

Abstract
Objective:
We sought to determine whether APOE genotype influences a previously observed decline in serum total cholesterol (TC) and low-density lipoprotein (LDL) levels preceding primary intracerebral hemorrhage (ICH), as a potential demonstration of nonamyloid mechanisms of APOE in ICH risk.
Methods:
We performed a single-center retrospective longitudinal analysis using patients with known APOE genotype drawn from an ongoing cohort study of ICH. Serum lipid measurements for TC, triglycerides (TGs), LDL, and high-density lipoprotein (HDL) collected within 2 years before and after index ICH were extracted from electronic medical records. Piecewise linear mixed-effects models were used to compare APOE allele–specific effects on temporal serum lipid trends in ICH. Demographics, medical history, medications, and health maintenance data were included as fixed effects. Inter- and intraindividual variations in lipid levels were modeled as random effects.
Results:
A total of 124 ICH cases were analyzed. APOE ε4 carriers had greater rates of decline in serum TC and LDL within 6 months preceding ICH (TC: −7.30 mg/dL/mo, p = 0.0035; LDL: −8.44 mg/dL/mo, p = 0.0001). Conversely, serum TC and LDL levels in APOE ε2 carriers were unchanged within the same time period. APOE genotype had no associations with serum HDL or TG trends.
Conclusions:
APOE allele status predicts serum TC and LDL changes preceding acute ICH. Our results have implications for ongoing efforts in dissecting the role of dyslipidemia in cerebrovascular disease risk. APOE genotype–specific influence on lipid trends provides a clue for one mechanism by which APOE may influence risk of ICH. Further characterization of the metabolic roles of APOE is needed to improve the understanding of APOE biology in cerebrovascular disease risk.
Primary intracerebral hemorrhage (ICH) accounts for 10%–15% of all strokes1 but is the most severe form of acute cerebrovascular disease, with 90-day mortality rates of 40%–50% and with fewer than a third of survivors regaining functional independence by 12 months.2,3 Previous studies have established ε2/ε4 alleles of the APOE gene as potent determinants of ICH risk, severity, and outcome.4–6 APOE ε2 and ε4 are associated with increased risk of ICH occurring in the lobar regions of the brain, whereas APOE ε4, but not ε2, is associated with risk of nonlobar ICH.4,7 Separately, several epidemiologic studies have also observed an association between serum lipid levels and ICH risk and outcome.8–16 Hypercholesterolemia has been associated with reduced risk of ICH,8–13 fewer cerebral microbleeds, and improved outcome after ICH.15,16 However, despite known functions of APOE gene products in lipid transport and regulating circulating lipid levels,17 the biological mechanisms mediating the roles of APOE and serum lipids on ICH risk remain unclear. A previous finding that serum low-density lipoprotein (LDL) mediates APOE ε4–associated nonlobar ICH risk7 suggests that the effect of APOE on ICH may be at least in part because of its effect on lipids.
We have recently demonstrated that ICH is preceded by declines in serum total cholesterol (TC) and LDL levels.18 We hypothesized that APOE genotype may influence these temporal lipid trends in ICH and tested this hypothesis by investigating APOE allele–specific effects on changes in serum lipid trends over time in a cohort of ICH patients with longitudinal lipid data.
METHODS
Study design.
Patients were drawn from an ongoing prospective longitudinal cohort study of primary ICH at Massachusetts General Hospital (MGH)19 (figure 1). All aspects of this study were approved by the MGH Institutional Review Board (IRB), and written informed consent was obtained from all patients or their legal guardians before study participation.
Figure 1. Study cohort and analysis plan.

MGH-ICH Database = Massachusetts General Hospital-based patients in the Genetics of Cerebral Hemorrhage on Anticoagulation Study; ICH = intracerebral hemorrhage.
Patient selection.
Individuals enrolled in the MGH longitudinal ICH study presenting to the MGH Emergency Department between June 1993 and June 2014 were screened for eligibility for the present study based on the following: (1) availability of APOE genotype, (2) survival up to 2 years after ICH, and (3) possession of at least 3 serum lipid values for each lipid fraction of interest including TC, LDL, triglycerides (TGs), and high-density lipoprotein (HDL) drawn >6 months apart within 24 months before and after the date of acute ICH. Patients with recurrent ICH or other non-ICH hospitalization events during the time period of interest were excluded to minimize confounding by variations in serum lipid levels during periods of acute illness.20,21
Data collection.
All included individuals had serum lipid values (TC, TG, LDL, and HDL) extracted via semiautomated review of hospital electronic medical records (EMRs). Because ICH events are observed at random, to make comparisons for variations in serum lipid trends before and after ICH, individual ICH events were time-locked into a balanced design with equal time periods of 24 months before and after the acute event. Serum lipid values were obtained for 6-month time intervals (4 intervals before and 4 intervals after the ICH event). Serum lipid values were segregated into their respective time intervals based on timing of the measurement in relation to the date of ICH occurrence. Means were obtained for multiple serum lipid measurements within the same time interval.
Clinical data were recorded at the time of index presentation including information on demographics, medical history (including hypertension and diabetes mellitus), pre-ICH medication use, and health maintenance (including cigarette smoking and alcohol use). Statin use within 24 months pre-ICH was determined at the time of ICH presentation using a patient-based questionnaire and scored as “yes/no” binary variables. Validity and reliability of statin use response in the questionnaire were internally assessed by comparisons with medication history obtained from hospital EMR search. A 10% random sampling of the enrolled cohort demonstrated 100% concordance in patients who were statin naive. ICH location was assigned as either lobar or nonlobar based on admission CT scans. Nonlobar ICH was restricted to hemorrhage locations involving the brainstem, thalamus, or basal ganglia, whereas lobar ICH included hemorrhages originating from the cortical-subcortical regions.
Genotyping.
Study individuals had APOE genotype determined previously from blood samples.4 In brief, peripheral whole blood was collected from study individuals at the time of consent. DNA was isolated from fresh or frozen blood, quantified using a quantification kit (Qiagen, Valencia, CA), and normalized to a concentration of 30 ng/μL. APOE genotypes were determined by genotyping 2 variants in APOE, rs7412 and rs429358, via 2 separate assays4 with the subsequent allelic reads from both assays then combined for translation to APOE genotypes (ε3ε3, ε3ε4, ε4ε4, ε3ε2, ε2ε2, and ε2ε4).
Standard protocol approvals, registrations, and patient consents.
This study was performed with approval of the MGH IRB. ICH patients provided informed consent for study participation.
Statistical methods.
Study individuals were grouped by APOE ε2 and ε4 carrier status (having either 1 or 2 allelic copies of ε2 and ε4, respectively) with APOE ε3ε3 individuals serving as a reference cohort as the ε3 allele is not associated with ICH risk.4 Patients with ε2ε4 APOE genotype (n = 5) were excluded because of an inability to assign a single carrier status. Comparisons of differences in cohort characteristics between ε2 or ε4 carriers and the reference ε3ε3 group were made by univariate analyses using unpaired t test, Mann-Whitney rank sum test, or Fisher exact test, as appropriate. Continuous numeric variables were expressed as mean ± SD.
Piecewise linear mixed-effects (PLME) random-coefficient models were used to evaluate APOE allele–specific effects on temporal variation in serum lipid trends in ICH patients within prespecified time periods, which are fixed in relation to acute ICH occurrence.22 This allowed for modeling of change in serum lipid trends in predetermined time intervals of interest to differ within and between ε2 or ε4 carriers and noncarriers. In a previous case-control analysis comparing ICH patients and non-ICH controls, we demonstrated significant decline in serum lipid trends in the 6-month interval immediately preceding the occurrence of ICH, which was not observed in non-ICH controls.18 Accordingly, fixed knots were placed at the date of acute ICH and the date 6 months before acute ICH to mark transitions in time periods of interest corresponding to the time period 6–24 months pre-ICH (P1), the time period 0–6 months immediately pre-ICH (P2), and time period 0–24 months post-ICH (P3).
Separate multivariate linear mixed models were constructed for ε2 and ε4 carriers, with carrier status and covariates whose p values were <0.20 on univariate analyses or with known potential to influence serum lipid levels included as fixed effects. The final multivariate model was adjusted for variables: age, sex, race, pre-ICH history of hypertension, statin use (yes/no), smoking history (ever smoked), and ICH location. Interindividual and intraindividual variation in serum lipid levels were modeled as random effects. Model validity was examined using a likelihood ratio test. Unstructured covariance was used as the covariance model. Comparisons of the significance in change in serum lipid trends (slope) at the time period of interest, P2 (0–6 months pre-ICH), by APOE allele carrier status were made using the Wald test. Subgroup analyses stratified by ICH location (lobar and nonlobar) for ε2 and ε4 carrier status were separately performed but not shown because of insufficient statistical power. Significance threshold was set at p < 0.05 (2-tailed) for univariate analyses and at p < 0.0125 (Bonferroni correction for 4 tests) for individual serum lipid fraction mixed-model analyses. All statistical analyses were performed using STATA 10.0 (StataCorp LP, College Station, TX).
RESULTS
Cohort characteristics.
A total of 212 ICH patients enrolled between June 1993 and June 2014 with longitudinal serum lipid levels measured that met our inclusion criteria; 129 of these patients were genotyped for APOE. The 83 ICH patients removed because of the absence of APOE genotype (figure 1) did not differ in clinical characteristics from the group of patients who ultimately were included in our analyses (table e-1 at Neurology.org/ng). After removing the 5 patients with APOE ε2/ε4, we analyzed 124 individuals including 19 ε2 carriers, 39 ε4 carriers, and 66 ε3/ε3 patients (table 1). Compared with the reference group (APOE ε3ε3), ε2 carriers were less likely to have a pre-ICH history of hypertension, and ε4 carriers were more likely to be smokers and have ICH located in the lobar region (all p < 0.05). There were no differences in the rates of statin use between the 3 groups. APOE allelic frequencies in our analysis cohort were consistent with previously observed population estimates for North American Caucasians.23
Table 1.
Baseline characteristics of the study cohort

APOE alleles and serum lipid levels in ICH patients.
We first sought to confirm previously observed effects of APOE on serum lipid levels, as seen in previous population-level genome-wide association studies of lipids.17 Comparisons of mean serum levels of TC, TG, LDL, and HDL before ICH by APOE allele status revealed an expected allelic dose-dependent increase in mean serum TC and LDL levels in ε4 carriers compared with noncarriers, whereas levels of both lipid fractions were decreased in ε2 carriers compared with noncarriers (TC: 217.21 ± 58.37 mg/dL in ε4 carriers, 157.67 ± 41.12 mg/dL in ε2 carriers; LDL: 130.43 ± 48.67 mg/dL in ε4 carriers, 69.90 ± 19.86 mg/dL in ε2 carriers). No associations were observed for serum TG and HDL levels, consistent with known absence of APOE effects on these lipid fractions.
APOE alleles influence 24-month pre-ICH serum lipid trends.
Temporal lipid patterns in our analysis cohort revealed a decline in both serum TC and LDL levels beginning several months preceding acute ICH occurrence consistent with observed trends seen previously in a larger cohort18 (figure 2). Subgroup analysis by APOE carrier status revealed distinct differences in temporal serum lipid trends, visualized using Loess smoothed curves, during this time period. APOE ε4 carriers experienced an overall decline in serum TC and LDL levels in the 24 months pre-ICH. In contrast, both serum TC and LDL trends remained relatively flat in non-ε4 carriers during the same time period preceding ICH (figure 3). Comparisons of serum lipid trends by APOE allele demonstrated an overall decline in serum TC and LDL levels during the pre-ICH time period in ε4 carriers compared with noncarriers (p = 0.049 and p = 0.014, respectively), whereas no changes were observed in ε2 carriers.
Figure 2. Temporal trends in individual serum lipid fractions in ICH patients.

(A–D) Loess smoothed curves of serum lipid levels (mg/dL) against time (in months) before and after ICH. Gray areas indicate standard error (SE). Time period of interest is indicated by shaded boxes (P2 0–6 months pre-ICH). *p < 0.0125, rate of change of serum lipids by Wald test for the time period P2. ICH = intracerebral hemorrhage.
Figure 3. Temporal trends in individual serum lipid fractions in ICH patients by APOE allele carrier status.

(A–D) Loess smoothed curves of serum lipid levels (mg/dL) against time (in months) before and after ICH. Gray areas indicate standard error (SE). Time period of interest is indicated by shaded boxes (P2 0–6 months pre-ICH). *p < 0.0125, rate of change of serum lipids by Wald test for time period P2 in APOE ε2 or ε4 carrier status compared with reference (APOE ε3/ε3). ICH = intracerebral hemorrhage.
APOE alleles influence differential change in serum lipid trends immediately preceding ICH occurrence.
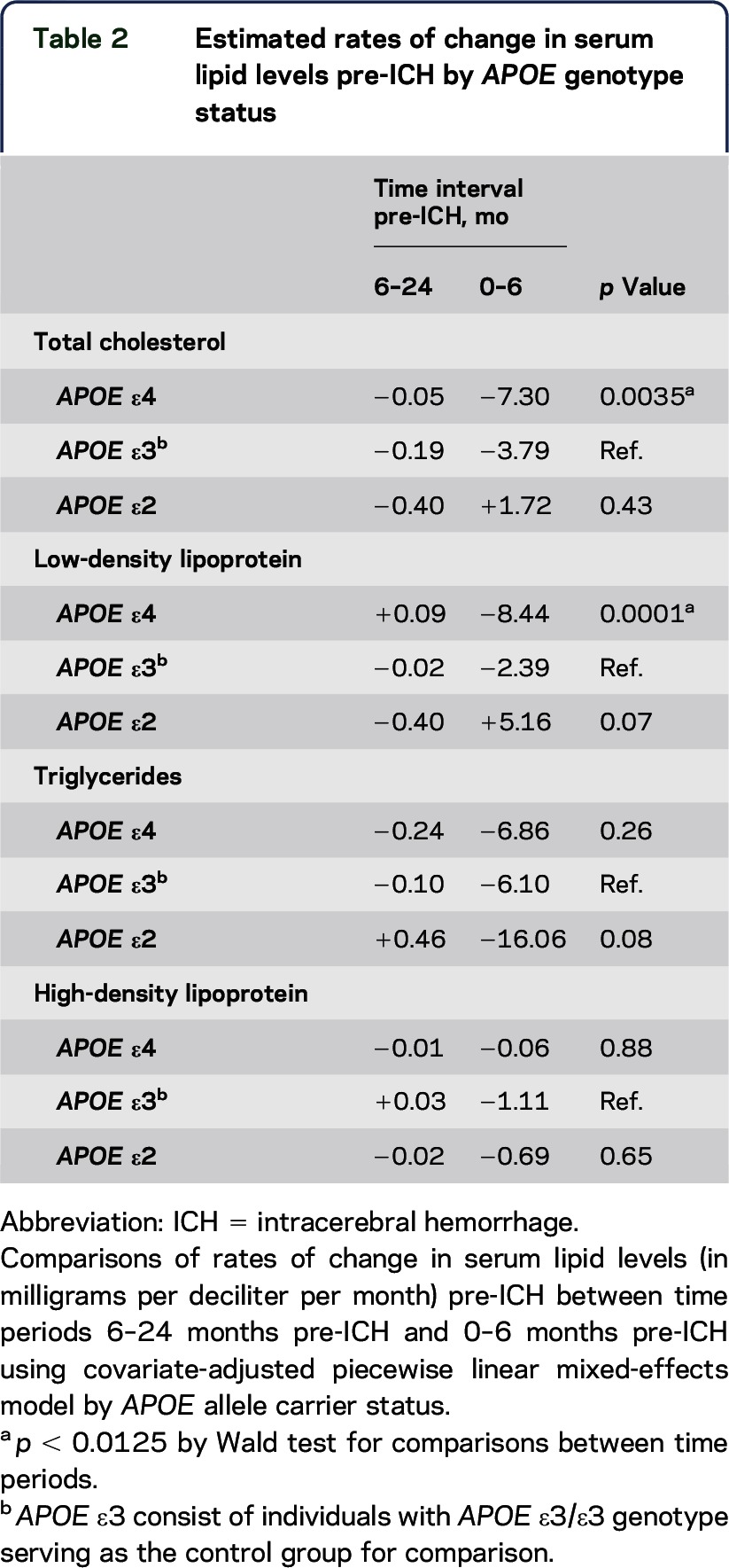
Visual inspection of the Loess smoothed curves also revealed distinct differences in temporal serum lipid trends in the 6-month time period immediately preceding ICH occurrence by APOE allele status. Subacute declines in mean serum TC and LDL levels beginning around 6 months pre-ICH were observed only in ε4 carriers (figure 3). This accelerated decline in the 6-month period before ICH is consistent with our previous report in a larger cohort.18 Covariate-adjusted PLME was used to compare differences in serum lipid trends by APOE genotype in the immediate 0- to 6-month interval pre-ICH and in the antecedent interval (6–24 months pre-ICH) (table 2). APOE ε4 carriers experienced acceleration in the rates of decline in serum TC and LDL levels in the 6 months before an acute ICH event compared with trends in the antecedent 18-month time interval. The observed association remained significant after the inclusion of potential confounders in multivariate analysis, including hypertension (table 2). Comparatively, serum TC trends were unchanged in the same 6-month time period immediately pre-ICH in both APOE ε2 carriers and APOE ε3/ε3 individuals. In contrast, APOE ε2 carriers experienced an increase in serum LDL levels in the immediate 0- to 6-month interval pre-ICH that did not surpass statistical thresholds, whereas serum LDL levels remained flat in APOE ε3/ε3 individuals within the same time period. Neither temporal serum HDL nor TG trends differed by APOE genotype.
Table 2.
Estimated rates of change in serum lipid levels pre-ICH by APOE genotype status
APOE alleles do not influence temporal serum lipid trends post-ICH.
Serum lipid levels remained largely depressed for TC, LDL, and HDL in the 48 months post-ICH with no difference in serum lipid trends by APOE carrier status during that time. Serum TG trends post-ICH demonstrate variability similar to the pre-ICH period, but no differences were observed between APOE ε2 and ε4 carriers or individuals with APOE ε3/ε3 genotype within the time period examined (figure 3).
DISCUSSION
Our results demonstrate that temporal variation in serum lipids in ICH patients are influenced by APOE allele status and differ from APOE associations with steady-state serum lipid levels.17 APOE ε4 carriers experienced drops in TC and LDL levels in the 24-month period before their ICH, in comparison with non-APOE ε4 carriers. Furthermore, in the 6-month period immediately preceding ICH, APOE ε4 carriers displayed increased rates of decline in serum TC and LDL levels. This observation of genotype-specific differences in temporal serum lipid trends builds on our previous observation of subacute decline in serum TC and LDL levels before acute ICH and suggests that APOE gene products may exert at least some of their effect on risk of ICH through modulation of serum lipids.
Demonstration of APOE epsilon allele–specific effects on serum lipid trends in ICH patients raises several hypotheses regarding the role of APOE in ICH risk. Both ε2 and ε4 are risk factors for lobar ICH, in part, through their effects on amyloid processing.4 APOE ε4 is also independently associated with increased risk of nonlobar ICH,4 presumably through nonamyloid-related mechanisms given that cerebral amyloid angiopathy is almost universally absent from the deeper small vessels.24 A growing body of evidence supports the association of hypocholesterolemia with elevated risk of ICH7–14 and with progression of ICH-related phenotypes such as cerebral microbleeds.14,25 This association has been hypothesized to be the result of loss in vascular integrity in low circulating cholesterol states, which predisposes toward vessel rupture in ICH, although the complex role of lipids in cellular biology, inflammation, and signalling,26–29 in addition to cell membrane integrity, makes it difficult to attribute the observed associations to any one mechanism.30–33
Known APOE effects in lipid metabolism and vascular amyloid deposition raise the possibility that APOE may influence ICH risk through amyloid and nonamyloid effects. Our results seem to support the hypothesis of a nonamyloid role of APOE in ICH risk through the observed APOE genotype–specific associations with subacute serum lipid changes before primary ICH. Given that APOE ε4 associates with higher average TC and LDL levels, our results also raise the hypothesis that the rate of change in serum levels, rather than the baseline average, is an important determinant of ICH risk imparted by the APOE ε4 genotype. Furthermore, our demonstration of APOE allele–specific effects on temporal serum lipid trends in ICH corroborates the notion of divergent mechanistic pathways between ε2 and ε4 alleles in the pathophysiology of ICH.34,35 Our observations of similar APOE allele–specific serum TC and LDL trends pre-ICH in subgroup analyses stratified by ICH location (lobar and nonlobar) likewise support such a notion, but our study was insufficiently powered to detect statistically significant differences because of the small sample size.
However, caution must be exercised in making mechanistic links because of the biological complexity of the underlying disease and APOE pleiotropy. Based on our observation, we can only speculate as to whether serum lipid declines directly drive the process of increased vessel wall vulnerability, leading ultimately to vessel rupture and ICH or serve as a surrogate marker of a separate process affecting cerebral small vessels. Active inflammation is associated with lower serum TC and LDL levels,36 whereas an APOE genotype–specific elevation in proinflammatory response has been observed in APOE ε4 carriers compared with APOE ε2 and APOE ε3 carriers in transgenic murine models.37 Thus, it is possible that the influence of APOE polymorphisms on temporal lipid trends in ICH may instead reflect APOE genotype–specific differences in innate inflammatory processes in the cerebral small vessels.
A strength of this study is the use of a unique data set combining both APOE genotype and longitudinal lipid data in a rigorously phenotyped ICH cohort. The additional information conferred by serum lipid variations over time both before and after primary ICH revealed APOE genotype–specific associations distinct from known steady-state relationships. This in turn allowed for the dissection of lipid-dependent associations of APOE in primary ICH risk.
There are limitations to our study. First, we had to exclude almost 40% of eligible cases identified because of the absence of APOE genotype data. It should be noted, however, that the analysis cohort remained representative of the larger ICH cohort, with no differences in any covariates of interest to suggest a sampling bias. Furthermore, distribution of the APOE alleles in our small study was also consistent with frequency estimates in the population at large. We were also unable to account completely for selection bias arising from subject-specific indications for serial serum lipid measurements, although, in our particular study cohort, previous analysis showed no differences in clinical characteristics between ICH patients with and without serum lipid data.18 A third limitation was our relatively small sample size, particularly with regard to the total number of APOE ε2 individuals, which may influence our ability to more accurately assess association with serum lipid trends in those individuals. We attempted to address this by using mixed-effect modeling to increase statistical power through additional use of interindividual change with time. Nevertheless, future studies incorporating a longitudinal design with available APOE genotypes and relatively frequently recorded lipid levels will be necessary to validate and confirm these results. Fourth, we were unable to exhaustively address the broad range of all the possible external environmental factors that can influence biological variation in serum lipid levels.37 We did attempt to minimize potential confounding by including these measures, where available, by including covariates of age, statin, and alcohol use in our models and using a longitudinal trial design of sufficiently long duration (4 years), which limits the effect of seasonal variations38 in serum lipids. In addition, we were also unable to account for either statin dose or type and potential intermittent use. However, because the decline in both serum TC and LDL levels immediately preceding ICH occurrence were previously noted to be independent of statin use,18 the differential degree of lipid lowering conferred by nuances in statin use is unlikely to contribute substantial confounding. Fifth, although there is a high likelihood that LDL may be a major mediator of TC effects seen in APOE ε4 carriers, our study design prohibits formal mediation analysis because of violation of several assumptions needed for establishing a correctly specified mediation model. Finally, although the demonstration of temporal changes in serum lipids preceding ICH strongly suggests a correlation between serum lipid changes and ICH development, we are unable to confirm a causal relationship because of the retrospective study design.
APOE ε4 strongly predicts pre-ICH trends in serum TC and LDL levels and the acute decline in serum TC and LDL in the 6-month period before acute ICH. Our results have implications for ongoing efforts in dissecting the role of dyslipidemia in cerebrovascular disease risk and provide novel insight regarding nonamyloid APOE mechanisms in ICH risk.
Supplementary Material
GLOSSARY
- HDL
high-density lipoprotein
- ICH
intracerebral hemorrhage
- LDL
low-density lipoprotein
- PLME
piecewise linear mixed-effects
- TC
total cholesterol
- TG
triglyceride
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
Dr. C.-L. Phuah participated in study design, data acquisition, statistical analyses, drafting, and revision of the manuscript. M.R. Raffeld and A.M. Ayres were responsible for data collection. Drs. A. Viswanathan, M. Edip Gurol, S.M. Greenberg, and J. Rosand participated in the final editing of the manuscript. Dr. C.D. Anderson participated in the study design and funding and in the revision of the manuscript.
STUDY FUNDING
The authors' work on this study was supported by funding from the NIH (see Disclosures). No funding entities were involved in study design, data collection, analysis, interpretation, writing of the report, or in the decision to submit the results for publication.
DISCLOSURE
Dr. C.-L. Phuah and M.R. Raffeld report no disclosures. A.M. Ayres has received research support from NIH. Dr. M. Edip Gurol has received research support from NIH–National Institute of Neurological Disorders and Stroke. Dr. A. Viswanathan has served on a data safety monitoring board for Roche Pharmaceuticals; has served as a consultant for Athena Diagnostics; and is supported by NIH–National Institute of Neurological Disorders and Stroke K23 AG028726. Dr. S.M. Greenberg has served on the MRI Review Committee of Hoffman-Laroche and the data safety monitoring board of Quintiles; has received travel funding/speaker honoraria from the Cerebral Amyloid Angiopathy Conference and the American Academy of Neurology; has served on the editorial boards of Stroke, Frontiers in Stroke, Cerebrovascular Disease, Neurology, and Alzheimer's Disease and Other Dementias; has received publishing royalties from UpToDate and MedLink; and is supported by NIH–National Institute of Neurological Disorders and Stroke U10 NS077360, R01 AG026484, and R01 NS070834. Dr. A. Biffi reports no disclosures. Dr. J. Rosand has served on the editorial boards of Lancet Neurology and Stroke and is supported by NIH–National Institute of Neurological Disorders and Stroke U01 NS069208, R01 NS073344, and R01 NS059727. Dr. C.D. Anderson has received research support from Biogen Idec, Inc., and the American Brain Foundation and is supported by NIH–National Institute of Neurological Disorders and Stroke K23 NS 086873. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Krishnamurthi RV, Moran AE, Forouzanfar MH, et al. The global burden of hemorrhagic stroke: a summary of findings from the GBD 2010 study. Glob Heart 2014;9:101–106. [DOI] [PubMed] [Google Scholar]
- 2.Poon MT, Fonville AF, Al-Shahi Salman R. Long-term prognosis after intracerebral haemorrhage: systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2014;85:660–667. [DOI] [PubMed] [Google Scholar]
- 3.van Asch CJ, Luitse MJ, Rinkel GJ, van der Tweel I, Algra A, Klijn CJ. Incidence, case fatality, and functional outcome of intracerebral haemorrhage over time, according to age, sex, and ethnic origin: a systematic review and meta-analysis. Lancet Neurol 2010;9:167–176. [DOI] [PubMed] [Google Scholar]
- 4.Biffi A, Sonni A, Anderson CD, et al. Variants at APOE influence risk of deep and lobar intracerebral hemorrhage. Ann Neurol 2010;68:934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Devan WJ, Falcone GJ, Anderson CD, et al. Heritability estimates identify a substantial contribution to risk and outcome of intracerebral hemorrhage. Stroke 2013;44:1578–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brouwers HB, Biffi A, Ayres AM, et al. Apolipoprotein E genotype predicts hematoma expansion in lobar intracerebral hemorrhage. Stroke 2012;43:1490–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raffeld MR, Biffi A, Battey TWK, et al. APOE ε4 and lipid levels affect risk of recurrent nonlobar intracerebral hemorrhage. Neurology 2015;85:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woo D, Deka R, Falcone GJ, et al. Apolipoprotein E, statins, and risk of intracerebral hemorrhage. Stroke 2013;44:3013–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Dong Y, Qi X, Huang C, Hou L. Cholesterol levels and risk of hemorrhagic stroke: a systematic review and meta-analysis. Stroke 2013;44:1833–1839. [DOI] [PubMed] [Google Scholar]
- 10.Collins R, Armitage J, Parish S, Sleight P, Peto R. Effects of cholesterol-lowering with simvastatin on stroke and other major vascular events in 20536 people with cerebrovascular disease or other high-risk conditions. Lancet 2004;363:757–767. [DOI] [PubMed] [Google Scholar]
- 11.Segal AZ, Chiu RI, Eggleston-Sexton PM, Beiser A, Greenberg SM. Low cholesterol as a risk factor for primary intracerebral hemorrhage: a case-control study. Neuroepidemiol 1999;18:185–193. [DOI] [PubMed] [Google Scholar]
- 12.Woo D, Kissela BM, Khoury JC, et al. Hypercholesterolemia, HMG-CoA reductase inhibitors, and risk of intracerebral hemorrhage: a case-control study. Stroke 2004;35:1360–1364. [DOI] [PubMed] [Google Scholar]
- 13.Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005;366:1267–1278. [DOI] [PubMed] [Google Scholar]
- 14.Wieberdink RG, Poels MM, Vernooij MW, et al. Serum lipid levels and the risk of intracerebral hemorrhage: the Rotterdam Study. Arteriocler Thromb Vasc Biol 2011;31:2982–2989. [DOI] [PubMed] [Google Scholar]
- 15.Rodriguez-Luna D, Rubeira M, Ribo M, et al. Serum low-density lipoprotein cholesterol level predicts hematoma growth and clinical outcome after acute intracerebral hemorrhage. Stroke 2011;42:2447–2452. [DOI] [PubMed] [Google Scholar]
- 16.Mustanoja S, Strbian D, Putaala J, et al. Association of prestroke statin use and lipid levels with outcome of intracerebral hemorrhage. Stroke 2013;44:2330–2332. [DOI] [PubMed] [Google Scholar]
- 17.Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010;466:707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Phuah CL, Raffeld MR, Ayres AM, et al. Subacute decline in serum lipids precedes the occurrence of primary intracerebral hemorrhage. Neurology 2016;86:2034–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O'Donnell HC, Rosand J, Knudsen KA, et al. Apolipoprotein E genotype and the risk of recurrent lobar intracerebral hemorrhage. N Engl J Med 2000;342:240–245. [DOI] [PubMed] [Google Scholar]
- 20.Man EB, Bettcher PG, Cameron CM, Peters JP. Plasma amino acids, nitrogen and serum lipids of surgical patients. J Clin Invest 1946;25:701–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gore JM, Goldberg RJ, Matsumoto AS, Castelli WP, McNamara AB, Dalen JE. Validity of serum total cholesterol level obtained within 24 hours of acute myocardial infarction. Am J Cardiol 1984;54:722–725. [DOI] [PubMed] [Google Scholar]
- 22.Naumova EN, Must A, Laird NM. Tutorial in biostatistics: evaluating the impact of “critical periods” in longitudinal studies of growth using piecewise mixed effects models. Int J Epidemiol 2001;30:1332–1341. [DOI] [PubMed] [Google Scholar]
- 23.Ordovas JM, Litwack-Klein L, Wilson PWF, Schaefer MM, Schaefer EJ. Apolipoprotein E isoform phenotyping methodology and population frequency with identification of ApoEl and ApoE5 isoforms. J Lipid Res 1987;28:371–380. [PubMed] [Google Scholar]
- 24.Woo D, Sauerbeck LR, Kissela BM, et al. Genetic and environmental risk factors for intracerebral hemorrhage: preliminary results of a population-based study. Stroke 2002;33:1190–1196. [DOI] [PubMed] [Google Scholar]
- 25.Romero JR, Preis SR, Beiser A, et al. Risk factors, stroke prevention treatments, and prevalence of cerebral microbleeds in the Framingham Heart Study. Stroke 2014;45:1492–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stables MJ, Gilroy DW. Old and new generation lipid mediators in acute inflammation and resolution. Prog Lipid Res 2011;50:35–51. [DOI] [PubMed] [Google Scholar]
- 27.Phillips MC. Molecular mechanisms of cellular cholesterol efflux. J Biol Chem 2014;289:24020–24029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ledeen RW, Wu G. Nuclear sphingolipids: metabolism and signaling. J Lipid Res 2008;49:1176–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohanian J, Ohanian V. Sphingolipids in mammalian cell signalling. Cell Mol Life Sci 2001;58:2053–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ooneda G, Yoshida Y, Suzuki K, Sekiguchi T. Morphogenesis of plasmatic arterionecrosis as the cause of hypertensive intracerebral hemorrhage. Virchows Arch A Pathol Pathol Anat 1973;361:31–38. [DOI] [PubMed] [Google Scholar]
- 31.Russell RW. How does blood pressure cause stroke? Lancet 1974;2:1283–1285. [DOI] [PubMed] [Google Scholar]
- 32.Konishi M, Terao A, Doi M, et al. Osmotic resistance and cholesterol content of the erythrocyte membrane in cerebral hemorrhage. Igaku No Ayumi 1982;120:30–32. [Google Scholar]
- 33.Kroes J, Ostwald R. Eryrthocyte membranes—effect of increased cholesterol content on permeability. Biochim Biophys Acta 1971;249:647–650. [DOI] [PubMed] [Google Scholar]
- 34.McCarron MO, Nicoll JA, Stewart J, et al. The apolipoprotein E epsilon2 allele and the pathological features in cerebral amyloid angiopathy-related hemorrhage. J Neuropathol Exp Neurol 1999;58:711–718. [DOI] [PubMed] [Google Scholar]
- 35.Greenberg SM, Vonsattel JP, Segal AZ, et al. Association of apolipoprotein E epsilon2 and vasculopathy in cerebral amyloid angiopathy. Neurology 1998;50:961–965. [DOI] [PubMed] [Google Scholar]
- 36.McGullicuddy FC, de la Llera Moy M, Hinkle CC, et al. Inflammation impairs reverse cholesterol transport in vivo. Circulation 2009;119:1135–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jofre-Monseny L, Loboda A, Wagner AE, et al. Effects of apoE genotype on macrophage inflammation and heme oxygenase-1 expression. Biochem Biophys Res Commun 2007;357:319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Durrington PN. Biological variation in serum lipid concentrations. Scand J Clin Lab Invest Suppl 1990;198:86–91. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.