

Abstract
Most autotransporter passenger domains, regardless of their diversity in function, fold or are predicted to fold as right-handed β-helices carrying various loops that are presumed to confer functionality. Our goal here was to identify the subdomain (loop) or amino acid sequence of the Pet passenger domain involved in the receptor binding site on the host cell for Pet endocytosis. Here, we show that d1 and d2 subdomains, as well as the amino acid sequence linking the subdomain d2 and the adjacent β-helix (PDWET), are not required for Pet secretion through the autotransporter system and that none of our deletion mutants altered the predicted long right-handed β-helical structure. Interestingly, Pet lacking the d2 domain (PetΔd2) was unable to bind on the epithelial cell surface, in contrast to Pet lacking d1 (PetΔd1) subdomain or PDWET sequences. Moreover, the purified d1 subdomain, the biggest subdomain (29.8 kDa) containing the serine protease domain, was also unable to bind the cell surface. Thus, d2 sequence (54 residues without the PDWET sequence) was required for Pet binding to eukaryotic cells. In addition, this d2 sequence was also needed for Pet internalization but not for inducing cell damage. In contrast, PetΔd1, which was able to bind and internalize inside the cell, was unable to cause cell damage. Furthermore, unlike Pet, PetΔd2 was unable to bind cytokeratin 8, a Pet receptor. These data indicate that the surface d2 subdomain is essential for the ligand-receptor (Pet-Ck8) interaction for Pet uptake and to start the epithelial cell damage by this toxin.
INTRODUCTION
The type V secretion system or autotransporter (AT) protein, including several variants, such as Va, Vb, Vc, Vd, and Ve, is the most common mechanism used to release virulence factors by Gram-negative bacteria (1, 2). The AT proteins promote their own secretion through the inner and outer membranes by using two preprotein processing domains: the signal sequence and the translocation unit (2). The precursor protein contains an N-terminal signal sequence which mediates Sec-dependent protein export into the periplasm, a passenger domain encoding the effector function, and a C-terminal domain mediating the translocation of the passenger domain across the outer membrane (2). The type Va secretion system is responsible for releasing a growing family of high-molecular-weight serine proteases into the external milieu, known as serine protease ATs of the Enterobacteriaceae (SPATE) family (1). The SPATE superfamily of virulence factors has been phylogenetically divided into two distinct classes based on the amino acid sequence of the passenger domain: class 1 SPATE are cytotoxic, whereas class 2 SPATE are lectin-like immunomodulators. Independent of their substrate or cleavage sites, class 1 SPATE have a common ability to cause cytopathic effects in cultured cells and display enterotoxin activity (3–7). Pet, Sat, EspC, and SigA class 1 SPATE show a higher identity/similarity (50 to 70%) than any other member of their class, which may explain the comparable protease strengths on their shared biological substrate, the actin-binding protein α-fodrin (α-spectrin) (3, 4, 8, 9).
Despite their diversity in function, most AT passenger domains fold or are predicted to fold as right-handed β-helices (10–14). Usually, the β-helix forms a backbone to which additional, functional subdomains are attached. This β-helical structure is capped by a C-terminal region, which emerges into the medium first (15). This region forms a structure unique to the AT family and is known as the junction or “autochaperone” (AC) due to its inferred role in folding the entire passenger. Although the domain organization and, to a certain degree, the three-dimensional structure of ATs is conserved, their sequences show only weak homology (16). Thus, all AT proteins are highly modular proteins, and it is clear that there is considerable variation in the passenger domains themselves (17), clearly suggesting that their different subdomains have a specialized functional role. These loops have very little sequence similarity but show similar positions and interactions (18). Pertactin from Bordetella pertussis, the first AT structure solved by X-ray crystallography, is an adherent factor for host cells by using a three-residue “RGD motif” projecting from the β-helix, which enables it to bind integrins. Additionally, just after the RGD motif is found a 5-fold repeat of the sequence GGXXP, which may also have been involved in binding to host cells (10). In the VacA p55 structure from Helicobacter pylori, the loops of the β-helix define different target cell specificity (13), and two long C-terminal loops denote the receptor binding site for all VacA subtypes, whereas residues near the N terminus have been shown to be involved in cell vacuolization and membrane depolarization but not in cell binding (19, 20). The SPATE, unlike pertactin and VacA, are more highly decorated with surface structures, including domain 1 (d1), the largest one containing the serine protease motif.
Pet is the most studied prototype class 1 SPATE (21). Pet is a 108-kDa heat-labile enterotoxin (5) and cytotoxin (22), phenotypes that are dependent on the proteolytic activity mediated by the catalytic serine protease motif in the Pet passenger domain (5, 22). Cell intoxication by Pet is associated with the ability of Pet to cleave the actin-binding protein α-fodrin and results in the rounding and detachment of cells from the substratum, cytoskeleton contraction, the loss of actin stress fibers, and the release of focal contacts (8). In addition to its serine protease motif, Pet intoxication requires toxin endocytosis for reaching the intracellular target (23). Pet binds cytokeratin 8 on the epithelial cell surface and is internalized by receptor-mediated endocytosis using clathrin-coated vesicles (24). Once inside the cell, Pet is retrograde trafficked by vesicle carriers from the cell surface to endosomes, from the endosomes to the Golgi apparatus, and from the Golgi apparatus to the endoplasmic reticulum (ER). Finally, it escapes from the ER using the sec translocon to gain the cytosol (25), where it comes into close contact with its α-fodrin substrate to initiate the cascade of events that lead to cell detachment (8). In this work, we identified the subdomain of the Pet passenger domain involved in the receptor binding site on host cells for Pet endocytosis by using subdomain deletion mutants and by detecting their binding to the epithelial cell surface, cell internalization, cell damage, and the ability to serve as a ligand for cytokeratin 8, a Pet receptor.
MATERIALS AND METHODS
Bacterial strains and plasmids.
Escherichia coli HB101 was used to express the minimal pet clone (pCEFN1) as previously described (5), as well as the different constructions derived from this clone (pCEFN4, pCEFN5, and pCEFN6) (Table 1). The minimal Pet clone contains the encoded region of pet, which was cloned into the BamHI and KpnI sites of pSPORT1. The XL1-Blue strain was used for genetic manipulations and BL21(DE3)pLysS for expression of Pet domain 1 (d1). All the strains were routinely cultivated in Luria-Bertani (LB) broth or LB agar supplemented with ampicillin (100 μg/ml) or chloramphenicol (35 μg/ml) as required. All strains were kept at −78°C in LB medium with 15% glycerol.
TABLE 1.
Bacterial strains, plasmids, and primers used in this study
| Strain or plasmid | Description and/or primer (sequence [5′-3′]) | Source or reference |
|---|---|---|
| Strains | ||
| HB101 | K-12/B hybrid | 5 |
| BL21(DE3)pLysS | Expression strain | Invitrogen |
| Plasmids | ||
| pCEFN1 | Minimal pet clone, cloned into the BamHI and KpnI sites of pSPORT1 | 5 |
| pCEFN4 | PetΔd1 derived from pCEFN1 | This study |
| AvrII-PetΔd1-forward (GTACCTAGGAGCTCTATAACTATTGGCAATACAACTCAAG) | ||
| AvrII-PetΔd1-reverse (CGCCTAGGAGATCTATTGGCGGCATATATTATATTAGTATAACTAA) | ||
| pCEFN5 | PetΔd2 derived from pCEFN1 | This study |
| BglII-PetΔd2-forward (GGAAGATCTACCAGAAAATTTAGATTCGACAATCTG) | ||
| BglII-PetΔd2-reverse (GGAAGATCTGCCCTGCATTACCAGAGGGGC) | ||
| pCEFN6 | PetΔPDWET derived from pCEFN1 | This study |
| BglII-PetΔPDWET-forward (CGCGCGAGATCTAGAAAATTTAGATTCGAC) | ||
| BglII-PetΔPDWET-reverse (CGCAGATCTCTGGGACAAATCGGAAA) | ||
| pLCh1 | Pet d1 domain cloned into pRSET-A | This study |
| BglII-dom1-forward (GAAGATCTGCCAATATGGATATATCTAAAGCATGGGC) | ||
| KpnI-dom1-reverse (GGGGTACCCTTACCACCACCAATGGTAGCAG) |
Molecular cloning and constructions for recombinant proteins.
All genetic manipulations were performed according to habitual methods (26). Plasmid DNA was extracted using the Wizard Plus SV Minipreps DNA purification system (Promega, Madison, WI). Purification of DNA fragments and their incision from agarose gels was performed using a QIAquick gel extraction kit (Qiagen, Inc.). Plasmid DNA was inserted into E. coli HB101, XL1-Blue, or BL21(DE3)pLysS by chemically transforming (calcium chloride) competent bacteria.
pCEFN4 (PetΔd1), pCEFN5 (PetΔd2), and pCEFN6 (PetΔPDWET) were derived from pCEFN1 (Pet) (Table 1) and were obtained by PCR using the following oligonucleotide sequences: PetΔd1, 5′-GTACCTAGGAGCTCTATAACTATTGGCAATACAACTCAAG-3′ (forward) and 5′-CGCCTAGGAGATCTATTGGCGGCATATATTATATTAGTATAACTAA-3′ (reverse), both containing AvrII sites; PetΔd2, 5′-GGAAGATCTACCAGAAAATTTAGATTCGACAATCTG-3′ (forward) and 5′-GGAAGATCTGCCCTGCATTACCAGAGGGGC-3′ (reverse), both containing BglII sites; and PetΔPDWET, 5′-CGCGCGAGATCTAGAAAATTTAGATTCGAC-3′ (forward) and 5′-CGCAGATCTCTGGGACAAATCGGAAA-3′ (reverse), both containing BglII sites.
For each construction, deletions were performed by ligation of PCR amplification products obtained by the use of forward and reverse oligonucleotides that hybridize with adjacent sequences at the end and just before the region to be deleted, respectively (Table 1). Thus, the pet gene and pSPORT1 (where pet is cloned) were amplified completely except for the sequence to be deleted. Therefore, we obtained the desired deletion construction by ligating the amplification products, which were expressed in E. coli HB101. Penicillin-resistant transformant clones were confirmed by using restriction enzymes and the PDWET deletion by sequencing.
For cloning the Pet d1 domain (pRSET-A-d1), d1 was amplified by PCR using pCEFN1 as the template and the oligonucleotides 5′-GAAGATCTGCCAATATGGATATATCTAAAGCATGGGC-3′ (forward) and 5′-GGGGTACCCTTACCACCACCAATGGTAGCAG-3′ (reverse) containing BglII and KpnI sites (Table 1). The amplification product (822 bp) was purified from an agarose gel and cloned between BglII and KpnI sites in the pRSET-A vector, resulting in pRSET-A-d1, now called pLCh1. This plasmid was used to transform E. coli XL1-Blue, and the transforming plasmid was detected by PCR using restriction enzymes.
Structure models.
Prediction models for the proteins PetΔd1, PetΔd2, and PetΔPDWET were performed using the crystallographic structure of Pet (PBD 4OM9) (27) and the RaptorX structure prediction server (28). The PBD files were modeled using the SWISS PDB viewer 4.1 software.
Expression and secretion of mature proteins.
To obtain Pet, PetΔd1, PetΔd2, and PetΔPDWET, bacterial strains were grown overnight at 37°C with shaking (160 rpm) in LB broth containing ampicillin. The cultures were then centrifuged at 3,124 × g at 4°C for 30 min. The supernatants were filtered through 0.22-μm-pore-size filters and concentrated 100-fold through Amicon Ultra filters with a cutoff of 50 or 100 kDa (Millipore) to obtain the secreted proteins. Proteins were quantified by the Bradford method and analyzed by 10% SDS–PAGE gels and Western blotting with anti-Pet antibodies and goat anti-rabbit IgG conjugated to horseradish peroxidase (HRP; Invitrogen). The reaction was detected using an Immobilon Western Chemiluminescent HRP substrate kit (Millipore).
Pet d1 protein was obtained as follows. A colony of E. coli BL21(pLCh1) cells was grown in LB broth supplemented with ampicillin and chloramphenicol at 37°C overnight. The overnight culture was inoculated into LB broth containing ampicillin at an optical density at 600 nm (OD600) of 0.1. Cultures were grown at 37°C with vigorous shaking until achieving an OD600 of 0.7 to 0.8 and then were induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 5 h. Cultures were centrifuged at 3,124 × g at 4°C for 20 min. Each pellet was resuspended in 1 ml of lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 5 mM imidazole [pH 8]) supplemented with 0.05 g of sarcosyl and mixed using a vortex mixer. Samples were sonicated twice at an amplitude of 50% for 15 s and then centrifuged at 9,000 × g at 4°C for 15 min. Each pellet was resuspended in lysis buffer and sonicated three times at 70% amplitude for 45 s. The samples were centrifuged at 20,598 × g at 4°C for 15 min, the supernatants were diluted to reach 10 ml in lysis buffer, and the mixture was incubated 1 h with shaking at 4°C. The d1 domain with the His tag was purified by affinity chromatography in nickel-nitrilotriacetic acid (nickel-NTA) agarose columns according to the manufacturer's instructions (Qiagen). The d1 domain protein was dialyzed against phosphate-buffered saline (PBS). The protein concentration was determined by the Bradford method and was analyzed by SDS-PAGE and Western blotting as indicated above.
Cell culture.
HEp-2 cells (from human larynx) and HT-29 cells (human colorectal adenocarcinoma) were propagated in humidified 5% CO2–95% air at 37°C in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum (HyClone, Logan, UT), 1% nonessential amino acids, 5 mM l-glutamine, penicillin (100 U/ml), and streptomycin (100 μg/ml). Subcultures were serially propagated after harvesting with 10 mM EDTA and 0.25% trypsin (Gibco-BRL) in PBS (pH 7.4). For fluorescence experiments, subconfluent cells were resuspended with EDTA-trypsin, plated in eight-well Lab-Tek slides (VWR, Bridgeport, NJ), and allowed to grow ∼24 h to 70% confluence before use.
Fluorescence assays.
Pet or Pet constructions were directly diluted into tissue culture medium without antibiotics or serum at a final concentration of 20 μg/ml, which was adjusted by densitometry using the purified Pet protein as a standard and then added to the target cells (HEp-2 and HT-29 cells) at a final volume of 250 μl per well in eight-well Lab-Tek slides. After the incubation for the indicated times in a humidified atmosphere of 5% CO2–95% air at 37°C, the medium was aspirated, the cells were washed twice with PBS, and 2% formalin in PBS was added for 20 min at room temperature. Fixed cells were either not permeabilized or permeabilized by adding 0.2% Triton X-100 in PBS for 5 min at room temperature. Actin filaments were visualized in the permeabilized cells by incubation with TRITC (tetramethyl rhodamine isothiocyanate)-phalloidin at 0.05 μg/ml for 30 min at room temperature (29), and nuclear DNA was stained with TO-PRO-3 (Invitrogen). Pet proteins were visualized by incubation with rabbit anti-Pet antibodies for 1 h at room temperature, followed by incubation with the secondary antibody (fluorescein isothiocyanate-goat anti-rabbit IgG) for 1 h at room temperature. Slides were mounted on Gelvatol, covered with a glass coverslip, and examined under a Leica TCS SP5 confocal microscope at a magnification of ×100 or ×63.
ELISA.
Pet or Pet constructions binding to recombinant Ck8 (rCk8) were immunodetected with a standard enzyme-linked immunosorbent assay (ELISA) method (30). Recombinant Ck8 (bioWORLD, Dublin, OH) at 30 nM in coating buffer (0.5 M sodium bicarbonate, 0.5 M sodium carbonate [pH 9.5]) was adsorbed to 96-well plates overnight at 4°C. Pet or Pet constructions (300 nM) bound to rCK8 were detected by ELISA using primary antibodies against Pet and the HRP-conjugated secondary antibody (goat anti-rabbit poly-HRP; Thermo Scientific). Bovine serum albumin (BSA; 150 nM) was also adsorbed to 96-well plates and used as an irrelevant antigen. The absorbance values for the enzymatic reactions at 490 nm were registered in a model 550 ELISA microplate reader (Bio-Rad).
RESULTS
Pet protein lacking d1 and d2 domains are expressed and extracellularly secreted by E. coli.
Pet protein is formed by the three main domains of the AT family (type Va secretion system): the signal sequence, the passenger domain, and the translocation unit (β-barrel), comprising residues 1 to 52, residues 53 to 1018, and residues 1019 to 1295, respectively (Fig. 1A). According to E. coli AT hemoglobin protease (Hbp) nomenclature, the passenger domain can be divided into at least seven subdomains: the d1 domain, a huge β-helix, which is interrupted by d2 and d4 domains, followed by the d5 domain, the autochaperone domain, and an α-helix domain (Fig. 1A). In order to determine which domain in the passenger domain is involved in Pet binding to host cells, three Pet deletion mutants were initially constructed: PetΔd1, PetΔd2, and PetΔPDWET (Fig. 1A). All the constructions were analyzed in silico to predict their three-dimensional structures by comparison to the native Pet protein (27). According to the models generated by using RaptorX structure prediction, none of the deletion mutants was predicted to have lost its hallmark feature shared by several AT proteins, characterized by a central β-helical stem decorated with several discursive subdomains, except for the respective deleted subdomain (Fig. 1B).
FIG 1.

Schematic representation of Pet protein and constructions containing deletions in specific domains of Pet protein. (A) Using Hbp nomenclature, a representation of the different domains harbored by Pet protein was prepared. Inside the passenger domain (residues 52 to 1018) at least seven domains can be identified. To determine which domain is involved in the binding to host cells, three mutants were constructed by deleting the d1 domain, the d2 domain, and five residues (PDWET) shared by the d2 domain and the β-helix. (B) Three-dimensional models of native Pet and the various constructions. The d1, β-helix, d2, and d4 domains are colored blue, green, red, and magenta, respectively, as indicated in the schematic representations. Models were generated using RaptorX structure prediction and are based on the structure resolved for the Pet protein.
All the constructions deleted by using inverse PCR from pCEFN1 (containing the pet gene in pSPORT1) were expressed in E. coli HB101. Deletions in d1 and d2 domains and in the residues 625PDWET630 were not required for Pet secretion into the extracellular medium, since all the constructions were expressed and secreted by the host bacteria, as detected in concentrated supernatants (50-kDa-retention filter) by Western blotting with anti-Pet antibodies (Fig. 2A). Pet protein was detected as a band of ∼104 kDa, whereas the deletion mutants were also detected as bands at the expected molecular masses: PetΔd1 of 75 kDa lacking the initial largest loop (residues 52 to 326), which contains the serine protease motif (GDSGS260P), PetΔd2 of 97 kDa lacking the second loop (residues 571 to 629), and PetΔPDWET of 103 kDa lacking four residues of the last amino acids of the d2 domain and the first one (T) of the next β-helix fragment. The PDWET sequence has been speculated to mediate interactions with other molecules, either of the host organism or of the parent bacterium (31).
FIG 2.
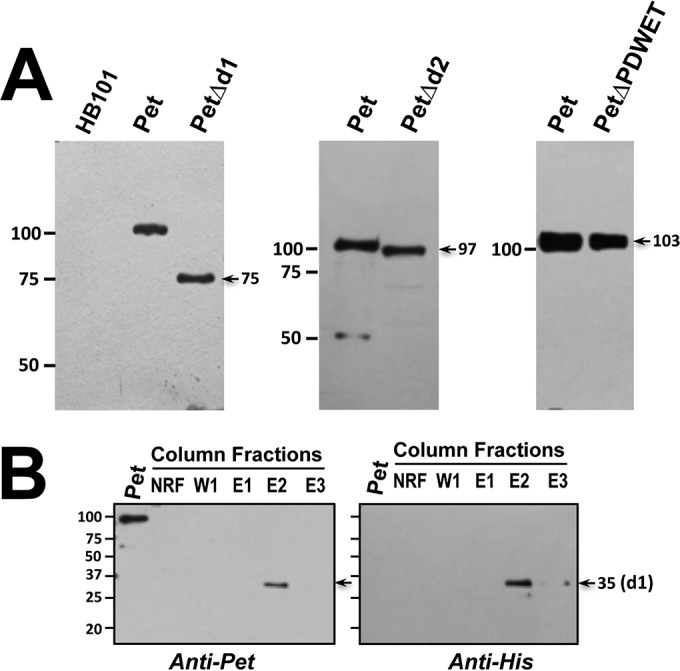
Expression of Pet protein and its deletion mutants. (A) Supernatants from overnight cultures of HB101(pCEFN1), HB101(pCEFN4), HB101(pCEFN5), and HB101(pCEFN6) were concentrated and fractionated with a 50-kDa retention filter to obtain Pet, PetΔd1, PetΔd2, and PetΔPDWET proteins. These four proteins were detected by Western blotting with antibodies against Pet. The sizes of marker proteins are indicated in kilodaltons on the left, and the sizes of the constructs are indicated on the right. (B) To express the d1 domain, the clone BL21(pLCh1), containing the Pet d1 domain and a His tag, was lysed, and the protein was purified using nickel-NTA agarose columns. Eluted fractions were analyzed by Western blotting with either anti-Pet or anti-His antibodies. NRF, nonretained fraction; W1, first wash; E1, E2, and E3, elutions 1, 2, and 3, respectively. These fractions were eluted with 200 mM imidazole.
Since the d1 domain is the largest one (35 kDa) and contains the serine protease motif, it is reasonable to think that this domain could contain a sequence for Pet binding to host cells. Thus, in addition to constructing the mutant with a mutation in the d1 domain, we constructed a clone [BL21(pLCh1)] containing the d1 domain by cloning this domain into pRSET-A, which has a His tag and was expressed in BL21(DE3)pLysS. The Pet d1 protein was efficiently purified using nickel-NTA agarose columns. As shown in Fig. 2B, Pet d1 (35 kDa) was eluted in the fraction E2 and detected by Western blotting with anti-Pet antibodies, as well as native Pet (104 kDa), which was used as a positive control, whereas the same band of 35 kDa was detected with anti-His antibodies but not native Pet, since it does not have a His tag.
The Pet d2 domain contains the site of Pet binding to the plasma membrane of epithelial cells.
In order to determine the site of Pet binding to epithelial cells, native Pet (used as a positive control), PetΔd1, PetΔd2, PetΔPDWET, and Pet d1 (20 μg/ml) were incubated with epithelial cells, which had been slightly prefixed to avoid Pet endocytosis, and then processed (but not permeabilized) and analyzed by confocal microscopy. Nonpermeabilized cells and the use of maximum projections of confocal optical sections (about 20 optical sections of 0.3 μm) allowed us to detect the binding of Pet to the cell surface as previously published (24), whereas the use of EspC, another SPATE that is known to lack a membrane receptor, was used as a negative control (32). Using this approach, we were able to find Pet, PetΔd1, and PetΔPDWET bound to both the HEp-2 and HT-29 epithelial cell lines. However, neither Pet d1 nor PetΔd2 was able to bind to these epithelial cells, thus confirming the data obtained using PetΔd1, which was able to bind to epithelial cells in the absence of the d1 domain (Fig. 3A and 5A).
FIG 3.

The d2 domain contains the site for binding to host cells. (A) HEp-2 cells were slightly fixed with 0.25% paraformaldehyde (PFA) for 1 h and then washed and treated with Pet or its mutants (PetΔd1, PetΔd2, or PetΔPDWET) or Pet d1 at 20 μg/ml for 30 min. After this treatment, the cells were washed again and fixed properly with 4% PFA but not permeabilized. Nuclear DNA was stained with TO-PRO 3 (blue), and Pet was detected with an anti-Pet polyclonal antibody, followed by a secondary antibody, anti-rabbit IgG coupled to fluorescein (green). EspC protein is another autotransporter protein which has no receptor on the epithelial cell surface and was used as a negative control. The slides were observed by confocal microscopy. (B) The Pet receptor binding site was detected in the d2 domain, an exposed segment highlighted in red containing 55 amino acids (from residues 571 to 625).
FIG 5.

Confirmation of the phenotypic features induced by Pet and its deletion constructions on a human-derived intestinal cell culture line. (A) Cell binding. HT-29 cells were slightly fixed with 0.25% PFA for 1 h and then washed and treated with Pet or its mutants (PetΔd1, PetΔd2, or PetΔPDWET) or Pet d1 at 20 μg/ml for 30 min. After this treatment, the cells were washed and prepared for confocal microscopy as mentioned in the legend for Fig. 3A. (B) Internalization and cell damage. HT-29 cells were treated with Pet or its mutants (PetΔd1, PetΔd2, or PetΔPDWET) or Pet d1 at 20 μg/ml for 3.5 h. The cells were washed, fixed, permeabilized, and stained with rhodaminated phalloidin for F actin (red), TO-PRO 3 for DNA (blue), and anti-Pet antibodies, followed by a secondary anti-rabbit IgG antibody labeled with fluorescein (green). The slides were observed using confocal microscopy.
Thus, the d2 domain of Pet contains the Pet binding site to host cells except the PDWET sequence, which is not involved in Pet binding to host cells. Thus, the second loop of Pet's structure, which includes 54 amino acid residues, is involved in the interaction of Pet with the host cell surface (Fig. 3B). Interestingly, this sequence of 54 residues in Pet is different than that from other SPATE in this d2 domain; its closest homologs are Sat and EspP, or a protein with which it shares an intracellular target, such as EspC, or the most separated homologs such as SepA and Pic, which is the prototypical member of another SPATE subdivision (class 2 SPATE) (Fig. 3B).
The Pet d2 domain is required for Pet internalization and cell damage.
We have previously demonstrated that Pet binding to plasma membrane of epithelial cells is preceded by Pet internalization into host cells (23, 24). We sought to determine the relationship between the passenger subdomains and Pet binding and internalization. To do this, epithelial cells were treated with native Pet, PetΔd1, PetΔd2, PetΔPDWET, or Pet d1 (20 μg/ml), processed, and analyzed by confocal microscopy. As previously reported, Pet protein was detected inside the epithelial cells (HEp-2 and HT-29 cells) after 3.5 h of incubation (Fig. 4 and 5B). Similarly, PetΔd1 and PetΔPDWET were also detected inside the epithelial cells, a finding consistent with their ability to bind to the epithelial cell surface. Interestingly, PetΔd2 was unable to get inside epithelial cells, which was consistent with previous results showing that this deletion mutant is also unable to bind to epithelial cells. Also consistently, Pet d1 was unable to enter cells or to bind to epithelial cells (Fig. 4 and 5B). Analyses of middle cuts and z projections confirmed the presence of Pet and these mutants inside the epithelial cells (data not shown).
FIG 4.

The d2 domain is required for Pet internalization and epithelial cell damage. HEp-2 cells were treated with Pet or its mutants (PetΔd1, PetΔd2, or PetΔPDWET) or Pet d1 at 20 μg/ml for 3.5 h. The cells were washed, fixed, permeabilized, and stained with rhodaminated phalloidin for F actin (red), TO-PRO 3 for DNA (blue), and anti-Pet antibodies, followed by a secondary anti-rabbit IgG antibody labeled with fluorescein (green). The slides were observed using confocal microscopy.
Once Pet is inside the cells, it leads to cell damage by cleaving the protein associated with the actin cytoskeleton, fodrin (8). Using experiments similar to those performed to observe Pet internalization, the cells were examined for cell damage induced by Pet, PetΔd1, PetΔd2, PetΔPDWET, or Pet d1. As expected, internalized native Pet caused cell damage, which was characterized by the loss of actin stress fibers and cell rounding, leading to cell detachment (Fig. 4 and 5B). Interestingly, as mentioned above, PetΔd1 was able to bind to epithelial cells and get inside them, highlighting the importance of d2 domain, but it was unable to cause cell damage, because it lacks the first loop where the serine protease motif is located (Fig. 4 and 5B), indicating that these two activities are in separated domains. As expected based on the data described above, PetΔd2 was unable to cause cell damage, since it was unable to bind to epithelial cells and to get inside them. PetΔPDWET was able to cause cell damage consistent with its ability to bind to and internalize epithelial cells, whereas Pet d1, which contains the serine protease motif, was unable to cause cell damage because it is unable to bind to or enter the epithelial cells. Altogether, these results indicate that the Pet d2 domain is required for the binding and internalization of epithelial cells; thus, its absence prevented cell damage. On the other hand, d1 is required for cell damage because it contains the serine protease motif, but it is not required for the binding and internalization of epithelial cells, since its absence allows Pet binding and internalization but prevents cell damage.
The Pet d2 domain is involved in the recognition of the Pet receptor, cytokeratin 8.
We have previously reported that Ck8 is a receptor for Pet on the epithelial cell surface, and it is required for Pet binding and internalization (24). In order to determine whether the Pet d2 domain is involved in the interaction with Ck8, as the sequence recognized for the ligand-receptor interaction, recombinant Ck8 was immobilized on ELISA plates and incubated with PetΔd2, PetΔd1, or native Pet as a positive control, while BSA, an irrelevant protein, was used as a negative control. Either native Pet or PetΔd1 was able to significantly bind to Ck8 compared to PetΔd2. In fact, native Pet was able to bind to Ck8 83% more efficiently than PetΔd2, and this truncated protein bound similarly to an irrelevant protein, BSA (Fig. 6). These data clearly indicate that the Pet d2 domain is involved in the recognition of the host cell surface, specifically the Pet receptor, Ck8, which allows Pet uptake by epithelial cells.
FIG 6.
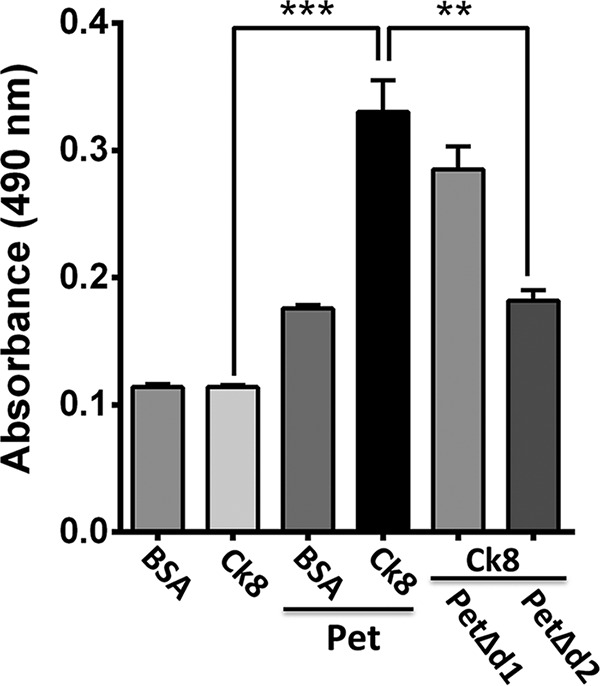
The d2 domain binds to recombinant cytokeratin 8, a cell receptor for Pet. ELISA plates coated with recombinant Ck8 (30 nM) were incubated for 2 h with native Pet, PetΔd1, or PetΔd2 (300 nM) and then washed with PBS to remove nonspecific binding. Finally, the amount of Ck8-bound Pet or PetΔd2 was determined with rabbit anti-Pet antibodies, followed by anti-rabbit secondary antibody conjugated to HRP, with the reading made at 490 nm. Wells coated with BSA were used as a negative control. Ck8 and BSA without Pet, PetΔd1, or PetΔd2 were used as control experiments for the primary antibody specificity. The results are expressed as means ± the standard errors of the means from at least three independent experiments. Statistical analysis were performed using the unpaired t test (**, P < 0.01; ***, P < 0.001).
DISCUSSION
Pet toxin is secreted using the type Va secretion system. Pet toxin is the mature active protein, and the precursor protein contains an N-terminal sequence that mediates the Sec-dependent protein export into the periplasm, a passenger domain encoding the mature protein, and a C-terminal domain mediating the translocation of the passenger domain across the outer membrane for its secretion to the extracellular medium (2). Furthermore, the whole Pet protein is a modular molecule because the passenger domain is subsequently divided into at least seven subdomains: d1, β-helix, d2, d4, d5, AC, and α-helix (33). Thus, each module appears to be involved in a specific function of Pet. Here, by using mutants with deletions in specific subdomains, we have found that subdomain 2 (d2) is involved in Pet uptake by epithelial cells.
The α-helix domain is a short α-helical peptide that connects the passenger domain to the characteristic β-barrel (33); in fact, the cleavage of the passenger domain of the SPATE from their cognate β-barrel is effected by nucleophilic attack of β-barrel residues on a single conserved asparagine residue in the α-helix (34). The autochaperone (AC) domain has been implicated in the secretion of passenger domains, where contemporaneous folding of the β-helix and a Brownian ratchet mechanism provide the vectorial impetus for the secretion (35, 36). However, it has been recently demonstrated that the AC domain is not essential for the secretion but is essential for the folding (33). The β-helix is the structural hallmark of the AT proteins, and this β-helix structure carries various loops (subdomains), which are presumed to confer functionality. In the case of Pet, the functional loops of the d5, d4, and d2 subdomains have not been elucidated. On the other hand, the d1 domain is the largest surface structure, which contains the serine protease motif of the SPATE family. In the case of Pet, a mutation on the serine of the active site gives a stable folded mutant protein with no cytotoxic effects (22). The active site in this motif is broadly conserved, and the active site serine was recognized from the sequence before the structures were solved (22, 37).
Pet binding to host cells is a key event, since it leads to Pet endocytosis, intracellular trafficking, and cell intoxication by the cleavage of fodrin, a protein associated with the actin cytoskeleton, resulting in cell contraction and detachment. To determine which subdomain of the passenger domain is involved in the binding to host cells, three mutants were initially constructed by deleting the d1 domain, the d2 domain, and five residues (PDWET) shared by the d2 domain and the adjacent β-helix. These domains are likely to be involved in Pet uptake by host cells; d1 is the largest domain (29.8 kDa) and contains the serine protease motif, which has its intracellular target (8), and d2 shares core residues with the NIDO domain of human proteins of the basal membrane. The link between the d2 domain and connective tissue suggests binding to host cells (31). Adjacent to the d2 domain, there is a region ∼50 residues in length that shows a higher variation than other parts of the protein. Part of this region in Pet has been named domain 2A and may be related to the cell uptake by that protein (31). The last few residues of domain 2A (PDWET) are completely conserved, and, like the d2 domain, this region appears to mediate interactions with other molecules, either from the host organism or the parent bacterium (31); however, to date no functional studies of this region have been published. Interestingly, deletion mutations in these three regions (the d1 and d2 domains and PDWET) do not block the secretion of these mutated proteins into the extracellular medium. Indeed, our models generated by using RaptorX structure prediction of these mutated proteins show that the three mutants must conserve their central β-helical stem decorated with the several discursive subdomains but without the respective mutated sequences. Although the use of structural prediction programs is not a guarantee of a lack of disruption of the structure, there was a correlation between the structural prediction and the secretion of the truncated proteins; the central β-helical stem is required for the autotransporter protein secretion (33).
By using slightly prefixed cells without permeabilization, an approach that we have previously used to identify a Pet receptor on the epithelial cell surface (24), we were able to discern that the d2 domain is required for Pet biding to epithelial cells. We therefore decided to investigate whether the sequence PDWET would be involved in this effect; PDWE are the last residues of the d2 domain, and T is the first residue of the adjacent β-helix domain (33). In the case of Hbp in these residues, the glutamine side chain of Gln 640 is the center of a network of hydrogen bonds involving aspartate (D) 642 and lying over tryptophan (W) 643, and this region has been suggested to mediate interactions with other molecules, either from the host organism or from the parent bacterium (31). Interestingly, the PDWET sequence was not required for Pet binding on the host cell surface, but other parts of the d2 domain (residues 571 to 625) were required.
More remarkable still, functional analyses showed that Pet lacking the d2 domain is unable to get inside the cells, and so it is unable to intoxicate epithelial cells, whereas PetΔPDWET is able to get inside the cells, causing cell damage. In fact, the d2 domain mutant phenotype acts as a knockdown of the Pet receptor in similar experiments (24). Interestingly, Pet lacking the d1 domain easily gets inside the cells, but, once internalized, it is unable to cause cell damage even though this construction has the complete d2 domain, clearly confirming previous results, i.e., the requirement of the d2 domain to be taken up by the cells and the important role of the serine protease motif (included in the d1 domain), as previously reported using a site-directed mutation in this motif (PetS260I) in similar experiments (8, 22). In contrast, a minimal clone containing the d1 domain was unable to get inside the cells and thus unable to cause cell damage.
We have previously shown that Pet binds to gastrointestinal epithelial cell membranes but not to epithelial kidney cells and that this binding to the cell surface depends on cytokeratin 8 (24). Here, we used two human epithelial cell lines from the gastrointestinal tract, HEp-2 cells and HT-29 cells. HEp-2 cells are not from an intestinal lineage but do provide an appropriate acute model to quickly detect morphological changes. In addition, HEp-2 cells grow faster than HT-29 cells, which were the intestinal cells used to confirm the initial results. By using these cell lines, we found that the d2 domain, among others in the passenger domain, is the sequence recognized by receptors on the epithelial cell surface, and it is separated from the catalytic active site found in the d1 domain. These results support the hypothesis that these subdomains projecting from the β-helix as loops, which are considerably variable in different passenger domains (17), have a specialized functional role for each one (18). In other, non-SPATE autotransporters, the RGD motif projecting from the β-helix is an adherent factor in pertactin from B. pertussis, whereas for VacA from H. pylori these loops define different target cell specificities (10), as well as separated subdomain activities: the C-terminal loops for receptor binding and residues near the N terminus for cell vacuolization and membrane depolarization (19, 20). We have shown here that Pet demonstrates separated activities in specific subdomains, but other related activities were observed in a specific subdomain (see Table 2 for a summary of the biological activities related to specific subdomains). In addition, the SPATE are more highly decorated with surface structures, and this feature may increase the specialized or specific functional roles. Indeed, according to our alignment of various SPATE, the d2 domain sequence in Pet is different than its closest homologs (class 1 SPATE) or its most separated homologs (class 2 SPATE), even though some of these conserve the sequences at the beginning and the end of the d2 domain (including PDWE). Class 2 SPATE had less identity with Pet in the d2 domain than class 1 SPATE; EspC, which does not have a cellular surface receptor but gets inside the cells, was also different in its d2 domain than the Pet protein.
TABLE 2.
Summary of the phenotype induced by Pet and its truncated proteins
| Phenotype | Resulta for: |
|||
|---|---|---|---|---|
| Pet | PetΔd1 | PetΔd2 | PetΔPDWET | |
| Secretion | ++++ | +++ | +++ | +++ |
| Protease activity | ++++ | − | ++++ | ++++ |
| Binding to epithelial cells | ++++ | +++ | − | ++ |
| Internalization | ++++ | +++ | − | ++ |
| Cell damage | ++++ | − | − | ++++ |
| Binding to Ck8 | ++++ | +++ | − | +++ |
++++, 100% of the phenotype induced by Pet; +++, 75 to 80% of Pet-induced phenotype; ++, 40 to 50% of Pet-induced phenotype; −, absence of Pet-induced phenotype.
We have recently shown that Ck8 acts as a Pet receptor, and we have shown here that the d2 domain is the ligand part of Pet that recognizes Ck8 on the cell surface, allowing receptor-mediated endocytosis, the retrograde transport, to reach the cytosol and its intracellular target, fodrin, which leads in turn to cytoskeleton disruption and cell detachment.
ACKNOWLEDGMENTS
This study was supported by grants from the Consejo Nacional de Ciencia y Tecnología (CONACYT; 44660-M and 128490) to F.N.-G.
REFERENCES
- 1.Ruiz-Perez F, Nataro JP. 2014. Bacterial serine proteases secreted by the autotransporter pathway: classification, specificity, and role in virulence. Cell Mol Life Sci 71:745–770. doi: 10.1007/s00018-013-1355-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henderson IR, Navarro-Garcia F, Desvaux M, Fernandez RC, Ala'Aldeen D. 2004. Type V protein secretion pathway: the autotransporter story. Microbiol Mol Biol Rev 68:692–744. doi: 10.1128/MMBR.68.4.692-744.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Hasani K, Navarro-Garcia F, Huerta J, Sakellaris H, Adler B. 2009. The immunogenic SigA enterotoxin of Shigella flexneri 2a binds to HEp-2 cells and induces fodrin redistribution in intoxicated epithelial cells. PLoS One 4:e8223. doi: 10.1371/journal.pone.0008223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maroncle NM, Sivick KE, Brady R, Stokes FE, Mobley HL. 2006. Protease activity, secretion, cell entry, cytotoxicity, and cellular targets of secreted autotransporter toxin of uropathogenic Escherichia coli. Infect Immun 74:6124–6134. doi: 10.1128/IAI.01086-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eslava C, Navarro-Garcia F, Czeczulin JR, Henderson IR, Cravioto A, Nataro JP. 1998. Pet, an autotransporter enterotoxin from enteroaggregative Escherichia coli. Infect Immun 66:3155–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mellies JL, Navarro-Garcia F, Okeke I, Frederickson J, Nataro JP, Kaper JB. 2001. espC pathogenicity island of enteropathogenic Escherichia coli encodes an enterotoxin. Infect Immun 69:315–324. doi: 10.1128/IAI.69.1.315-324.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henderson IR, Hicks S, Navarro-Garcia F, Elias WP, Philips AD, Nataro JP. 1999. Involvement of the enteroaggregative Escherichia coli plasmid-encoded toxin in causing human intestinal damage. Infect Immun 67:5338–5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Canizalez-Roman A, Navarro-Garcia F. 2003. Fodrin CaM-binding domain cleavage by Pet from enteroaggregative Escherichia coli leads to actin cytoskeletal disruption. Mol Microbiol 48:947–958. doi: 10.1046/j.1365-2958.2003.03492.x. [DOI] [PubMed] [Google Scholar]
- 9.Navarro-Garcia F, Serapio-Palacios A, Vidal JE, Salazar MI, Tapia-Pastrana G. 2014. EspC promotes epithelial cell detachment by enteropathogenic Escherichia coli via sequential cleavages of a cytoskeletal protein and then focal adhesion proteins. Infect Immun 82:2255–2265. doi: 10.1128/IAI.01386-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emsley P, Charles IG, Fairweather NF, Isaacs NW. 1996. Structure of Bordetella pertussis virulence factor P.69 pertactin. Nature 381:90–92. doi: 10.1038/381090a0. [DOI] [PubMed] [Google Scholar]
- 11.Kajava AV, Steven AC. 2006. The turn of the screw: variations of the abundant beta-solenoid motif in passenger domains of type V secretory proteins. J Struct Biol 155:306–315. doi: 10.1016/j.jsb.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 12.Otto BR, Sijbrandi R, Luirink J, Oudega B, Heddle JG, Mizutani K, Park SY, Tame JR. 2005. Crystal structure of hemoglobin protease, a heme binding autotransporter protein from pathogenic Escherichia coli. J Biol Chem 280:17339–17345. doi: 10.1074/jbc.M412885200. [DOI] [PubMed] [Google Scholar]
- 13.Gangwer KA, Mushrush DJ, Stauff DL, Spiller B, McClain MS, Cover TL, Lacy DB. 2007. Crystal structure of the Helicobacter pylori vacuolating toxin p55 domain. Proc Natl Acad Sci U S A 104:16293–16298. doi: 10.1073/pnas.0707447104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson TA, Qiu J, Plaut AG, Holyoak T. 2009. Active-site gating regulates substrate selectivity in a chymotrypsin-like serine protease: the structure of Haemophilus influenzae immunoglobulin A1 protease. J Mol Biol 389:559–574. doi: 10.1016/j.jmb.2009.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Junker M, Besingi RN, Clark PL. 2009. Vectorial transport and folding of an autotransporter virulence protein during outer membrane secretion. Mol Microbiol 71:1323–1332. doi: 10.1111/j.1365-2958.2009.06607.x. [DOI] [PubMed] [Google Scholar]
- 16.Dautin N. 2010. Serine protease autotransporters of enterobacteriaceae (SPATEs): biogenesis and function. Toxins 2:1179–1206. doi: 10.3390/toxins2061179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yen YT, Kostakioti M, Henderson IR, Stathopoulos C. 2008. Common themes and variations in serine protease autotransporters. Trends Microbiol 16:370–379. doi: 10.1016/j.tim.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 18.Nishimura K, Yoon YH, Kurihara A, Unzai S, Luirink J, Park SY, Tame JR. 2010. Role of domains within the autotransporter Hbp/Tsh. Acta Crystallogr D Biol Crystallogr 66:1295–1300. doi: 10.1107/S0907444910036966. [DOI] [PubMed] [Google Scholar]
- 19.Ivie SE, McClain MS, Torres VJ, Algood HM, Lacy DB, Yang R, Blanke SR, Cover TL. 2008. Helicobacter pylori VacA subdomain required for intracellular toxin activity and assembly of functional oligomeric complexes. Infect Immun 76:2843–2851. doi: 10.1128/IAI.01664-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sewald X, Fischer W, Haas R. 2008. Sticky socks: Helicobacter pylori VacA takes shape. Trends Microbiol 16:89–92. doi: 10.1016/j.tim.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Navarro-Garcia F. 2010. Enteroaggregative Escherichia coli plasmid-encoded toxin. Future Microbiol 5:1005–1013. doi: 10.2217/fmb.10.69. [DOI] [PubMed] [Google Scholar]
- 22.Navarro-Garcia F, Sears C, Eslava C, Cravioto A, Nataro JP. 1999. Cytoskeletal effects induced by pet, the serine protease enterotoxin of enteroaggregative Escherichia coli. Infect Immun 67:2184–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Navarro-Garcia F, Canizalez-Roman A, Burlingame KE, Teter K, Vidal JE. 2007. Pet, a non-AB toxin, is transported and translocated into epithelial cells by a retrograde trafficking pathway. Infect Immun 75:2101–2109. doi: 10.1128/IAI.01515-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nava-Acosta R, Navarro-Garcia F. 2013. Cytokeratin 8 is an epithelial cell receptor for Pet, a cytotoxic serine protease autotransporter of Enterobacteriaceae. mBio 4:e00838-13. doi: 10.1128/mBio.00838-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Navarro-Garcia F, Canizalez-Roman A, Vidal JE, Salazar MI. 2007. Intoxication of epithelial cells by plasmid-encoded toxin requires clathrin-mediated endocytosis. Microbiology 153:2828–2838. doi: 10.1099/mic.0.2007/007088-0. [DOI] [PubMed] [Google Scholar]
- 26.Maniatis T, Fritsch EF. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 27.Domingo Meza-Aguilar J, Fromme P, Torres-Larios A, Mendoza-Hernandez G, Hernandez-Chinas U, Arreguin-Espinosa de Los Monteros RA, Eslava Campos CA, Fromme R. 2014. X-ray crystal structure of the passenger domain of plasmid encoded toxin (Pet), an autotransporter enterotoxin from enteroaggregative Escherichia coli (EAEC). Biochem Biophys Res Commun 445:439–444. doi: 10.1016/j.bbrc.2014.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kallberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J. 2012. Template-based protein structure modeling using the RaptorX web server. Nat Protoc 7:1511–1522. doi: 10.1038/nprot.2012.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knutton S, Phillips AD, Smith HR, Gross RJ, Shaw R, Watson P, Price E. 1991. Screening for enteropathogenic Escherichia coli in infants with diarrhea by the fluorescent-actin staining test. Infect Immun 59:365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salazar-Gonzalez H, Navarro-Garcia F. 2011. Intimate adherence by enteropathogenic Escherichia coli modulates TLR5 localization and proinflammatory host response in intestinal epithelial cells. Scand J Immunol 73:268–283. doi: 10.1111/j.1365-3083.2011.02507.x. [DOI] [PubMed] [Google Scholar]
- 31.Nishimura K, Tajima N, Yoon YH, Park SY, Tame JR. 2010. Autotransporter passenger proteins: virulence factors with common structural themes. J Mol Med 88:451–458. doi: 10.1007/s00109-010-0600-y. [DOI] [PubMed] [Google Scholar]
- 32.Vidal JE, Navarro-Garcia F. 2006. Efficient translocation of EspC into epithelial cells depends on enteropathogenic Escherichia coli and host cell contact. Infect Immun 74:2293–2303. doi: 10.1128/IAI.74.4.2293-2303.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sevastsyanovich YR, Leyton DL, Wells TJ, Wardius CA, Tveen-Jensen K, Morris FC, Knowles TJ, Cunningham AF, Cole JA, Henderson IR. 2012. A generalized module for the selective extracellular accumulation of recombinant proteins. Microb Cell Fact 11:69. doi: 10.1186/1475-2859-11-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tajima N, Kawai F, Park SY, Tame JR. 2010. A novel intein-like autoproteolytic mechanism in autotransporter proteins. J Mol Biol 402:645–656. doi: 10.1016/j.jmb.2010.06.068. [DOI] [PubMed] [Google Scholar]
- 35.Oliver DC, Huang G, Nodel E, Pleasance S, Fernandez RC. 2003. A conserved region within the Bordetella pertussis autotransporter BrkA is necessary for folding of its passenger domain. Mol Microbiol 47:1367–1383. doi: 10.1046/j.1365-2958.2003.03377.x. [DOI] [PubMed] [Google Scholar]
- 36.Renn JP, Clark PL. 2008. A conserved stable core structure in the passenger domain beta-helix of autotransporter virulence proteins. Biopolymers 89:420–427. doi: 10.1002/bip.20924. [DOI] [PubMed] [Google Scholar]
- 37.Kostakioti M, Stathopoulos C. 2004. Functional analysis of the Tsh autotransporter from an avian pathogenic Escherichia coli strain. Infect Immun 72:5548–5554. doi: 10.1128/IAI.72.10.5548-5554.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]