

Abstract
Synapses are the principal sites for chemical communication between neurons and are essential for performing the dynamic functions of the brain. In Alzheimer’s disease and related tauopathies, synapses are exposed to disease modified protein tau, which may cause the loss of synaptic contacts that culminate in dementia. In recent decades, structural, transcriptomic and proteomic studies suggest that Alzheimer’s disease represents a synaptic disorder. Tau neurofibrillary pathology and synaptic loss correlate well with cognitive impairment in these disorders. Moreover, regional distribution and the load of neurofibrillary lesions parallel the distribution of the synaptic loss. Several transgenic models of tauopathy expressing various forms of tau protein exhibit structural synaptic deficits. The pathological tau proteins cause the dysregulation of synaptic proteome and lead to the functional abnormalities of synaptic transmission. A large body of evidence suggests that tau protein plays a key role in the synaptic impairment of human tauopathies.
Keywords: Alzheimer’s disease, Synaptic loss, Tau protein, Neurofibrillary degeneration, Tauopathies, Tau mislocalization, Transgenic models
Introduction
Cognitive functions such as learning and memory depend on synaptic efficiency in certain regions of the brain [1]. In human neurodegenerative diseases, synapses are exposed to pathologically modified proteins aggregated in the intracellular and extracellular space. The protein aggregates may induce the loss of synaptic connections in vulnerable brain areas. The recurrent dysregulation of synaptic proteins [2], rapid N-methyl-d-aspartate receptors (NMDAR) endocytosis and regression of dendritic spines [3] are the forerunners in imposing synaptic impairment in these disorders.
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disorder with an estimated 35 million people affected worldwide [4]. The risk factors for AD include lower mental and physical activity during old age, head trauma, cardiovascular diseases, diabetes, obesity and smoking [5]. Histological examinations of an AD brain uncover two classical hallmarks, namely neurofibrillary tangles - composed of tau protein and senile plaques consisting of amyloid-beta (Aβ) protein [6, 7]. A very small proportion of AD cases have genetic dispositions which are categorized as familial AD [8]. However, the majority of AD cases are idiopathic, meaning that the cause of the illness is unknown. [9]. Despite scientific and pharmaceutical advancements, AD still poses as an epidemiological challenge for the future [10]. Therapeutic intervention for a neurodegenerative disease is best performed before irreversible memory loss and tissue damage occurs [11]. Therefore, investigating and understanding early pathological changes in AD would prove to be beneficial.
It has been suggested that AD may represent a synaptic disorder [12–14]. Synaptic impairment occurs very early in AD and correlates well with the severity of dementia [15–18]. Furthermore, at least certain components of the synaptic loss in AD occur regionally and are disproportionately large in the hippocampus [2].
Several studies have demonstrated that the degree of synaptic impairment and loss is linked with tangle pathology [19–21]. Therefore, studies on the involvement of tau protein in synaptic damage have received increased importance in recent years [22]. Identification of the molecular mechanisms underlying tau mediated synaptic damage in AD signifies an important step in the development of therapeutic agents that can prevent or delay the onset or progression of the disease.
Tau physiology and function
Tau protein belongs to the family of microtubule-associated proteins (MAP). It is localized mainly in the neurons of both vertebrates and certain invertebrates. In the human brain, tau proteome consists of six isoforms ranging from 352 to 441 aa [23]. The tau proteins are further classified by the presence of three repeat (3R) or four repeat (4R) regions in the C-terminal and the presence or absence of one (29 aa) or two (58 aa) inserts in the N-terminal region [24–26]. It is suggested that the repeat regions aa 244–368 of tau bind to microtubules directly [27] and the aa domains 151–243 and 369–400 surrounding the repeat region enhance the affinity of microtubule binding of tau.
Tau is involved in retrograde and anterograde transport by differential interaction with dynein and kinesin motor proteins [28]. Tau interacts with actin and spectrin proteins, this allows microtubules to interconnect with other cytoskeletal components and restrict the flexibility of the microtubules [29]. Furthermore, the N-terminal domain also interacts with the SRC homology 3 (SH3) domain of phospholipase C-γ (PLC-γ) and mediates the generation of arachidonic acid [30]. These results suggest that tau may modulate microtubule flexibility and also alter cell shape and structure. It is reported that the phosphorylation of tau protein in early developmental stages is slightly upregulated to provide optimal flexibility to the growth cones [31]. In addition, tau interacts with embryonic ectoderm development (Eed) protein to facilitate nuclear transport of Eed suggesting a possible role of tau in embryonic development [32]. Studies also suggest a role of tau protein in metabolic rate depression in hibernating animals [33].
Recent studies demonstrate the role of tau protein in long term potentiation [34] and long term depression [35, 36]. Tau to dendrite conglomeration is also necessary for targeting of fyn kinase to the postsynaptic compartment [37] and subsequent phosphorylation of NMDAR subunit NR2B in dendrites [38], and in the initiation of myelination [39]. To sum up, tau protein plays a diverse role in neuronal activity including cytoskeleton organization, signaling and synaptic plasticity.
Neurofibrillary tangles and tau protein
In 1906, Alzheimer first reported the presence of neurofibrillary tangles (NFT) in a woman suffering from dementia [40]. These structures are observed mainly in the glutamatergic pyramidal neurons of the hippocampus and the entorhinal cortex, supra and infragranular layers of association cortical areas, cholinergic neurons of nucleus basalis of Meynert and noradrenergic neurons in the locus coeruleus [6, 23]. Electron microscopy images of NFT were first studied by Kidd and were referred to as longitudinally arranged fibrillar bundles [41]. Additionally, diffraction pattern revealed the presence of a double helical stack of cytoskeletal protofilaments [42]. NFT predominantly composed of paired helical filaments (PHF) are morphologically described as helical ribbons being 8–24 nm in width, with 80-nm periodic twists in AD [43]. Several decades later it was established that tau was one of the main components of NFT [44–47]. However, it was not until 1988 that tau protein was proved to be the major and integral part of the PHFs in AD [48, 49].
Tau is an intrinsically disordered protein [50]. In diseased brains, tau protein undergoes numerous pathological alterations leading to aberrant conformational modifications which liberate tau from the microtubules [51–53]. Although tau phosphorylation is observed in normal human brains, the degree and extent of tau phosphorylation is severe in AD [54, 55]. Phosphorylation is one of the crucial post-translational modifications of tau protein in AD brains [24]. In AD, tau protein is hyperphosphorylated at 19 aa residues [56]. Numerous studies have reported the phosphorylation of tau in the binding domain, which hinders tau binding to microtubules such as Ser 262 [57], Thr 231 [58], Thr 212 and Ser 214 [59]. The liberated tau proteins then aggregate into PHF and deposit intracellularly into NFT. Studies have further shown that isolated PHF exhibit either α-helical or β-sheeted structures in various conditions [60, 61] and intermediary conformations during structural transition [62].
Truncation of tau transforms physiological tau to pathological forms that are vulnerable to oligomerization [63]. Biochemical characterization of PHF revealed a 12 kDa pronase resistant fragment decorated by antibody MN423 [64]. Epitope mapping suggests that MN423 recognizes truncated tau at Glu391 of the PHF core, suggesting that tau truncation is a disease associated process [65]. Later it was shown that the truncation at Glu391 enhanced the rate of tau filament formation [66]. Furthermore, it was also shown that truncation of tau may be involved in the evolution of NFT in AD brains [67–69].
Tau proteome in human tauopathies
Biochemical and proteomic studies demonstrate the existence of different pathological tau compositions in tauopathies. Based on the type of tau isoforms involved, tauopathies are classified into several classes [70]. In class I tauopathies, the aggregation of all 6 tau isoforms in equal ratios is observed [70, 71]. Biochemically, tau triplets of 60, 64 and 69 kDa, and additional minor bands of 72/74 kDa are characteristic for AD, some cases of frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP 17), Niemann-Pick disease, type C, Down syndrome and dementia pugilistica [70].
In class II tauopathies, insoluble tau doublets of 64 and 69 kDa predominantly composed of 4R tau isoforms are observed. This class includes progressive supranuclear palsy (PSP), corticobasal degeneration (CBD) and some specific cases of FTDP-17 [72, 73]. Class III tauopathies are characterized by the presence of pathological tau doublets of 60 kDa and 64 kDa with predominant 3R tau isoforms (lacking the exon 10) [70]. Pick’s disease is the sole neurodegenerative tauopathy assigned to this class [72]. Class IV tauopathy is represented by a single neurological disorder – myotonic dystrophy of type I or Steinert’s disease (DM1), in which a major insoluble tau band of 60 kDa, and minor 64 and 69 kDa bands are identified.
Tau synaptic proteome
The localization of tau in the axonal and somatodendritic compartments has drawn considerable interest in the last decade. It has been suggested that tau protein is localized mainly in the axons [74] due to the presence of an axonal targeting sequence [75]. However, some recent studies indicate a wide spread distribution of tau protein in other compartments including the nucleus [76] and dendrites [37, 77]. Interestingly, tau protein has been detected in the total synaptosomes isolated from a rat brain [38]. Our study demonstrated that tau protein in the rat brain was mainly distributed in presynaptic fractions while in postsynaptic densities it was almost absent [78] (Fig. 1). Isolation and evaluation of synaptic fractions from human and dogs also revealed identical patterns of tau protein distribution (Fig. 1). These results suggest that tau is mostly located in the presynaptic component, which supports the notion that tau is predominantly distributed in axonal compartment.
Figure 1.

Tau synaptic proteome in physiological conditions. Pre- and postsynaptic fractions were isolated as previously published [78]. Synaptic fractions from rat, dog and humans show that tau protein is predominantly distributed in the presynaptic fraction (pre), while in postsynaptic fraction is observed in traces (post).
On the other hand, it has been shown that tau protein migrates to dendrites following synaptic activation and is phosphorylated at various sites [38, 79]. In synapses, tau protein interacts with actin [80], microfilaments [81], postsynaptic density protein 95 (PSD-95), NMDAR [38] and kinases such as fyn [37]. Likewise, tau protein is necessary for dendritic targeting of fyn kinase [37]. Besides, the loss of tau protein in dendrites resulted in a decreased spine density [82]. Independent results from tau knockout mice show that tau protein is essential for NMDA-dependent long term potentiation (LTP) [34] and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)- dependent long term depression (LTD) [35, 36]. Furthermore, selective phosphorylation of tau protein was observed following NMDAR activation which in turn regulates tau interaction with fyn kinase [38]. These results establish a profound role of tau protein in the neuronal dendrites.
Synaptic impairment in Alzheimer’s disease
In AD, cognitive decline best correlates with synaptic loss and synaptic failure [6, 12, 83–85]. Synaptic degeneration is a slow process, which begins as a reversible functionallyresponsive stage marked by deregulation of synaptic function, and then culminating into irreparable loss of synapses [86]. Furthermore, the degree of synaptic reorganization in AD is also perturbed due to defective microtubule re-organization, impaired actin dynamics, and re-entry into the cell cycle [87–89].
Loss of synapses in the limbic cortex is the basis for cognitive deficits in AD brains [12, 13]. It is suggested that dementia in AD is a combined manifestation of the disruption of neuritic substructures and the loss of synaptic terminals in neocortical and subcortical regions in the brain [90]. Initial investigations in the field of synaptic impairment in AD involved morphological studies for synaptic loss and damage in various brain areas [16], both in early and late stages of AD [2, 91–95]. During early stages, an increase in glutamatergic and cholinergic synapses was observed [96, 97]. However, as the disease progressed there was a rampant change in the density of these synapses. Synaptic loss occurs in early pathological stages with almost 45% fewer synapses in mild AD [18, 98]. A significant decrease in the synapses/ neurons ratio by up to 48% in hippocampus and 56% in cerebellum was reported [99]. In AD brains, synaptic loss is seen in the cortical areas [13, 100], predominantly in the frontal (45% reduction) and temporal cortex (25–36% reduction) [15, 90, 101]. Furthermore, the entorhinal cortex and locus coeruleus also display a loss of synapses [18, 90, 102–104]. In addition, cognitive disabilities in mild cognitive impairment were associated with decreased levels of glutamatergic synapses [97, 105]. The levels of glutamatergic synapses strongly correlated with clinical dementia in patients with mild and severe AD [97]. Interestingly, an enlarged average area of surviving synapses in AD was also observed [99]. However, this increase led to an overall reduction in synaptic surface per μm3 of tissue indicating that structural changes and concomitant functional changes play a crucial role in synaptic pathology in AD.
The mechanisms of synaptic damage in AD are still unclear. It is suggested that abnormal processing of growth associated proteins may be responsible for synaptic damage in CNS of AD brains [106, 107]. Ultrastructural investigations revealed pathological accumulation of cytoskeletal proteins and lysosomal structures in the synapses of AD patients [106, 108, 109]. Additionally, accumulation of both Aβ and tau protein in the synaptosomes from AD brains [110] present cues for synaptic pathogenesis and its possible relation to either the abnormal function of synaptic proteins or direct toxic effects at the synaptic sites or both [111]. Several factors may be attributed to the changes in the synapses of AD brains: 1) decreased mRNA levels of synaptic proteins [85, 112]; 2) selective degradation of proteins; for example, the presence of caspases was observed in synaptosomes isolated from AD brains [110, 113]; 3) decreased transporter proteins in the synapses [114]; 4) abnormal function of synaptic proteins [111,115]; 5) abnormal deposition of proteins leading to diminished synaptic activity; for example, tau protein was shown to interact with synaptic proteins in vivo [37, 38].
Role of tau protein in synaptic pathology
Several studies have focused on Aβ as the trigger for synaptic damage in AD and suggest that tau protein is downstream of Aβ in AD pathology [7, 116, 117]. Interestingly, it was shown that the loss of neocortical synaptic inputs in AD brain could be independent from amyloid deposits [107,118]. In addition, neurodegeneration in AD is not a direct result of extracellular Aβ neurotoxicity [119]. Therefore, Aβ pathology may or may not be a direct causal agent for synapse loss in AD [120]. Conversely, limited studies focusing on tau as the candidate mediating synaptic protein loss and damage have been reported. Several factors point towards a prominent role of tau protein in mediating synaptic pathology: 1) the progression of tau pathology correlates well with the cognitive decline in human AD [121]; tangle pathology also showed stronger correlation with synapse density and Blessed score of cognitive impairment in AD [122], 2) synapse loss parallels tangle formation and occurs in the same regions in AD brains [13, 15, 20, 21], 3) higher tangle count is associated with lower levels of presynaptic proteins in AD [91]; furthermore, neurons containing NFT are responsible for selective synaptic deficits [123], 4) NFT-bearing neurons demonstrated a 35–57% reduction in synaptophysin mRNA in AD brain [85], and even more importantly 5) synaptic deficits are observed in frontotemporal lobar degeneration (FTLD), PSP, and Niemann-Pick disease type C (NP-C), which are independent of any Aβ pathology [124–128]. All these evidences suggest a well-established relationship between synaptic damage and tau pathology.
Insights on tau mediated pathology in synapses from tau transgenic models
Tau transgenic models have been widely used to examine disease pathogenesis of tau protein. Behavioral and cognitive functional deficits can be easily studied in these animals due to the availability of lab scale methodologies such as Morris maze test, object recognition test and many others neurobehavioral tests [129]. Transgenic models used for the study of the tau neurodegenerative cascade express human wild-type tau, mutant tau linked to FTDP-17 or structurally modified tau species derived from AD [130]. Tau transgenic lines are driven by constitutive or inducible promoters to regulate the expression of the exogenous protein [131, 132]. Several of these tau transgenic models exhibit deregulation in synaptic proteome, impairment of synaptic transmission, loss of synapses and dendritic loss (Table 1).
Table 1.
A summary of transgenic tauopathy models, form of tau protein expressed and their effect on synapse structure and function.
| Tau type | Host | Human Tau form | Line | Structural changes | Electrophysiological abnormalities | Proteomic changes | Reference |
|---|---|---|---|---|---|---|---|
| Normal | 6 tau isoforms | hTau mice | ↓ Synaptophysin, synaptojanin, mGluR, synaptobrevin, syntaxin, PSD-95, TrkB | 173 | |||
| ↓ HFS-induced LTP in Schaffer collateral fibers | 136 | ||||||
| Tau 2N/4R isoform | Wtau-Tg | ↓ Synapse density | ↓ PSD-95 | 133 | |||
| Tau.4R mice | ↓ Length of mushroom spines, ↑ spine density | 134 | |||||
| Mutated | Mice | Tau 40 with P301L mutation | JNPL3 | ↓ Density of synaptic boutons, synaptic stripping | 147 | ||
| ↑ Late phase-LTP | 148 | ||||||
| Tau-4R-P301L mice | ↓ Length of mushroom spines, ↑ spine density | 134 | |||||
| rTg4510 | ↓ Dendritic complexity and length, ↓ spine density | 144 | |||||
| ↓ Dendritic diameters, ↓ cortical spine | ↑ sEPSC | 145 | |||||
| ↓ Mushroom spines, dendritic regression | ↑ Action potential firing rates, ↑ sEPSC | 146 | |||||
| ↓ AMPAR and NMDAR | 77 | ||||||
| ↓ Apical dendritic spines, ↓ synapses | 141 | ||||||
| ↓ Synapses, ↓ dendritic spines | 142 | ||||||
| ↑ Hippocampal vGLUT1, GLT-1 | 151 | ||||||
| rTgTauEC | ↓ Synaptic vesicles | ↓ Synaptophysin, synapsin, spinophilin | 149 | ||||
| ↑ Axonal excitability, ↓ PTP | 150 | ||||||
| Tau 40 with P301S mutation | PS19 mice | ↓ Hippocampal synapse | ↓ LTP | 138 | |||
| ↓ Synapse density in CA3 | ↓ Glutamate levels in hippocampus and thalamus | 139 | |||||
| P301S Tau x YFP-H mice | ↓ Spine density in cortical layer V | 140 | |||||
| Mutated Tau 43 K257T/P301S | DM-htau tg mice | ↓ Maintenance of LTP in the dentate gyrus | 155 | ||||
| Tau 34 G272V/P301S mutation | THY-Tau22 | ↓ EPSP | 153 | ||||
| Tau 40 with deletion of K280 | hTau40DK280 | ↓ Dendritic spines | 157 | ||||
| ↓ Dendritic spines | ↓ Hippocampal LTP | ↓ AMPAR, NMDAR, synaptophysin | 174 | ||||
| Deletion/ Truncated | Mice | ↓ LTP | ↓ Synaptophysin, NMDAR1, PSD-95, drebrin | 156 | |||
| Δtau 244–372 with deletion of K280 | RDTau (244-372) DK280 | ↓ LTP | 160 | ||||
| ↓ Spinophillin | 132 | ||||||
| ↓ Synaptic vesicles density | ↓ LTP of the mossy fiber tract | 158 | |||||
| Δtau aa1-255 | Δtau74 | ↓ Dendritic targeting of Fyn kinase | 37 | ||||
| Rat | Δtau 3R aa151-391 | SHR24 | ↓ Synaptic vesicle density, microtubule bundling in the presynapses | ↓ Synaptophysin, neurofilament H and M, ↑ tubulin proteins | 78 |
Legends: 2N, 2 inserts; 3R, 3 repeat; 4R, 4 repeat; DM, AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors; Double mutation; LTP, Long-term potentiation; EC, Entorhinal cortex; sEPSP, Synaptic excitatory postsynaptic potentials; GLT-1, Glutamate transporter 1 (ortholog of EAAT2); hTau, human tau; HFS, High frequency stimulation; NMDAR, N-methyl-D-aspartate receptors; PS1, Presenilin 1; PP, Perforant path; PSD-95, Postsynaptic density protein 95; PTP, Post-tetanic potentiation; YFP, Yellow fluorescent protein; vGLUT, Vesicular glutamate transporter.
Structural alterations and electrophysiological changes
Transgenic tauopathy models recapitulate several AD like morphological changes in the synapses. Transgenic tau lines expressing human 6 tau isoforms or human full length tau protein (hTau2N/4R) display loss of synapses and mushroom spines [133–135]. More specifically, mice lines expressing 6 human tau isoforms in tau knockout background exhibit more thin spines rather than mushroom like spines [135]. Interestingly, an initial decline in mushroom spine volume at 3 months of age was reversed after 6 months, indicating a certain degree of compensatory mechanism [135]. Despite an increase in mushroom spine volume, the older animals still displayed diminished LTP and spatial memory deficits [136]. Interestingly, the effect of htau40 in spine reduction was rescued by double transfection of the cells with MARK2 (phosphorylates tau in repeat region KXGS) indicating that phosphorylation of tau at this site is crucial for tau release from microtubules [137].
Several mice models expressing FTDP-17 tau mutations have been developed which demonstrate synaptic deficiency. For instance, mice expressing P301S mutation show hippocampal synaptic loss [138], mainly in the CA3 region [139]. More specifically, a progressive loss of spines in layer V of the neocortex along with reduced LTP was observed in these mice [140]. Similarly, mice expressing human mutant tau with P301L mutation also exhibit loss of synapses in this subset of neurons [141–143] and a loss of dendritic spines [77]. In addition, remnant dendritic spines exhibit deficits in dendritic diameter and length [144,145]. Finally, synaptic hyperexcitability in the form of increase in depolarization, action potential and synaptic excitatory postsynaptic potentials (sEPSP) were observed in these animals [146]. Comparable effects were also observed in other mice strains expressing P301L mutant tau with a different genetic background (JNPL13) [147], mainly as increases in long-phase LTP [148]. The animals also exhibited improved learning and memory processes. Interestingly, mice models expressing P301L mutated tau protein residing solely in the entorhinal cortex (rTgTauEC) exhibited loss of synaptic vesicles [149]. In contrast, the presynaptic alteration enhanced axonal excitability in these animals, but reduced LTP [150]. However, signs of cognitive insufficiency were absent or mild in these animals. Interestingly, in early stages the P301L tau mutant mice showed elevated levels of dendritic spine when compared to wild type mice (tau-P301L mice) [143]. The P301L tau mutant mice also exhibited increased levels of vesicular glutamate transporter 1 (vGLUT1) indicating a compensatory mechanism [151]. Although in older animals the deleterious repercussions of P301L mutant tau expression are widely noted.
Unlike previously reported tau lines, young mice expressing G272V/P301S double mutant tau showed no overt synaptic pathology [152]. However, older animals displayed a decrease in excitatory postsynaptic potentiation (EPSP) [153]. Organotypic sections from THY-Tau22 mice (G272V and P301S mutations) showed that the reduction in synaptic activity is induced by brain-derived neurotrophic factor [154]. Likewise, the double mutated tau mice model expressing K257T/P301S also displayed impairment in sustenance of LTP [155].
Sydow et al. [156] generated on/off mouse model (TauRDΔPP (244–372) ΔK280 mice) expressing the tau repeat domain with pro-aggregant mutation (where point mutation at lysine 280 drives aggregation of tau). The mice showed aggregation of endogenous and recombinant tau, tau missorting into the dendrites and synaptic loss [157]. Moreover, organotypic slides from mice showed marked reduction in synaptic boutons, diminished dendritic density and altered morphology [158–159]. Furthermore, diminished calcium influx after membrane depolarization was observed suggesting altered calcium dynamics in these neurons. Transgenic mice also showed marked deficits in LTP in CA1 and CA3 hippocampal regions [156, 160].
Studies from transgenic tau models clearly show that tau protein can damage the structural and functional properties of synapses leading to the impairment of LTP and/or LTD.
Proteomic changes in tau transgenic models
The synaptic impairment in AD is mainly characterized by the dysregulation of synaptic proteins at proteomic and transcriptomic levels [91, 161, 162]. Massive loss in components of the synaptic and dense core vesicles is prominent in AD [163, 164]. More specifically, proteins regulating synaptic plasticity are reduced in the AD brain [165]. Moreover, stage dependent decline in synaptic protein levels have also been observed [91].
Interestingly, synaptic fractions from human AD displayed elevated levels of tau protein in the postsynaptic density suggesting tau mislocalization and missorting [166]. Synaptic activity induced the physiological release of tau protein from synapses [167], a process which is aggravated in synaptosomes from AD brains [168], suggesting a massive deregulation in synaptic machinery in AD brains. Interestingly, C-terminally truncated tau was more prominent in the presynapses of AD brain (about 75–85%) with low levels of N- and C-terminal double truncated tau species [168]. Remarkably, predominant tau species in the synapses of AD brains were insoluble indicating tau aggregation. These results demonstrate that tau mislocalization and truncation exacerbate synaptic dysfunction in AD brains.
It is still uncertain how the tau isoforms in the synaptic compartments of diseased brain vary when compared to the healthy individuals. In murine neurons expressing human tau proteins, the three repeat tau isoform shows both neuronal body and synapse like distribution, while four repeat tau isoform was more “synapse like” in distribution [169]. However, based on these pieces of evidence, it is hypothesized that regional specific changes in the tau isoforms may contribute to pathogenesis of human tauopathies [169]. Therefore altered distribution of tau isoforms may be one of the causative factors for deficits in the synapses in the tauopathy brain.
Although several studies have evaluated the deregulation of synaptic proteins, less is known about the pathological tau proteome in the presynaptic and postsynaptic compartments. We evaluated the synaptic tau proteome in a rat model of tauopathy expressing human truncated tau [78]. Transgenic rat models fully recapitulate the human neurodegenerative tau cascade [170, 171]. Like humans, rats also express six tau isoforms in the CNS and can mimic changes in tau proteome as in humans [172]. Synaptic tau proteome was significantly altered in transgenic rats [78]. Tau protein in the presynaptic compartment was elevated in transgenic rats expressing human truncated tau (Fig. 2). In the postsynaptic fraction, expression of human truncated tau protein induced missorting and mislocalization of endogenous tau (Fig. 2). These results are consistent with an earlier report showing mislocalization of tau in postsynaptic density of AD brains [166]. These results establish that physiological distribution of tau protein is perturbed in AD brains contributing to synaptic degeneration.
Figure 2.
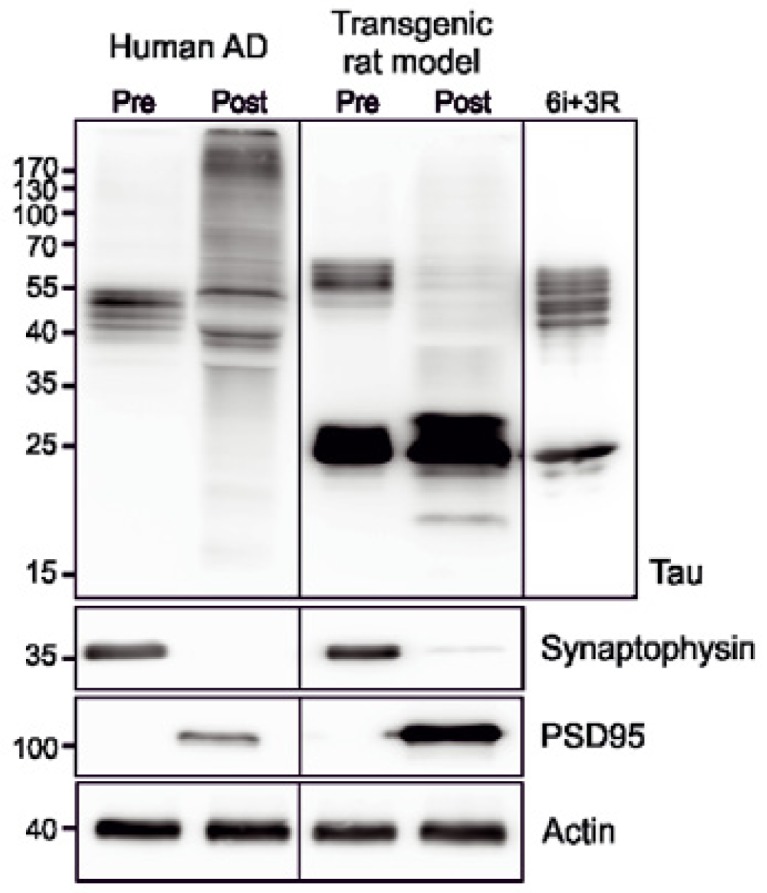
Tau mislocalization in disease conditions. In Alzheimer’s disease (Human AD) and transgenic rat model expressing human truncated tau protein aa151-391 (Transgenic rat model) tau protein is mislocalized to the postsynaptic fraction (Post). Synaptic fractions were isolated as previously reported [78]. Recombinant 6 human tau isoforms (6i) and truncated 3 repeat tau protein (3R) were used as controls.
Evaluation of truncated tau proteome in the presynaptic compartments of transgenic rats revealed phosphorylation of human truncated tau at residues T205, S214, S262 and S356. Conversely, truncated tau in the postsynaptic compartment was phosphorylated mainly at T212. This pattern of tau distribution elicited specific pathological changes in these rats. Elevated levels of α- and β-tubulin proteins and specific increase in glutamylated and detyrosinated tubulin was observed in the presynaptic terminals of these animals. Electron microscopy revealed microtubule bundling and diminished levels of synaptic vesicles in the presynaptic terminals of these animals. In the postsynaptic compartment, expression of human truncated tau protein led to a decrease in neurofilament proteins. However, no change in the levels of tubulin or MAP 2B/2C was observed. These evidences suggest that different phospho-tau species elicits specific pathological effects in the presynaptic and postsynaptic compartments of transgenic rat model [78].
Deregulation of synaptic proteins is widely observed in transgenic models of tauopathies. Loss of synaptophysin, a synaptic vesicle protein, is commonly observed in tau transgenic models [78, 149, 173, 174]. In addition, other synaptic proteins synapsin, synaptojanin, synaptobrevin are reportedly decreased in these animals. Furthermore, expression of tau also deregulated dendritic proteins PSD95 and spinophillin suggesting postsynaptic deficits in these animals [132, 149]. In mice models, tau protein induces synaptic impairment by diminishing the trafficking of metabotropic glutamate receptors (mGluR) [156,173], AMPA and NMDA receptors [77], which contribute to LTP deficits.
Recent evidence speculates a role of tau protein in synaptic signaling and point towards functional reduction of tau within synaptic contacts in transgenic tau models. All of this evidence points to a more intricate role of tau protein in the synapses. Furthermore, synaptic tau proteome alterations may be one of the key pathological steps in AD and other tauopathies.
Conclusion
Outcomes from numerous studies indicate a vital role of physiological tau protein in the synaptic biology including induction of LTP, LTD and dendritic activity (Fig. 3). Besides, cues from transgenic rodent tau models clearly demonstrate deleterious effects of pathological tau protein in the synapses (Fig. 3). A strong knowledge on tau driven synaptic damage will be essential in order to understand and intervene early pathological events in AD. We emphasize the role of disease modified tau protein in inducing synaptic impairment and comprehensively assemble multiple evidence of synaptic damage from transgenic tau models expressing various forms of tau protein. Put together, unlike previously assumed, tau protein may have a significantly larger role in imparting synaptic instability and cognitive deficits in AD and other tauopathies.
Figure 3.

Tau protein in the physiology and pathology of neuronal synapses. Tau protein performs physiological functions in synapses (in light green). In diseased conditions, misfolded phosphorylated and truncated tau proteins impair several pre- and postsynaptic machineries to perturb synaptic function (dark green). Misfolded tau impairs synaptic vesicle transport and release, deregulates several synaptic proteins and alters synaptic and dendritic morphology. The cumulative effect of synaptic tau in disease condition results in reduction of synaptic plasticity and induction of excitotoxicity and postsynaptic long-term depression.
Acknowledgment
Conflict of interest statement: The authors declare that they have no competing interests regarding this manuscript. The work was supported by research grants APVV 0206-11 and EU structural fund 26240220046.
References
- 1.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 2.Honer WG, Dickson DW, Gleeson J, Davies P. Regional synaptic pathology in Alzheimer’s disease. Neurobiol Aging. 1992;13:375–382. doi: 10.1016/0197-4580(92)90111-a. [DOI] [PubMed] [Google Scholar]
- 3.Danysz W, Parsons CG. Alzheimer’s disease β-amyloid glutamate, NMDA receptors and memantine - searching for the connections. Br J Pharmacol. 2012;167:324–352. doi: 10.1111/j.1476-5381.2012.02057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anstey KJ, Cherbuin N, Herath PM. Development of a new method for assessing global risk of Alzheimer’s disease for use in population health approaches to prevention. Prev Sci. 2013;14:411–421. doi: 10.1007/s11121-012-0313-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mosconi L, McHugh PF. Let food be thy medicine: diet, nutrition, and biomarkers’ risk of Alzheimer’s disease. Curr Nutr Rep. 2015;4:126–135. doi: 10.1007/s13668-014-0111-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 7.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 8.Sepulveda-Falla D, Barrera-Ocampo A, Hagel C, Korwitz A, Vinueza-Veloz MF, Zhou K, et al. Familial Alzheimer’s disease-associated presenilin-1 alters cerebellar activity and calcium homeostasis. J Clin Invest. 2014;124:1552–1567. doi: 10.1172/JCI66407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168–174. doi: 10.1001/archpsyc.63.2.168. [DOI] [PubMed] [Google Scholar]
- 10.Schneider LS, Mangialasche F, Andreasen N, Feldman H, Giacobini E, Jones R, et al. Clinical trials and late-stage drug development for Alzheimer’s disease: an appraisal from 1984 to 2014. J Intern Med. 2014;275:251–283. doi: 10.1111/joim.12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mallucci GR. Prion neurodegeneration: starts and stops at the synapse. Prion. 2009;3:195–201. doi: 10.4161/pri.3.4.9981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 13.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 14.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 15.Davies CA, Mann DM, Sumpter PQ, Yates PO. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J Neurol Sci. 1987;78:151–164. doi: 10.1016/0022-510x(87)90057-8. [DOI] [PubMed] [Google Scholar]
- 16.Scheff SW, Price DA. Alzheimer’s disease-related synapse loss in the cingulate cortex. J Alzheimers Dis. 2001;3:495–505. doi: 10.3233/jad-2001-3509. [DOI] [PubMed] [Google Scholar]
- 17.Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, et al. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology. 2001;56:127–129. doi: 10.1212/wnl.56.1.127. [DOI] [PubMed] [Google Scholar]
- 18.Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68:1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- 19.Falke E, Nissanov J, Mitchell TW, Bennett DA, Trojanowski JQ, Arnold SE. Subicular dendritic arborization in Alzheimer’s disease correlates with neurofibrillary tangle density. Am J Pathol. 2003;163:1615–1621. doi: 10.1016/S0002-9440(10)63518-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, et al. Early Aβ accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 21.Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lansdall CJ. An effective treatment for Alzheimer’s disease must consider both amyloid and tau. Biosci Horiz. 2014;7:hzu002. [Google Scholar]
- 23.Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 24.Ihara Y, Nukina N, Miura R, Ogawara M. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J Biochem. 1986;99:1807–1810. doi: 10.1093/oxfordjournals.jbchem.a135662. [DOI] [PubMed] [Google Scholar]
- 25.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 26.Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 1989;8:393–399. doi: 10.1002/j.1460-2075.1989.tb03390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Preuss U, Biernat J, Mandelkow EM, Mandelkow E. The ‘jaws’ model of tau-microtubule interaction examined in CHO cells. J Cell Sci. 1997;110:789–800. doi: 10.1242/jcs.110.6.789. [DOI] [PubMed] [Google Scholar]
- 28.Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–1089. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matus A. Stiff microtubules and neuronal morphology. Trends Neurosci. 1994;17:19–22. doi: 10.1016/0166-2236(94)90030-2. [DOI] [PubMed] [Google Scholar]
- 30.Hwang SC, Jhon DY, Bae YS, Kim JH, Rhee SG. Activation of phospholipase C-γ by the concerted action of tau proteins and arachidonic acid. J Biol Chem. 1996;271:18342–18349. doi: 10.1074/jbc.271.31.18342. [DOI] [PubMed] [Google Scholar]
- 31.Mattsson N, Savman K, Osterlundh G, Blennow K, Zetterberg H. Converging molecular pathways in human neural development and degeneration. Neurosci Res. 2010;66:330–332. doi: 10.1016/j.neures.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 32.Lee G, Kwei SL, Newman ST, Lu M, Liu Y. A new molecular interactor for tau protein. Soc Neurosci Abstr. 1996;22:975. [Google Scholar]
- 33.Stieler JT, Bullmann T, Kohl F, Tøien ⊘, Brückner MK, Härtig W, et al. The physiological link between metabolic rate depression and tau phosphorylation in mammalian hibernation. PLoS One. 2011;6:e14530. doi: 10.1371/journal.pone.0014530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahmed T, Van der Jeugd A, Blum D, Galas MC, D’Hooge R, Buée L. Cognition and hippocampal synaptic plasticity in mice with a homozygous tau deletion. Neurobiol Aging. 2014;35:2474–2478. doi: 10.1016/j.neurobiolaging.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 35.Kimura T, Whitcomb DJ, Jo J, Regan P, Piers T, Heo S, et al. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos Trans R Soc B. 2014;369:20130144. doi: 10.1098/rstb.2013.0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Regan P, Piers T, Yi JH, Kim DH, Huh S, Park SJ, et al. Tau phosphorylation at serine 396 residue is required for hippocampal LTD. J Neurosci. 2015;35:4804–4812. doi: 10.1523/JNEUROSCI.2842-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 38.Mondragón-Rodríguez S, Trillaud-Doppia E, Dudilot A, Bourgeois C, Lauzon M, Leclerc N, et al. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J Biol Chem. 2012;287:32040–32053. doi: 10.1074/jbc.M112.401240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klein C, Kramer EM, Cardine AM, Schraven B, Brandt R, Trotter J. Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J Neurosci. 2002;22:698–707. doi: 10.1523/JNEUROSCI.22-03-00698.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amihăesei IC, Cojocarut E, Mungiu OC. Alzheimer - certitudes and hypotheses. Rev Med Chir Soc Med Nat Iasi. 2013;117:119–126. [PubMed] [Google Scholar]
- 41.Kidd M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature. 1963;197:192–193. doi: 10.1038/197192b0. [DOI] [PubMed] [Google Scholar]
- 42.Crowther RA, Wischik CM. Image reconstruction of the Alzheimer paired helical filament. EMBO J. 1985;4:3661–3665. doi: 10.1002/j.1460-2075.1985.tb04132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ksiezak-Reding H, Morgan K, Mattiace LA, Davies P, Liu WK, Yen SH, et al. Ultrastructure and biochemical composition of paired helical filaments in corticobasal degeneration. Am J Pathol. 1994;145:1496–1508. [PMC free article] [PubMed] [Google Scholar]
- 44.Brion JP, Couck AM, Passareiro H, Flament-Durand J. Neurofibrillary tangles of Alzheimer’s disease: an immunohistochemical and immunoelectron study. J Submicrosc Cytol. 1985;17:89–96. [PubMed] [Google Scholar]
- 45.Kosik KS, Joachim CL, Selkoe DJ. Microtubule-associated protein tau (τ) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci USA. 1986;83:4044–4048. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (τ) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986;261:6084–6089. [PubMed] [Google Scholar]
- 48.Wischik CM, Novak M, Thøgersen HC, Edwards PC, Runswick MJ, Jakes R, et al. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci USA. 1988;85:4506–4510. doi: 10.1073/pnas.85.12.4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wischik CM, Novak M, Edwards PC, Klug A, Tichelaar W, Crowther RA. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci USA. 1988;85:4884–4888. doi: 10.1073/pnas.85.13.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Skrabana R, Skrabanova M, Csokova N, Sevcik J, Novak M. Intrinsically disordered tau protein in Alzheimer’s tangles: a coincidence or a rule? Bratisl Lek Listy. 2006;107:354–358. [PubMed] [Google Scholar]
- 51.Gong CX, Liu F, Grundke-Iqbal I, Iqbal K. Post-translational modifications of tau protein in Alzheimer’s disease. J Neural Transm. 2005;112:813–838. doi: 10.1007/s00702-004-0221-0. [DOI] [PubMed] [Google Scholar]
- 52.Pevalova M, Filipcik P, Novak M, Avila J, Iqbal K. Post-translational modifications of tau protein. Bratisl Lek Listy. 2006;107:346–353. [PubMed] [Google Scholar]
- 53.Martin L, Latypova X, Terro F. Post-translational modifications of tau protein: implications for Alzheimer’s disease. Neurochem Int. 2011;58:458–471. doi: 10.1016/j.neuint.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 54.Ksiezak-Reding H, Liu WK, Yen SH. Phosphate analysis and dephosphorylation of modified tau associated with paired helical filaments. Brain Res. 1992;597:209–219. doi: 10.1016/0006-8993(92)91476-u. [DOI] [PubMed] [Google Scholar]
- 55.Köpke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke-Iqbal I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem. 1993;268:24374–24384. [PubMed] [Google Scholar]
- 56.Augustinack JC, Schneider A, Mandelkow EM, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 103:26–35. doi: 10.1007/s004010100423. 200. [DOI] [PubMed] [Google Scholar]
- 57.Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. 1993;11:153–163. doi: 10.1016/0896-6273(93)90279-z. [DOI] [PubMed] [Google Scholar]
- 58.Goedert M, Jakes R, Crowther RA, Cohen P, Vanmechelen E, Vandermeeren M, et al. Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer’s disease: identification of phosphorylation sites in tau protein. Biochem J. 1994;301:871–877. doi: 10.1042/bj3010871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng-Fischhöfer Q, Biernat J, Mandelkow EM, Illenberger S, Godemann R, Mandelkow E. Sequential phosphorylation of Tau by glycogen synthase kinase-3β and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur J Biochem. 1998;252:542–552. doi: 10.1046/j.1432-1327.1998.2520542.x. [DOI] [PubMed] [Google Scholar]
- 60.von Bergen M, Barghorn S, Li L, Marx A, Biernat J, Mandelkow EM, et al. Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local β-structure. J Biol Chem. 2001;276:48165–48174. doi: 10.1074/jbc.M105196200. [DOI] [PubMed] [Google Scholar]
- 61.Goux WJ. The conformations of filamentous and soluble tau associated with Alzheimer paired helical filaments. Biochemistry. 2002;41:13798–13806. doi: 10.1021/bi016079h. [DOI] [PubMed] [Google Scholar]
- 62.Hiraoka S, Yao TM, Minoura K, Tomoo K, Sumida M, Taniguchi T, Ishida T. Conformational transition state is responsible for assembly of microtubule-binding domain of tau protein. Biochem Biophys Res Commun. 2004;315:659–663. doi: 10.1016/j.bbrc.2004.01.107. [DOI] [PubMed] [Google Scholar]
- 63.Kovacech B, Skrabana R, Novak M. Transition of tau protein from disordered to misordered in Alzheimer’s disease. Neurodegener Dis. 2010;7:24–27. doi: 10.1159/000283478. [DOI] [PubMed] [Google Scholar]
- 64.Novak M, Wischik CM, Edwards PC, Panell R, Milstein C. Characterization of the first monoclonal antibody against the pronase resistant core of the Alzheimer PHF. Prog Clin Biol Res. 1989;317:755–761. [PubMed] [Google Scholar]
- 65.Novak M, Kabat J, Wischik CM. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer’s disease paired helical filament. EMBO J. 1993;12:365–370. doi: 10.1002/j.1460-2075.1993.tb05665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Abraha A, Ghoshal N, Gamblin TC, Cryns V, Berry RW, Kuret J, et al. C-terminal inhibition of tau assembly in vitro and in Alzheimer’s disease. J Cell Sci. 2000;113:3737–3745. doi: 10.1242/jcs.113.21.3737. [DOI] [PubMed] [Google Scholar]
- 67.Ghoshal N, García-Sierra F, Fu Y, Beckett LA, Mufson EJ, Kuret J, et al. Tau-66: evidence for a novel tau conformation in Alzheimer’s disease. J Neurochem. 2001;77:1372–1385. doi: 10.1046/j.1471-4159.2001.00346.x. [DOI] [PubMed] [Google Scholar]
- 68.Basurto-Islas G, Luna-Muñoz J, Guillozet-Bongaarts AL, Binder LI, Mena R, García-Sierra F. Accumulation of aspartic acid 421- and glutamic acid 391-cleaved tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J Neuropathol Exp Neurol. 2008;67:470–483. doi: 10.1097/NEN.0b013e31817275c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bondareff W, Mountjoy CQ, Roth M, Hauser DL. Neurofibrillary degeneration and neuronal loss in Alzheimer’s disease. Neurobiol Aging. 1989;10:709–715. doi: 10.1016/0197-4580(89)90007-9. [DOI] [PubMed] [Google Scholar]
- 70.Sergeant N, Delacourte A, Buee L. Tau protein as a differential biomarker of tauopathies. Biochem Biophys Acta. 2005;1739:179–219. doi: 10.1016/j.bbadis.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 71.Williams DR. Tauopathies: classification and clinical update on neurodegenerative diseases associated with microtubule-associated protein tau. Intern Med J. 2006;36:652–660. doi: 10.1111/j.1445-5994.2006.01153.x. [DOI] [PubMed] [Google Scholar]
- 72.Buée L, Delacourte A. Comparative biochemistry of tau in progressive supranuclear palsy corticobasal degeneration, FTDP-17 and Pick’s disease. Brain Pathol. 1999;9:681–693. doi: 10.1111/j.1750-3639.1999.tb00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fu YJ, Nishihira Y, Kuroda S, Toyoshima Y, Ishihara T, Shinozaki M, et al. Sporadic four-repeat tauopathy with frontotemporal lobar degeneration, Parkinsonism, and motor neuron disease: a distinct clinicopathological and biochemical disease entity. Acta Neuropathol. 2010;120:21–32. doi: 10.1007/s00401-010-0649-2. [DOI] [PubMed] [Google Scholar]
- 74.Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–1378. doi: 10.1083/jcb.101.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aronov S, Aranda G, Behar L, Ginzburg I. Axonal tau mRNA localization coincides with tau protein in living neuronal cells and depends on axonal targeting signal. J Neurosci. 2001;21:6577–6587. doi: 10.1523/JNEUROSCI.21-17-06577.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sultan A, Nesslany F, Violet M, Bégard S, Loyens A, Talahari S, et al. Nuclear tau, a key player in neuronal DNA protection. J Biol Chem. 2011;286:4566–4575. doi: 10.1074/jbc.M110.199976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hoover BR, Reed MN, Jianjun Su, Penrod RD, Kotilinek LA, Grant MK, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jadhav S, Katina S, Kovac A, Kazmerova Z, Novak M, Zilka N. Truncated tau deregulates synaptic markers in rat model for human tauopathy. Front Cell Neurosci. 2015;9:24. doi: 10.3389/fncel.2015.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Frandemiche ML, De Seranno S, Rush T, Borel E, Elie A, Arnal I, et al. Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-β oligomers. J Neurosci. 2014;34:6084–6097. doi: 10.1523/JNEUROSCI.4261-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zmuda JF, Rivas RJ. Actin disruption alters the localization of tau in the growth cones of cerebellar granule neurons. J Cell Sci. 2000;113:2797–2809. doi: 10.1242/jcs.113.15.2797. [DOI] [PubMed] [Google Scholar]
- 81.DiTella M, Feiguin F, Morfini G, Cáceres A. Microfilament-associated growth cone component depends upon tau for its intracellular localization. Cell Motil Cytoskeleton. 1994;29:117–130. doi: 10.1002/cm.970290204. [DOI] [PubMed] [Google Scholar]
- 82.Chen Q, Zhou Z, Zhang L, Wang Y, Zhang YW, Zhong M, et al. Tau protein is involved in morphological plasticity in hippocampal neurons in response to BDNF. Neurochem Int. 2012;60:233–242. doi: 10.1016/j.neuint.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 83.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 84.Blennow K, Bogdanovic N, Alafuzoff I, Ekman R, Davidsson P. Synaptic pathology in Alzheimer’s disease: relation to severity of dementia, but not to senile plaques, neurofibrillary tangles, or the ApoE4 allele. J Neural Transm. 1996;103:603–618. doi: 10.1007/BF01273157. [DOI] [PubMed] [Google Scholar]
- 85.Callahan LM, Vaules WA, Coleman PD. Progressive reduction of synaptophysin message in single neurons in Alzheimer disease. J Neuropathol Exp Neurol. 2002;61:384–395. doi: 10.1093/jnen/61.5.384. [DOI] [PubMed] [Google Scholar]
- 86.Rapoport SI. In vivo PET imaging and postmortem studies suggest potentially reversible and irreversible stages of brain metabolic failure in Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci. 1999;249:S46–S55. doi: 10.1007/pl00014174. [DOI] [PubMed] [Google Scholar]
- 87.Kowall NW, Kosik KS. Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer’s disease. Ann Neurol. 1987;22:639–643. doi: 10.1002/ana.410220514. [DOI] [PubMed] [Google Scholar]
- 88.McKee AC, Kowall NW, Kosik KS. Microtubular reorganization and dendritic growth response in Alzheimer’s disease. Ann Neurol. 1989;26:652–659. doi: 10.1002/ana.410260511. [DOI] [PubMed] [Google Scholar]
- 89.Penzes P, Vanleeuwen JE. Impaired regulation of synaptic actin cytoskeleton in Alzheimer’s disease. Brain Res Rev. 2011;67:184–192. doi: 10.1016/j.brainresrev.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Masliah E, Terry RD, Alford M, DeTeresa R, Hansen LA. Cortical and subcortical patterns of synaptophysinlike immunoreactivity in Alzheimer’s disease. Am J Pathol. 1991;138:235–246. [PMC free article] [PubMed] [Google Scholar]
- 91.Honer WG. Pathology of presynaptic proteins in Alzheimer’s disease: more than simple loss of terminals. Neurobiol Aging. 2003;24:1047–1062. doi: 10.1016/j.neurobiolaging.2003.04.005. [DOI] [PubMed] [Google Scholar]
- 92.Sze CI, Bi H, Kleinschmidt-DeMasters BK, Filley CM, Martin LJ. Selective regional loss of exocytotic presynaptic vesicle proteins in Alzheimer’s disease brains. J Neurol Sci. 2000;175:81–90. doi: 10.1016/s0022-510x(00)00285-9. [DOI] [PubMed] [Google Scholar]
- 93.Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, et al. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology. 2001;56:127–129. doi: 10.1212/wnl.56.1.127. [DOI] [PubMed] [Google Scholar]
- 94.Reddy PH, Mani G, Park BS, Jacques J, Murdoch G, Whetsell W, Jr, Kaye J, Manczak M. Differential loss of synaptic proteins in Alzheimer’s disease: implications for synaptic dysfunction. J Alzheimers Dis. 2005;7:103–117. doi: 10.3233/jad-2005-7203. [DOI] [PubMed] [Google Scholar]
- 95.Counts SE, Nadeem M, Lad SP, Wuu J, Mufson EJ. Differential expression of synaptic proteins in the frontal and temporal cortex of elderly subjects with mild cognitive impairment. J Neuropathol Exp Neurol. 2006;65:592–601. doi: 10.1097/00005072-200606000-00007. [DOI] [PubMed] [Google Scholar]
- 96.DeKosky ST, Ikonomovic MD, Styren SD, Beckett L, Wisniewski S, Bennett DA, et al. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol. 2002;51:145–155. doi: 10.1002/ana.10069. [DOI] [PubMed] [Google Scholar]
- 97.Bell KFS, Bennett DA, Cuello AC. Paradoxical upregulation of glutamatergic presynaptic boutons during mild cognitive impairment. J Neurosci. 2007;27:10810–10817. doi: 10.1523/JNEUROSCI.3269-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging. 2006;27:1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 99.Bertoni-Freddari C, Fattoretti P, Solazzi M, Giorgetti B, Di Stefano G, Casoli T, et al. Neuronal death versus synaptic pathology in Alzheimer’s disease. Ann NY Acad Sci. 2003;1010:635–638. doi: 10.1196/annals.1299.116. [DOI] [PubMed] [Google Scholar]
- 100.Hof P, Morrison J. The cellular basis of cortical disconnection and related dementing conditions. In: Terry R, Katzman R, Bick K, editors. Alzheimer’s disease. Raven Press; New York, USA: 1994. pp. 197–230. [Google Scholar]
- 101.Scheff SW, DeKosky ST, Price DA. Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol Aging. 1990;11:29–37. doi: 10.1016/0197-4580(90)90059-9. [DOI] [PubMed] [Google Scholar]
- 102.Masliah E, Mallory M, Hansen L, DeTeresa R, Alford M, Terry R. Synaptic and neuritic alterations during the progression of Alzheimer’s disease. Neurosci Lett. 1994;174:67–72. doi: 10.1016/0304-3940(94)90121-x. [DOI] [PubMed] [Google Scholar]
- 103.Scheff SW, Sparks L, Price DA. Quantitative assessment of synaptic density in the entorhinal cortex in Alzheimer’s disease. Ann Neurol. 1993;34:356–361. doi: 10.1002/ana.410340309. [DOI] [PubMed] [Google Scholar]
- 104.Wakabayashi K, Honer WG, Masliah E. Synapse alterations in the hippocampal-entorhinal formation in Alzheimer’s disease with and without Lewy body disease. Brain Res. 1994;667:24–32. doi: 10.1016/0006-8993(94)91709-4. [DOI] [PubMed] [Google Scholar]
- 105.Masliah E, Alford M, DeTeresa R, Mallory M, Hansen L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann Neurol. 1996;40:759–766. doi: 10.1002/ana.410400512. [DOI] [PubMed] [Google Scholar]
- 106.Masliah E, Hansen L, Albright T, Mallory M, Terry RD. Immunoelectron microscopic study of synaptic pathology in Alzheimer’s disease. Acta Neuropathol. 1991;81:428–433. doi: 10.1007/BF00293464. [DOI] [PubMed] [Google Scholar]
- 107.Masliah E, Mallory M, Hansen L, DeTeresa R, Terry RD. Quantitative synaptic alterations in the human neocortex during normal aging. Neurology. 1993;43:192–197. doi: 10.1212/wnl.43.1_part_1.192. [DOI] [PubMed] [Google Scholar]
- 108.Gonatas NK, Anderson WW, Evangelista I. The contribution of altered synapses in the senile plaque: an electron microscopic study in Alzheimer’s disease. J Neuropathol Exp Neurol. 1967;26:25–39. doi: 10.1097/00005072-196701000-00003. [DOI] [PubMed] [Google Scholar]
- 109.Masliah E, Mallory M, Deerinck T, DeTeresa R, Lamont S, Miller A, et al. Re-evaluation of the structural organization of neuritic plaques in Alzheimer’s disease. J Neuropathol Exp Neurol. 1993;52:135–142. doi: 10.1097/00005072-199311000-00009. [DOI] [PubMed] [Google Scholar]
- 110.Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Synaptic changes in Alzheimer’s disease: increased amyloid-β and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol. 2004;165:1809–1817. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Masliah E, Terry R. The role of synaptic proteins in the pathogenesis of disorders of the central nervous system. Brain Pathol. 1993;3:77–85. doi: 10.1111/j.1750-3639.1993.tb00728.x. [DOI] [PubMed] [Google Scholar]
- 112.Callahan LM, Vaules WA, Coleman PD. Quantitative decrease in synaptophysin message expression and increase in cathepsin D message expression in Alzheimer disease neurons containing neurofibrillary tangles. J Neuropathol Exp Neurol. 1999;58:275–287. doi: 10.1097/00005072-199903000-00007. [DOI] [PubMed] [Google Scholar]
- 113.Louneva N, Cohen JW, Han LY, Talbot K, Wilson RS, Bennett DA, et al. Caspase-3 is enriched in postsynaptic densities and increased in Alzheimer’s disease. Am J Pathol. 2008;173:1488–1495. doi: 10.2353/ajpath.2008.080434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jacob CP, Koutsilieri E, Bartl J, Neuen-Jacob E, Arzberger T, Zander N, et al. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J Alzheimers Dis. 2007;11:97–116. doi: 10.3233/jad-2007-11113. [DOI] [PubMed] [Google Scholar]
- 115.Masliah E, Mallory M, Alford M, DeTeresa R, Iwai A, Saitoh T. Molecular mechanisms of synaptic disconnection in Alzheimer’s disease. In: Hyman BT, Duyckaerts C, editors. Connections, cognition and Alzheimer’s disease. Springer; Berlin, Germany: 1997. pp. 121–140. [Google Scholar]
- 116.Delacourte A, Sergeant N, Champain D, Wattez A, Maurage CA, Lebert F, et al. Nonoverlapping but synergetic tau and APP pathologies in sporadic Alzheimer’s disease. Neurology. 2002;59:398–407. doi: 10.1212/wnl.59.3.398. [DOI] [PubMed] [Google Scholar]
- 117.Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 118.Sadigh-Eteghad S, Sabermarouf B, Majdi A, Talebi M, Farhoudi M, Mahmoudi J. Amyloid-beta: a crucial factor in Alzheimer’s disease. Med Princ Pract. 2015;24:1–10. doi: 10.1159/000369101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Carter J, Lippa CF. Beta-amyloid, neuronal death and Alzheimer’s disease. Curr Mol Med. 2001;1:733–737. doi: 10.2174/1566524013363177. [DOI] [PubMed] [Google Scholar]
- 120.Musiek ES, Holtzman DM. Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nat Neurosci. 2015;18:800–806. doi: 10.1038/nn.4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Delacourte A. Tauopathies: recent insights into old diseases. Folia Neuropathol. 2005;43:244–257. [PubMed] [Google Scholar]
- 122.Masliah E, Ellisman M, Carragher B, Mallory M, Young S, Hansen L, et al. Three-dimensional analysis of the relationship between synaptic pathology and neuropil threads in Alzheimer disease. J Neuropathol Exp Neurol. 1992;51:404–414. doi: 10.1097/00005072-199207000-00003. [DOI] [PubMed] [Google Scholar]
- 123.Callahan LM, Coleman PD. Neurons bearing neurofibrillary tangles are responsible for selected synaptic deficits in Alzheimer’s disease. Neurobiol Aging. 1995;16:311–314. doi: 10.1016/0197-4580(95)00035-d. [DOI] [PubMed] [Google Scholar]
- 124.Brun A, Liu X, Erikson C. Synapse loss and gliosis in the molecular layer of the cerebral cortex in Alzheimer’s disease and in frontal lobe degeneration. Neurodegeneration. 1995;4:171–177. doi: 10.1006/neur.1995.0021. [DOI] [PubMed] [Google Scholar]
- 125.Liu X, Brun A. Regional and laminar synaptic pathology in frontal lobe degeneration of non-Alzheimer type. Int J Geriatr Psychiatry. 1996;11:47–55. [Google Scholar]
- 126.Bigio EH, Vono MB, Satumtira S, Adamson J, Sontag E, Hynan LS, et al. Cortical synapse loss in progressive supranuclear palsy. J Neuropathol Exp Neurol. 2001;60:403–410. doi: 10.1093/jnen/60.5.403. [DOI] [PubMed] [Google Scholar]
- 127.Suzuki K, Parker CC, Pentchev PG, Katz D, Ghetti B, D’Agostino AN, et al. Neurofibrillary tangles in Niemann-Pick disease type C. Acta Neuropathol. 1995;89:227–238. doi: 10.1007/BF00309338. [DOI] [PubMed] [Google Scholar]
- 128.Suzuki M, Desmond TJ, Albin RL, Frey KA. Cholinergic vesicular transporters in progressive supranuclear palsy. Neurology. 2002;58:1013–1018. doi: 10.1212/wnl.58.7.1013. [DOI] [PubMed] [Google Scholar]
- 129.Picciotto MR, Wickman K. Using knockout and transgenic mice to study neurophysiology and behavior. Physiol Rev. 1998;78:1131–1163. doi: 10.1152/physrev.1998.78.4.1131. [DOI] [PubMed] [Google Scholar]
- 130.Zilka N, Korenova M, Novak M. Misfolded tau protein and disease modifying pathways in transgenic rodent models of human tauopathies. Acta Neuropathol. 2009;118:71–86. doi: 10.1007/s00401-009-0499-y. [DOI] [PubMed] [Google Scholar]
- 131.Probst A, Gotz J, Wiederhold KH, Tolnay M, Mistl C, Jaton AL, et al. Axonopathy and amyotrophy in mice transgenic for human fourrepeat tau protein. Acta Neuropathol. 2000;99:469–481. doi: 10.1007/s004010051148. [DOI] [PubMed] [Google Scholar]
- 132.Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, et al. The potential for β-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous tau in inducible mouse models of tauopathy. J Neurosci. 2008;28:737–748. doi: 10.1523/JNEUROSCI.2824-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kimura T, Yamashita S, Fukuda T, Park JM, Murayama M, Mizoroki T, et al. Hyperphosphorylated tau in parahippocampal cortex impairs place learning in aged mice expressing wild-type human tau. EMBO J. 2007;26:5143–5152. doi: 10.1038/sj.emboj.7601917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kremer A, Maurin H, Demedts D, Devijver H, Borghgraef P, Van Leuven F. Early improved and late defective cognition is reflected by dendritic spines in Tau.P301L mice. J Neurosci. 2011;31:18036–18047. doi: 10.1523/JNEUROSCI.4859-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dickstein DL, Brautigam H, Stockton SD, Schmeidler J, Hof PR. Changes in dendritic complexity and spine morphology in transgenic mice expressing human wild-type tau. Brain Struct Funct. 2010;214:161–179. doi: 10.1007/s00429-010-0245-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Polydoro M, Acker CM, Duff K, Castillo PE, Davies P. Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J Neurosci. 2009;29:10741–10749. doi: 10.1523/JNEUROSCI.1065-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Thies E, Mandelkow EM. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/ Par-1. J Neurosci. 2007;27:2896–2907. doi: 10.1523/JNEUROSCI.4674-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 139.Crescenzi R, DeBrosse C, Nanga RP, Reddy S, Haris M, Hariharan H, et al. In vivo measurement of glutamate loss is associated with synapse loss in a mouse model of tauopathy. Neuroimage. 2014;101:185–192. doi: 10.1016/j.neuroimage.2014.06.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hoffmann NA, Dorostkar MM, Blumenstock S, Goedert M, Herms J. Impaired plasticity of cortical dendritic spines in P301S tau transgenic mice. Acta Neuropathol Commun. 2013;1:82. doi: 10.1186/2051-5960-1-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kopeikina KJ, Polydoro M, Tai HC, Yaeger E, Carlson GA, Pitstick R, et al. Synaptic alterations in the rTg4510 mouse model of tauopathy. J Comp Neurol. 2013;521:1334–1353. doi: 10.1002/cne.23234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kopeikina KJ, Wegmann S, Pitstick R, Carlson GA, Bacskai BJ, Betensky RA, et al. Tau causes synapse loss without disrupting calcium homeostasis in the rTg4510 model of tauopathy. PLoS One. 2013;8:e80834. doi: 10.1371/journal.pone.0080834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Boekhoorn K, Terwel D, Biemans B, Borghgraef P, Wiegert O, Ramakers GJ, et al. Improved long-term potentiation and memory in young tau-P301L transgenic mice before onset of hyperphosphorylation and tauopathy. J Neurosci. 2006;26:3514–3523. doi: 10.1523/JNEUROSCI.5425-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rocher AB, Crimins JL, Amatrudo JM, Kinson MS, Todd-Brown MA, Lewis J, et al. Structural and functional changes in tau mutant mice neurons are not linked to the presence of NFTs. Exp Neurol. 2010;223:385–393. doi: 10.1016/j.expneurol.2009.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Crimins JL, Rocher AB, Peters A, Shultz P, Lewis J, Luebke JI. Homeostatic responses by surviving cortical pyramidal cells in neurodegenerative tauopathy. Acta Neuropathol. 2011;122:551–564. doi: 10.1007/s00401-011-0877-0. [DOI] [PubMed] [Google Scholar]
- 146.Crimins JL, Rocher AB, Luebke JI. Electrophysiological changes precede morphological changes to frontal cortical pyramidal neurons in the rTg4510 mouse model of progressive tauopathy. Acta Neuropathol. 2012;124:777–795. doi: 10.1007/s00401-012-1038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Katsuse O, Lin WL, Lewis J, Hutton ML, Dickson DW. Neurofibrillary tangle-related synaptic alterations of spinal motor neurons of P301L tau transgenic mice. Neurosci Lett. 2006;409:95–99. doi: 10.1016/j.neulet.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 148.Levenga J, Krishnamurthy P, Rajamohamedsait H, Wong H, Franke TF, Cain P, et al. Tau pathology induces loss of GABAergic interneurons leading to altered synaptic plasticity and behavioral impairments. Acta Neuropathol Commun. 2013;1:34. doi: 10.1186/2051-5960-1-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Harris JA, Koyama A, Maeda S, Ho K, Devidze N, Dubal DB, et al. Human P301L-mutant tau expression in mouse entorhinal-hippocampal network causes tau aggregation and presynaptic pathology but no cognitive deficits. PLoS One. 2012;7:e45881. doi: 10.1371/journal.pone.0045881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Polydoro M, Dzhala VI, Pooler AM, Nicholls SB, McKinney AP, Sanchez L. Soluble pathological tau in the entorhinal cortex leads to presynaptic deficits in an early Alzheimer’s disease model. Acta Neuropathol. 2014;127:257–270. doi: 10.1007/s00401-013-1215-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hunsberger HC, Rudy CC, Batten SR, Gerhardt GA, Reed MN. P301L tau expression affects glutamate release and clearance in the hippocampal trisynaptic pathway. J Neurochem. 2015;132:169–182. doi: 10.1111/jnc.12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Van der Jeugd A, Ahmed T, Burnouf S, Belarbi K, Hamdame M, Grosjean ME, et al. Hippocampal tauopathy in tau transgenic mice coincides with impaired hippocampus-dependent learning and memory, and attenuated late-phase long-term depression of synaptic transmission. Neurobiol Learn Mem. 2011;95:296–304. doi: 10.1016/j.nlm.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 153.Schindowski K, Bretteville A, Leroy K, Bégard S, Brion JP, Hamdane M, et al. Alzheimer’s disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am J Pathol. 2006;169:599–616. doi: 10.2353/ajpath.2006.060002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Burnouf S, Martire A, Derisbourg M, Laurent C, Belarbi K, Leboucher A, et al. NMDA receptor dysfunction contributes to impaired brain-derived neurotrophic factor-induced facilitation of hippocampal synaptic transmission in a tau transgenic model. Aging Cell. 2013;12:11–23. doi: 10.1111/acel.12018. [DOI] [PubMed] [Google Scholar]
- 155.Rosenmann H, Grigoriadis N, Eldar-Levy H, Avital A, Rozenstein L, Touloumi O, et al. A novel transgenic mouse expressing double mutant tau driven by its natural promoter exhibits tauopathy characteristics. Exp Neurol. 2008;212:71–84. doi: 10.1016/j.expneurol.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 156.Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, et al. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic tau mutant. J Neurosci. 2011;31:2511–2525. doi: 10.1523/JNEUROSCI.5245-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Eckermann K, Mocanu MM, Khlistunova I, Biernat J, Nissen A, Hofmann A, et al. The beta-propensity of tau determines aggregation and synaptic loss in inducible mouse models of tauopathy. J Biol Chem. 2007;282:31755–31765. doi: 10.1074/jbc.M705282200. [DOI] [PubMed] [Google Scholar]
- 158.Decker JM, Krüger L, Sydow A, Zhao S, Frotscher M, Mandelkow E, et al. Pro-aggregant tau impairs mossy fiber plasticity due to structural changes and Ca++ dysregulation. Acta Neuropathol Commun. 2015;3:23. doi: 10.1186/s40478-015-0193-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Messing L, Decker JM, Joseph M, Mandelkow E, Mandelkow EM. Cascade of tau toxicity in inducible hippocampal brain slices and prevention by aggregation inhibitors. Neurobiol Aging. 2013;34:1343–1354. doi: 10.1016/j.neurobiolaging.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, et al. Reversibility of tau-related cognitive defects in a regulatable FTD mouse model. J Mol Neurosci. 2011;45:432–437. doi: 10.1007/s12031-011-9604-5. [DOI] [PubMed] [Google Scholar]
- 161.Heffernan JM, Eastwood SL, Nagy Z, Sanders MW, McDonald B, Harrison PJ. Temporal cortex synaptophysin mRNA is reduced in Alzheimer’s disease and is negatively correlated with the severity of dementia. Exp Neurol. 1998;150:235–239. doi: 10.1006/exnr.1997.6772. [DOI] [PubMed] [Google Scholar]
- 162.Coleman PD, Yao PJ. Synaptic slaughter in Alzheimer’s disease. Neurobiol Aging. 2003;24:1023–1027. doi: 10.1016/j.neurobiolaging.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 163.Lassmann H, Weiler R, Fischer P, Bancher C, Jellinger K, Floor E, et al. Synaptic pathology in Alzheimer’s disease: immunological data for markers of synaptic and large dense-core vesicles. Neuroscience. 1992;46:1–8. doi: 10.1016/0306-4522(92)90003-k. [DOI] [PubMed] [Google Scholar]
- 164.Lassmann H, Fischer P, Jellinger K. Synaptic pathology of Alzheimer’s disease. Ann NY Acad Sci. 1993;695:59–64. doi: 10.1111/j.1749-6632.1993.tb23028.x. [DOI] [PubMed] [Google Scholar]
- 165.Hatanpää K, Isaacs KR, Shirao T, Brady DR, Rapoport SI. Loss of proteins regulating synaptic plasticity in normal aging of the human brain and in Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:637–643. doi: 10.1097/00005072-199906000-00008. [DOI] [PubMed] [Google Scholar]
- 166.Tai HC, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol. 2012;181:1426–1435. doi: 10.1016/j.ajpath.2012.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yamada K, Holth JK, Liao F, Stewart FR, Mahan TE, Jiang H, et al. Neuronal activity regulates extracellular tau in vivo. J Exp Med. 2014;211:387–393. doi: 10.1084/jem.20131685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sokolow S, Henkins KM, Bilousova T, Gonzalez B, Vinters HV, Miller CA, et al. Pre-synaptic C-terminal truncated tau is released from cortical synapses in Alzheimer’s disease. J Neurochem. 2015;133:368–379. doi: 10.1111/jnc.12991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.McMillan P, Korvatska E, Poorkaj P, Evstafjeva Z, Robinson L, Greenup L, et al. Tau isoform regulation is region- and cell-specific in mouse brain. J Comp Neurol. 2008;511:788–803. doi: 10.1002/cne.21867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zilka N, Filipcik P, Koson P, Fialova L, Skrabana R, Zilkova M, et al. Truncated tau from sporadic Alzheimer’s disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett. 2006;580:3582–3588. doi: 10.1016/j.febslet.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 171.Filipcik P, Zilka N, Bugos O, Kucerak J, Koson P, Novak P, Novak M. First transgenic rat model developing progressive cortical neurofibrillary tangles. Neurobiol Aging. 2012;33:1448–1456. doi: 10.1016/j.neurobiolaging.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 172.Hanes J, Zilka N, Bartkova M, Caletkova M, Dobrota D, Novak M. Rat tau proteome consists of six tau isoforms: implication for animal models of human tauopathies. J Neurochem. 2009;108:1167–1176. doi: 10.1111/j.1471-4159.2009.05869.x. [DOI] [PubMed] [Google Scholar]
- 173.Alldred MJ, Duff KE, Ginsberg SD. Microarray analysis of CA1 pyramidal neurons in a mouse model of tauopathy reveals progressive synaptic dysfunction. Neurobiol Dis. 2012;45:751–762. doi: 10.1016/j.nbd.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Van der Jeugd A, Hochgräfe K, Ahmed T, Decker JM, Sydow A, Hofmann A, et al. Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human tau. Acta Neuropathol. 2012;123:787–805. doi: 10.1007/s00401-012-0987-3. [DOI] [PMC free article] [PubMed] [Google Scholar]