

Abstract
Macrophages populate every tissue of the body and play vital roles in homeostasis, pathogen elimination, and tissue healing. These cells possess the ability to adapt to a multitude of abruptly changing and complex environments. Furthermore, different populations of resident tissue macrophages each exhibit their own defining gene signatures. The enhancer repertoire of these cells underlies both the cellular identity of a given subset of resident macrophage population and their ability to dynamically alter, in an efficient manner, their gene expression programs in response to internal and external signals. Notably, transcription is pervasive at active enhancers and enhancer RNAs, or eRNAs, are tightly correlated to regulated transcription of protein-coding genes. Furthermore, selection and establishment of enhancers is a dynamic and plastic process in which activation of intracellular signaling pathways by factors present in a macrophage’s environment play a determining role. Here, we review recent studies providing insights into the distinct mechanisms that contribute to the selection and function of enhancers in macrophages, and the relevance of studying these mechanisms to gain a better understanding of complex human diseases.
Introduction
Macrophages are fundamental effectors of the innate immune system (Geissmann et al. 2010). Their capacity to abruptly and dramatically alter their gene expression output to quickly adapt to sudden changes in their environment, as it occurs during infections or tissue injuries, makes them one of the most dynamic cell types transcriptionally. In addition, each subset of resident tissue macrophages performs dedicated and specialized functions, as illustrated, for example, by the role of microglia in promoting adult neurogenesis (Sierra et al. 2010) and that of large peritoneal macrophages in regulating production in IgA by B-1 cells (Okabe and Medzhitov 2014). Although the overall transcriptome signature across different macrophage populations displays substantial similarities that define a core macrophage identity (Gautier et al. 2012), specific populations also exhibit striking differences in expression of subsets of genes that are presumably linked to their tissue-specific functions. For example, microglia express ~900 mRNAs at more than a 16-fold higher level than observed in large peritoneal macrophages, and a similar number of genes are expressed at more than a 16-fold higher level in large peritoneal macrophages than microglia (Gosselin et al. 2014). As macrophages can originate from different precursors (Ginhoux et al. 2010; Hoeffel et al. 2012; Schulz et al. 2012; Guilliams et al. 2013; Epelman et al. 2014; Ginhoux and Jung 2014), an important question is the extent to which the characteristics of different tissue resident populations of macrophages reflect the specific tissue environment or their developmental histories.
From a transcriptional perspective, the concept of subset identity is rather intriguing as macrophages, for any given individual, share the same genome and express to a great extent the same array of transcription factors (TFs). The recent development of massively parallel sequencing assays has made it possible to characterize transcription factor binding and chromatin features on a genome-wide level. A major concept to emerge from these approaches is that each cell type selects a specific repertoire of discrete DNA regulatory elements termed enhancers that are critical to that cell’s identity and determine its ability to respond to internal and external signals (Heinz et al. 2015). Here, we discuss mechanisms that underlie the selection and activation of enhancers and their relationship to the development of tissue-specific macrophage phenotypes.
Enhancers as determinants of cell-specific and signal-dependent gene expression
Enhancers were initially identified as discreet regions of DNA that increase transcriptional activity of promoters from a distance (Banerji et al. 1981). Systematic analysis of the genome led to the recognition that enhancers are marked by high abundance of mono-methylation at histone H3 lysine 4 and concomitantly low levels of tri-methylation (i.e. H3K4me1high/H3K4me3low) (Heintzman et al. 2007). Promoters, on the other hand, display an opposite molecular phenotype (i.e. H3K4me1low/H3K4me3high). Both promoters, and enhancers are also marked by high abundance of di-methylation at histone H3 lysine 4 (H3K4me2high). Active enhancers and promoters also exhibit enrichment of acetylation at histone H3 lysine 27 (H3K27ac) (Creyghton et al. 2010). Further, enhancers can be actively repressed with high levels of tri-methylation of histone H3 lysine 27 (H3K27me3) (Barski et al. 2007; Calo and Wysocka 2013). These, and other features, enable enhancer-like regions to be identified systematically in different cells and tissues by chromatin precipitation coupled to massively parallel sequencing (ChIP-Seq) and other sequencing based methods. Using these approaches, the mouse and human genomes have been estimated to contain several hundred thousand enhancers, the great majority of which are present in cell-restricted patterns (Consortium 2012).
More recently the concept of stretch or super-enhancers (SE) were introduced (Hnisz et al. 2013; Whyte et al. 2013), large genomic regions, usually one order of magnitude larger than traditional enhancers with an unusually strong enrichment for the binding of transcriptional co-activators. Most cells exhibit between 300 – 800 SEs, a substantial fraction of which are associated with genes encoding cell-type specific TFs and other genes important for that cell’s identity and function (Hnisz et al. 2013; Adam et al. 2015).
Between 35,000 and 45,000 H3K4me1high/H3K4me3low enhancer regions exist in mouse macrophages (Ghisletti et al. 2010; Heinz et al. 2010), substantially exceeding the ~12,000 active promoters in these cells. Of note, DNA sequences at enhancers in macrophages exhibit a robust enrichment for motifs recognized by TFs relevant to macrophage ontogeny and functions. Among these, the motif recognized by the ETS family member PU.1 is predominant, which is concordant with the observation that PU.1 binding sites genome-wide (i.e. the PU.1 cistrome) extensively coincide with H3K4me1high/H3K4me3low regions in macrophages. Motifs associated with C/EBP, AP-1, IRF, NF-κb, and LXR transcription factors are also prevalent within enhancers in macrophages (Barish et al. 2010; Ghisletti et al. 2010; Heinz et al. 2010; Gosselin et al. 2014; Lavin et al. 2014).
Such co-occurrences of motifs belonging to different families of TFs indicate that enhancers provide sites for the integration at the genomic level of TFs regulated by cell intrinsic and/or extrinsic signals. Furthermore, given the preponderance of enhancers compared to promoters, these interactions between TFs and DNA are more likely to occur at enhancers than at promoters. Therefore, this characteristic enables a key effector mechanism by which enhancers can synchronize the transcriptional output of a macrophage in accordance with its cellular activity and metabolism.
A collaborative/hierarchical model for enhancer selection and activation
Enhancer selection is mediated by the activities of so-called pioneer factors, a subset of which function as lineage-determining transcription factors (LDTFs) (McPherson et al. 1993; Bossard and Zaret 1998; Lee et al. 2005; Heinz et al. 2010). Various examples of LDTFs include PU.1 in macrophages (Heinz et al. 2010) and FoxA1 in breast cancer cells (Lupien et al. 2008). A defining feature of pioneering TFs is their ability to recognize and bind their DNA recognition motifs in closed chromatin, thus effectively competing with nucleosomes to create nucleosome-free regions. Transcription factor binding is then followed by modifications of the histone tails of the enhancer-associated nucleosomes located in the vicinity of the nucleosome-free regions.
All transcription factors recognize short DNA sequences (about 6 – 12 base pairs) and their motifs exhibit varying levels of degeneracy (D’haeseleer 2006). ChIP-Seq experiments allow the identification of regions in the genome that are bound by TFs (Johnson et al. 2007). Analyses of a wide variety of different TFs have shown that only a small subset of possible binding sites is occupied by the TF (Carr and Biggin 1999; Iyer et al. 2001; Yang et al. 2006). For example, given the size of the mouse genome, there are potentially between 650,000 and 1.4 million sites where PU.1 can bind DNA on a spectrum of different stringencies (Heinz et al. 2013; Barozzi et al. 2014), yet only about up to 45,000 of those are selected in differentiated macrophages (Heinz et al. 2010).
There are likely a multitude of factors contributing to these restricted, yet functional, binding events of LDTFs. Among these factors, the collaborative activity between LDTFs and other TFs appear to be fundamental to a significant proportion of actual LDTF binding (Figure 1A). For example the transcription factor PU.1 is an important LDTF in macrophages and B cells. ChIP-Seq experiments showed that macrophage-specific enhancers are enriched for binding motifs of PU.1 and macrophage-specific collaborative factors C/EBP and AP-1 family members, whereas in B cells specific enhancers are enriched for PU.1 and B cell-specific factors E2A, EBF1, and Oct2 (Ghisletti et al. 2010; Heinz et al. 2010). Furthermore, macrophage-specific enhancers are depleted of the DNA binding motifs for B cell specific LDTFs and vice versa (Heinz et al. 2010).
Figure 1.
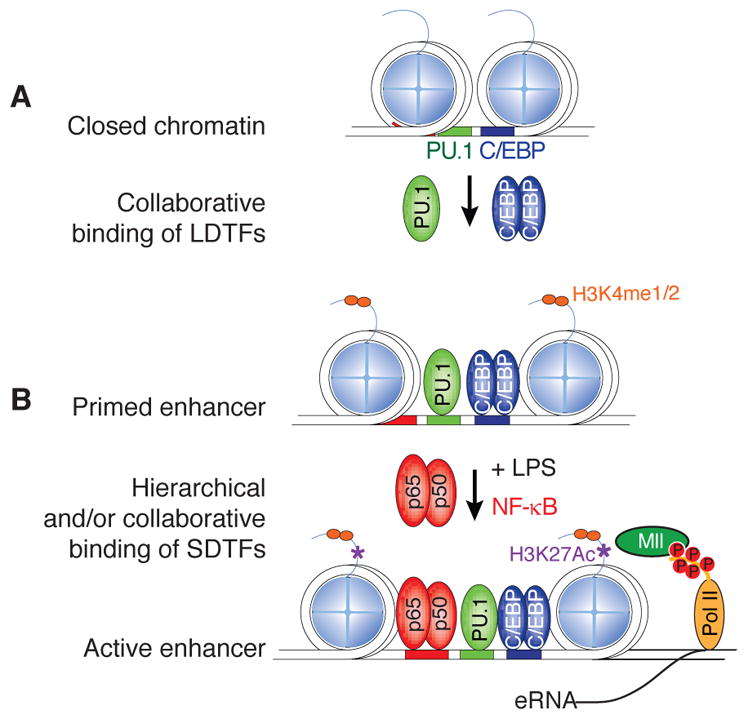
Schematic overview of a ‘collaborative/hierarchical’ model for macrophage enhancer selection and function. Panel A. Enhancers are initially selected by ‘collaborative’ interactions of lineage-determining transcription factors (LDTFs) at regions of the genome that contain closely spaced binding sites for these factors. The term collaborative is meant to indicate that each factor requires the other to effectively compete with nucleosomes and establish a nucleosome-free region. Panel B. Pre-existing enhancers are primary sites of action of signal-dependent transcription factors (SDTFs), here illustrated for the p65 component of NFκB following macrophage activation by LPS. The dependence of SDTFs on pre-existing binding of LDTFs establishes a hierarchical relationship that enables cell-specific functions of widely expressed transcription factors.
Using natural genetic variation across different mouse strains as a means to achieve ‘mutagenesis’ screening in vivo, we provided functional evidence supporting the involvement of collaborative binding of TFs in the selection of actual LDTF binding in macrophages (Heinz et al. 2013). These studies demonstrated that mutations in a C/EBP motif abrogates not only C/EBP binding, but also impairs the neighboring PU.1-DNA interactions. Similar observations were made in the case of mutations in AP-1 motifs. Importantly, such mutations also disrupted the molecular phenotype of the associated enhancer (i.e. decreased abundance of H3K4me2 mark at mutated enhancers), thus making the PU.1 – C/EBP and PU.1 – AP-1 collaborative activity a critical determinant of enhancer selection in macrophages. Lastly, we note that the natural genetic variation approach was recently extended to human white adipose tissue cells to demonstrate that mutations in C/EBP motifs disrupted nearby PPARγ binding in those cells (Soccio et al. 2015).
Macrophages, among many other cell types, possess the ability to quickly adapt their gene expression outcome after a change in environment. Further, the same signal can lead to the induction of the same single-dependent transcription factor (SDTF) but different transcriptional outcomes in different cell types. Recent studies showed, that in more than 60% SDTF bind to enhancers already established by LDTF, explaining how the same broadly expressed SDTF can exert cell specific functions and responses (Figure 1B). Indeed, investigating the role of natural genetic variation between inbreed mouse strains on binding of NF-κB p65, showed that 34% of mouse strain specific p65 binding was due to mutations in LDTF motifs, whereas only 9% was due to mutations in the p65 binding motif itself (Heinz et al. 2013).
The nature of biochemical events associated with the collaborative model of actual LDTFs binding events is not well understood. Typically the motifs of the collaborative factors can be found in close proximity to each other in sequence (< 100bp), but most of these are not found at a distance of less than 20bp (Kazemian et al. 2013), implicating a collaborative model that is not strictly dependent on protein-protein interactions. Importantly however, these restrictions address three key issues with regards to enhancer selection. One, it eliminates, for a given LDTF, numerous potential binding sites in the genome (i.e. those without nearby motifs for collaborative factors). In addition, it provides a molecular and epigenetic mechanism whereby the same LDTF can set-up very different enhancer repertoires in different cell types, as it is the case of PU.1 in macrophages and B cells (mentioned above). Finally it explains how broadly expressed SDTF can result in different cell-specific transcriptional outcomes (mentioned above)
De novo/latent enhancers
Although the majority of signal-dependent responses are mediated by activation of pre-existing enhancers and promoters, some signals can also be associated with the selection of new enhancers. For instance, TLR4 activation in mouse macrophages leads to a gain of de novo H3K4me1 marks at 1,000 to 3,000 discrete regions in the genome compared to unstimulated macrophages (Kaikkonen et al. 2013; Ostuni et al. 2013). Similar epigenomic remodeling occurred in response to the activation of other cytokine signaling pathways, including the interferon γ (IFN-γ) - STAT1 and the interleukin 4 (IL-4) – STAT6 axes (Ostuni et al. 2013). These new enhancer-like regions have been referred to as ‘de novo’ or ‘latent-enhancers’. In the case of de novo enhancers selected in response to TLR4 activation, the p65 component of NFκB was observed to participate in collaborative binding interactions with PU.1 (Kaikkonen et al. 2013; Ostuni et al. 2013). Importantly, many of these latent enhancers appear to persist in time and correlative evidence suggests that they may possess some functionality. Indeed, 72 hours following an initial IL-4 stimulation, a second stimulation leads to quicker activation of the de novo gained enhancers, as assessed by H3K27ac, compared to the first stimulation (Kaikkonen et al. 2013; Ostuni et al. 2013).
Active enhancers are sites of transcriptional initiation
DNA regulatory elements exist in one state at any given time: active (H3K4me1high, H3K4me2high, H3K27achigh), primed (H3K4me1high, H3K4me2high), actively silenced (DNA-methylation) or poised/repressed (H3K27me3high). Intriguingly, compelling evidence suggests that RNA polymerase II-mediated transcription occurs at active enhancers, yielding enhancer RNAs (eRNAs). Indeed, upon binding of activating SDTFs at enhancers, including nuclear receptors and NF-κB, eRNAs are rapidly synthesized and their expression levels correlate strongly with corresponding signal-dependent transcriptional changes of nearby genes (Ernst et al. 2011; Hah et al. 2011; Kaikkonen et al. 2013; Step et al. 2014). Similarly, eRNA transcription correlates positively with enhancer – promoter spatial interactions; enhancers that interact frequently with promoters through chromatin looping exhibit higher levels of eRNA transcripts (Li et al. 2013). Although the functional significance of enhancer transcription remains poorly understood, studies in macrophages suggest two potential roles. First, enhancer transcription at newly selected signal-dependent enhancers was linked to the deposition of H3K4me1/2 at these locations (Kaikkonen et al. 2013; Ostuni et al. 2013). Thus enhancer transcription may be important in initiating and/or maintaining the histone signature characteristic of enhancers. Second, at least some eRNAs appear to contribute to enhancer function. With regards to this latter point, we recently found that the Rev-Erb nuclear receptors actively repress gene transcription in mouse macrophages by inhibiting eRNA transcription of target enhancers (Lam et al. 2013). In particular, binding of Rev-erbs at enhancers regulating Mmp9 and Cx3cr1 gene expression represses enhancer activity and eRNA transcription, and is associated with low levels of Mmp9 and Cx3cr1 mRNAs. Presumably, this likely occurs through the recruitment of the NCoR – HDAC3 repressor complex (Zamir et al. 1996; Yin and Lazar 2005). However, in the absence of Rev-Erb factors, these enhances are de-repressed and transcribe high levels of eRNAs which translate into aberrant increase of Mmp9 and Cx3cr1 gene expression.
Cell identity and enhancer repertoire is linked to integration of environment signal with genome
The observation that treatment of macrophages with TLR4 agonists or IL-4 in vitro could drive selection of new enhancers (Kaikkonen et al. 2013; Ostuni et al. 2013) suggested a basis by which macrophages residing in different tissues acquire very different patterns of gene expression. Indeed, in spite of the fact that every subset of resident tissue macrophages in the body depends on PU.1 activity (McKercher et al. 1996; DeKoter et al. 1998; Schulz et al. 2012), gene expression in these cells varies greatly from one subset to another (Gautier et al. 2012). However, each subset requires its own set of signaling factors and pathways to function optimally. This entails that the corresponding resident macrophage population is exposed to a defined set of signals in its environment, and thus display a defined set of activated SDTFs at any given time (Figure 2).
Figure 2.
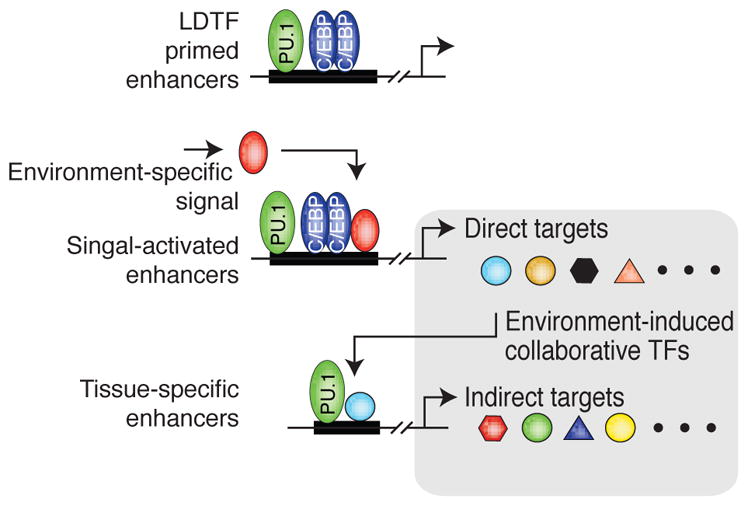
Schematic overview of the interplay between primed enhancers established by lineage-determining transcription factors (LDTF) and activation by signals from the environment. Environment-specific signals activate a subset of enhancers that are primed in all macrophages. This leads to the expression of environment-induced genes, a subset of which encode collaborative transcription factors (TFs). These TFs in turn drive the establishment of tissue-specific enhancers in collaboration with already expressed LDTFs, such as PU.1. The combination of direct and indirect responses contributes to the identity and function of macrophages residing in that particular tissue environment.
We and others recently provided extensive evidence supporting the hypothesis that tissue-derived factors play an essential role in defining the identity of its resident macrophage population (Gosselin et al. 2014; Lavin et al. 2014). For instance, comparing the enhancer landscape of microglia to that of large peritoneal macrophages revealed that 60% of the active enhancers are shared, but 40% are macrophage subset specific (Gosselin et al. 2014). Cell-type specificity is more striking when considering SEs, consistent with work by other groups suggesting that these regulatory elements are critical to cell identity (Hnisz et al. 2013; Gosselin et al. 2014; Adam et al. 2015).
By removing macrophages from their environment and putting them into tissue-culture condition about 50% of the enhancers remained unchanged, but only 26% of SE still met the SE criteria (Gosselin et al. 2014). This further supports the hypothesis that SEs are uniquely regulated genomic regions that are sensitive to environment and determine the fate of the cell type.
Computational analyses demonstrated that macrophage subset-specific enhancers display very distinct enrichment of DNA motifs recognized by SDTFs. These include SMAD motifs in microglia-specific enhancer, GATA6 and RARβ in large peritoneal macrophages, PPARγ in alveolar macrophages, LXRα in Kupffer cells, etc. (Gosselin et al. 2014; Lavin et al. 2014). Of note, natural genetic variation analyses implicated SMAD and GATA6 as collaborative factors with PU.1 to select microglia-specific and large peritoneal macrophage-specific enhancers, respectively (Gosselin et al. 2014).
These analyses suggested that different combinations of SDTFs might cause the subset-specificity of the enhancer repertoire of different populations of resident-tissue macrophages. Lavin and colleagues (2014) provided a compelling demonstration of the role of environment in supplying key signals to activate SDTFs. Indeed, isolating peritoneal macrophages and transplanting then into the alveolar cavity of the lungs led to a loss of peritoneal macrophage-associated enhancer repertoire and, concomitantly, a gain of an alveolar macrophage-enhancer repertoire. These effects were paralleled by a respective loss of GATA6 motif enrichment and a gain of a PPARγ motif signature in the reconfigured enhancer landscape.
We provided strong additional evidence for the role of the environment in driving macrophage phenotype while exploring the molecular mechanisms of macrophage subset identity. Tissue macrophages extracted and transferred to an in vitro culture environment changed their gene expression program dramatically, resulting in a loss of their original gene signature identity. Importantly, the culture conditions led to a down-regulation of a significant number of TFs, many of which may act as potential collaborative factors with PU.1. However, supplementing macrophages in vitro with key stimulating ligands has profound effects of their identity. Indeed, supplementing large peritoneal macrophages in culture condition with retinoic acid (RA) maintained a significant proportion of the features of their original in vivo enhancer repertoire and gene expression program. On the other hand, added Tgf-β1 to the media polarized peritoneal macrophages in culture towards a microglia in vivo phenotype. These observations are highly consistent with previous studies that reported that RA and Tgf-β1 are key factors for the ontogeny and/or functions of large peritoneal macrophages and microglia, respectively (Makwana et al. 2007; Butovsky et al. 2014; Okabe and Medzhitov 2014).
These latter experiments suggested that a macrophage’s subset identity is the product of an elaborate molecular cascade. Indeed, the Rarb gene locus is primed in microglia, but display a SE phenotype in large peritoneal macrophages, which is concordant with the observation that RA promotes Rarβ expression in a positive feedback manner (de The et al. 1990). Of importance, high expression of Rarβ then promotes expression of numerous TFs, including Gata6, Nfe2, Bhlhe40, etc, which can in turn function in a collaborative manner with PU.1 to precisely expand the functional enhancer repertoire and gene expression program of large peritoneal macrophages. Therefore, such chain of events, involving the integration of tissue environment-derived factors with DNA, provides an elegant and highly specific mechanism for the establishment of the identity of the different resident tissue macrophage subsets (Figure 2).
Intersection of epigenomic concepts with GWAS data
The technical and conceptual advances in epigenomics described above provide a powerful means to contextualize work in the field of medical genetic research on a genome-wide level. A significant dividend of the efforts to understand how enhancer selection proceeds and more general principals of epigenomic control of gene regulation is already the appreciation that natural genetic variation impacts gene expression through effects on DNA regulatory elements (Dimas et al. 2009; Pomerantz et al. 2009; Ernst et al. 2011; Harismendy et al. 2011; Maurano et al. 2012; Sanyal et al. 2012; Schaub et al. 2012; Hnisz et al. 2013; Farh et al. 2015; Marson et al. 2015). Importantly, these studies also highlighted that the majority of single nucleotide polymorphisms (SNPs) found to be associated with disease risk by genome-wide association studies (GWAS) are in non-coding regions of the genome and frequently map within regulatory elements active predominantly in cell types relevant to the disease (Dimas et al. 2009; Hu et al. 2011). For example, SNPs that confer susceptibility for autoimmune diseases like multiple sclerosis and rheumatoid arthritis are prevalent within T cell enhancers (Okada et al. 2014). Furthermore, risk SNPs are enriched in regions of TF binding, suggesting that disruption of TF-DNA interactions may be a primary mode of actions for non-coding SNPs that are clinically relevant (Ernst et al. 2011; Maurano et al. 2012; Kilpinen et al. 2013) (Figure 3A). Of note, such a mode of action for SNPs has also been reported for other complex diseases, including metabolic disorders (Soccio et al. 2015) and coronary artery diseases (Harismendy et al. 2011). Several studies also showed that sequence variation associated with disease is especially enriched in SE of disease-relevant cell types (Hnisz et al. 2013; Parker et al. 2013). SEs are very sensitive to perturbations (Siersbaek et al. 2014) and changes in environment (Gosselin et al. 2014; Adam et al. 2015) and are therefore a good target for disease-associated SNPs.
Figure 3.

Implications of mechanisms of enhancer selection and function for interpretation of genome-wide association studies (GWAS). Panel A. Schematic overview illustrating how SNPs identified in GWAS and associated with expression quantitative trait loci (eQTL) can affect enhancers in a cell-type specific manner. Panel B. Schematic overview how studies identifying disease-associated genomic variations in mouse can be helpful in understanding human disease through conserved sequence.
Although very informative, these early studies could not discriminate between actual causal SNPs and neutral/non-causal SNPs that emerged as potential candidate because of linkage disequilibrium. However, according to recent estimates, 90% of causal SNPs associated with autoimmune diseases map onto non-coding regions of the genome, and 60% of those localize within immune-cell enhancers (Farh et al. 2015). Unexpectedly, data from this study also indicated that disruptive SNPs might not necessarily interfere with TF-DNA interactions. Indeed, SNPs did not map within TF motifs, and were instead located in their vicinity. Thus, the mechanism disrupted by causal SNPs is difficult to interpret by current paradigms in epigenomics.
This latter observation by Farh and colleagues (2015) points to a critical role for the “DNA context” within enhancers in regulating their functions optimally. For example chromosomal rearrangements of only one enhancer can cause dysregulation of two specific acute myeloid leukemia (AML) predisposition genes (Groschel et al. 2014). Evidence also suggests that SNPs lying outside of TF motifs can affect chromatin looping (Grubert et al. 2015). Thus, although enhancers serve as platforms for the integration of SDTFs binding with DNA and the transcription machinery, their proper functions may require more than simple local coordinated enrichment of DNA motif recognized by TF. The nature of the additional mechanism(s) involved in enhancer functions remain quite elusive and may relate to transcriptional processes and/or eRNA synthesis. We note also that our comprehension of the biochemical principles guiding TF-DNA interactions remains rudimentary. In this regard we recently reported that under certain circumstances DNA variance within a TF motif appear to have limited influence in driving differential TF-DNA interactions at a similar regulatory elements between two different strain of mice, as is the case of recruitment of the RelA/p65 subunit of NF-κB at macrophage enhancers following TLR4 activation (Heinz et al. 2013).
Conservation and relevance to human diseases
DNA sequences at enhancers and other regulatory elements display a relatively high level of conservation across mammalian species (Goke et al. 2011; Cheng et al. 2014) (Figure 3B), although it has to be noted that the same enhancer potentially can have different regulatory outcomes (Yan et al. 1997). Nevertheless, leveraging the conservation between species can inform about the etiology and/or maintenance/pathological mechanisms that contribute to diseases.
Studies using a mouse model of Alzheimer’s disease (AD) that display chronic neurodegeneration provide an elegant example of such application of epigenomics (Gjoneska et al. 2015). In such model, as neurons die an accompanying activity of de novo enhancers emerge at the whole-tissue level. Of note, these enhancers exhibit a significant enrichment for the DNA motif recognized by PU.1, and the DNA sequences encompassing these de novo-PU.1 enhancers are well conserved in humans. Strikingly however, they also display features of active and/or primed enhancers in CD14+ human monocytes, a cell type previously implicated in the etiology of AD by independent studies (Bradshaw et al. 2013).
That a complex amalgam of signals triggered by neuronal death and distress elicits an immune response that implicates DNA sequences and regulatory elements that are conserved across species in related myeloid cells is quite provocative. Furthermore, refinements of the acquisition of data in this model, for example by extracting specifically brain microglia as the brain degenerates, can potentially lead to clearer resolution of the data. This could prove to be helpful to deconvulate the signaling pathways whose activity become integrated with that of PU.1 to activate the de novo regulatory elements. Finally, we note that previous expression quantitative trait loci (eQTLs) studies associated monocytes, but not T cells, with neurodegenerative diseases, including Alzheimer’s and Parkinson’s diseases (Raj et al. 2014). Overall, this raises the intriguing possibility that the role of candidate mutations arising GWAS and eQTL studies can be interrogated in complex settings using the relevant model of human disease (Figure 3).
Conclusions and perspectives
Through epigenomics and next-generation sequencing technologies, we now possess powerful tools to explore and appreciate the mechanism through which a cell’s environment influences how genetics information is interpreted by that cell to achieve its identity and function. In spite of recent discoveries, however, many outstanding questions remain. With regards to macrophages, it is not clear whether different developmental origin (i.e. primitive vs. definitive hematopoiesis) can have residual effects on their transcriptional programs. Specifically, do enhancers relevant to monocyte but not to resident tissue macrophage become silent, or are they erased, as monocytes differentiate, for example, into Kupffer cells, cardiac macrophages or resident peritoneal macrophage? On the other hand, for macrophage populations that do not depend on monocytes to be maintained like microglia, how does the aging process affect their epigenome and/or ability to optimally regulate their gene expression programs? Finally, the ability to infer signaling pathways activity from activation or inhibition of DNA regulatory elements is a powerful lever to gain a better understanding of signals that promote homeostasis or drive disease mechanism. Therefore, application of these tools will help gain a better understanding of macrophage activation and functions as it applies to any in vivo contexts, including human diseases.
Acknowledgments
We would like to thank Leslie Van Ael for her assistance with figure and manuscript preparation. These studies were primarily supported by NIH grants DK091183, CA17390, and DK063491 and the San Diego Center for Systems Biology (GM085764). David Gosselin is supported by fellowship from Multiple Sclerosis Society of Canada.
References
- Adam RC, Yang H, Rockowitz S, Larsen SB, Nikolova M, Oristian DS, Polak L, Kadaja M, Asare A, Zheng D, et al. Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice. Nature. 2015;521:366–370. doi: 10.1038/nature14289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji J, Rusconi S, Schaffner W. Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell. 1981;27:299–308. doi: 10.1016/0092-8674(81)90413-x. [DOI] [PubMed] [Google Scholar]
- Barish GD, Yu RT, Karunasiri M, Ocampo CB, Dixon J, Benner C, Dent AL, Tangirala RK, Evans RM. Bcl-6 and NF-kappaB cistromes mediate opposing regulation of the innate immune response. Genes & development. 2010;24:2760–2765. doi: 10.1101/gad.1998010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barozzi I, Simonatto M, Bonifacio S, Yang L, Rohs R, Ghisletti S, Natoli G. Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers. Mol Cell. 2014;54:844–857. doi: 10.1016/j.molcel.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Bossard P, Zaret KS. GATA transcription factors as potentiators of gut endoderm differentiation. Development. 1998;125:4909–4917. doi: 10.1242/dev.125.24.4909. [DOI] [PubMed] [Google Scholar]
- Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, Rosenkrantz LL, Imboywa S, Lee M, Von Korff A, et al. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat Neurosci. 2013;16:848–850. doi: 10.1038/nn.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013;49:825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr A, Biggin MD. A comparison of in vivo and in vitro DNA-binding specificities suggests a new model for homeoprotein DNA binding in Drosophila embryos. The EMBO Journal. 1999;18 doi: 10.1093/emboj/18.6.1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Ma Z, Kim BH, Wu W, Cayting P, Boyle AP, Sundaram V, Xing X, Dogan N, Li J, et al. Principles of regulatory information conservation between mouse and human. Nature. 2014;515:371–375. doi: 10.1038/nature13985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’haeseleer P. What are DNA sequence motifs? Nature Biotechnology. 2006;24:423– 425. doi: 10.1038/nbt0406-423. [DOI] [PubMed] [Google Scholar]
- de The H, Vivanco-Ruiz MM, Tiollais P, Stunnenberg H, Dejean A. Identification of a retinoic acid responsive element in the retinoic acid receptor beta gene. Nature. 1990;343:177–180. doi: 10.1038/343177a0. [DOI] [PubMed] [Google Scholar]
- DeKoter RP, Walsh JC, Singh H. PU.1 regulates both cytokine-dependent proliferation and differentiation of granulocyte/macrophage progenitors. EMBO J. 1998;17:4456–4468. doi: 10.1093/emboj/17.15.4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H, Ingle C, Beazley C, Gutierrez Arcelus M, Sekowska M, et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40:91–104. doi: 10.1016/j.immuni.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, Shoresh N, Whitton H, Ryan RJ, Shishkin AA, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518:337–343. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisletti S, Barozzi I, Mietton F, Polletti S, De Santa F, Venturini E, Gregory L, Lonie L, Chew A, Wei CL, et al. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010;32:317–328. doi: 10.1016/j.immuni.2010.02.008. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nature reviews Immunology. 2014;14:392–404. doi: 10.1038/nri3671. [DOI] [PubMed] [Google Scholar]
- Gjoneska E, Pfenning AR, Mathys H, Quon G, Kundaje A, Tsai LH, Kellis M. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature. 2015;518:365–369. doi: 10.1038/nature14252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goke J, Jung M, Behrens S, Chavez L, O’Keeffe S, Timmermann B, Lehrach H, Adjaye J, Vingron M. Combinatorial binding in human and mouse embryonic stem cells identifies conserved enhancers active in early embryonic development. PLoS Comput Biol. 2011;7:e1002304. doi: 10.1371/journal.pcbi.1002304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. 2014;159:1327–1340. doi: 10.1016/j.cell.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groschel S, Sanders MA, Hoogenboezem R, de Wit E, Bouwman BA, Erpelinck C, van der Velden VH, Havermans M, Avellino R, van Lom K, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157:369–381. doi: 10.1016/j.cell.2014.02.019. [DOI] [PubMed] [Google Scholar]
- Grubert F, Zaugg JB, Kasowski M, Ursu O, Spacek DV, Martin AR, Greenside P, Srivas R, Phanstiel DH, Pekowska A, et al. Genetic Control of Chromatin States in Humans Involves Local and Distal Chromosomal Interactions. Cell. 2015 doi: 10.1016/j.cell.2015.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, Deswarte K, Malissen B, Hammad H, Lambrecht BN. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. The Journal of experimental medicine. 2013;210:1977–1992. doi: 10.1084/jem.20131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, Kraus WL. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell. 2011;145:622–634. doi: 10.1016/j.cell.2011.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harismendy O, Notani D, Song X, Rahim NG, Tanasa B, Heintzman N, Ren B, Fu XD, Topol EJ, Rosenfeld MG, et al. 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature. 2011;470:264–268. doi: 10.1038/nature09753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, Allison KA, Kaikkonen MU, Orozco LD, Glass CK. Effect of natural genetic variation on enhancer selection and function. Nature. 2013;503:487–492. doi: 10.1038/nature12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16:144–154. doi: 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffel G, Wang Y, Greter M, See P, Teo P, Malleret B, Leboeuf M, Low D, Oller G, Almeida F, et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. The Journal of experimental medicine. 2012;209:1167–1181. doi: 10.1084/jem.20120340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Kim H, Stahl E, Plenge R, Daly M, Raychaudhuri S. Integrating autoimmune risk loci with gene-expression data identifies specific pathogenic immune cell subsets. Am J Hum Genet. 2011;89:496–506. doi: 10.1016/j.ajhg.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer VR, Horak CE, Scafek CS, Botsteink D, Snyder M, Brown PO. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature. 2001;409 doi: 10.1038/35054095. [DOI] [PubMed] [Google Scholar]
- Johnson SD, Mortazavi AM, RM, Wold B. Genome-Wide Mapping of in Vivo Protein-DNA Interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- Kaikkonen MU, Spann NJ, Heinz S, Romanoski CE, Allison KA, Stender JD, Chun HB, Tough DF, Prinjha RK, Benner C, et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol Cell. 2013;51:310–325. doi: 10.1016/j.molcel.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemian M, Pham H, Wolfe SA, Brodsky MH, Sinha S. Widespread evidence of cooperative DNA binding by transcription factors in Drosophila development. Nucleic Acids Res. 2013;41:8237–8252. doi: 10.1093/nar/gkt598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpinen H, Waszak SM, Gschwind AR, Raghav SK, Witwicki RM, Orioli A, Migliavacca E, Wiederkehr M, Gutierrez-Arcelus M, Panousis NI, et al. Coordinated effects of sequence variation on DNA binding, chromatin structure, and transcription. Science. 2013;342:744–747. doi: 10.1126/science.1242463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam MT, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y, Benner C, Kaikkonen MU, Kim AS, Kosaka M, et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature. 2013;498:511–515. doi: 10.1038/nature12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CS, Friedman JR, Fulmer JT, Kaestner KH. The initiation of liver development is dependent on Foxa transcription factors. Nature. 2005;435:944–947. doi: 10.1038/nature03649. [DOI] [PubMed] [Google Scholar]
- Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498:516–520. doi: 10.1038/nature12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, Carroll JS, Liu XS, Brown M. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132:958–970. doi: 10.1016/j.cell.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makwana M, Jones LL, Cuthill D, Heuer H, Bohatschek M, Hristova M, Friedrichsen S, Ormsby I, Bueringer D, Koppius A, et al. Endogenous transforming growth factor beta 1 suppresses inflammation and promotes survival in adult CNS. J Neurosci. 2007;27:11201–11213. doi: 10.1523/JNEUROSCI.2255-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson A, Housley WJ, Hafler DA. Genetic basis of autoimmunity. J Clin Invest. 2015;125:2234–2241. doi: 10.1172/JCI78086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKercher SR, Torbett BE, Anderson KL, Henkel GW, Vestal DJ, Baribault H, Klemsz M, Feeney AJ, Wu GE, Paige CJ, et al. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. 1996;15:5647–5658. [PMC free article] [PubMed] [Google Scholar]
- McPherson CESE, Friedman DS, Zaret KS. An Active Tissue-Specific Enhancer and Bound Transcription Factors Existing in a Precisely Positioned Nucleosomal Array. Cell. 1993;75:387–398. doi: 10.1016/0092-8674(93)80079-t. [DOI] [PubMed] [Google Scholar]
- Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell. 2014;157:832–844. doi: 10.1016/j.cell.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Yoshida S, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506:376–381. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, Natoli G. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152:157–171. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- Parker SC, Stitzel ML, Taylor DL, Orozco JM, Erdos MR, Akiyama JA, van Bueren KL, Chines PS, Narisu N, Program NCS, et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci U S A. 2013;110:17921–17926. doi: 10.1073/pnas.1317023110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz MM, Ahmadiyeh N, Jia L, Herman P, Verzi MP, Doddapaneni H, Beckwith CA, Chan JA, Hills A, Davis M, et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat Genet. 2009;41:882–884. doi: 10.1038/ng.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj T, Rothamel K, Mostafavi S, Ye C, Lee MN, Replogle JM, Feng T, Lee M, Asinovski N, Frohlich I, et al. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science. 2014;344:519–523. doi: 10.1126/science.1249547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012;22:1748–1759. doi: 10.1101/gr.136127.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, Tsirka SE, Maletic-Savatic M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7:483–495. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siersbaek R, Rabiee A, Nielsen R, Sidoli S, Traynor S, Loft A, La Cour Poulsen L, Rogowska-Wrzesinska A, Jensen ON, Mandrup S. Transcription factor cooperativity in early adipogenic hotspots and super-enhancers. Cell Rep. 2014;7:1443–1455. doi: 10.1016/j.celrep.2014.04.042. [DOI] [PubMed] [Google Scholar]
- Soccio RE, Chen ER, Rajapurkar SR, Safabakhsh P, Marinis JM, Dispirito JR, Emmett MJ, Briggs ER, Fang B, Everett LJ, et al. Genetic Variation Determines PPARgamma Function and Anti-diabetic Drug Response In Vivo. Cell. 2015;162:33–44. doi: 10.1016/j.cell.2015.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Step SE, Lim HW, Marinis JM, Prokesch A, Steger DJ, You SH, Won KJ, Lazar MA. Anti-diabetic rosiglitazone remodels the adipocyte transcriptome by redistributing transcription to PPARgamma-driven enhancers. Genes & development. 2014;28:1018–1028. doi: 10.1101/gad.237628.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Jones CA, Sigmund CD, Gross KW, Catanzaro DF. Conserved Enhancer Elements in Human and Mouse Renin Genes Have Different Transcriptional Effects in As4.1 Cells. Circulation Research. 1997;81:558–566. doi: 10.1161/01.res.81.4.558. [DOI] [PubMed] [Google Scholar]
- Yang A, Zhu Z, Kapranov P, McKeon F, Church GM, Gingeras TR, Struhl K. Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol Cell. 2006;24:593–602. doi: 10.1016/j.molcel.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Yin L, Lazar MA. The orphan nuclear receptor Rev-erbalpha recruits the N-CoR/histone deacetylase 3 corepressor to regulate the circadian Bmal1 gene. Molecular endocrinology. 2005;19:1452–1459. doi: 10.1210/me.2005-0057. [DOI] [PubMed] [Google Scholar]
- Zamir I, Harding HP, Atkins GB, Horlein A, Glass CK, Rosenfeld MG, Lazar MA. A nuclear hormone receptor corepressor mediates transcriptional silencing by receptors with distinct repression domains. Molecular and cellular biology. 1996;16:5458–5465. doi: 10.1128/mcb.16.10.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]