Rα2. The data represent two independent experiments performed in triplicate. The error bars indicate SEM. *, P < 0.05; NS, not significant.
Pseudomonas stimulation of Chil1-deficient macrophages induces exaggerated expression of TNF-α.
During P. aeruginosa pneumonia, numerous immune cell types are activated and recruited to the lungs for host defense. Alveolar macrophages are one of the first cell types to recognize P. aeruginosa infection, and Chil1 is reported to regulate their activity in other models (8, 38). We sought to determine if Chil1 regulates macrophage production of TLR- and inflammasome-related cytokines. We primed WT or Chil1-deficient BMDMs with LPS for 2.5 h and then stimulated them with PA103 at an MOI of 20:1 for 15 min and 45 min. PA103 stimulation of Chil1-deficient BMDMs produced significantly more TNF-α than PA103-stimulated WT BMDMs (Fig. 6A).
FIG 6.

Chil1 expression limits TNF-α production that is independent of ExoU or ExoT. Bone marrow-derived macrophages were primed with LPS for 2.5 h and then stimulated with PA103 (MOI, 20:1), DUT (MOI, 20:1), or PBS control for the times indicated, and TNF-α production was determined by ELISA. (A) LPS priming alone induced more TNF-α in Chil1-deficient BMDMs than in WT BMDMs. (B) The addition of PA103 or DUT (MOI 20:1 for 45 min) to LPS-primed BMDMs did not induce additional TNF-α expression. The data represent two or three independent experiments performed in triplicate. The error bars indicate SEM. *, P < 0.05; NS, not significant.
PA103 possesses a T3SS that introduces the bacterium-derived proteins ExoU and ExoT into host cells, which inhibits inflammasome activation and regulates TNF-α production (15, 39, 40). WT and Chil1-deficient BMDMs were primed with LPS and stimulated with either PA103 or DUT (PA103 with genetic absence of ExoU and ExoT) at a multiplicity of infection of 20:1 for 45 min. Stimulation of LPS-primed BMDMs with PA103 or DUT induced similar amounts of TNF-α in WT BMDMs (Fig. 6B). Priming of Chil1-deficient BMDMs with LPS induced more TNF-α than LPS priming of WT BMDMs. After priming with LPS, neither stimulation with PA103 nor stimulation with DUT induced additional TNF-α production from Chil1-deficient or WT BMDMs compared to LPS stimulation alone (Fig. 6B). Priming of Chil1-deficient BMDMs with LPS with subsequent stimulation with either PA103 or DUT induced more TNF-α than WT BMDMs that were LPS primed and stimulated with PA103 or DUT (Fig. 6B). These data suggest that Chil1 plays a role in regulating TNF-α production in macrophages.
Pseudomonas stimulation of Chil1-deficient macrophages induces exaggerated expression of IL-1β production.
Macrophage-mediated IL-1β production is induced by NLRC4 inflammasome recognition of the P. aeruginosa T3SS needle complex (17). In order to understand if Chil1 limits IL-1β production via an NLRC4-dependent inflammasome mechanism, we infected LPS-primed BMDMs with PA103, DUT (which lacks ExoU and ExoT and does not inhibit the inflammasome), or PA103 PSCI (which lacks the T3SS needle complex and thus does not induce the inflammasome). PA103-stimulated BMDMs from Chil1-deficient mice induced more IL-1β than PA103-stimulated WT BMDMs (Fig. 7A). The expression of the P. aeruginosa-derived proteins ExoU and ExoT, which enter host cells via expression of the T3SS complex, inhibits NLRC4-dependent IL-1β production (15). We hypothesized that Chil1 may regulate macrophage-mediated IL-1β production. To test this hypothesis, we stimulated LPS-primed BMDMs with DUT and observed that, as expected, more IL-1β was produced than by PA103-stimulated WT BMDMs. Of interest, the greater increase in IL-1β production by both IL-13Rα2-deficient and Chil1-deficient BMDMs than by similarly stimulated WT BMDMs remained (Fig. 7A). Next, we sought to determine the role the T3SS needle complex plays in Chil1 regulation of IL-1β production. Interestingly, stimulation of Chil1-deficient BMDMs with PSCI (lacking the T3SS needle complex) induced slightly more IL-1β production than WT and IL-13Rα2-deficient BMDMs (Fig. 7A). Given that infection of Chil1-deficient BMDMs with PA103 (which inhibits NLRC4) and PA103 PSCI (which does not induce NLRC4) induces more IL-1β production than infection of BMDMs from WT mice, we concluded that Chil1 regulates IL-1β production by at least an NLRC4-independent mechanism. Together, these data suggest that Chil1 regulates IL-1β production in macrophages. We are actively investigating the various inflammasome complexes that Chil1 regulates during P. aeruginosa infection.
FIG 7.

Chil1 regulates macrophage-derived IL-1β production and is partially IL-13Rα2 dependent. Bone marrow-derived macrophages were primed with LPS for 2.5 h and then stimulated with PA103, DUT, or PSCI (MOI, 20:1) or with PBS control for 45 min, and IL-1β production was determined by ELISA. (A) BMDMs deficient in either Chil1 or IL-13Rα2 produced more IL-1β after stimulation than stimulated WT BMDMs. DUT stimulation of WT, Chil1-deficient, and IL-13Rα2-deficient BMDMs led to more IL-1β production than PA103 stimulation in WT, Chil1-deficient, and IL-13Rα2-deficient BMDMs. Stimulation of WT BMDMs with PSCI limited IL-1β production compared to Chil1-deficient BMDMs. (B and C) Bone marrow-derived macrophages were cultured with PBS or rmChil1 (1,000 ng) for 2 h and then primed with LPS for 2.5 h, followed by stimulation with PA103 (MOI, 20:1) for 45 min, and IL-1β production was measured by ELISA. (B) rmChil1 prevents excessive IL-1β production by WT, Chil1-deficient, and IL-13Rα2-deficient BMDMs. The addition of rmChil1 limited IL-1β production by WT, Chil1-deficient, and IL-13Rα2-deficient BMDMs when stimulated with either PA103 or DUT compared with PBS control. (C) rmChil1 limited IL-1β production with PA103 and DUT stimulation of LPS-primed BMDMs. The data represent two independent experiments performed in triplicate. The error bars indicate SEM. *, P < 0.05; $, P < 0.05 between PA103 stimulation with and without rmChil1; &, P < 0.05 between DUT stimulation with and without rmChil1.
Recombinant Chil1 partially rescues IL-1β production by macrophage-mediated BMDMs.
In an attempt to demonstrate that Chil1 exerts its anti-inflammatory effect via its receptor, IL-13Rα2, we measured the production of IL-1β from LPS-primed PA103- or DUT-stimulated BMDMs for 45 min in the presence or absence of rmChil1 (500 ng/ml for 2 h prior to priming with LPS). The addition of rmChil1 decreased IL-1β production by WT and Chil1-deficient BMDMs compared to PA103 stimulation in the absence of rmChil1 (Fig. 7B). Interestingly, the addition of rmChil1 to PA103-stimulated IL-13Rα2-deficient BMDMs only partially decreased IL-1β production (Fig. 7B). This suggests the possibility that Chil1 signals through both IL-13Rα2-dependent and IL-13Rα2-independent mechanisms. Again, DUT stimulation of LPS-primed BMDMs from WT, Chil1-deficient, and IL-13Rα2-deficient cells induced more IL-1β production than PA103 stimulation of WT, Chil1-deficient, and IL-13Rα2-deficient BMDMs (Fig. 7C). The addition of rmChil1 lessens IL-1β production from WT, Chil1-deficient, and IL-13Rα2-deficient BMDMs stimulated with either PA103 or DUT (Fig. 7C). To determine if the Chil1-dependent regulation of IL-1β expression is specific to PA103, we primed BMDMs from WT and Chil1-deficient mice with LPS and then infected them with Pseudomonas strain PAK. Similar results were observed, with more IL-1β production from infected BMDMs from Chil1-deficient mice than from BMDMs from WT mice (data not shown). These findings show that administration of rmChil1 is able to downmodulate IL-1β production by regulating inflammasome activation, as measured by IL-1β production.
DISCUSSION
Here, we sought to determine the role that Chil1 plays during P. aeruginosa pneumonia. The survival of pneumonia pathogens involves a balance between two distinct processes, host resistance and host tolerance (sometimes called host resilience). Host resistance is the efficient killing and clearance of infectious organisms, while host tolerance includes mechanisms that prevent microbially induced collateral damage to host tissue and mechanisms that promote the repair of unavoidable host tissue damage (10, 12). We hypothesized that Chil1 expression promotes host resistance and host tolerance by inhibiting inflammatory myeloid-derived cell response and cell death during P. aeruginosa pneumonia. To test this hypothesis, we measured host tolerance and resistance by determining host survival, pulmonary inflammatory-cell recruitment, and bacterial loads in WT and Chil1-deficient mice. These investigations demonstrated that infected WT mice had prolonged survival compared to infected Chil1-deficient mice and that infected WT mice had less neutrophil recruitment than infected Chil1-deficient mice. WT mice infected with PA103 were able to maintain their weight closer to baseline than infected Chil1-deficient mice, which was independent of temperature fluctuations. Similar observations have been demonstrated during Burkholderia (also known as Pseudomonas) pseudomallei infection when WT mice were infected with the prototypical B. pseudomallei strain and a more virulent B. pseudomallei strain (41, 42). As in our P. aeruginosa infection model, the physiologic characteristics noted in B. pseudomallei infection were associated with more IL-1β and TNF-α production in mice infected with the more virulent B. pseudomallei strain than in mice infected with the less virulent strain of B. pseudomallei. Despite the differences in cytokine expression during B. pseudomallei infection, the surviving mice did not demonstrate correlation of weight loss and temperature changes. The early and intermediate clinical features in the B. pseudomallei model include early decreased grooming, followed by a rough coat with decreased motility. Late clinical features include cessation of grooming, abnormal posturing, and nasal and ocular discharge (42). These similar clinical features may play a role in our model. The idea that metabolic derangements lead to decreased host health in infected Chil1-deficient mice compared to infected WT mice is an intriguing possibility and merits further investigation.
Neutrophil recruitment plays a key role in bacterial resistance during P. aeruginosa infection. The important role neutrophils play in P. aeruginosa pneumonia has been demonstrated clinically by increased mortality of neutropenic patients infected with P. aeruginosa and increased mortality in neutropenic-mouse models of infection with P. aeruginosa (43, 44). Although Chil1-deficient mice had more neutrophils recruited to the airways, this did not seem to impact their host resistance, as WT and Chil1-deficient mice had similar bacterial loads. The role of Chil1 in response to P. aeruginosa infection differed from the role Chil1 plays during pneumococcal infection (26, 27). The absence of Chil1 during pneumococcal infection results in more neutrophil recruitment, as well as a greater bacterial burden (27). This suggests that Chil1 regulates the inflammatory response differently during P. aeruginosa and pneumococcal infection by promoting or maintaining P. aeruginosa bacterial killing or clearance.
P. aeruginosa is a complex extracellular Gram-negative rod-shaped bacterium that expresses TLR agonists, as well as a type III secretion system. P. aeruginosa-induced immune activation leads to the production of the TLR-associated cytokine TNF-α (37). TNF-α is expressed from bone marrow-derived cells and plays a role in promoting host resistance to P. aeruginosa infection (37). During P. aeruginosa infection in our model, the absence of Chil1 resulted in increased TNF-α measured in the BAL fluid. In addition, macrophages deficient in Chil1 produced more TNF-α upon LPS stimulation alone. However, no further elevation in TNF-α levels was observed upon subsequent treatment with P. aeruginosa in WT or Chil1-deficient BMDMs.
Several NLRs have been identified as playing a role in various disease states (45–47). Inflammasome activation is a well-recognized mechanism by which a host responds to microbial attack. P. aeruginosa pneumonia is known to activate NLRs. The best-characterized NLR during P. aeruginosa infection is NLRC4. Inflammasome activation promotes host resistance by increasing bacterial killing. The Pseudomonas T3SS needle complex induces IL-1β production via activation of NLRC4, while expression of P. aeruginosa-derived proteins that pass through the T3SS needle complex inhibits inflammasome activation (17, 19, 48, 49). While there are similar expression levels of pro-IL-1β in the lungs of infected WT and Chil1-deficient mice, we observed that P. aeruginosa infection of Chil1-deficient mice induced more active IL-1β in BAL fluid than infection of WT mice. This led us to hypothesize that Chil1 regulates IL-1β production by regulating inflammasome activation, because NLRC4 plays a significant role in recognizing the P. aeruginosa needle complex. In an attempt to determine if Chil1 regulates inflammasome-dependent IL-1β production, we stimulated bone marrow-derived macrophages with a P. aeruginosa T3SS-expressing strain, as well as P. aeruginosa strains deficient in the T3SS needle complex or the secreted T3SS proteins ExoU and ExoT. Our data indicate that Chil1 regulates IL-1β production in macrophages. The inflammasome(s) that Chil1 targets during P. aeruginosa infection is not known, but it may act through the regulation of Nlrp3-mediated tissue injury, as was demonstrated in pneumococcal infection (27), or through direct regulation of NLRC4. This is an active area of investigation in our laboratory.
The 18-glycosyl hydrolase protein family is composed of true chitinases and chitinase-like proteins (20, 21, 50). True chitinases possess the ability to cleave their natural ligand, chitin, while chitinase-like proteins can bind to chitin but lack the ability to cleave chitin due to point mutations in the cleavage site (20, 50). This evolutionarily conserved family of proteins is known to inhibit innate immune activation while promoting T helper type 2 (Th2) responses that can contribute to tissue healing and fibrosis. The mechanism by which Chil1 augments host tolerance was recently recognized to be through its ability to form a heterodimer with IL-13 and IL-13Rα2, leading to antiapoptosis by subsequent mitogen-activated protein kinase (MAPK) and AKT cellular signaling activation (26). Our investigations revealed that Chil1 signals through an IL-13Rα2-dependent and an IL-13Rα2-independent mechanism. The IL-13Rα2-independent signaling mechanism remains elusive, but it may involve the pro-Th2 receptor CRTH2 or another, unidentified pathway (25).
As demonstrated above, our investigations are the first to show that Chil1 is an important immune regulator during P. aeruginosa pneumonia. These data add to our understanding of Chil1 biology during innate immune activation in Gram-negative infections. We demonstrated that Chil1 expression promotes host tolerance by decreasing the expression of the TLR-associated cytokine TNF-α, as well as the inflammasome-associated cytokine IL-1β. Our investigations focused on Chil1 as a regulator of inflammasome signaling and showed that Chil1 regulates inflammasome activation and is partially dependent on signaling through the Chil1 receptor, IL-13Rα2.
In summary, our investigation has added to the scientific understanding of the mechanisms by which a host balances host tolerance and resistance during acute P. aeruginosa infection. Our data demonstrate that Chil1 can limit the immune response to P. aeruginosa while maintaining the ability to clear the pathogen. Macrophages contribute to TNF-α and IL-1β production early in P. aeruginosa infection. Additional investigation into the role of Chil1 in regulating inflammasome activation during P. aeruginosa infection is warranted. Moreover, studies on the involvement of cells other than macrophages that contribute to Chil1, TLR-associated cytokines, and the inflammasome during P. aeruginosa infection would further increase our understanding of the role of Chil1 in the anti-P. aeruginosa host response in the lung.
ACKNOWLEDGMENTS
We thank Susan Ardito for editorial assistance.
The study was supported by grants from the NHLBI (HL126094 and HL103770) (C.S.D.C.) and T32 HL007778 (C.R.M). The publication was also made possible by CTSA grant number UL1 TR000142 (C.S.D.C. and C.R.M.) from the National Center for Advancing Translational Science (NCATS), a component of the National Institutes of Health (NIH).
The funding bodies had no part in study design, collection, analysis, and interpretation of the data, or writing or submission of the manuscript.
Footnotes
C.R.M. and J.W. are co-first authors.
REFERENCES
- 1.Centers for Disease Control and Prevention. 2010. Deaths: final data for 2010. National Center for Health Statistics, Hyattsville, MD: http://www.cdc.gov/nchs/data/nvsr/nvsr61/nvsr61_04.pdf. [Google Scholar]
- 2.Falcone M, Venditti M, Shindo Y, Kollef MH. 2011. Healthcare-associated pneumonia: diagnostic criteria and distinction from community-acquired pneumonia. Int J Infect Dis 15:e545–e550. doi: 10.1016/j.ijid.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 3.Murray TS, Egan M, Kazmierczak BI. 2007. Pseudomonas aeruginosa chronic colonization in cystic fibrosis patients. Curr Opin Pediatr 19:83–88. doi: 10.1097/MOP.0b013e3280123a5d. [DOI] [PubMed] [Google Scholar]
- 4.Novosad SA, Barker AF. 2013. Chronic obstructive pulmonary disease and bronchiectasis. Curr Opin Pulm Med 19:133–139. doi: 10.1097/MCP.0b013e32835d8312. [DOI] [PubMed] [Google Scholar]
- 5.Dermengiu D, Curca GC, Ceausu M, Hostiuc S. 2013. Particularities regarding the etiology of sepsis in forensic services. J Forensic Sci 58:1183–1188. doi: 10.1111/1556-4029.12222. [DOI] [PubMed] [Google Scholar]
- 6.Meduri GU, Reddy RC, Stanley T, El-Zeky F. 1998. Pneumonia in acute respiratory distress syndrome. A prospective evaluation of bilateral bronchoscopic sampling. Am J Respir Crit Care Med 158:870–875. [DOI] [PubMed] [Google Scholar]
- 7.Strowig T, Henao-Mejia J, Elinav E, Flavell R. 2012. Inflammasomes in health and disease. Nature 481:278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 8.Lavoie EG, Wangdi T, Kazmierczak BI. 2011. Innate immune responses to Pseudomonas aeruginosa infection. Microbes Infect 13:1133–1145. doi: 10.1016/j.micinf.2011.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadikot RT, Blackwell TS, Christman JW, Prince AS. 2005. Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med 171:1209–1223. doi: 10.1164/rccm.200408-1044SO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ayres JS, Schneider DS. 2012. Tolerance of infections. Annu Rev Immunol 30:271–294. doi: 10.1146/annurev-immunol-020711-075030. [DOI] [PubMed] [Google Scholar]
- 11.Ayres JS. 2013. Inflammasome-microbiota interplay in host physiologies. Cell Host Microbe 14:491–497. doi: 10.1016/j.chom.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 12.Schneider DS, Ayres JS. 2008. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat Rev Immunol 8:889–895. doi: 10.1038/nri2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quinton LJ, Mizgerd JP. 2015. Dynamics of lung defense in pneumonia: resistance, resilience, and remodeling. Annu Rev Physiol 77:407–430. doi: 10.1146/annurev-physiol-021014-071937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soares MP, Ribeiro AM. 2015. Nrf2 as a master regulator of tissue damage control and disease tolerance to infection. Biochem Soc Trans 43:663–668. doi: 10.1042/BST20150054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hauser AR. 2009. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol 7:654–665. doi: 10.1038/nrmicro2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. 2011. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 17.Cunha LD, Zamboni DS. 2013. Subversion of inflammasome activation and pyroptosis by pathogenic bacteria. Front Cell Infect Microbiol 3:76. doi: 10.3389/fcimb.2013.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Angot A, Vergunst A, Genin S, Peeters N. 2007. Exploitation of eukaryotic ubiquitin signaling pathways by effectors translocated by bacterial type III and type IV secretion systems. PLoS Pathog 3:e3. doi: 10.1371/journal.ppat.0030003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson DM, Feix JB, Monroe AL, Peterson FC, Volkman BF, Haas AL, Frank DW. 2013. Identification of the major ubiquitin-binding domain of the Pseudomonas aeruginosa ExoU A2 phospholipase. J Biol Chem 288:26741–26752. doi: 10.1074/jbc.M113.478529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee CG, Da Silva CA, Dela Cruz CS, Ahangari F, Ma B, Kang MJ, He CH, Takyar S, Elias JA. 2011. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu Rev Physiol 73:479–501. doi: 10.1146/annurev-physiol-012110-142250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee CG, Hartl D, Lee GR, Koller B, Matsuura H, Da Silva CA, Sohn MH, Cohn L, Homer RJ, Kozhich AA, Humbles A, Kearley J, Coyle A, Chupp G, Reed J, Flavell RA, Elias JA. 2009. Role of breast regression protein 39 (BRP-39)/chitinase 3-like-1 in Th2 and IL-13-induced tissue responses and apoptosis. J Exp Med 206:1149–1166. doi: 10.1084/jem.20081271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johansen JS, Krabbe KS, Moller K, Pedersen BK. 2005. Circulating YKL-40 levels during human endotoxaemia. Clin Exp Immunol 140:343–348. doi: 10.1111/j.1365-2249.2005.02763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ober C, Tan Z, Sun Y, Possick JD, Pan L, Nicolae R, Radford S, Parry RR, Heinzmann A, Deichmann KA, Lester LA, Gern JE, Lemanske RF Jr, Nicolae DL, Elias JA, Chupp GL. 2008. Effect of variation in CHI3L1 on serum YKL-40 level, risk of asthma, and lung function. N Engl J Med 358:1682–1691. doi: 10.1056/NEJMoa0708801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Otsuka K, Matsumoto H, Niimi A, Muro S, Ito I, Takeda T, Terada K, Yamaguchi M, Matsuoka H, Jinnai M, Oguma T, Nakaji H, Inoue H, Tajiri T, Iwata T, Chin K, Mishima M. 2012. Sputum YKL-40 levels and pathophysiology of asthma and chronic obstructive pulmonary disease. Respiration 83:507–519. doi: 10.1159/000330840. [DOI] [PubMed] [Google Scholar]
- 25.Zhou Y, He CH, Herzog EL, Peng X, Lee CM, Nguyen TH, Gulati M, Gochuico BR, Gahl WA, Slade ML, Lee CG, Elias JA. 2015. Chitinase 3-like-1 and its receptors in Hermansky-Pudlak syndrome-associated lung disease. J Clin Invest 125:3178–3192. doi: 10.1172/JCI79792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He CH, Lee CG, Dela Cruz CS, Lee CM, Zhou Y, Ahangari F, Ma B, Herzog EL, Rosenberg SA, Li Y, Nour AM, Parikh CR, Schmidt I, Modis Y, Cantley L, Elias JA. 2013. Chitinase 3-like 1 regulates cellular and tissue responses via IL-13 receptor alpha2. Cell Rep 4:830–841. doi: 10.1016/j.celrep.2013.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dela Cruz CS, Liu W, He CH, Jacoby A, Gornitzky A, Ma B, Flavell R, Lee CG, Elias JA. 2012. Chitinase 3-like-1 promotes Streptococcus pneumoniae killing and augments host tolerance to lung antibacterial responses. Cell Host Microbe 12:34–46. doi: 10.1016/j.chom.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsuura H, Hartl D, Kang MJ, Dela Cruz CS, Koller B, Chupp GL, Homer RJ, Zhou Y, Cho WK, Elias JA, Lee CG. 2011. Role of breast regression protein-39 in the pathogenesis of cigarette smoke-induced inflammation and emphysema. Am J Respir Cell Mol Biol 44:777–786. doi: 10.1165/rcmb.2010-0081OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sohn MH, Kang MJ, Matsuura H, Bhandari V, Chen NY, Lee CG, Elias JA. 2010. The chitinase-like proteins breast regression protein-39 and YKL-40 regulate hyperoxia-induced acute lung injury. Am J Respir Crit Care Med 182:918–928. doi: 10.1164/rccm.200912-1793OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bardoel BW, Kenny EF, Sollberger G, Zychlinsky A. 2014. The balancing act of neutrophils. Cell Host Microbe 15:526–536. doi: 10.1016/j.chom.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 31.Lovewell RR, Patankar YR, Berwin B. 2014. Mechanisms of phagocytosis and host clearance of Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol 306:L591–L603. doi: 10.1152/ajplung.00335.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawai T, Akira S. 2007. TLR signaling. Semin Immunol 19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 33.Branger J, Knapp S, Weijer S, Leemans JC, Pater JM, Speelman P, Florquin S, van der Poll T. 2004. Role of Toll-like receptor 4 in gram-positive and gram-negative pneumonia in mice. Infect Immun 72:788–794. doi: 10.1128/IAI.72.2.788-794.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raoust E, Balloy V, Garcia-Verdugo I, Touqui L, Ramphal R, Chignard M. 2009. Pseudomonas aeruginosa LPS or flagellin are sufficient to activate TLR-dependent signaling in murine alveolar macrophages and airway epithelial cells. PLoS One 4:e7259. doi: 10.1371/journal.pone.0007259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. 2007. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med 204:3235–3245. doi: 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mijares LA, Wangdi T, Sokol C, Homer R, Medzhitov R, Kazmierczak BI. 2011. Airway epithelial MyD88 restores control of Pseudomonas aeruginosa murine infection via an IL-1-dependent pathway. J Immunol 186:7080–7088. doi: 10.4049/jimmunol.1003687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gosselin D, DeSanctis J, Boule M, Skamene E, Matouk C, Radzioch D. 1995. Role of tumor necrosis factor alpha in innate resistance to mouse pulmonary infection with Pseudomonas aeruginosa. Infect Immun 63:3272–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hashimoto S, Pittet JF, Hong K, Folkesson H, Bagby G, Kobzik L, Frevert C, Watanabe K, Tsurufuji S, Wiener-Kronish J. 1996. Depletion of alveolar macrophages decreases neutrophil chemotaxis to Pseudomonas airspace infections. Am J Physiol 270:L819–L828. [DOI] [PubMed] [Google Scholar]
- 39.Bleves S, Viarre V, Salacha R, Michel GP, Filloux A, Voulhoux R. 2010. Protein secretion systems in Pseudomonas aeruginosa: a wealth of pathogenic weapons. Int J Med Microbiol 300:534–543. doi: 10.1016/j.ijmm.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 40.Wangdi T, Mijares LA, Kazmierczak BI. 2010. In vivo discrimination of type 3 secretion system-positive and -negative Pseudomonas aeruginosa via a caspase-1-dependent pathway. Infect Immun 78:4744–4753. doi: 10.1128/IAI.00744-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ceballos-Olvera I, Sahoo M, Miller MA, Del Barrio L, Re F. 2011. Inflammasome-dependent pyroptosis and IL-18 protect against Burkholderia pseudomallei lung infection while IL-1beta is deleterious. PLoS Pathog 7:e1002452. doi: 10.1371/journal.ppat.1002452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Massey S, Yeager LA, Blumentritt CA, Vijayakumar S, Sbrana E, Peterson JW, Brasel T, LeDuc JW, Endsley JJ, Torres AG. 2014. Comparative Burkholderia pseudomallei natural history virulence studies using an aerosol murine model of infection. Sci Rep 4:4305. doi: 10.1038/srep04305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang CI, Kim SH, Park WB, Lee KD, Kim HB, Kim EC, Oh MD, Choe KW. 2005. Clinical features and outcome of patients with community-acquired Pseudomonas aeruginosa bacteraemia. Clin Microbiol Infect 11:415–418. doi: 10.1111/j.1469-0691.2005.01102.x. [DOI] [PubMed] [Google Scholar]
- 44.Koh AY, Priebe GP, Ray C, Van Rooijen N, Pier GB. 2009. Inescapable need for neutrophils as mediators of cellular innate immunity to acute Pseudomonas aeruginosa pneumonia. Infect Immun 77:5300–5310. doi: 10.1128/IAI.00501-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lamkanfi M, Dixit VM. 2012. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol 28:137–161. doi: 10.1146/annurev-cellbio-101011-155745. [DOI] [PubMed] [Google Scholar]
- 46.Schroder K, Tschopp J. 2010. The inflammasomes. Cell 140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 47.Skeldon A, Saleh M. 2011. The inflammasomes: molecular effectors of host resistance against bacterial, viral, parasitic, and fungal infections. Front Microbiol 2:15. doi: 10.3389/fmicb.2011.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sato H, Frank DW. 2014. Intoxication of host cells by the T3SS phospholipase ExoU: PI(4,5)P2-associated, cytoskeletal collapse and late phase membrane blebbing. PLoS One 9:e103127. doi: 10.1371/journal.pone.0103127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kazmierczak BI, Engel JN. 2002. Pseudomonas aeruginosa ExoT acts in vivo as a GTPase-activating protein for RhoA, Rac1, and Cdc42. Infect Immun 70:2198–2205. doi: 10.1128/IAI.70.4.2198-2205.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hartl D, He CH, Koller B, Da Silva CA, Homer R, Lee CG, Elias JA. 2008. Acidic mammalian chitinase is secreted via an ADAM17/epidermal growth factor receptor-dependent pathway and stimulates chemokine production by pulmonary epithelial cells. J Biol Chem 283:33472–33482. doi: 10.1074/jbc.M805574200. [DOI] [PMC free article] [PubMed] [Google Scholar]