

The P. syringae type III effector protein HopG1 is necessary for disease-related changes in plant actin cytoskeleton organization.
Abstract
The plant cytoskeleton underpins the function of a multitude of cellular mechanisms, including those associated with developmental- and stress-associated signaling processes. In recent years, the actin cytoskeleton has been demonstrated to play a key role in plant immune signaling, including a recent demonstration that pathogens target actin filaments to block plant defense and immunity. Herein, we quantified spatial changes in host actin filament organization after infection with Pseudomonas syringae pv. tomato DC3000 (Pst DC3000), demonstrating that the type-III effector HopG1 is required for pathogen-induced changes to actin filament architecture and host disease symptom development during infection. Using a suite of pathogen effector deletion constructs, coupled with high-resolution microscopy, we found that deletion of hopG1 from Pst DC3000 resulted in a reduction in actin bundling and a concomitant increase in the density of filament arrays in Arabidopsis, both of which correlate with host disease symptom development. As a mechanism underpinning this activity, we further show that the HopG1 effector interacts with an Arabidopsis mitochondrial-localized kinesin motor protein. Kinesin mutant plants show reduced disease symptoms after pathogen infection, which can be complemented by actin-modifying agents. In total, our results support a model in which HopG1 induces changes in the organization of the actin cytoskeleton as part of its virulence function in promoting disease symptom development.
The actin cytoskeleton supports the function of a diverse suite of cellular processes, including those associated with plant growth and development, cell shape and movement, and response to stress (Day et al., 2011). At a mechanistic level, this model underpinning these associations posits that actin filaments are constantly rearranged via fast assembly and disassembly (Staiger et al., 2009; Smertenko et al., 2010; Staiger et al., 2010). The characterization of this incessant remodeling forms the basis for the favored hypothesis that actin functions as a surveillance platform, rapidly reorganizing and supporting countless cellular processes required for response to numerous internal and external stimuli. Upon perception, rapid, yet highly stimulus-specific changes in actin filament organization occur, and from this, regulated changes in cell signaling are initiated (Thomas, 2012; Porter and Day, 2013). In this regard, it is not surprising that critical cellular responses associated with stress signaling, including abiotic (Rodríguez-Milla and Salinas, 2009) and biotic processes (Day et al., 2011), are impacted by the activity and organization of the host actin cytoskeleton.
In plants, immune signaling is broadly classified based on the activity of two primary nodes: pathogen-associated molecular pattern (PAMP)-triggered immunity (PTI), and effector-triggered immunity (Chisholm et al., 2006). In the case of PTI, signaling is activated after recognition of PAMPs (e.g. flagellin, chitin) by plasma membrane-localized pattern recognition receptors (PRRs), which leads to the initiation of downstream signaling cascades, including MAPK signaling, the generation of reactive oxygen species, and the transcriptional reprogramming of pathogen-responsive genes (Zhang et al., 2010; Porter et al., 2012). To counter PTI, bacterial phytopathogens employ a virulence-associated type-III secretion system (T3SS) to deliver type-III effector (T3E) proteins into host cells (Dou and Zhou, 2012). In the case of the bacterial pathogen Pseudomonas syringae pv. tomato DC3000 (Pst DC3000), approximately 28 T3E proteins have been identified, a number of which have been demonstrated to be important for pathogen virulence (Xin and He, 2013). However, the mode of action as well as the breadth of host cellular targets of many of these effectors remains largely undefined. Current models predict that effectors target basic physiological processes, including those specifically associated with defense activation, to disarm the host immune system; ultimately, this enhances pathogen infection and proliferation (Chisholm et al., 2006). To counter the activity of T3Es, plants have evolved a second node of immune signaling, effector-triggered immunity, which results in the activation of an enhanced PTI-like response, initiated via the direct or indirect recognition of pathogen effectors by host resistance (R) proteins (Chisholm et al., 2006).
The link between the actin cytoskeleton and the plant immune system has been best defined through the analysis of PTI. For example, several studies have shown that perturbation to the host cell through the application of pharmacological agents that modify the stochastic behavior of the actin cytoskeleton (e.g. cytochalasin-d, jasplakinolide) also impact immune signaling and pathogen infection (Takemoto et al., 1999; Yun et al., 2003; Shimada et al., 2006; Miklis et al., 2007; Tian et al., 2009; Wang et al., 2009). Collectively, these studies have not only demonstrated a requirement for cytoskeletal rearrangement at the site of pathogen penetration, but that actin filament organization is required for the function of numerous intracellular processes, including trafficking, the production of metabolites associated with resistance signaling, and response to mechanical force. Additional work has further demonstrated that after perturbation to the naïve cycling of the plant actin cytoskeleton, many of the early events required for the activation of PTI, including attenuation of signaling through endocytosis of the PRR (Beck et al., 2012), MAPK signaling (Porter et al., 2012), the generation of reactive oxygen (Liu et al., 2012), callose deposition (Cai et al., 2011), and the activation of transcription (Moes et al., 2013), are blocked.
As a progenitor for many of these initial responses, recent work has identified a rapid, transient, reorganization of the host actin cytoskeleton concomitant with the activation of PTI (Henty-Ridilla et al., 2013). Using PAMP-PRR activation as a tractable marker to monitor the induction of defense signaling, Henty-Ridilla et al. (2013) provides strong evidence in support of a role for the remodeling of the cytoskeleton in host epidermal cells as an early (approximately 1–2 h post-inoculation; hpi) prerequisite for the activation of PTI (Henty-Ridilla et al., 2013). However, the specific roles of individual, or groups of, Pst DC3000 T3Es in modulating these responses remain undefined. Additionally, the role of the actin cytoskeleton in the latter stages of the host-pathogen interaction has not been investigated.
In this study, we conducted a reverse genetic screen, coupled to a quantitative microscopy-based approach, to identify and define the interaction(s) between the Arabidopsis actin cytoskeleton and the constellation of 28 P. syringae T3E proteins. To do this, we utilized a previously characterized collection of Pst DC3000 effector deletion constructs (Kvitko et al., 2009) to quantify pathogen-induced changes in cytoskeletal organization. Using this approach, we defined the minimal set of T3Es required to alter filament array architecture in host epidermal cells during infection, including those required for the onset of disease. Using this as a foundation, we next generated a series of single effector deletion constructs, and through a series of comprehensive complementation assays, we identified the P. syringae effector protein HopG1 as necessary for changes in host actin cytoskeletal organization. In combination with genetic screens for virulence, we show that HopG1 impacts the organization of the actin cytoskeleton, and that this modulation is required for the development of disease symptoms during pathogen infection. Lastly, we identified an association between HopG1 and a mitochondrial-localized kinesin motor protein. We posit that this interaction—and putative virulence targeting of kinesin by HopG1—leads to an increase in host actin filament bundling, which in turn leads to the induction of disease symptoms and pathogen proliferation in Arabidopsis.
RESULTS
P. syringae Type III Effectors Modulate Arabidopsis Actin Cytoskeletal Organization
To determine whether T3Es from Pst DC3000 induce changes in host actin cytoskeletal organization during infection, and to characterize this response as a function of pathogen virulence and disease symptom development, we imaged epidermal pavement cells in Arabidopsis ‘Columbia’ (wild-type Col-0) expressing the fluorescent actin marker GFP-fABD2 (Sheahan et al., 2004) during infection with Pst DC3000. In brief, 10-d-old Col-0/GFP-fABD2 cotyledons were collected using laser-scanning confocal microscopy and quantitatively assessed using an undirected approach to identify pathogen-induced changes in host actin filament architecture 24 h after inoculation. As previously described in Henty-Ridilla et al. (2013), images were evaluated using two quantitative parameters for actin organization: bundling (skewness) and filament density (percent occupancy).
Arabidopsis epidermal cells showed two distinct populations of actin configurations: single actin filaments (i.e. dimmer, thinner ephemeral filamentous structures) and actin bundles (i.e. brighter, thicker filamentous structures). After mock (MgCl2) inoculation of Arabidopsis cotyledons, we observed dense filamentous structures in Arabidopsis epidermal cells; in contrast, after inoculation with the virulent pathogen Pst DC3000, we observed an increase in the population of bundled structures (i.e. increased skewness), with a concomitant reduction in filament abundance (i.e. decreased density; Fig. 1A). Quantitative analysis of actin filament architecture (i.e. the extent of filament bundling and filament density) supports these visual observations (Fig. 1A; third and fourth panels).
Figure 1.

Modulation of host actin cytoskeletal organization by P. syringae is T3SS system-dependent. Actin filament organization in 10-d-old Col-0/GFP-fABD2 cotyledons after pathogen inoculation was monitored at 24 h by laser scanning confocal microscopy. A, Pst DC3000; B, Pst DC3000 D28E; C, Pst DC3000 ΔhrcC. Seedlings were inoculated with Pst DC3000 strains at a concentration of 3 × 107 colony-forming units (cfu) ml−1. Micrographs shown are representative of collected images. Error bars represent mean ± se from the skewness and density values of three independent biological replicates (n = 87–90 z-series projections per treatment). Statistical significance was determined using a Student’s t-test, as compared to mock treatment. **, P < 0.001. Bar = 20 μm.
Next, to determine whether modulation of the actin cytoskeleton during infection correlates with the presence of a functional T3SS and its repertoire of effectors, we inoculated 10-d-old Col-0/GFP-fABD2 seedlings with a Pst DC3000 strain lacking the entire suite of 28 effector genes [i.e. Pst DC3000 D28E; (Cunnac et al., 2011)], as well as parallel inoculations with a Pst DC3000 strain lacking the T3SS (i.e. Pst DC3000 ΔhrcC). As predicted, cotyledons treated with Pst DC3000 D28E (Fig. 1B) or Pst DC3000 ΔhrcC (Fig. 1C) showed no significant difference in either actin filament parameter (i.e. bundling or density) compared to mock-inoculated cotyledons. This data suggest a requirement for the one, or multiple, T3Es on pathogen-induced changes in the host actin cytoskeleton.
To define pathogen-induced changes in host cytoskeletal organization during infection, we first assessed the contribution of PAMP perception and signaling on the organization of the actin cytoskeleton. First, as a marker for the induction of PTI, we evaluated the expression of a conserved plant defense marker, FRK1 mRNA accumulation, after elicitation with the PAMP flg22. As expected, we observed the rapid induction of the FRK1 marker gene at 6 h (Supplemental Fig. S1A). In parallel treatments, we quantitatively evaluated extent of actin bundling and filament density at 24 h after flg22 treatment. As expected, we observed no change in filament architecture (i.e. actin bundling) at 24 h (Supplemental Fig. S1B). This is important, as it distinguished the 24 h time-point from previously described changes associated with PTI (Henty-Ridilla et al., 2013), and therefore potentially specific to effector-induced changes in actin cytoskeletal organization. Based on these data, we posit that the increase in actin filament bundling and decrease in overall actin filament abundance at 24 hpi is mediated by Pst DC3000 in a T3E-dependent manner.
P. syringae DC3000 Effector Polymutants Display a Varying Degree of Virulence and In Planta Growth
To define which Pst DC3000 T3Es are necessary for pathogen-induced modulation of the cytoskeleton, we utilized a collection of T3E gene polymutants to dissect the relative contribution of individual, or suites of, pathogen effectors to changes in host cytoskeletal organization (Wei et al., 2007; Kvitko et al., 2009). To establish this approach as a robust, genetically tractable pathosystem, we first monitored the growth of each of the polymutant strains in 10-d-old wild-type Col-0 cotyledons at 24 h after dip-inoculation. Similar to previous reports using Nicotiana benthamiana as a host, the Pst DC3000 polymutant strains varied in in planta growth in Arabidopsis by up to two orders of magnitude (Fig. 2A). The remaining effector gene polymutants showed little (i.e. <1 log) impact on in planta pathogen growth at 24 hpi (Fig. 2A). To ensure that the activity (i.e. in planta growth, disease symptom elicitation) of the Pst DC3000 clones used for pathogen growth and actin architecture comparisons were consistent across experiments, irrespective of pathogen genotype, we tested two independent Pst DC3000 strains. We observed no differences in the behavior of Pst DC3000 containing the empty plasmid vector pVSP61 (Loper and Lindow, 1987) versus the behavior of wild-type Pst DC3000 (i.e. no pVSP61 plasmid) in growth assays (Supplemental Fig. S2A) or changes in host actin architecture (Supplemental Fig. S2B).
Figure 2.

P. syringae T3E polymutants display differential activities with regard to manipulation of the Arabidopsis actin cytoskeleton. A, Type three-secretion system effector Pst DC3000 polymutants show a differential growth response in Col-0. Bacterial growth of Pst DC3000 and the suite of Pst DC3000 effector polymutants were enumerated at 24 h after dip-inoculation with Pst DC3000 polymutant strains at a concentration of 3 × 107 cfu ml−1. Shaded boxes indicate the presence of the described effector gene clusters. Error bars, representing mean ± se, were calculated from five (n = 15) biological replicates for Pst DC3000, and from two (n = 6) for each of the Pst DC3000 polymutants. Bacterial growth was enumerated based on cfu per mg fresh weight. Statistical significance was determined using a one-way ANOVA, Tukey test; P < 0.01. B, Pst DC3000 in planta growth correlates with the degree of modulation of host actin architecture. Actin skewness and density values were determined in Col-0/GFP-fABD2 seedlings at 24 hpi. Representative micrographs (left) from n = 80–90 are shown. Quantification of actin bundling (center) and density (right) indicates a bacterial density-dependent alteration in host actin cytoskeleton architecture after inoculation. Wild-type Col-0 plants and Col-0/GFP-fABD2 plants were dip-inoculated with Pst DC3000 at concentrations of 3 × 107 cfu ml−1, 3 × 106 cfu ml−1, and 106 cfu ml−1. Mock = MgCl2 dip-inoculated. **, P < 0.001. Bar = 20 μm. C, In planta bacterial growth enumeration of Pst DC3000 D28E and the ∆hrcC mutant. Bacterial growth was enumerated at 24 hpi with Pst DC3000 at three separate initial inoculation concentrations; concentrations of 3 × 107 cfu ml−1, 3 × 106 cfu ml−1, and 106 cfu ml−1. Pst DC3000 D28E and the ∆hrcC mutant were inoculated at 3 × 107 cfu ml−1. Bacterial growth assays were repeated twice. Error bars, representing mean ± se, were calculated from three (n = 9) technical replicates. Statistical significance was determined using a one-way ANOVA; P < 0.01.
T3E Gene Clusters Enable Pst DC3000 to Differentially Promote and Suppress Host Actin Cytoskeletal Bundling and Disassembly
Previous work has shown that after pathogen inoculation, the host responds to certain immune system signals in a pathogen density-dependent manner (Mishina and Zeier, 2007; Jing et al., 2011). To determine whether pathogen-induced changes in actin cytoskeletal organization are influenced in a density-dependent manner, we assessed changes in actin bundling and filament density using different pathogen inoculation concentrations. As anticipated, we were able to identify a correlation between changes in actin architecture and the infecting inoculum concentration of Pst DC3000 (Fig. 2B). As shown, at higher inoculum concentrations (e.g. 3 × 107 cfu ml−1), we observed an increase in actin bundling/skewness, versus when we inoculated plants with a lower inoculum concentration (e.g. 106 cfu ml−1). Based on these data, we calibrated the individual infecting doses of Pst DC3000 such that in planta bacterial growth at 24 hpi would attain similar levels as each of the various polymutants; this would permit us to compensate for bacterial density-dependent induced changes in host cytoskeletal architecture in our analyses (Fig. 2A). Thus, by pairing the behavior of actin organization in response to the polymutants to that of Pst DC3000 with similar levels of growth, we were able to account for growth-dependent effects in relation to T3E-mediated effects. Additionally, we observed, similar to previously described data (Kvitko et al., 2009; Cunnac et al., 2011; Kvitko and Collmer, 2011), that the Pst D28E and Pst ∆hrcC strains showed a reduction in planta growth compared with wild-type Pst DC3000 (Fig. 2C). This observation will be exploited to further calibrate our analysis of pathogen-induced changes in actin filament organization, below.
To identify and define Pst DC3000 effectors that induce marked changes in actin cytoskeletal organization independent of bacterial growth, we next utilized the suite of CUCPB polymutant strains to quantify changes in actin skewness and density during infection. As shown in Fig. 3 and Supplemental Fig. S3A, we observed a significant change in the ratio of the two populations of actin filaments in a pathogen genotype-dependent manner. For example, we observed a significant decrease in actin filament bundling, with a corresponding increase in actin filament density, in plants inoculated with CUCPB5459 (ΔI, II, IV, IX, X; Fig. 3A), CUCPB5529 (ΔhopQ1-1, IX; Fig. 3B), and CUCPB5452 (ΔII, IV, IX, X; Supplemental Fig. S3A). In all cases, changes in actin cytoskeletal organization were quantified relative to plants inoculated with the paired calibrated dose of Pst DC3000. In plants infected with additional effector polymutants, the responses were similar to those infected with paired calibrated doses of wild-type Pst DC3000 (Supplemental Fig. S3, B–F). This observation supports the hypothesis that most of the T3E genes are not necessary for Pst DC3000 to cause changes to the overall organization of the host actin cytoskeleton. In contrast, only one deleted cluster of T3E genes was common to the polymutants that showed significant differences in actin architecture relative to those infected with calibrated doses of Pst DC3000 (Fig. 3, A and B; Supplemental Fig. S3A). This polymutant, consisting of cluster IX T3Es, encompasses hopAA1-2, hopV1, hopAO1, and hopG1. Interestingly, as shown in Supplemental Fig. S3, D–F, we did not observe a change in the extent of actin bundling or filament density after inoculation with CUCPB5500, CUCPB5518, and CUCPB5539, which also lack cluster IX effectors. Not surprisingly, the most affected polymutant strains with respect to diminished in planta growth (i.e. CUCPB5500, CUCPB5518, and CUCPB5539), lacked the greatest number of effector genes. Interestingly, we found that the commonality among each of these polymutants was the deletion of the conserved effector locus [CEL; (Alfano et al., 2000; Collmer et al., 2000)], which harbors the effector genes hopN1, hopAA1-1, hopM1, and avrE (∆CEL mutant, Fig. 3C; ∆avrE and ∆hopM1, Supplemental Fig. S4). The simplest explanation for this observation is that each of these three polymutants also lacks the CEL, whereas CUCPB5459, CUCPB5529, and CUCPB5452 retain this locus. To test this, we quantified changes in actin architecture in plants infected with the ∆CEL mutant (Fig. 3C). Relative to a calibrated dose of Pst DC3000, plants showed an opposite change in actin organization, displaying an increase in filament bundling and a decrease in filament abundance after pathogen infection (Fig. 3C; Supplemental Fig. S4). Taken together, these data show that Pst DC3000 harbors two T3Es with opposing effects on actin cytoskeletal organization in host cells.
Figure 3.

Actin bundling in Arabidopsis is suppressed by the Pst DC3000 cluster IX polymutant. Actin filament architecture in 10-d-old Col-0/GFP-fABD2 cotyledons was quantified at 24 h after dip-inoculation with the Pst DC3000 polymutants. A, CUCPB5459 (ΔI, II, IV, IX, X); B, ΔIX (CUCPB5529); and C, ΔCEL (CUCPB5115). Seedlings were dip-inoculated with Pst DC3000 strains at a concentration of 3 × 107 cfu ml−1 for analyses shown in A and B. In C, seedlings were dip-inoculated with Pst DC3000 at a concentration of 106 cfu ml−1 and with Pst DC3000 ΔCEL at 3 × 107 cfu ml−1. Representative confocal micrographs are shown. Values for actin skewness and density are shown as relative comparisons to Pst DC3000-inoculated plants. Error bars represent mean ± se from the skewness and density values of three independent biological repeats (n = 80–90). Statistical significance was determined using a Student’s t-test, as compared to Pst DC3000 treatment. *, P < 0.01; **, P < 0.001. Bar = 20 μm.
HopG1 Induces Actin Filament Bundling in Arabidopsis
To identify the effector gene(s) in cluster IX necessary for the observed changes in host actin array organization during Pst DC3000 infection, we individually transformed and expressed in the Pst DC3000 ΔIX polymutant each of the deleted T3E genes, hopAA1-2, hopAO1, hopG1, and hopV1 (Fig. 4A), as well as hopQ1-1. As above, the in planta growth of all ΔIX polymutant-complemented strains in wild-type Col-0 at 24 hpi was assessed (Supplemental Fig. S5). First, we observed that the independent introduction of the T3E genes hopAO1 (Fig. 4B), hopV1 (Fig. 4C), and hopAA1-2 (Fig. 4D) failed to induce changes to the actin cytoskeleton to levels similar to a calibrated dose of Pst DC3000. Furthermore, we introduced hopQ1-1 into the Pst DC3000 cluster IX polymutant (i.e. CUCPB5529) to determine whether HopQ1-1 impacts actin cytoskeletal organization; as previously described in Wei et al. (2007), CUCPB5529 contains not only a deletion of the cluster IX T3E genes, but also hopQ1-1. As predicted, we did not observe a significant change in actin remodeling as compared to Pst DC3000 (Fig. 4E). However, when hopG1 was reintroduced into the cluster IX polymutant, it restored the ability of the polymutant to induce Pst DC3000-like changes in the host actin cytoskeletal array (Fig. 4F). These data provide the first evidence that HopG1 is required for Pst DC3000-induced changes to the host actin cytoskeleton 24 hpi.
Figure 4.

The T3E HopG1 induces actin bundling and a concomitant reduction in actin filament array density in Arabidopsis epidermal cells. Changes in actin filament organization in 10-d-old Col-0/GFP-fABD2 cotyledons was quantified at 24 h after dip-inoculation with Pst DC3000 and each of the cluster IX single effector mutant-complemented CUCPB5529 (i.e. cluster ΔIX) lines. A, Illustration of the cluster IX region of Pst DC3000. Scale bar at right = 1 kb. B, ΔIX + phopAO1; C, ΔIX + phopV1; D, ΔIX + phopAA1-2; E, ΔIX + phopQ1-1; and F, ΔIX + phopG1. Representative micrographs are shown. Initial pathogen inoculums for Pst DC3000 were 3 × 107 cfu ml−1. Actin skewness and density values were determined in 10-d-old Col-0/GFP-fABD2 seedlings at 24 h after dip-inoculation with Pst DC3000. Error bars represent mean ± se from the skewness and density values of three independent biological repeats (n = 80–90). Statistical significance was determined using a Student’s t-test, as compared to Pst DC3000 treatment. *, P < 0.01; **, P < 0.001. Bar = 20 μm.
To provide conclusive evidence demonstrating that HopG1 is required for Pst DC3000-induced changes in cytoskeletal organization, we constructed and tested a single hopG1 deletion mutant (ΔhopG1), and again quantified changes in actin bundling and filament density at 24 hpi. In support of the requirement for HopG1, we observed a decrease in actin bundling and an increase in host actin filament density in plants infected with Pst DC3000 ΔhopG1, relative to plants infected with Pst DC3000 (Fig. 5A). Complementation of Pst DC3000 ΔhopG1 with a plasmid expressing a native promoter-driven hopG1 construct (i.e. phopG1) restored wild-type actin filament architecture in Arabidopsis (Fig. 5B). In support of our hypothesis that HopG1 function does not impact the in planta growth of Pst DC3000, and therefore, supports our argument that changes in actin filament organization through HopG1 are independent of pathogen dose, we did not observe a change in bacterial growth (i.e. in planta) in the ΔhopG1 mutant or the phopG1 complementation strain (Fig. 5C).
Figure 5.

Pathogen-induced actin bundling is suppressed in the hopG1 single mutant. Actin filament organization in 10-d-old Col-0/GFP-fABD2 cotyledons was observed at 24 h after dip-inoculation with the Pst DC3000, ΔhopG1, and Pst DC3000 ΔhopG1 + phopG1. A, Pst DC3000 ΔhopG1 and B, Pst DC3000 ΔhopG1 + phopG1. Pst DC3000 strains were inoculated at a concentration of 3 × 107 cfu ml−1. Representative micrographs are shown. Actin skewness and density values were determined in 10-d-old Col-0/GFP-fABD2 seedlings at 24 h after dip-inoculation with Pst DC3000. Error bars represent mean ± se from the skewness and density values of seven independent biological repeats (n = 202–204) for A and three independent biological repeats (n = 71–74) for B. Statistical significance was determined using a Student’s t-test, as compared to Pst DC3000 wild type. **P < 0.001. Bar = 20 μm. C, In planta bacterial growth enumeration of Pst DC3000, the Pst DC3000 ΔhopG1 mutant, and the Pst DC3000 ΔhopG1 mutant complemented with hopG1 (i.e. ΔhopG1 phopG1). Bacterial growth was enumerated at 24 h after dip-inoculation at 3 × 107 cfu mL−1. Bacterial growth assays were repeated twice. Error bars, representing mean ± se, were calculated from three (n = 6) technical replicates. Statistical significance was determined using a one-way ANOVA, Tukey test; P < 0.01.
HopG1 Promotes Disease Symptom Development Through the Modulation of the Host Actin Cytoskeleton
HopG1 is localized within a genomic cluster (i.e. cluster IX) of virulence factors and T3E genes that have recently been characterized as being associated with the induction of chlorosis (Fig. 4A) (Worley et al., 2013). To determine whether HopG1 function is associated with the induction of chlorosis during Pst DC3000 infection of Arabidopsis, 10-d-old seedlings were dip-inoculated with Pst DC3000, the original effector polymutant lacking HopG1 (i.e. ΔIX), the complemented polymutant (i.e. ΔIX phopG1), and Pst DC3000 ΔhopG1. As shown in Figure 6A, inoculation of wild-type Col-0 seedlings with Pst DC3000 resulted in pronounced chlorosis 3 d post-inoculation (dpi), while inoculation with the ΔIX polymutant showed a significant reduction in disease symptom development. Complementation of the ΔIX polymutant with native promoter-expressed hopG1 (i.e. ΔIX phopG1) resulted in a restoration of the chlorosis-inducing ability of the pathogen. As predicted, the Pst DC3000 ΔhopG1 bacterium was reduced in its ability to induce chlorosis during infection. These data are consistent with a role for HopG1 in the induction of host chlorosis associated with pathogen infection.
Figure 6.

HopG1 is required for Pst DC3000-induced actin-dependent chlorosis. A, Pst DC3000 induces chlorosis in Arabidopsis at 3 dpi. Inoculation of 10-d-old wild-type Col-0 seedlings with Pst DC3000, Pst DC3000 ΔIX, Pst DC3000 ΔIX phopG1, and Pst DC3000 ΔhopG1 at a concentration of 3 × 107 cfu ml−1 revealed a requirement for HopG1 in chlorosis elicitation. B, Actin stabilization inhibits HopG1-induced chlorosis. Five-week-old plants were co-infiltrated with either MgCl2, cytochalasin-d, or jasplakinolide and Pst DC3000, the cluster IX polymutant (± HopG1), or Pst DC3000 ΔhopG1; plants were monitored for the induction of senescence-associated chlorosis at 3 dpi. Leaves inoculated with wild-type Pst DC3000 alone showed visible signs of chlorosis at 3 dpi. C, Bacterial growth assays of Pst DC3000 and the cluster IX variants 10-d-old Col-0 plants at 0–3 dpi. Pst DC3000 strains were inoculated at a concentration of 3 × 107 cfu ml−1. Bacterial growth assays were repeated at least three times. Error bars, representing mean ± se, were calculated from three (n = 9) technical replicates of three independent biological repeats. Bacterial growth was enumerated based on cfu per mg fresh weight of plant tissue. D, The actin-stabilizing and barbed-end binding agents, jasplakinolide and cytochalasin-d, respectively, do not affect the growth of Pst DC3000 or the cluster IX mutant variants in wild-type Col-0. Five-week-old Col-0 leaves were co-infiltrated with cytochalasin-d or jasplakinolide, and Pst DC3000 strains at a concentration of 2 × 105 cfu mL−1, and bacterial growth was enumerated at 3 dpi. Bacterial growth assays were repeated three times. Error bars, representing mean ± se, were calculated from three (n = 9) technical replicates of three independent biological repeats. Statistical significance was determined using a one-way ANOVA, Tukey test; P < 0.01. CD, cytochalasin-d; f.w., fresh weight; Jasp, jasplakinolide; Mock, MgCl2.
Next, to define the relationship among host actin cytoskeletal dynamics, HopG1, and pathogen-induced chlorosis and disease symptom development, we co-inoculated plants with Pst DC3000 and in combination with the actin cytoskeleton-modifying agents cytochalasin-d (inhibitor of actin polymerization) and jasplakinolide (inducer of actin polymerization). In support of a role for the actin-HopG1-disease signaling node, co-infiltration of wild-type Col-0 with jasplakinolide and Pst DC3000 led to a reduction in chlorosis, whereas co-infiltration with cytochalasin-d and Pst DC3000 resulted in enhanced chlorosis (Fig. 6B). These data suggest that actin destabilization is required for pathogen induction of host disease symptoms, and to some degree, may indicate an additive effect of cytochalasin-D and HopG1. As a control, wild-type Col-0 plants co-inoculated with cytochalasin-d and the cluster IX effector polymutant showed levels of chlorosis similar to that observed when plants were infected with Pst DC3000 alone. Conversely, co-infiltration of wild-type Col-0 with jasplakinolide and the cluster IX effector polymutant resulted in no change in chlorosis, further supporting our hypothesis that HopG1 induces host actin destabilization, and that this activity is required for the subsequent induction of host chlorosis and senescence-associated phenotypes. Co-infiltration of wild-type Col-0 plants with jasplakinolide and Pst DC3000 resulted in a significant reduction in pathogen-induced chlorosis, further supporting our hypothesis of the role of actin destabilization through HopG1. Infiltration of wild-type Col-0 plants with cytochalasin or jasplakinolide, alone, did not induce chlorosis (Supplemental Fig. S6).
Finally, to rule out the possibility that pathogen dose is a primary contributor to host symptom development during infection, we enumerated the in planta growth of each of the HopG1-relevant Pst DC3000 strains tested (i.e. ΔIX, ΔIX + phopG1, and ΔhopG1), and as shown in Figure 6C, all of the strains grew to similar levels. Thus, we can conclude that the observed differences in chlorosis are not the result of differences in pathogen growth at 24 hpi, but rather the presence of hopG1. Similarly, pathogen growth among the four strains was not affected by the presence of the actin cytoskeletal inhibitors cytochalasin-d and jasplakinolide (Fig. 6D). These data demonstrate that the presence of HopG1 in Pst DC3000 is required for the induction of disease symptoms in an actin-associated manner.
HopG1 Interacts with a Mitochondrial-Localized Kinesin Motor Protein and Is Associated with the Actin Cytoskeleton
An interaction between HopG1 and a mitochondria-localized kinesin (Itoh et al., 2001) was previously identified from a comprehensive yeast two-hybrid screen to identify immune-associated protein-protein interactions between Pst DC3000 and Arabidopsis (Mukhtar et al., 2011). To confirm that HopG1 does in fact interact with kinesin, we performed co-immunprecipitation assays of T7-epitope-tagged HopG1 and HA-epitope-tagged kinesin using Agrobacterium-mediated transient expression in N. benthamiana. We detected a specific interaction between the T3E HopG1 and the mitochondrial-localized kinesin motor protein (Fig. 7A). These data suggest that targeting of HopG1 to mitochondria (Block et al., 2010) may function as a virulence mechanism to block actin-cytoskeletal motor function through modulation of kinesin activity.
Figure 7.

HopG1 induces host disease symptoms through the function of a mitochondrial-localized kinesin. A, The Pst DC3000 T3E HopG1 interacts with the N-terminal half of an Arabidopsis kinesin (At4g39050). Epitope-tagged fusions to the open-reading frame of HopG1 (T7) and the first 978 nucleotides of kinesin (HA) were transiently expressed (40 h) in N. benthamiana and protein extracts were isolated and analyzed for a specific interaction between HopG1 and kinesin using co-immunoprecipitation assays. B, Kinesin and HopG1 are associated with the host actin cytoskeleton. Epitope-tagged fusions to the first 978 nucleotides of kinesin (HA) and the open-reading frame of HopG1 (T7) were transiently expressed (40 h) in N. benthamiana and total protein extracts were analyzed by co-immunoprecipitation analysis for a specific interaction among kinesin, HopG1, and the actin cytoskeleton (left blot), and an interaction between kinesin and the actin cytoskeleton (right blot). C, A mitochondrial-localized kinesin is required for Pst DC3000-induced symptom development. Five-week-old plants were co-infiltrated with either MgCl2, cytochalasin-d, or jasplakinolide and Pst DC3000 at a concentration of 2 × 105 cfu ml−1, and plants were monitored for the induction of chlorosis. Wild-type Col-0 plants inoculated with Pst DC3000 showed visible signs of disease at 3 dpi. When plants were co-inoculated with Pst DC3000 and cytochalasin-d, pathogen-induced chlorosis phenotype was enhanced; in the presence of jasplakinolide, the chlorosis phenotype was reduced. In the Arabidopsis kinesin mutant, inoculation with Pst DC3000 showed a significantly reduced chlorosis phenotype (compared to wild-type Col-0), while co-inoculation with Pst DC3000 and cytochalasin-d resulted in the elicitation of a pronounced disease phenotype. Co-inoculation of the kinesin mutant with Pst DC3000 and jasplakinolide showed a marked reduction in disease symptoms. D, Bacterial growth assays of Pst DC3000 in wild-type Col-0 and the kinesin mutant. Pst DC3000 strains were inoculated at a concentration of 3 × 107 cfu ml−1. Bacterial growth assays were performed two times with similar results. Error bars, representing mean ± se, were calculated from three (n = 6) technical replicates of two independent biological repeats. In planta pathogen growth was enumerated based on cfu per cm2 plant tissue. Statistical significance was assessed using a Student’s t-test. E, The actin-stabilizing and barbed-end binding agents, jasplakinolide and cytochalasin-d, respectively, do not affect the in planta growth of Pst DC3000 in wild-type Col-0 or the kinesin mutant. Five-week-old wild-type Col-0 leaves were co-infiltrated with cytochalasin-d or jasplakinolide and Pst DC3000 (2 × 105 cfu ml−1), and bacterial growth was enumerated at 3 dpi. Bacterial growth assays were performed two times. Error bars, representing mean ± se, were calculated from three (n = 6) technical replicates of two independent biological repeats. Statistical significance was determined using a one-way ANOVA, Tukey test; P < 0.01. CD, cytochalasin-d; f.w., fresh weight; Jasp, jasplakinolide; Mock, MgCl2.
To determine whether HopG1 is associated with the actin cytoskeleton, either directly or via association with kinesin, we next performed co-immunoprecipitation experiments using transient expression of HopG1, together with kinesin, in N. benthamiana. As shown in Figure 7B (left blot), when transiently expressed kinesin and HopG1 were immunoprecipitated with a monoclonal antibody specific to actin, we were able to pull down both kinesin and HopG1, together, as well as a protein complex associated with actin. Furthermore, when kinesin alone was transiently expressed in N. benthamiana and isolated total proteins were immunoprecipitated with an actin monoclonal antibody, we were successful in detecting kinesin (Fig. 7B, right blot), supporting the hypothesis that kinesin is associated with actin. We were unable to detect an association between HopG1 and actin when HopG1 alone was transiently expressed (data not shown). In total, these data support our hypothesis that HopG1’s association with actin is likely indirect, via its interaction with kinesin.
The Arabidopsis Kinesin Mutant Shows Reduced Susceptibility to Pst DC3000
To define the relationship and activity of HopG1 association with kinesin, we next assessed the ability of Pst DC3000 to induce chlorosis and disease symptom development in a kinesin mutant plant (SALK_130788; Supplemental Fig. S7A). As shown in Figure 7C, we observed a significant reduction in pathogen-induced chlorosis in the kinesin mutant line as compared to wild-type Col-0. Interestingly, the disease chlorosis phenotype in the Arabidopsis kinesin mutant after inoculation with Pst DC3000 could be recovered by co-infiltration of plants with cytochalasin-d, similar to co-inoculation of wild-type Col-0 with the Pst DC3000 hopG1 single mutant and cytochalasin-d (Fig. 6B). We did not observe any phenotypic differences in leaves from the kinesin mutant inoculated with cytochalasin-d or jasplakinolide alone (Supplemental Fig. S7B). Consistent with the data presented in Fig. 6, C and D, we observed no impact on Pst DC3000 growth in the presence or absence of the actin cytoskeletal inhibitors in the Arabidopsis kinesin mutant (Fig. 7, D and E). Interestingly, treatment of the kinesin mutant with flg22 showed no significant changes in defense marker gene expression or MAPK phosphorylation as compared to wild-type Col-0 (Supplemental Fig. S8). As a function of the link between HopG1, kinesin, and host susceptibility, we also observed no change in the in planta bacterial growth in the kinesin mutant inoculated with the Pst DC3000 ΔhopG1 (Supplemental Fig. S9). In total, these data further support the hypothesis that HopG1 functions in an actin-associated manner to induce host chlorosis. From this, we posit a working model whereby HopG1 targeting of kinesin is a mechanism to induce actin filament bundling during infection, which, in turn, stimulates host chlorosis and disease symptom development.
DISCUSSION
In this study, we present a comprehensive analysis aimed at defining the link between the modulation of the Arabidopsis actin cytoskeleton and the function of the T3E repertoire. Through this work, we have identified the effector protein HopG1 as a specific inducer of changes in host cytoskeletal architecture, and using a combination of genetic- and cell biology-based approaches, we demonstrate that modulation of the host cytoskeleton, through the action of HopG1, is required for disease symptom development.
In plants, the actin cytoskeleton has been demonstrated to specifically respond to numerous stimuli and processes associated with defense signaling during pathogen infection (Miklis et al., 2007; Tian et al., 2009; Wang et al., 2009; Porter et al., 2012). Similarly, recent evidence supports a role for plant pathogenic effectors as possessing specific functions associated with directly, and/or indirectly, modulating the host cytoskeleton during immune signaling (Lee et al., 2012; Kang et al., 2014). It is hypothesized that these activities function to block defense signaling and disease elicitation. Herein, our aim was to identify and characterize the role of the T3E constellation from Pst DC3000 on the modulation of the host actin cytoskeleton during pathogen infection. Using quantitative imaging of pathogen-infected cells we analyzed the abundance of actin and the extent of actin filament bundling in the cortex of Arabidopsis epidermal cells. Using this approach, we observed an increase in filament bundling and an overall reduction in filament density at 24 h after inoculation with the virulent bacterial phytopathogen Pst DC3000 (Henty-Ridilla et al., 2013), and demonstrate that this response was dependent upon the delivery of T3Es into the host cell. Based on these observations, we posit that pathogen-induced alterations to host cytoskeletal architecture represent a virulence mechanism to dampen host immunity. Indeed, the data presented herein support the hypothesis that the reorganization of the actin cytoskeleton in response to bacterial phytopathogen perception is not only part of the initial host immune response (i.e. PTI), but also provides support that manipulation of this activity by a pathogen is a virulence mechanism whose function is to modulate disease progression.
One of the outstanding questions with regard to phytopathogen manipulation of the host actin cytoskeleton is if pathogen targeting is associated with a virulence function specifically directed at blocking host immune signaling, or, if pathogens hijack actin-associated immune signaling processes to alter broader host responses. To address this, and to further differentiate the early [i.e. PTI; 3–6 h; (Henty-Ridilla et al., 2013)] and late (i.e. effector-triggered susceptibility; 24 hpi) cortical actin remodeling events, we employed a suite of Pst DC3000 mutants (Wei et al., 2007; Kvitko et al., 2009) to dissect the contribution of effector activity on host cytoskeletal organization during infection. Using this approach, we correlated the presence or absence of combinatorial suites of T3Es with pathogen proliferation, the onset of disease symptoms, and changes in host filament architecture. Through this, we determined that the ability of Pst DC3000 to manipulate actin architecture is encoded within two distinct effector gene regions. The first, residing within the CEL cluster, displayed an activity associated with the inhibition of filament bundling. We identified this response as being associated with the conserved effector AvrE (Supplemental Fig. S4). The second effector (i.e. HopG1), localized within cluster IX, revealed an effector activity consistent with enhanced filament bundling, destabilization of individual filaments, and reduced array density. Precedence for similar virulence activities associated with the manipulation of host actin cytoskeletal architecture by bacterial pathogenic effectors exists, and work in this area has led to the characterization of the virulence activities of numerous T3Es (Shao, 2008; McGhie et al., 2009). For example, analysis of the human bacterial pathogen Salmonella enterica has led to the identification of what is now one of the best-characterized virulence strategies of pathogen modulation of host signaling through opposing effector functions (for review, see Galan and Zhou, 2000; Patel and Galán, 2005). In short, work in this area has defined the activities of the T3Es SipC (G-actin polymerization) and SipA (F-actin stabilization), whose synchronous activities function to modulate host filament dynamics to dampen immune signaling. As a well-defined mechanism of attenuation, the S. enterica T3E, SptP, functions to reset host filament dynamics after pathogen invasion, effectively resetting the homeostatic behavior of the host cytoskeleton. Through these effector activities, such a mechanism would serve to aid in bacterial invasion of the host cell (e.g. block phagocytosis and vesicle trafficking), while functioning to limit cellular damage.
Previous work suggests that HopG1 delivery and its subsequent localization to the mitochondria correlates with the initiation of hallmark host defense responses, including the generation of reactive oxygen species, the suppression of programmed cell death, and the induction of chlorosis and disease symptoms (Jamir et al., 2004; Block et al., 2010; Cunnac et al., 2011). In the case of the latter, the work presented herein uncovering a link between HopG1 and host symptom development provides a foundation to further define the relationship among host actin cytoskeletal modulation, pathogen targeting of the immune network, and onset of disease. As a function of pathogen virulence, the data presented herein describe the targeting of the plant actin cytoskeleton by a phytopathogenic bacterial effector protein leading to the development of disease symptoms after infection.
Previous work by Mukhtar et al. (2011) identified an association between HopG1 and an Arabidopsis kinesin motor protein through a comprehensive yeast two-hybrid screen aimed at identifying putative virulence- and resistance-associated interactions during host infection by Pst DC3000. As a putative mechanism underpinning the relevance of this association, work by Block et al. (2010) showed that transiently expressed HopG1 localized to the plant mitochondria, a phenomenon hypothesized to be associated with a mechanism underpinning pathogen-induced cell death. However, a putative mechanism for HopG1 virulence, as well as the consequence of kinesin association, remained undefined. As a possible mechanism to describe the activity and localization of HopG1, we identified a specific in planta interaction between the T3E and the mitochondrial-localized kinesin motor protein, demonstrating that this interaction, together with the function of the host actin cytoskeleton, leads to the elicitation of pathogen-induced senescence. As a broader function of the role of kinesin in actin cytoskeletal dynamics, a study by Iwai et al. (2004) showed that in Dictyostelium discoideum, a kinesin-related protein likely functions as a biochemical link between the organization of the actin filament array and the microtubule network. However, the precise function of kinesin as a regulator of actin filament dynamics still remains largely undefined. To this end, a recent study by Kong et al. (2015) suggested a possible mechanism through the use of live cell imaging to demonstrate that a kinesin-like calmodulin-binding protein is required for proper microtubule-actin filament assembly. In short, this data suggests that kinesin(s) may fulfill a function ensuring that the cytoskeleton is properly arranged to support cell shape and development. While a specific role for kinesin during pathogen defense signaling has not been described, it is interesting to speculate that one potential role of this class of motor proteins is to integrate the functions of myosin and actin to support cellular trafficking during pathogen invasion, and ultimately, to support immune signaling.
As described, our data support the hypothesis that the activity of kinesin is linked to the virulence function of HopG1 and the modulation of host immunity. Based on the results presented herein, we posit that localization of HopG1 to Arabidopsis mitochondria, where it associates with kinesin, results in an increase in actin bundling via the activity of kinesin. Support for such a mechanism lies in previous studies that have demonstrated a relationship between actin filament bundling and the induction of cell death, a process that has been observed in both plant and animal systems (Gourlay and Ayscough, 2005a, 2005b; Franklin-Tong and Gourlay, 2008; Chang et al., 2015). Based on these data, coupled with our current observations presented herein, we propose a mechanism through which modulation of the actin cytoskeleton organization by HopG1 leads to the induction of host senescence, a process that results in pathogen-induced susceptibility and disease symptom development. What remains to be defined is the consequence of the HopG1-kinesin association; at this stage, we hypothesize that HopG1 modifies kinesin function to promote host actin cytoskeletal bundling, which in turn leads to the modulation of cellular processes associated with the development of disease symptoms. Indeed, our data support such as a mechanism, and moreover, provide evidence that this activity results in the induction of host disease symptoms. Similar to that previously described in mammalian cells (Weinrauch and Zychlinsky, 1999), we hypothesize that such a mechanism would serve to promote pathogen infection, survival, and proliferation. To further define the host defense mechanism(s) through which kinesin functions, we also tested a suite of previously described PTI signaling genes for their response to pathogen perception; however, we observed little to no change in the response(s) elicited in the kinesin mutant relative to wild-type Col-0. We posit that this further strengthens our hypothesis that the role of kinesin-actin-HopG1 signaling node is primarily associated with effector-triggered susceptibility.
In the case of plant-pathogen interactions, the induction of symptom development would also serve the additional purpose of promoting the release of metabolites into the extracellular space of the host cell, thereby providing the pathogen with enhanced access to nutrients. While the molecular and biochemical mechanism(s) associated with the induction of disease symptoms by pathogens in their hosts is a poorly understood process, work in this area has established a foundation supporting the hypothesis that symptom development is a process that is required for pathogen egress, facilitating pathogen survival, escape, and spread (Butt et al., 1998; Mecey et al., 2011). The data presented herein describe a link among HopG1, the induction of changes in host actin cytoskeletal architecture, and the elicitation of disease symptoms. Precedence for such a mechanism comes from work in yeast models, which have demonstrated that rearrangements to actin filament architecture have been shown to stimulate apoptosis (Gourlay and Ayscough, 2005a, 2005b), further suggesting that actin may be a significant player in the modulation of programmed cell death-associated processes. Previous work demonstrating mitochondrial association places HopG1 in a cellular location to facilitate such a process. This work provides evidence supporting a role for the mitochondria and the host actin cytoskeleton in pathogen-induced chlorosis and the promotion of disease symptom development, a key virulence function of plant pathogens (Kim et al., 2008). In total, the data presented herein not only define the hijacking of the host actin cytoskeleton function by the T3E HopG1, but also provide a foundation to further describe the relationship(s) between the modulation of the host actin cytoskeleton and the regulation of numerous physiological processes, including those required for cell death, immunity, and pathogen survival.
MATERIALS AND METHODS
Plant Growth
Arabidopsis Col-0 plants were grown in a model no. FLX-37 growth chamber (BioChambers, Winnipeg, Manitoba, Canada) at 20°C under a 12 h/12 h light/dark cycle with 60% relative humidity and a light intensity of 120 μmol photons m−2 s−1.
Pathogen Inoculation and Measurement of Bacterial Growth
Pseudomonas syringae pv. tomato DC3000 (Pst DC3000) strains relevant to this study are shown in Supplemental Table S1, and were grown as previously described in Tian et al. (2009). Full-length Pst DC3000 T3E genes were cloned in a Gateway-compatible pBBR-MCS2 vector and mobilized into Pst DC3000 via triparental mating or electroporation as previously described in Chang et al. (2005) and Choi et al. (2006). For bacterial growth curve analyses, plants were dip-inoculated as described in Kunkel et al. (1993). Samples were collected at 0 and 24 h post-inoculation. Bacterial growth assays on mature leaves were performed as previously described in Tian et al. (2009). For growth assays performed on 10-d-old seedlings, 12 individual seedlings (approximately 20 mg) were surface-sterilized in 10% bleach for 30 s, followed by two washes with sterile dH2O. After washing, seedlings were transferred to sterile microfuge tubes containing 10 mm MgCl2 + 0.02% Tween 20, and shaken at 28°C for 1 h. Samples were further processed as described above.
Actin Cytoskeletal Inhibitors and Flg22 Treatment
Cytochalasin-d (Sigma-Aldrich, St. Louis, MO) and jasplakinolide (Calbiochem/EMD Millipore, Darmstadt, Germany) were dissolved in DMSO and stored as 10-mm and 1-mm stock solutions, respectively, at −20°C. Working concentrations for cytochalasin-d and jasplakinolide were 10 µm and 0.5 µm, respectively. Cytochalasin-d and jasplakinolide treatments were performed by hand-infiltration using a needleless syringe. Flg22 was spray-inoculated onto plants at a final concentration of 10 µm.
RNA Isolation and Quantitative Real-Time PCR
Extraction of total RNA from Arabidopsis leaves was performed using the PrepEase Plant RNA Spin kit (USB Affymetrix, Cleveland, OH), as previously described in Knepper et al. (2011). One μg of total RNA was used for first-strand cDNA synthesis using the First-Strand cDNA Synthesis kit (USB Affymetrix). All primers used for quantitative real-time PCR (qRT-PCR) are listed in Supplemental Table S2. HotStart SYBR Green qPCR Master Mix 2X (USB Affymetrix) was used to perform qRT-PCR with a Mastercycler EP Realplex System (Eppendorf AG, Hamburg, Germany). Ubiquitin (UBQ10) was used as an internal control for amplification. This after-equation was used to calculate fold expression:
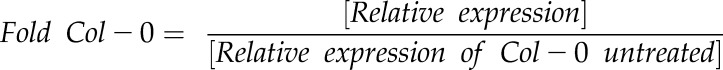 |
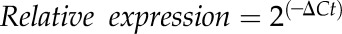 , where
, where 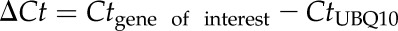 .
.
Construction of the hopG1 Mutant
The ΔhopG1 pK18mobsacB-derivative deletion construct pMSPP001 was created by PCR amplification of a hopG1 upstream flanking region with primers BKO1 and BKO2 followed by cloning into pCR2.1-TOPO (Invitrogen, Carlsbad, CA). The hopG1 flank was then PCR-amplified from pCR2.1 using the DNA primers M13R and BKO2 to append the flank with a SpeI site. The amplicon was restriction-digested with SpeI and SalI and ligated with the SpeI-and SalI-digested backbone of pCPP5917 (pK18mobsacB::ΔhopAA1-2-hopG1::FRT-SpR-FRT) (Kvitko et al., 2009). This replaces the hopAA1-2 flanking region and FRT-SpR-FRT cassette with the hopG1 upstream flanking region. HopG1 was deleted from Pst DC3000 to recover PSPRL011 using previously described methods from Kvitko and Collmer (2011). Briefly, E. coli RHO5 (Kvitko et al., 2012) pMSPP001 and Pst DC3000 were conjugated in a biparental mating to recover kanamycin-resistant (KmR) single-crossover pMSP001 merodiploids. Resolved KmS double-crossover strains were recovered by Suc counter-selection and hopG1 deletions were confirmed by PCR with primers BKO52 and BKO53.
Confocal Microscopy and Actin Filament Analysis
To evaluate pathogen-induced changes in actin organization, skewness and density analyses were performed according to Higaki et al. (2010) with slight modification. All quantitative image analyses were performed as undirected single-blind experiments. Actin filament bundling and percent occupancy calculations were performed as previously described in Henty-Ridilla et al. (2013), using skewness and density as defined metrics to describe actin filament organization. Fields of epidermal pavement cells were imaged using a laser confocal scanning microscope to obtain 25 z-series sections at 0.5-μm intervals. Laser confocal scanning microscopy was performed with a 60×/1.42 PlanApo N objective on a model no. FV1000D microscope (Olympus, Melville, NY). Serial optical sections were projected with maximum intensity and analyzed with the software ImageJ (National Institutes of Health, Bethesda, MD) using algorithms previously described in Higaki et al. (2010). Gaussian blur and high-band pass filters were applied to stack images before generating projection images for density analysis. Statistical analyses were performed using a Student’s t-test.
HopG1 and Kinesin Cloning
The open reading frame of HopG1 was amplified using the gene-specific DNA primers HopG1-XhoIF and HopG1-SpeIT7-R (Supplemental Table S2) by PCR, and subcloned into pENTR/D-TOPO (Invitrogen), then moved into the binary plant expression vector pEarleyGate-203 carrying a c-Myc tag at the N terminus using Gateway cloning (LR Clonase II; Thermo Fisher Scientific, Waltham, MA). The 5′-terminal half (i.e. nucleotides 1–978) of kinesin (At4g39050), which contains a sequence encoding a putative mitochondria-localization signal, was amplified from reverse-transcribed cDNA using the gene-specific primers Kinesin-F and Kinesin-R (Supplemental Table S2) and subcloned into pENTR/D-TOPO. The resultant DNA fragment was cloned using Gateway LR Clonase II into the Gateway destination vector pEarleyGate-101 (Thermo Fisher Scientific), yielding a C-terminal fusion with YFP-HA. The Arabidopsis kinesin mutant (SALK_130788) was received from the Arabidopsis Biological Resource Center (The Ohio State University, Columbus, OH).
Agrobacterium-Mediated Protein Expression and Co-Immunoprecipitation Assays
Sequence-confirmed binary vector constructs were electroporated into Agrobacterium tumefaciens strains C58C1 and GV3101 and maintained on Luria-Bertani plates containing rifampicin (C58C1; 50 µm/ml), kanamycin (C58C1 and GV3101; 100 µm/ml), gentamycin (GV3101; 50 µm/ml), and tetracycline (C58C1; 10 µm/ml). For transient expression in Nicotiana benthamiana, A. tumefaciens containing expression constructs of interest were preincubated in induction media [10 mm MES (pH 5.6), 10 mm MgCl2, 150 mm acetosyringone (Sigma-Aldrich)] for 2 h at room temperature before inoculation into 5-week-old N. benthamiana leaves using a needleless syringe. Inoculated plants were kept at 26°C for 40 h (12 h light/12 h dark), after which time 15 leaf discs (1 cm2/each) were harvested. For co-immunprecipitation experiments, 15−1 cm2 leaf discs were harvested and processed as previously described in Knepper et al. (2011). In short, total protein extracts were isolated by grinding leaf discs in liquid nitrogen into a fine powder, and then transferred to a chilled mortar and further homogenized in homogenization buffer [50 mm HEPES (pH 7.5), 50 mm NaCl, 10 mm EDTA, 0.2% Triton X-100, 1 cOmplete, Mini, EDTA-Free Protease Inhibitor Cocktail Tablet (1 tablet/50 ml homogenization buffer; Sigma-Aldrich)]. Samples were first tumbled end-over-end at 4°C for 1 h with either 5 μl of anti-HA monoclonal antibody or anti-T7 monoclonal antibody, followed by the addition of 50 μl protein G sepharose 4 fast flow (GE Healthcare Life Sciences, Little Chalfont, Buckinghamshire, UK) and tumbled end-over-end at 4°C for 4 h. After 4 h, samples were centrifuged at 15,000g for 15 s, and the pellets were washed (three times) with wash buffer [50 mm HEPES (pH 7.5), 50 mm NaCl, 10 mm EDTA (EDTA), 0.1% Triton X-100, 1 cOmplete, Mini, EDTA-Free Protease Inhibitor Cocktail Tablet (1 tablet/50 ml homogenization buffer; Sigma-Aldrich)]. For SDS-PAGE (10% Bis-Tris) analysis, 25 μl of the input sample, 10 μl of the IP sample, and 20 μl of the co-immunprecipitation samples were separated, and resolved samples were transferred to nitrocellulose (GE Healthcare Life Sciences). Signals were detected using Super Signal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific), using anti-HA-HRP (Roche, Basel, Switzerland), anti-T7-HRP (Novagen, Madison, WI), or monoclonal anti-actin (Sigma-Aldrich).
Supplemental Data
The following supplemental materials are available.
Supplemental Table S1. Pst DC3000 strains relevant to this study.
Supplemental Table S2. DNA primers used for cloning and qRT-PCR analysis.
Supplemental Figure S1. FRK1 mRNA expression is induced in 10-d-old wild-type Col-0 seedlings expressing GFP-fABD2 when spray-inoculated with flg22, but changes in filament architecture are not.
Supplemental Figure S2. Arabidopsis seedlings inoculated with Pst DC3000 EV and Pst DC3000 wild type show a similar actin skewness/density response.
Supplemental Figure S3. Actin skewness and density in Arabidopsis inoculated with Pst DC3000 mutants lacking clustered T3E genes.
Supplemental Figure S4. The type-III secreted effector AvrE suppresses actin bundling and a concomitant increase in filament density in Arabidopsis epidermal cells.
Supplemental Figure S5. Pst DC3000 complemented cluster IX polymutants for in planta growth.
Supplemental Figure S6. Cytochalasin-d and jasplakinolide do not induce chlorosis in wild-type Col-0 plants.
Supplemental Figure S7. Genetic (qRT-PCR) and actin pharmacological evaluation of the Arabidopsis kinesin mutant, SALK_130788.
Supplemental Figure S8. qRT-PCR analysis of defense signaling marker genes from wild-type Col-0 and the Arabidopsis kinesin mutant (SALK_130788).
Supplemental Figure S9. The Pst DC3000 Δhopg1 mutant does not show an altered in planta growth phenotype in the kinesin mutant (SALK_130788) as compared to wild-type Col-0.
Supplementary Material
Acknowledgments
We thank Amy Baetsen-Young and Alex Corrion for critical reading of the manuscript, as well as suggestions made during its development. Melinda Frame (MSU Center for Advanced Microscopy) is acknowledged for technical assistance and guidance. The authors would like to acknowledge Alan Collmer for providing the P. syringae CUCPB clones used in this study.
Glossary
- CEL
conserved effector locus
- PAMP(s)
pathogen-associated molecular pattern(s)
- PRR(s)
pathogen receptive gene(s)
- PTI
PAMP-triggered immunity
- T3SS
type-III secretion system
- T3E(s)
type-III effector(s)
Footnotes
Articles can be viewed without a subscription.
References
- Alfano JR, Charkowski AO, Deng WL, Badel JL, Petnicki-Ocwieja T, van Dijk K, Collmer A (2000) The Pseudomonas syringae Hrp pathogenicity island has a tripartite mosaic structure composed of a cluster of type III secretion genes bounded by exchangeable effector and conserved effector loci that contribute to parasitic fitness and pathogenicity in plants. Proc Natl Acad Sci USA 97: 4856–4861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M, Zhou J, Faulkner C, MacLean D, Robatzek S (2012) Spatio-temporal cellular dynamics of the Arabidopsis flagellin receptor reveal activation status-dependent endosomal sorting. Plant Cell 24: 4205–4219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block A, Guo M, Li G, Elowsky C, Clemente TE, Alfano JR (2010) The Pseudomonas syringae type III effector HopG1 targets mitochondria, alters plant development and suppresses plant innate immunity. Cell Microbiol 12: 318–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt A, Mousley C, Morris K, Beynon J, Can C, Holub E, Greenberg JT, Buchanan-Wollaston V (1998) Differential expression of a senescence-enhanced metallothionein gene in Arabidopsis in response to isolates of Peronospora parasitica and Pseudomonas syringae. Plant J 16: 209–221 [DOI] [PubMed] [Google Scholar]
- Cai G, Faleri C, Del Casino C, Emons AM, Cresti M (2011) Distribution of callose synthase, cellulose synthase, and sucrose synthase in tobacco pollen tube is controlled in dissimilar ways by actin filaments and microtubules. Plant Physiol 155: 1169–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JH, Urbach JM, Law TF, Arnold LW, Hu A, Gombar S, Grant SR, Ausubel FM, Dangl JL (2005) A high-throughput, near-saturating screen for type III effector genes from Pseudomonas syringae. Proc Natl Acad Sci USA 102: 2549–2554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang X, Riemann M, Liu Q, Nick P (2015) Actin as deathly switch? How auxin can suppress cell-death related defence. PLoS One 10: e0125498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisholm ST, Coaker G, Day B, Staskawicz BJ (2006) Host-microbe interactions: shaping the evolution of the plant immune response. Cell 124: 803–814 [DOI] [PubMed] [Google Scholar]
- Choi KH, Kumar A, Schweizer HP (2006) A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J Microbiol Methods 64: 391–397 [DOI] [PubMed] [Google Scholar]
- Collmer A, Badel JL, Charkowski AO, Deng WL, Fouts DE, Ramos AR, Rehm AH, Anderson DM, Schneewind O, van Dijk K, Alfano JR (2000) Pseudomonas syringae Hrp type III secretion system and effector proteins. Proc Natl Acad Sci USA 97: 8770–8777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunnac S, Chakravarthy S, Kvitko BH, Russell AB, Martin GB, Collmer A (2011) Genetic disassembly and combinatorial reassembly identify a minimal functional repertoire of type III effectors in Pseudomonas syringae. Proc Natl Acad Sci USA 108: 2975–2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day B, Henty JL, Porter KJ, Staiger CJ (2011) The pathogen-actin connection: a platform for defense signaling in plants. Annu Rev Phytopathol 49: 483–506 [DOI] [PubMed] [Google Scholar]
- Dou D, Zhou JM (2012) Phytopathogen effectors subverting host immunity: different foes, similar battleground. Cell Host Microbe 12: 484–495 [DOI] [PubMed] [Google Scholar]
- Franklin-Tong VE, Gourlay CW (2008) A role for actin in regulating apoptosis/programmed cell death: evidence spanning yeast, plants and animals. Biochem J 413: 389–404 [DOI] [PubMed] [Google Scholar]
- Galan JE, Zhou D (2000) Striking a balance: modulation of the actin cytoskeleton by Salmonella. Proc Natl Acad Sci USA 97: 8754–8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourlay CW, Ayscough KR (2005a) Identification of an upstream regulatory pathway controlling actin-mediated apoptosis in yeast. J Cell Sci 118: 2119–2132 [DOI] [PubMed] [Google Scholar]
- Gourlay CW, Ayscough KR (2005b) The actin cytoskeleton: a key regulator of apoptosis and ageing? Nat Rev Mol Cell Biol 6: 583–589 [DOI] [PubMed] [Google Scholar]
- Henty-Ridilla JL, Shimono M, Li J, Chang JH, Day B, Staiger CJ (2013) The plant actin cytoskeleton responds to signals from microbe-associated molecular patterns. PLoS Pathog 9: e1003290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higaki T, Kutsuna N, Sano T, Kondo N, Hasezawa S (2010) Quantification and cluster analysis of actin cytoskeletal structures in plant cells: role of actin bundling in stomatal movement during diurnal cycles in Arabidopsis guard cells. Plant J 61: 156–165 [DOI] [PubMed] [Google Scholar]
- Itoh R, Fujiwara M, Yoshida S (2001) Kinesin-related proteins with a mitochondrial targeting signal. Plant Physiol 127: 724–726 [PMC free article] [PubMed] [Google Scholar]
- Iwai S, Ishiji A, Mabuchi I, Sutoh K (2004) A novel actin-bundling kinesin-related protein from Dictyostelium discoideum. J Biol Chem 279: 4696–4704 [DOI] [PubMed] [Google Scholar]
- Jamir Y, Guo M, Oh HS, Petnicki-Ocwieja T, Chen S, Tang X, Dickman MB, Collmer A, Alfano JR (2004) Identification of Pseudomonas syringae type III effectors that can suppress programmed cell death in plants and yeast. Plant J 37: 554–565 [DOI] [PubMed] [Google Scholar]
- Jing B, Xu S, Xu M, Li Y, Li S, Ding J, Zhang Y (2011) Brush and spray: a high-throughput systemic acquired resistance assay suitable for large-scale genetic screening. Plant Physiol 157: 973–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Jelenska J, Cecchini NM, Li Y, Lee MW, Kovar DR, Greenberg JT (2014) HopW1 from Pseudomonas syringae disrupts the actin cytoskeleton to promote virulence in Arabidopsis. PLoS Pathog 10: e1004232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JG, Taylor KW, Hotson A, Keegan M, Schmelz EA, Mudgett MB (2008) XopD SUMO protease affects host transcription, promotes pathogen growth, and delays symptom development in xanthomonas-infected tomato leaves. Plant Cell 20: 1915–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knepper C, Savory EA, Day B (2011) Arabidopsis NDR1 is an integrin-like protein with a role in fluid loss and plasma membrane-cell wall adhesion. Plant Physiol 156: 286–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Z, Ioki M, Braybrook S, Li S, Ye ZH, Julie Lee YR, Hotta T, Chang A, Tian J, Wang G, Liu B (2015) Kinesin-4 functions in vesicular transport on cortical microtubules and regulates cell wall mechanics during cell elongation in plants. Mol Plant 8: 1011–1023 [DOI] [PubMed] [Google Scholar]
- Kunkel BN, Bent AF, Dahlbeck D, Innes RW, Staskawicz BJ (1993) RPS2, an Arabidopsis disease resistance locus specifying recognition of Pseudomonas syringae strains expressing the avirulence gene avrRpt2. Plant Cell 5: 865–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvitko BH, Bruckbauer S, Prucha J, McMillan I, Breland EJ, Lehman S, Mladinich K, Choi KH, Karkhoff-Schweizer R, Schweizer HP (2012) A simple method for construction of pir+ Enterobacterial hosts for maintenance of R6K replicon plasmids. BMC Res Notes 5: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvitko BH, Collmer A (2011) Construction of Pseudomonas syringae pv. tomato DC3000 mutant and polymutant strains. Methods Mol Biol 712: 109–128 [DOI] [PubMed] [Google Scholar]
- Kvitko BH, Park DH, Velásquez AC, Wei CF, Russell AB, Martin GB, Schneider DJ, Collmer A (2009) Deletions in the repertoire of Pseudomonas syringae pv. tomato DC3000 type III secretion effector genes reveal functional overlap among effectors. PLoS Pathog 5: e1000388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Hurley B, Felsensteiner C, Yea C, Ckurshumova W, Bartetzko V, Wang PW, Quach V, Lewis JD, Liu YC, Börnke F, Angers S, et al. (2012) A bacterial acetyltransferase destroys plant microtubule networks and blocks secretion. PLoS Pathog 8: e1002523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SG, Zhu DZ, Chen GH, Gao XQ, Zhang XS (2012) Disrupted actin dynamics trigger an increment in the reactive oxygen species levels in the Arabidopsis root under salt stress. Plant Cell Rep 31: 1219–1226 [DOI] [PubMed] [Google Scholar]
- Loper JE, Lindow SE (1987) Lack of evidence for in situ fluorescent pigment production by Pseudomonas syringae pv. syringae on bean leaf surface. Phytopathology 77: 1449–1454 [Google Scholar]
- McGhie EJ, Brawn LC, Hume PJ, Humphreys D, Koronakis V (2009) Salmonella takes control: effector-driven manipulation of the host. Curr Opin Microbiol 12: 117–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecey C, Hauck P, Trapp M, Pumplin N, Plovanich A, Yao J, He SY (2011) A critical role of STAYGREEN/Mendel’s I locus in controlling disease symptom development during Pseudomonas syringae pv tomato infection of Arabidopsis. Plant Physiol 157: 1965–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklis M, Consonni C, Bhat RA, Lipka V, Schulze-Lefert P, Panstruga R (2007) Barley MLO modulates actin-dependent and actin-independent antifungal defense pathways at the cell periphery. Plant Physiol 144: 1132–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishina TE, Zeier J (2007) Pathogen-associated molecular pattern recognition rather than development of tissue necrosis contributes to bacterial induction of systemic acquired resistance in Arabidopsis. Plant J 50: 500–513 [DOI] [PubMed] [Google Scholar]
- Moes D, Gatti S, Hoffmann C, Dieterle M, Moreau F, Neumann K, Schumacher M, Diederich M, Grill E, Shen WH, Steinmetz A, Thomas C (2013) A LIM domain protein from tobacco involved in actin-bundling and histone gene transcription. Mol Plant 6: 483–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtar MS, Carvunis AR, Dreze M, Epple P, Steinbrenner J, Moore J, Tasan M, Galli M, Hao T, Nishimura MT, Pevzner SJ, Donovan SE, et al. ; European Union Effectoromics Consortium (2011) Independently evolved virulence effectors converge onto hubs in a plant immune system network. Science 333: 596–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JC, Galán JE (2005) Manipulation of the host actin cytoskeleton by Salmonella—all in the name of entry. Curr Opin Microbiol 8: 10–15 [DOI] [PubMed] [Google Scholar]
- Porter K, Day B (2013) Actin branches out to link pathogen perception and host gene regulation. Plant Signal Behav 8: e23468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter K, Shimono M, Tian M, Day B (2012) Arabidopsis Actin-Depolymerizing Factor-4 links pathogen perception, defense activation and transcription to cytoskeletal dynamics. PLoS Pathog 8: e1003006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Milla MA, Salinas J (2009) Prefoldins 3 and 5 play an essential role in Arabidopsis tolerance to salt stress. Mol Plant 2: 526–534 [DOI] [PubMed] [Google Scholar]
- Shao F. (2008) Biochemical functions of Yersinia type III effectors. Curr Opin Microbiol 11: 21–29 [DOI] [PubMed] [Google Scholar]
- Sheahan MB, Staiger CJ, Rose RJ, McCurdy DW (2004) A green fluorescent protein fusion to actin-binding domain 2 of Arabidopsis fimbrin highlights new features of a dynamic actin cytoskeleton in live plant cells. Plant Physiol 136: 3968–3978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada C, Lipka V, O’Connell R, Okuno T, Schulze-Lefert P, Takano Y (2006) Nonhost resistance in Arabidopsis-Colletotrichum interactions acts at the cell periphery and requires actin filament function. Mol Plant Microbe Interact 19: 270–279 [DOI] [PubMed] [Google Scholar]
- Smertenko AP, Deeks MJ, Hussey PJ (2010) Strategies of actin reorganisation in plant cells. J Cell Sci 123: 3019–3028 [DOI] [PubMed] [Google Scholar]
- Staiger CJ, Poulter NS, Henty JL, Franklin-Tong VE, Blanchoin L (2010) Regulation of actin dynamics by actin-binding proteins in pollen. J Exp Bot 61: 1969–1986 [DOI] [PubMed] [Google Scholar]
- Staiger CJ, Sheahan MB, Khurana P, Wang X, McCurdy DW, Blanchoin L (2009) Actin filament dynamics are dominated by rapid growth and severing activity in the Arabidopsis cortical array. J Cell Biol 184: 269–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto D, Maeda H, Yoshioka H, Doke N, Kawakita K (1999) Effect of cytochalasin d on defense responses of potato tuber discs treated with hyphal wall components of Phytophthora infestans. Plant Sci 141: 219–226 [Google Scholar]
- Thomas C. (2012) Bundling actin filaments from membranes: some novel players. Front Plant Sci 3: 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian M, Chaudhry F, Ruzicka DR, Meagher RB, Staiger CJ, Day B (2009) Arabidopsis actin-depolymerizing factor AtADF4 mediates defense signal transduction triggered by the Pseudomonas syringae effector AvrPphB. Plant Physiol 150: 815–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wen Y, Berkey R, Xiao S (2009) Specific targeting of the Arabidopsis resistance protein RPW8.2 to the interfacial membrane encasing the fungal Haustorium renders broad-spectrum resistance to powdery mildew. Plant Cell 21: 2898–2913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CF, Kvitko BH, Shimizu R, Crabill E, Alfano JR, Lin NC, Martin GB, Huang HC, Collmer A (2007) A Pseudomonas syringae pv. tomato DC3000 mutant lacking the type III effector HopQ1-1 is able to cause disease in the model plant Nicotiana benthamiana. Plant J 51: 32–46 [DOI] [PubMed] [Google Scholar]
- Weinrauch Y, Zychlinsky A (1999) The induction of apoptosis by bacterial pathogens. Annu Rev Microbiol 53: 155–187 [DOI] [PubMed] [Google Scholar]
- Worley JN, Russell AB, Wexler AG, Bronstein PA, Kvitko BH, Krasnoff SB, Munkvold KR, Swingle B, Gibson DM, Collmer A (2013) Pseudomonas syringae pv. tomato DC3000 CmaL (PSPTO4723), a DUF1330 family member, is needed to produce L-allo-isoleucine, a precursor for the phytotoxin coronatine. J Bacteriol 195: 287–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin XF, He SY (2013) Pseudomonas syringae pv. tomato DC3000: a model pathogen for probing disease susceptibility and hormone signaling in plants. Annu Rev Phytopathol 51: 473–498 [DOI] [PubMed] [Google Scholar]
- Yun BW, Atkinson HA, Gaborit C, Greenland A, Read ND, Pallas JA, Loake GJ (2003) Loss of actin cytoskeletal function and EDS1 activity, in combination, severely compromises non-host resistance in Arabidopsis against wheat powdery mildew. Plant J 34: 768–777 [DOI] [PubMed] [Google Scholar]
- Zhang J, Li W, Xiang T, Liu Z, Laluk K, Ding X, Zou Y, Gao M, Zhang X, Chen S, Mengiste T, Zhang Y, et al. (2010) Receptor-like cytoplasmic kinases integrate signaling from multiple plant immune receptors and are targeted by a Pseudomonas syringae effector. Cell Host Microbe 7: 290–301 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.