

Abstract
Chronic lower respiratory tract infection with Pseudomonas aeruginosa is difficult to treat due to enhanced antibiotic resistance and decreased efficacy of drug delivery to destroyed lung tissue. To determine the potential for restorative immunomodulation therapies, we evaluated the effect of Toll-like receptor 4 (TLR4) stimulation on the host immune response to Pseudomonas infection in mice. We implanted sterile plastic tubes precoated with P. aeruginosa in the bronchi of mice, administered the TLR4/MD2 agonistic monoclonal antibody UT12 intraperitoneally every week, and subsequently analyzed the numbers of viable bacteria and inflammatory cells and the levels of cytokines. We also performed flow cytometry-based phagocytosis and opsonophagocytic killing assays in vitro using UT12-treated murine peritoneal neutrophils. UT12-treated mice showed significantly enhanced bacterial clearance, increased numbers of Ly6G+ neutrophils, and increased concentrations of macrophage inflammatory protein 2 (MIP-2) in the lungs (P < 0.05). Depletion of CD4+ T cells eliminated the ability of the UT12 treatment to improve bacterial clearance and promote neutrophil recruitment and MIP-2 production. Additionally, UT12-pretreated peritoneal neutrophils exhibited increased opsonophagocytic killing activity via activation of the serine protease pathway, specifically neutrophil elastase activity, in a TLR4-dependent manner. These data indicated that UT12 administration significantly augmented the innate immune response against chronic bacterial infection, in part by promoting neutrophil recruitment and bactericidal function.
INTRODUCTION
Pseudomonas aeruginosa is extremely difficult to eradicate once established in the lower respiratory tract (LRT) during chronic respiratory infection. Poor penetration of antibiotics into purulent airway secretions due to lung structure damage, acquired antibiotic resistance, defects in mucosal defenses, and the interference of bacterium-produced biofilms with phagocytic killing impede treatment (1). Chronic P. aeruginosa infection also represents an independent risk factor for accelerated loss of pulmonary function and decreased survival (2, 3).
Lipopolysaccharide (LPS) is a glycolipid component of the Gram-negative bacterial cell wall that is recognized by Toll-like receptor 4 (TLR4), which induces various host responses, including proinflammatory cytokine production, and has known immunomodulatory properties (4). Priming with LPS has been shown to improve murine responses to bacterial infections (5–7), and prior LPS exposure attenuates proinflammatory cytokine production in response to LPS or bacterial challenge, a phenomenon that has historically been referred to as endotoxin tolerance (8, 9). Several immunomodulators, such as the TLR4 agonist monophosphoryl lipid A (MPLA), promote host immunity and are used as vaccine adjuvants for humans (10). In animal models, MPLA enhances the action of macrophages, B cells, and other antigen-presenting cells and promotes the differentiation of Th1 and Th2 cells from naïve T cells (11). The administration of MPLA as an adjuvant for respiratory syncytial virus vaccine also resulted in increased virus-neutralizing antibody levels (12). For P. aeruginosa-infected animals in a mouse burn injury model, MPLA promoted the concentration of neutrophils at infection sites and enhanced their bacterium-eradicating effects (13).
Additionally, several studies have reported the therapeutic effects of TLR4 agonistic antibodies, such as UT12, against bacterial infections. UT12 can induce stimulatory signals comparable to those induced by LPS through TLR4/MD-2 and with 15- to 20-fold-greater potency by weight (14). Previously, we demonstrated that preadministration of UT12 increased protection against severe pneumococcal pneumonia induced by coinfection with influenza virus (15). As yet, however, the efficacy of TLR4 agonist therapy under conditions of established chronic infection and the associated mechanism remain unclear. In this study, we used a mouse model of chronic P. aeruginosa respiratory tract infection to demonstrate that UT12 administration alone promoted neutrophil concentration in the LRT and induced the activation of neutrophil bactericidal function, thereby enhancing the clearance of P. aeruginosa from the LRT. Few studies have definitively demonstrated the possibility of controlling chronic LRT infection (cLRTI) through the activation of innate immunity; therefore, this result offers the potential for future clinical applications.
MATERIALS AND METHODS
Reagents.
The TLR4/MD2 agonistic antibody UT12 was a gift from K. Fukudome (Saga Medical School, Saga, Japan). UT12 (1 mg/ml) was diluted with sterile saline to 10 μg/ml. Neutrophils were treated for 30 min at 37°C with the following inhibitors: 10 μM diphenyleneiodonium (DPI; Sigma-Aldrich, St. Louis, MO, USA), 500 μM 4-(2-aminoethyl)benzenesulfonyl fluoride HCl (AEBSF; Calbiochem, San Diego, CA, USA), chymostatin (CHYM; Sigma-Aldrich), and an elastase inhibitor (Calbiochem).
Mice.
Six-week-old specific-pathogen-free female mice (weight, 30 to 35 g) were purchased from Japan SLC (Hamamatsu, Japan). Tlr4−/− mice were purchased from Oriental BioService (Kyoto, Japan). All experiments were conducted according to the guidelines of the Laboratory Animal Center for Biomedical Research, Nagasaki University School of Medicine, Nagasaki, Japan.
Experimental murine model of cLRTI.
Chronic airway infection was induced in mice as described previously (16). Briefly, a clinical isolate of P. aeruginosa, strain S10, was cultured on a lysogeny broth agar plate (Becton Dickinson [BD] Microbiology Systems, Cockeysville, MD, USA) for 24 h. The intubation tube was immersed in the bacterial suspension, which was adjusted to 2 × 109 CFU/ml, for 3 days at 37°C and was then advanced through the vocal cords and into the trachea. Mice were sacrificed by CO2 asphyxiation, and the lungs were dissected under aseptic conditions and were suspended in 1 ml saline, followed by homogenization using an AS One (Osaka, Japan) homogenizer. P. aeruginosa was quantified by serial dilution plating of the homogenized lungs onto lysogeny broth agar plates, followed by incubation at 37°C for 18 h.
UT12 administration.
UT12 was delivered by intraperitoneal (i.p.) injection at weekly intervals (1 μg/week) for 4 weeks, beginning 1 week after intratracheal placement of the bacterium-coated tube. Saline was injected into the control group. Viable bacterial counts were evaluated beginning at 1 week after treatment (Fig. 1).
FIG 1.
Schedule of UT12 treatment for chronic lower respiratory tract infection by P. aeruginosa in mice.
BAL.
For bronchoalveolar lavage (BAL), after the chest was opened to expose the lungs, a cut-down disposable sterile plastic intravenous catheter was inserted into the trachea. BAL was performed in situ three times sequentially using 1 ml saline, and the recovered fluid (BALF) fractions were pooled for each animal (17).
Flow cytometry.
Cells were collected from BALF by centrifugation at 4°C and then resuspended in phosphate-buffered saline (PBS) with 1% bovine serum albumin (BSA). Nonspecific binding was blocked using a rat anti-mouse antibody directed against the FcγIII/II receptor (CD16/CD32; BD Biosciences), and the following rat anti-mouse cell surface antibodies were applied: fluorescein isothiocyanate (FITC)-conjugated anti-Ly6G (dilution, 1:300; BD Biosciences), phycoerythrin (PE)-conjugated anti-CD45 (1:400; BD Biosciences), and allophycocyanin-conjugated anti-F4/80 (1:400; eBioscience, San Diego, CA, USA). Then, the cells were incubated with the antibodies on ice in the dark for 45 min. All samples were resuspended in wash buffer and were then subjected to flow cytometry analysis on a BD FACSCanto flow cytometer (BD Biosciences). Data were analyzed using FlowJo for Mac, version 9.7 (FlowJo LLC, Ashland, OR, USA).
Cytokine and chemokine analysis by ELISA.
Concentrations of tumor necrosis factor alpha (TNF-α), interleukin 6 (IL-6), macrophage inflammatory protein 2 (MIP-2), and monocyte chemoattractant protein 1 (MCP-1) in aqueous lung extracts from homogenates at 1 week after treatment were assayed using mouse Quantikine ELISA (enzyme-linked immunosorbent assay) kits according to the manufacturer's protocol (R&D Systems, Minneapolis, MN, USA).
CD4+ T cell depletion.
To deplete CD4+ T cells in vivo, monoclonal antibodies (MAbs) against mouse CD4 (MAb GK1.5) were purified from the peritoneal fluid of BALB/cSlc-nu/nu mice as reported previously (18). Each animal was administered a total of 0.6 mg of MAb GK1.5 i.p. in six doses, given 1 day prior to UT12 treatment and 1, 3, 8, 12, and 18 days after UT12 treatment. Flow cytometry of splenocytes at the time of sacrifice confirmed CD4+ T cell depletion. At 1 week and 3 weeks after UT12 treatment, viable bacterial loads in homogenized lung extracts were determined, and cytokine analyses and BALF cell counts were performed.
Isolation of murine neutrophils.
Murine neutrophils were isolated as described previously (19). Briefly, mice were administered 10% casein in PBS (1 ml/dose) i.p. 24 h and again 2 h before cell harvest, and phagocytes were obtained by lavage of their peritoneal cavities with 8 ml/animal of phosphate-buffered saline [PBS] containing 20 mM EDTA. Cells were enriched for neutrophils via separation by Ficoll density gradient centrifugation according to the manufacturer's protocol (MP Biomedicals, Santa Ana, CA, USA).
OPH killing assay.
Opsonophagocytic (OPH) killing assays were conducted as described previously (20). Bacterial strains were grown to mid-log phase, washed with PBS, and resuspended in Hanks buffer containing Ca2+ and Mg2+ buffer (GIBCO; Thermo Scientific, Milford, MA, USA) plus 0.1% gelatin (+++ solution). Briefly, 1 × 103 bacterial cells (in 10 μl) were preopsonized with infant rabbit serum (10 μl; Pel-Freez Biologicals, Rogers, AR, USA) for 30 min at 37°C. For analysis of the bactericidal capacity of UT12, we used neutrophils pretreated with UT12 and either DPI, an inhibitor of NADPH oxidase; AEBSF, a serine protease inhibitor; CHYM, an inhibitor of proteases, including chymotrypsin-like serine protease and lysosomal cysteine protease; or a neutrophil elastase inhibitor. Neutrophils were then added to reaction mixtures (1 × 105 cells/reaction mixture in 40 μl) with +++ solution (110 μl) and were incubated for 45 min at 37°C with rotation. Reactions were stopped by incubation at 4°C, and counts of viable bacteria were determined.
Bacterial uptake assay.
Phagocytosis was assessed as described by Lu et al. (21). Briefly, log-phase bacteria were pelleted, mixed for 1 h at 4°C with 1 ml FITC (1 mg/ml; Sigma-Aldrich) dissolved in a buffer containing 0.05 M sodium carbonate and 0.1 M sodium chloride, and then washed three times with Hanks buffer to remove unbound FITC. OPH assays were performed as described above, and the results were analyzed by flow cytometry. Fifty thousand events were collected per sample, and a single gate was used to exclude cell debris and free bacteria.
Detection of anti-Pseudomonas IgG levels in serum and IgA levels in BALF by ELISA.
PBS-washed whole bacteria diluted with coating buffer (0.015 M Na2CO3 and 0.035 M NaHCO3) to a final optical density at 620 nm (OD620) of 0.1 in 100 μl were added to Immulon 2HB 96-well plates (Thermo Scientific) and were fixed by overnight incubation at 4°C. Plates were washed with PBS containing 0.05% Brij 35 (Sigma-Aldrich), blocked with 100 μl 1% bovine serum albumin in PBS for 1 h, and washed again. Serum or BALF in 1% bovine serum albumin was then added in 10-fold serial dilutions overnight at 4°C. Antigen-specific antibodies were detected by alkaline phosphatase (Sigma-Aldrich)-conjugated goat anti-mouse IgG (serum) or IgA (BALF) for 1.5 h and developed with p-nitrophenyl phosphate (Sigma-Aldrich). The absorbance at 415 nm was determined after a standardized period of 1 h. The bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL, USA) was used to determine protein concentrations in BALF samples (22).
Statistical analyses.
All data were analyzed using Prism 5 (GraphPad Software) and are expressed as means ± standard errors of the means (SEM). Differences between the treatment group and the control group were tested for significance using the Mann-Whitney U test. Differences among more than three groups were examined using analysis of variance, followed by Tukey's post hoc test. A P value of <0.05 was considered to indicate a statistically significant difference.
RESULTS
UT12 promotes the clearance of P. aeruginosa from the LRT.
In this study, we investigated whether treatment with the TLR4/MD2 agonistic MAb UT12 could activate host immunity against P. aeruginosa cLRTI. The number of viable bacterial cells in the lungs (expressed as log10 CFU per lung) was significantly lower in UT12-treated mice than in control mice at 14 days (3.36 ± 0.35 versus 4.43 ± 0.30 [P < 0.05]), 21 days (2.77 ± 0.15 versus 4.87 ± 0.24 [P < 0.05]), and 28 days (1.67 ± 0.27 versus 5.27 ± 0.29 [P < 0.01]) (Fig. 2A). MPLA, commercialized as a TLR4 agonist by Sigma-Aldrich, showed a similar therapeutic effect (Fig. 2B). Fluorescence-activated cell sorting (FACS) analysis of BALF to measure the kinetics of inflammatory cell recruitment to the LRT (Fig. 3) showed that the numbers of neutrophils in the LRT were significantly higher in UT12-treated mice than in untreated mice, whereas macrophage numbers remained unchanged. These data indicated that the reduction in viable P. aeruginosa counts associated with cLRTI following UT12 administration was mediated, in part, by the facilitation of neutrophil migration to the infection source.
FIG 2.

CFU counts in the lungs of mice treated with UT12 (A) or MPLA (B) compared with those for the control group. The numbers of viable bacteria in the lungs were significantly reduced over time. Data represent means ± SEM. Asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) from the control (n = 5).
FIG 3.

FACS analysis of the number of inflammatory cells in bronchoalveolar lavage fluid. Ly6G+ CD45+ neutrophil counts in the UT12-treated group were elevated over those in the control group at each time point (A), but F4/80+ macrophage counts were not (B). (i) Day 14; (ii) day 21; (iii) day 28. Data represent means ± SEM. An asterisk indicates a significant difference (*, P < 0.05) from the control (n = 5).
Kinetics of cytokine and chemokine production in the lung after UT12 administration.
We analyzed cytokine production levels at 1 week after UT12 administration, since TLR4 stimulation is responsible for cytokine overproduction. No significant difference, except for MIP-2 concentrations, was observed in the lung homogenates of UT12-treated mice, and the production of inflammatory cytokines, such as IL-6 and TNF-α, was not altered (Fig. 4). To identify the cells underlying the observed effects of UT12, we depleted CD4+ T cells, which secrete many types of cytokines and chemokines, by administration of MAb GK1.5. The UT12-mediated enhancement of both neutrophil migration and MIP-2 production was lost 1 week after UT12 treatment (Fig. 5A and B); however, the density of P. aeruginosa remained significantly reduced (Fig. 5C). UT12 efficacy under conditions of CD4+ T cell depletion were still observed 28 days after P. aeruginosa inoculation (data not shown), indicating that CD4+ T cells underlie UT12-mediated effects but that they play a minor role in the clearance of P. aeruginosa from the LRT.
FIG 4.
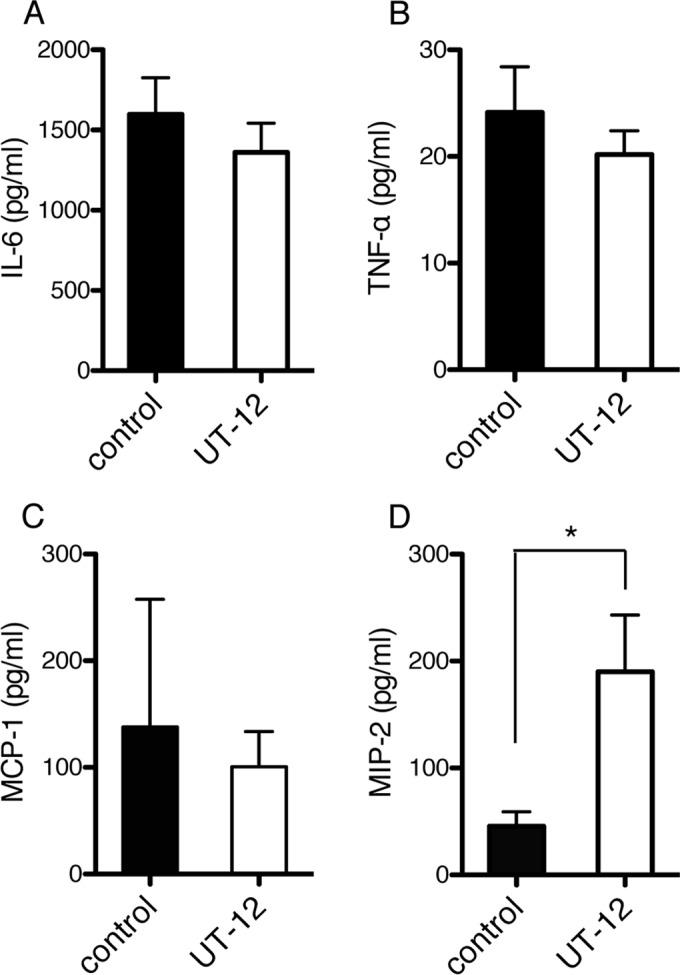
Levels of inflammatory cytokines and chemokines at 1 week after UT12 administration. No significant increase in the production of the inflammatory cytokine IL-6 (A) or TNF-α (B), or in the production of MCP-1 (C), was detected in the lung homogenates of UT12-treated mice relative to that for controls; however, increased MIP-2 levels (D) relative to those for controls were observed. Data represent means ± SEM. An asterisk indicates a significant difference (*, P < 0.05) from the control (n = 5).
FIG 5.

Influence of CD4+ T cell depletion on UT12-mediated protective effects and neutrophil migration. (A and B) The promotion of neutrophil migration (A) and MIP-2 production (B) mediated by UT12 was abolished by the administration of an anti-CD4 antibody. Data represent means ± SEM. (C) Bacterial counts in lungs were unchanged despite CD4+ T cell depletion, indicating the importance of Th cell-independent bactericidal effects. Asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) from the control (n, ∼5 to 8).
UT12 activates the OPH killing function in neutrophils.
To further investigate the effects of UT12 on neutrophil-dependent host protection, we examined neutrophil function using flow cytometry-based phagocytosis and OPH killing assays in vitro. FITC-bound P. aeruginosa was coincubated with murine peritoneal neutrophils, and phagocytic capacity was estimated by counting the number of FITC-Ly6G double-positive cells, which was significantly increased by UT12 treatment (Fig. 6A and B). Additionally, the significant reduction in the number of viable bacteria observed following coincubation with UT12-stimulated neutrophils was lost in Tlr4−/− mice (Fig. 6C). These data indicated that UT12 acts specifically on TLR4 to activate not only the migration of neutrophils but also their phagocytic and bactericidal functions.
FIG 6.

UT12 enhances the ability of neutrophils to phagocytose FITC-bound P. aeruginosa. (A and B) Representative results of FACS analyses of phagocytic reactions (A) and the percentage of FITC-positive Ly6G+ CD45+ cells (B) for neutrophils pretreated with UT12 and for control neutrophils. FACS plots are gated on CD45+ antigen-presenting cells (APCs). (C) Neutrophils were incubated with rabbit serum-opsonized P. aeruginosa, and survival was assessed following a 45-min incubation. The percentage of bacterial survival was calculated based on viable counts (CFU per milliliter) relative to counts for no-neutrophil controls. Counts of bacteria incubated with UT12-pretreated neutrophils from wild-type mice were significantly lower than counts of bacteria incubated with neutrophils from Tlr4−/− mice, indicating that the UT12-mediated enhancement of neutrophil bactericidal activity was dependent on TLR4. Data represent means ± SEM. An asterisk indicates a significant difference (*, P < 0.05) from the control.
UT12 activates the bactericidal effect of neutrophils via a serine protease-dependent pathway.
Neutrophils control bacterial infection through a combination of oxidative and nonoxidative (a mainly serine protease dependent pathway) mechanisms. To examine which bactericidal neutrophil components are activated by UT12, we pretreated neutrophils with various inhibitors, including DPI, AEBSF, CHYM, and a neutrophil elastase inhibitor, before the OPH assay. Figure 7 shows that the effect of UT12 in reducing the viable bacterial count disappeared only upon pretreatment of neutrophils with AEBSF or the neutrophil elastase inhibitor (Fig. 7B) and not upon pretreatment with DPI (Fig. 7A). This finding indicates that the nonoxidative pathway, specifically involving neutrophil elastase, might be important for UT12-mediated activation of the neutrophil-associated bactericidal effect. Taken together, although the mechanism has not been fully elucidated, UT12 improved bacterial clearance by prompting not only the migration of neutrophils but also their phagocytic killing function, mediated, at least in part, by neutrophil serine proteases via the TLR4 pathway.
FIG 7.

Serine proteases, especially neutrophil elastase, contribute to the UT12-mediated clearing of P. aeruginosa. Neutrophils were incubated with rabbit serum-opsonized P. aeruginosa, and survival was assessed following a 45-min incubation. The percentage of bacterial survival was calculated based on viable counts (CFU per milliliter) relative to those for no-neutrophil controls. Shown are the results obtained using neutrophils treated with the oxidative burst inhibitor DPI (A), the serine protease elastase inhibitor AEBSF, CHYM, or the neutrophil elastase inhibitor (B) before the OPH assay. The UT12-mediated reduction of viable bacterial counts was abolished by elastase inhibitor pretreatment, suggesting that the nonoxidative pathway, specifically involving neutrophil elastase, might be important for UT12-mediated bactericidal activation. An asterisk indicates a significant difference (*, P < 0.05) from non-UT12-treated neutrophils.
DISCUSSION
In this study, we found that the administration of TLR4/MD2 agonistic MAbs significantly improved airway clearance in a mouse model of cLRTI with P. aeruginosa. These antibodies both promoted neutrophil migration and activated nonoxidative bactericidal mechanisms. Chronic airway infection with P. aeruginosa is one of the best-known intractable infections that cannot be cured by normal antimicrobial chemotherapy. In clinical practice, tobramycin inhalation therapy not only is useful in improving lung function and reducing the frequency of hospitalization but also is well tolerated over long-term use (23). Additionally, the guidelines of the Cystic Fibrosis Foundation on the treatment of pulmonary exacerbations recommend this therapy for preserving lung function and preventing acute exacerbation of chronic airway infection (24). Nevertheless, once chronic, persistent P. aeruginosa infection becomes established, in most cases, complete bacterial eradication is extremely difficult to achieve. The activation of innate immunity by UT12 therapy might potentially be effective for treating intractable chronic infections that are difficult to cure with antibiotic treatment alone. TLR agonists are already in practical use as adjuvants for a range of vaccines against viruses and other infections. The TLR4 agonist MPLA is used as an adjuvant for the malaria vaccine (25), and its use in animal models as a booster for influenza (26) and HIV (27) vaccines, as well as its direct effects in a model of P. aeruginosa cutaneous-burn infection and bacteremia (13), has also been reported. Furthermore, we recently reported that UT12 inoculation improved the prognosis for secondary bacterial pneumonia following influenza (15). The anti-infective action of TLR agonists might involve immunological tolerance or activation, but the detailed mechanisms remain unknown. This study is the first to address the effect of TLR agonists on cLRTI caused by P. aeruginosa.
Neutrophils are an important part of the innate immune response against P. aeruginosa; however, host resistance is greatly diminished in neutrophil-deficient mice (28). In this study, among mice with persistent P. aeruginosa infection, those treated with UT12 exhibited significantly lower bacterial counts in the lungs over time than controls, whereas BALF neutrophil counts and MIP-2 production were significantly elevated. MIP-2 and keratinocyte chemoattractant are involved in neutrophil migration, and studies have found that mice inoculated with MIP-2- and keratinocyte chemoattractant-specific antibodies have higher rates of mortality from pneumonia and increased bacterial counts in the lungs (29). Romero et al. reported that MPLA promoted neutrophil migration at the site of infection and enhanced the clearance of P. aeruginosa in a murine model of systemic infection induced by cecal ligation and puncture (13). To identify the immune cells responsible for promoting UT12-mediated MIP-2 production, we carried out the same study using CD4+ T cell-depleted mice and found that both neutrophil counts and MIP-2 production diminished, as did the suppression of bacterial counts in the lungs. This suggested that UT12 acts primarily on CD4+ T cells to promote MIP-2 production and enhance neutrophil migration to the site of infection. We also investigated the effect of UT12 on humoral immunity, but UT12 administration did not activate the production of P. aeruginosa-specific antibody (as evidenced by BALF IgA and serum IgG levels [data not shown]). A previous study showed that the production of antigen-specific antibody was inhibited under the conditions of endotoxin tolerance induced by UT12 administration (30). Another reason why UT12 inhibited antigen-specific antibody production could be related to the reduction in the number of viable microbes during the observation period. Similarly, the reduction of viral titers by antiviral agents is responsible for the attenuation of antigen-specific IgA production in influenza virus-infected mice (31), suggesting that the inhibitory effect of UT12 on persistent P. aeruginosa infection observed in this study was dependent mainly on the activation of the innate immune system.
We hypothesized that since UT12 retained an effect on P. aeruginosa clearance even in the absence of CD4+ T cells, it likely enhanced neutrophil function beyond promoting migration. Accordingly, we isolated mouse peritoneal neutrophils and determined that UT12 pretreatment enhanced their phagocytic and bactericidal effects. Generally, neutrophils exert their bactericidal effect on pathogens via NADPH oxidase and serine proteases (neutrophil elastase, cathepsin G, and protease 3). Both serine protease-deficient and nitric oxide synthase-deactivated mice suffer more-serious pneumonia and have higher lung bacterial counts than wild-type (WT) mice (32, 33). Furthermore, nitric oxide reductase-deficient P. aeruginosa bacteria are rapidly killed (34). Using a range of different inhibitors, we found that only neutrophil elastase inhibition disrupted UT12-enhanced bactericidal effects, indicating the involvement of serine proteases, specifically neutrophil elastase, rather than the oxidative system. Serine proteases also affect host cells, causing organ damage; however, we did not observe signs of acute lung damage in pathological specimens from UT12-treated mice (data not shown). Neutrophil elastase might cause little tissue damage, although the inflammation with chronic infection was comparatively mild, without progression to neutrophil breakdown and excessive serine protease release. However, since all the mice died upon an increase in the UT12 dosage, the underlying mechanism should be investigated prior to clinical application.
This study had a number of limitations. First, we were unable to create a model of chronic persistent infection in Tlr4−/− mice, perhaps because TLR4 is considered essential for mucosal oversecretion, and its absence prevented the establishment of infection (35). Second, we were unable to carry out studies on neutrophil- and macrophage-deficient mice to identify the direct targets of UT12 action, because the mice died rapidly following peritoneal inoculation of various cell-specific antibodies. This suggested that chronic persistent infection progresses to fatal infection in the event of immune cell deficiency. Third, we did not investigate the action of UT12 on other components of innate immunity apart from neutrophils. Previous studies reported that the functions of several types of immune cells were modified via TLR4-dependent stimulation (30, 36–38). We were unable to ascertain whether the clinical efficacy of UT12 was due to its effect on neutrophils alone or resulted from its action on multiple immune response systems.
Our findings suggested that the TLR4/MD-2 agonistic antibody UT12 might be effective against chronic P. aeruginosa respiratory tract infection. Other pathogens that cause similar cLRTI include Acinetobacter spp. and Haemophilus influenzae, Staphylococcus aureus (including methicillin-resistant Staphylococcus aureus), and nontuberculous Mycobacterium spp. The activation of innate immunity by UT12 therapy, therefore, might potentially be effective for treating a spectrum of intractable chronic infections that are difficult to cure with antibiotic treatment alone, offering a new strategy for the treatment of infection.
ACKNOWLEDGMENT
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
REFERENCES
- 1.Sordé R, Pahissa A, Rello J. 2011. Management of refractory Pseudomonas aeruginosa infection in cystic fibrosis. Infect Drug Resist 4:31–41. doi: 10.2147/IDR.S16263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenfeld M, Gibson RL, McNamara S, Emerson J, Burns JL, Castile R, Hiatt P, McCoy K, Wilson CB, Inglis A, Smith A, Martin TR, Ramsey BW. 2001. Early pulmonary infection, inflammation, and clinical outcomes in infants with cystic fibrosis. Pediatr Pulmonol 32:356–366. doi: 10.1002/ppul.1144. [DOI] [PubMed] [Google Scholar]
- 3.Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL. 2002. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol 34:91–100. doi: 10.1002/ppul.10127. [DOI] [PubMed] [Google Scholar]
- 4.Broad A, Jones DE, Kirby JA. 2006. Toll-like receptor (TLR) response tolerance: a key physiological “damage limitation” effect and an important potential opportunity for therapy. Curr Med Chem 13:2487–2502. doi: 10.2174/092986706778201675. [DOI] [PubMed] [Google Scholar]
- 5.Ribes S, Ebert S, Regen T, Agarwal A, Tauber SC, Czesnik D, Spreer A, Bunkowski S, Eiffert H, Hanisch UK, Hammerschmidt S, Nau R. 2010. Toll-like receptor stimulation enhances phagocytosis and intracellular killing of nonencapsulated and encapsulated Streptococcus pneumoniae by murine microglia. Infect Immun 78:865–871. doi: 10.1128/IAI.01110-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphey ED, Fang G, Sherwood ER. 2008. Endotoxin pretreatment improves bacterial clearance and decreases mortality in mice challenged with Staphylococcus aureus. Shock 29:512–518. doi: 10.1097/SHK.0b013e318150776f. [DOI] [PubMed] [Google Scholar]
- 7.Wynn JL, Scumpia PO, Winfield RD, Delano MJ, Kelly-Scumpia K, Barker T, Ungaro R, Levy O, Moldawer LL. 2008. Defective innate immunity predisposes murine neonates to poor sepsis outcome, but is reversed by TLR agonists. Blood 112:1750–1758. doi: 10.1182/blood-2008-01-130500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cavaillon JM, Adrie C, Fitting C, Adib-Conquy M. 2003. Endotoxin tolerance: is there a clinical relevance? J Endotoxin Res 9:101–107. doi: 10.1177/09680519030090020501. [DOI] [PubMed] [Google Scholar]
- 9.Cross AS. 2002. Endotoxin tolerance—current concepts in historical perspective. J Endotoxin Res 8:83–98. doi: 10.1177/09680519020080020201. [DOI] [PubMed] [Google Scholar]
- 10.Thoelen S, Van Damme P, Mathei C, Leroux-Roels G, Desombere I, Safary A, Vandepapeliere P, Slaoui M, Meheus A. 1998. Safety and immunogenicity of a hepatitis B vaccine formulated with a novel adjuvant system. Vaccine 16:708–714. doi: 10.1016/S0264-410X(97)00254-5. [DOI] [PubMed] [Google Scholar]
- 11.De Becker G, Moulin V, Pajak B, Bruck C, Francotte M, Thiriart C, Urbain J, Moser M. 2000. The adjuvant monophosphoryl lipid A increases the function of antigen-presenting cells. Int Immunol 12:807–815. doi: 10.1093/intimm/12.6.807. [DOI] [PubMed] [Google Scholar]
- 12.Kamphuis T, Stegmann T, Meijerhof T, Wilschut J, de Haan A. 2013. A virosomal respiratory syncytial virus vaccine adjuvanted with monophosphoryl lipid A provides protection against viral challenge without priming for enhanced disease in cotton rats. Influenza Other Respir Viruses 7:1227–1236. doi: 10.1111/irv.12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Romero CD, Varma TK, Hobbs JB, Reyes A, Driver B, Sherwood ER. 2011. The Toll-like receptor 4 agonist monophosphoryl lipid A augments innate host resistance to systemic bacterial infection. Infect Immun 79:3576–3587. doi: 10.1128/IAI.00022-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohta S, Bahrun U, Shimazu R, Matsushita H, Fukudome K, Kimoto M. 2006. Induction of long-term lipopolysaccharide tolerance by an agonistic monoclonal antibody to the Toll-like receptor 4/MD-2 complex. Clin Vaccine Immunol 13:1131–1136. doi: 10.1128/CVI.00173-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanaka A, Nakamura S, Seki M, Fukudome K, Iwanaga N, Imamura Y, Miyazaki T, Izumikawa K, Kakeya H, Yanagihara K, Kohno S. 2013. Toll-like receptor 4 agonistic antibody promotes innate immunity against severe pneumonia induced by coinfection with influenza virus and Streptococcus pneumoniae. Clin Vaccine Immunol 20:977–985. doi: 10.1128/CVI.00010-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yanagihara K, Tomono K, Sawai T, Hirakata Y, Kadota J, Koga H, Tashiro T, Kohno S. 1997. Effect of clarithromycin on lymphocytes in chronic respiratory Pseudomonas aeruginosa infection. Am J Respir Crit Care Med 155:337–342. doi: 10.1164/ajrccm.155.1.9001333. [DOI] [PubMed] [Google Scholar]
- 17.Yanagihara K, Seki M, Cheng PW. 2001. Lipopolysaccharide induces mucus cell metaplasia in mouse lung. Am J Respir Cell Mol Biol 24:66–73. doi: 10.1165/ajrcmb.24.1.4122. [DOI] [PubMed] [Google Scholar]
- 18.Xing JZ, Yang X, Xu P, Ang WT, Chen J. 2012. Ultrasound-enhanced monoclonal antibody production. Ultrasound Med Biol 38:1949–1957. doi: 10.1016/j.ultrasmedbio.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 19.Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. 2010. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med 16:228–231. doi: 10.1038/nm.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vishwanath S, Ramphal R, Guay CM, DesJardins D, Pier GB. 1988. Respiratory-mucin inhibition of the opsonophagocytic killing of Pseudomonas aeruginosa. Infect Immun 56:2218–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu L, Ma Z, Jokiranta TS, Whitney AR, DeLeo FR, Zhang JR. 2008. Species-specific interaction of Streptococcus pneumoniae with human complement factor H. J Immunol 181:7138–7146. doi: 10.4049/jimmunol.181.10.7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roche AM, King SJ, Weiser JN. 2007. Live attenuated Streptococcus pneumoniae strains induce serotype-independent mucosal and systemic protection in mice. Infect Immun 75:2469–2475. doi: 10.1128/IAI.01972-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramsey BW, Pepe MS, Quan JM, Otto KL, Montgomery AB, Williams-Warren J, Vasiljev-K M, Borowitz D, Bowman CM, Marshall BC, Marshall S, Smith AL, for the Cystic Fibrosis Inhaled Tobramycin Study Group . 1999. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. N Engl J Med 340:23–30. [DOI] [PubMed] [Google Scholar]
- 24.Flume PA, Mogayzel PJ Jr, Robinson KA, Goss CH, Rosenblatt RL, Kuhn RJ, Marshall BC, Clinical Practice Guidelines for Pulmonary Therapies Committee . 2009. Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Am J Respir Crit Care Med 180:802–808. doi: 10.1164/rccm.200812-1845PP. [DOI] [PubMed] [Google Scholar]
- 25.Moon JJ, Suh H, Li AV, Ockenhouse CF, Yadava A, Irvine DJ. 2012. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proc Natl Acad Sci U S A 109:1080–1085. doi: 10.1073/pnas.1112648109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patil HP, Murugappan S, ter Veer W, Meijerhof T, de Haan A, Frijlink HW, Wilschut J, Hinrichs WLJ, Huckriede A. 2014. Evaluation of monophosphoryl lipid A as adjuvant for pulmonary delivered influenza vaccine. J Control Release 174:51–62. doi: 10.1016/j.jconrel.2013.11.013. [DOI] [PubMed] [Google Scholar]
- 27.Pouliot K, Buglione-Corbett R, Marty-Roix R, Montminy-Paquette S, West K, Wang S, Lu S, Lien E. 2014. Contribution of TLR4 and MyD88 for adjuvant monophosphoryl lipid A (MPLA) activity in a DNA prime-protein boost HIV-1 vaccine. Vaccine 32:5049–5056. doi: 10.1016/j.vaccine.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koh AY, Priebe GP, Ray C, Van Rooijen N, Pier GB. 2009. Inescapable need for neutrophils as mediators of cellular innate immunity to acute Pseudomonas aeruginosa pneumonia. Infect Immun 77:5300–5310. doi: 10.1128/IAI.00501-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsai WC, Strieter RM, Mehrad B, Newstead MW, Zeng X, Standiford TJ. 2000. CXC chemokine receptor CXCR2 is essential for protective innate host response in murine Pseudomonas aeruginosa pneumonia. Infect Immun 68:4289–4296. doi: 10.1128/IAI.68.7.4289-4296.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rachmawati NM, Fukudome K, Tsuneyoshi N, Bahrun U, Tsukamoto H, Yanagibashi T, Nagai Y, Takatsu K, Ohta S, Kimoto M. 2013. Inhibition of antibody production in vivo by pre-stimulation of Toll-like receptor 4 before antigen priming is caused by defective B-cell priming and not impairment in antigen presentation. Int Immunol 25:117–128. doi: 10.1093/intimm/dxs096. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi E, Kataoka K, Fujii K, Chida J, Mizuno D, Fukui M, Ito H, Fujihashi K, Kido H. 2010. Attenuation of inducible respiratory immune responses by oseltamivir treatment in mice with influenza A virus. Microbes Infect 12:778–783. doi: 10.1016/j.micinf.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 32.Hirche TO, Benabid R, Deslee G, Gangloff S, Achilefu S, Guenounou M, Lebargy F, Hancock RE, Belaaouaj A. 2008. Neutrophil elastase mediates innate host protection against Pseudomonas aeruginosa. J Immunol 181:4945–4954. doi: 10.4049/jimmunol.181.7.4945. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y, Li X, Carpinteiro A, Goettel JA, Soddemann M, Gulbins E. 2011. Kinase suppressor of Ras-1 protects against pulmonary Pseudomonas aeruginosa infections. Nat Med 17:341–346. doi: 10.1038/nm.2296. [DOI] [PubMed] [Google Scholar]
- 34.Kakishima K, Shiratsuchi A, Taoka A, Nakanishi Y, Fukumori Y. 2007. Participation of nitric oxide reductase in survival of Pseudomonas aeruginosa in LPS-activated macrophages. Biochem Biophys Res Commun 355:587–591. doi: 10.1016/j.bbrc.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 35.Chen L, Wang T, Zhang JY, Zhang SF, Liu DS, Xu D, Wang X, Chen YJ, Wen FQ. 2009. Toll-like receptor 4 relates to lipopolysaccharide-induced mucus hypersecretion in rat airway. Arch Med Res 40:10–17. doi: 10.1016/j.arcmed.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 36.Wolk K, Döcke WD, von Baehr V, Volk HD, Sabat R. 2000. Impaired antigen presentation by human monocytes during endotoxin tolerance. Blood 96:218–223. [PubMed] [Google Scholar]
- 37.Matsushita H, Ohta S, Shiraishi H, Suzuki S, Arima K, Toda S, Tanaka H, Nagai H, Kimoto M, Inokuchi A, Izuhara K. 2010. Endotoxin tolerance attenuates airway allergic inflammation in model mice by suppression of the T-cell stimulatory effect of dendritic cells. Int Immunol 22:739–747. doi: 10.1093/intimm/dxq062. [DOI] [PubMed] [Google Scholar]
- 38.del Fresno C, García-Rio F, Gómez-Piña V, Soares-Schanoski A, Fernández-Ruíz I, Jurado T, Kajiji T, Shu C, Marín E, Gutierrez del Arroyo A, Prados C, Arnalich F, Fuentes-Prior P, Biswas SK, López-Collazo E. 2009. Potent phagocytic activity with impaired antigen presentation identifying lipopolysaccharide-tolerant human monocytes: demonstration in isolated monocytes from cystic fibrosis patients. J Immunol 182:6494–6507. doi: 10.4049/jimmunol.0803350. [DOI] [PubMed] [Google Scholar]