

Abstract
Paraquat (PQ) is a commonly used herbicide that induces oxidative stress via reactive oxygen species (ROS) generation. This study aimed to investigate the effects of the antioxidant N-acetylcysteine (NAC) against PQ-induced oxidative stress in mice. Male Balb/C mice (24) were randomly divided into 4 groups and treated for 3 weeks: 1) control (saline), 2) NAC (0.5% in diet), 3) PQ (20 mg/kg, IP) and 4) combination (PQ + NAC). Afterwards mice were sacrificed and oxidative stress markers were analyzed. Our data showed no significant change in serum antioxidant capacity.
PQ enhanced lipid peroxidation (MDA) levels in liver tissue compared to control whereas NAC decreased MDA levels (p<0.05). NAC significantly increased MDA in brain tissue (p<0.05). PQ significantly depleted glutathione (GSH) levels in liver (p=0.001) and brain tissue (p<0.05) but non-significant GSH depletion in lung tissue. NAC counteracted PQ, showing a moderate increase GSH levels in liver and brain tissues. PQ significantly increased 8-oxodeoxyguanosine (8-OH-dG) levels (p<0.05) in liver tissue compared to control without a significant change in brain tissue. NAC treatment ameliorated PQ-induced oxidative DNA damage in the liver tissue. PQ significantly decreased the relative mtDNA amplification and increased the frequency of lesions in liver and brain tissue (p<0.0001), while NAC restored the DNA polymerase activity in liver tissue but not in brain tissue. In conclusion, PQ induced lipid peroxidation, oxidative nuclear DNA and mtDNA damage in liver tissues and depleted liver and brain GSH levels. NAC supplementation ameliorated the PQ-induced oxidative stress response in liver tissue of mice.
Keywords: Antioxidant Capacity, DNA Integrity, Glutathione levels, MDA levels, Mice, Oxidative Stress, Reactive Oxygen Species
1. Introduction
Oxidative stress occurs when the dynamics of reduction and oxidation (REDOX) balance between oxidants and antioxidants is shifted towards oxidant potentials [71]. Oxidative stress-related diseases include psoriasis, renal graft, retinal damage, atherosclerosis, asthma, Parkinson’s disease and multi-organ diseases such as diabetes and cancer [79]. An overwhelm number of studies converge that the mitochondria plays an important role on the onset and development of the aforementioned pathologies [35].
The mitochondria unlike other organelles, contains its own genome, mitochondria DNA (mtDNA) and is the largest supplier of ATP and the main source of reactive oxygen species (ROS) in aerobic organisms [10, 51]. Due to the electron transport chain (ETC) in the inner mitochondrial membrane, there is a high endogenous ROS production that disrupts the homeostasis, cell signaling and also leads to mitochondrial genome instability [23]. A compromised ETC increases the ROS levels introducing damage in lipids, DNA, and perturbs both the glutathione scavenging and antioxidant capacity [34]. Cells are constantly challenged by exogenous stressors from environmental conditions such as chemicals, drugs, and UV light exposure as well as by endogenous stressors including ROS and quinones. The repetitive exposure to environmental agents such as pesticides/herbicides exacerbates the generation of ROS, and down regulates the levels of antioxidant defenses. For example, Paraquat (PQ; 1,1-dimethyl-4,4-bipyridium dichloride), is a redox cycling compound that selectively produce ROS by uncoupling the ETC complex in the mitochondria [15]. This causes an increase in the intracellular oxidative microenvironment leading to the decline in the supply of cellular energy, inducing cell death, and depleting glutathione (GSH) [18, 40].
The mechanism of action of PQ toxicity consists in the generation of superoxide anions, which leads to the proliferation of ROS and the oxidation of NADPH, a major source of reducing equivalents required in biosynthetic processes [76]. ROS interferes with NADPH electron shuttling by diminishing the pool of available NADP+, whom itself is needed for the progression of metabolic REDOX reactions. The main organ affected by PQ are the lungs, in which the oxidant agent is transported in the Clara cells and alveolar type I and II epithelial cells, leading to a second proliferating phase defined by alveolitis and pulmonary edema [12]. Trace amounts of PQ can be detected in more than 100 crops (corn, tomatoes, olives, field beans, fruits) highlighting the need to study the effects of PQ that may be affecting the public health of the general population whom often misinformed on the proper use of PQ [61, 62]. PQ effects in rodent models showed that prolonged exposure leads to accumulation and persistent damage in brain lung and liver tissues providing insight of a better understanding of PQ actions [15, 17, 39, 40, 61, 62, 78].
N-acetylcysteine (NAC) is an oral supplement that have demonstrated high efficacy acting as a reducing agent. NAC is considered a cost-effective and approved therapy with a forty-year safety history towards a number of illnesses such as neurocognitive disorders, liver toxicity, heavy metal toxicity, acute respiratory syndrome, and chemotherapy induced toxicity [9, 63, 83]. NAC is the acetylated precursor of L-cysteine, who itself is a precursor of glutathione (GSH). NAC acts as an electron acceptor, preventing oxidative damage and increasing intracellular glutathione biosynthesis, which is a key component in the preservation of a positive REDOX potential [27]. NAC can permeate the cell membrane under physiological conditions [58,67]. According to previous studies, NAC crosses the Blood Brain Barrier (BBB) through deacetylation pathways and is capable of being actively transported through cysteine carrier mediated mechanism in kidneys, liver, adrenal glands, lungs, and brain tissues [73, 75]. Small molecules as NAC can diffuse across the cell membrane when the membrane loses its stability as a consequence of oxidative stress damage [43].
In vivo studies, showed the ability of NAC to penetrate the BBB by crossing the capillary wall and reaching brain tissues and extracellular space using radiolabeled NAC in a SAMP8 mice model [28]. NAC can stabilize to the mitochondria ETC acting upon the complexes I and IV of liver and brain mice tissue [19, 54]. In vitro studies showed that NAC supplementation preserved and protected the inner mitochondrial membrane leading to a greater amount of ATP production and decreasing ROS levels [5, 6, 33]. In lung tissue, NAC has been used as a therapeutic mucolytic agent providing protection in the surfactant surrounding the alveolar tissue by avoiding GSH depletion [31].
The present study was undertaken in order to investigate the antioxidant effects of NAC to afford protection in mice vital tissues that have been exposed to a well-known ROS generator, as PQ. Our central hypothesis stated that NAC reduces oxidative stress induced by PQ in liver, brain, and lung mice tissues.
2. Materials and Methods
2.1 Reagents
Paraquat dichloride and N-Acetylcysteine (NAC) were purchased from Sigma-Aldrich. Dulbecco’s Phosphate-Buffered Saline (DPBS) was purchased from Life Technologies. Colorimetric assay kits were purchased from Cayman chemicals. 8-oxodeoxyguanosine was measured using the Oxiselect ™ Oxidative DNA damage ELISA (Cell Biolab). PCR studies were performed with the GeneAmp High Fidelity PCR system (Applied Biosystems) and Quant-iT PicoGreen® dsDNA Assay Kit (Invitrogen).
2.2 Oxidative Stress Animal Model
Balb/C male mice (6–7 months old, weighing approximately 24–32g) were keep in a temperature-controlled room on a 12:12 hrs light:dark schedule with regular chow and water ad libitum. The food intake and the body weight of the animals were recorded weekly. The Institutional Animal Care and Use Committee (IACUC) approved all procedures in accordance with the Animal Welfare Assurance A3585-01.
2.3 Drug schedule and administration
N-Acetylcysteine (NAC) was administered in the form of supplemented rodent chow at 0.5% and DPBS saline buffer (1X) was used for sham treatments and as a vehicle for Paraquat treatment. The four experimental groups were as followed: Control (sham ip injections and regular chow), NAC (sham ip injections and supplemented chow), PQ (20 mg/kg ip injections of Paraquat and regular chow) PQ+NAC (20 mg/kg ip injections of Paraquat and supplemented chow). During a 3-week period, mice were given the aforementioned injections twice a week. All groups received their appropriate treatments simultaneously and at same time points.
2.4 Tissue and blood collection
Liver, brain and lung tissues were collected and cryopreserved using liquid nitrogen and stored at −80°C. The tissues were prepared individually accordingly to the manufacturer’s specifications for each assay. Mice were anesthetized using isoflurane (Sigma). Between 200–350μL of whole blood was extracted via heart-puncture using a 22G × 1.5 inches BD 3mL Syringe needle (Terumo, Co) and stored in 1.5mL collection tubes (non-anticoagulant tubes). Blood was allowed to clot for 30 min at 25°C and centrifuged at 2,000 × g for 15 min at 4°C. The top yellow serum was collected and stored on ice to avoid hemolysis.
2.4.1 Tissue homogenization for lipid peroxidation and total glutathione analysis
Liver, brain and lung tissues were homogenized manually using a TissueMiser (Fisher Scientific) in a solution of 1x PBS with EDTA (1mM, pH 7.4) over ice. According to the tissue weight, a total of 1 mL of PBS with EDTA solution was added to brain and liver tissues and a total of 250μL of PBS with EDTA was added to the lung tissue. The homogenates were centrifuged at 10,000 × g for 15 min at 4°C. The supernatant obtained were transferred to 0.5 mL microtubes and stored at −20°C. Lastly, supernatant volumes were aliquoted and equally divided further for lipid peroxidation and total glutathione analysis.
2.4.2 Tissue homogenization for mtDNA analysis
Frozen liver and brain tissues were homogenized with a Bullet Blender Machine (Next Advance, Inc.). The brain tissue samples were homogenized by the pink bead lysis/0.5 mm glass beads and the hepatic tissue samples were homogenized with green bead lysis/0.5 mm zirconium oxide beads. Homogenized tissues were lysed with Buffer G2 (Qiagen, Valencia).
2.5 Oxidative stress analysis
2.5.1 Antioxidant Capacity test
To evaluate the antioxidant capacity of serum we used an Antioxidant Assay Kit that relies on the ability of antioxidants in a sample to inhibit the oxidation of ABTS (2,2′-Azino-di-(3-ethylbenzthiazoline sulphonate) to ABTS+ by metmyoglobin. The serum samples were diluted 1:20 and absorbance was read at 405 nm using Dynex spectrophotometer (Life technologies). The assay was done at duplicates and the results were showed as the antioxidant capacity (mM), in terms of Trolox equivalents.
2.5.2 Glutathione (GSH) assay
Glutathione levels in liver, brain and lung tissues were measured using a Glutathione Assay Kit. These samples were deprotinated according to the manufacture’s recommendations, assayed in triplicates and analyzed at 405 nm using a Thermo Multiskan spectrophotometer microplate reader. The results were expressed as the total GSH levels (μM)/g of tissue. Individualized dilution factors were performed for liver tissue (1:50 dilution factor) and lung tissue (1:1, 1:2, 1:3, 1:3.5 dilution factors), whereas for brain tissue, undiluted sample were used. Samples were stored at −20°C until further use for glutathione analysis.
2.5.3 Lipid peroxidation
We evaluated the susceptibility of liver, brain, lung tissues to lipid peroxidation, induced by ROS acting upon polyunsaturated fatty acids, by using a TBARS assay to measure Malondialdehyde (MDA) levels. Homogenized tissues were centrifuged and supernatant were assayed in triplicates. All assays were analyzed using a spectrophotometer at 530–540 nm. The results were expressed as MDA (μM)/g of tissue.
2.5.4 Mitochondrial DNA (mtDNA) integrity analysis
Quantitative Polymerase Chain Reaction (QPCR) was used to determine the extent of mitochondrial damage. The rationale of the QPCR technique for quantitation of DNA damage is based on the ability of certain DNA lesions to disrupt the progression of the thermostable polymerase on the DNA template, resulting in a decrease of template amplification. To measure the relative amplification levels and the frequency of lesions in our samples, we selected two amplicons: a long amplicon (10kB) and a short amplicon (117bp). The long amplicon products are more susceptible to have a lesion that stops the thermostable DNA polymerase and only those DNA template not containing DNA lesions will amplify. Small amplicon products were relatively insensitive to DNA damage, which serves as an internal control. The amplification of both fragments was conducted using mitochondrial gene-specific primers. The primer nucleotide sequences for the amplification of the 10 kB mtDNA amplicon were the following: 5′-CCACTCCATCCACCACCATC-3′ (forward, genomic position 5733) and 5′-CCACAACACCCC CATTCCTC-3′ (reverse, genomic position 15733). For the short amplicon (117bp) the primer nucleotide sequences were: 5′-CCCAGCTACCATCATTCA-3′ (forward, genomic position 13603) and 5′-AGTGATGGTTTGGGATGGTTGATGT-3′ (reverse, genomic position 13719). The validation for the mtDNA analysis consisted in the optimization for the PCR profile and characterization of the amplicons of interest for liver and brain tissues. To guarantee that the amplification rate was at the exponential phase, during each set of amplifications, a 50% (internal quality control) parameter was included containing half of the amount of the non-damage template, which yielded a 50% reduction of the amplification signal.
2.5.4.1 DNA isolation and DNA quality assessment
DNA isolation was performed using the High Molecular Weight DNA Isolation kit, Qiagen Genomic Tip 20/G and Qiagen DNA Buffer according to the manufacturer’s instructions. DNA from liver and brain tissues homogenates were isolated for downstream analysis (PCR). Quality control was performed using a Nanodrop spectrophotometer (NanoDrop 2000, Thermo Fisher) with the following quality parameters (A260/280=1.9).
2.5.4.2 Characterization of amplicons
In order to confirm the amplicons integrity, two electrophoresis gels were run independently, according to their respective length. A 3% agarose gel was run for the small amplicon (117 bp) at 100V for 20 mins and a 0.8% agarose gel was run for the large amplicon (10 KB) at 100V for 1 hr. TAE (1X) buffer was used for running electrophoresis gels. Two ladders were used to characterize the small and large fragments, 50 bp DNA ladder (Novagen) and a 1 Kb ladder (Promega). An amount of 10 μL of PCR product and 3 μL of loading dye were used when running the agarose gels.
2.5.4.3 PCR conditions
For amplification of long and small segment a total of 5 μL DNA template was used for a total concentration of 20 μg/μL in a final volume of 50 μL. The 50% (internal quality control) amplification sample consisted of 5 μL DNA template was used for a total concentration of 10 μg/μL in a final volume of 50μL. The mtDNA damage was detected in liver and brain tissues by the GeneAmp® High Fidelity PCR system (Applied Biosystems). mtDNA specific primers were purchased from Integrated DNA Technology, Inc. (IDT) for each one of the amplicons. The Bio-Rad PCR thermal cycler was used to provide the specific thermal conditions for the study. Thermal cycles were determined to keep the reaction in the exponential phase of the PCR. The PCR thermal program for the amplification of 117 bp mtDNA fragment consisted of an initial denaturation for 1 min at 94°C, followed by 17 cycles of denaturation for 15 sec at 94°C, annealing/extension at 60°C for 12 min and a final extension at 72°C for 10 min. The same parameters were used for the amplification of 10 kB mtDNA fragment with a change in annealing/extension step consisting of 67°C for 12 min.
2.5.4.4 Fluorometric dsDNA quantitation assay
dsDNA PCR products were quantified by measuring the DNA fluorescence (RFU). The assay was performed according to the manufacturer instructions (Quant-iT PicoGreen® dsDNA Assay Kit, Invitrogen). To improve sensitivity of the lecture, black solid 96-well microplates (Corning Life Sciences) and fluorescence was determined by the BioTek Synergy HT at 485 nm (20 nm bandwidth excitation filter) and a 528 nm (20 nm bandwidth emission filter). Raw Fluoresence Units (RFU) values were used to calculate the relative amplification levels and the Poisson equation for frequency of lesions.
2.6 Detection of 8-oxodeoxyguanosine (8-OHdG) levels
We measured levels of 8-oxodeoxyguanosine to quantify DNA oxidation due to oxidative stress damage using DNA extracted from liver and brain tissues. The extracted DNA was digested into ssDNA by incubating the samples at 95°C for 5 mins followed by incubation on ice. The DNA denaturalization was performed with 5–20 units of nuclease P1 (Sigma Aldrich) for 2 hrs at 37°C in 20mM Sodium Acetate solution (pH 5.2). In addition, a total of 5–10 units of alkaline phosphatase (Cell Biolab) were added for 1 hr at 37°C at 100mM (pH 7.5). Final reaction mixture was centrifuged for 5 mins at 6,000 × g and supernatant was used for the Oxiselect ™ Oxidative DNA damage ELISA (Cell Biolab) to determine the extent of the oxidized DNA nucleoside, an ubiquitous oxidative stress marker. All experimental samples were read at 450 nm using a microplate reader (Life technologies). The standards and experimental samples were performed in duplicate and the final results were expressed as 8-OH-dG levels (μg/mL).
3. Results
Typically, low molecular weight molecules with high antioxidant capacity are transported in the serum to counteract oxidative stress. Such molecules are glutathione, ascorbic acid, uric acid, tocopherols, carotenoids, bilirubin, and amino acids such as methionine and cysteine. The total effect exerted by these molecules serves as a common indicator of the progression of oxidative stress. Under our experimental conditions, we aimed to determine the effects of NAC against PQ-induced oxidative stress in mice serum. Mice serum collected from our experimental groups were submitted to the quantification of the antioxidant capacity levels in order to screen if PQ decrease the antioxidant capacity levels and what effect NAC supplementation would have. As depicted in Fig. 1, One-way ANOVA showed no significant differences in the distribution of the antioxidant capacity levels among the experimental groups (p= 0.20). Our results suggested that neither PQ nor NAC modulate the antioxidant capacity levels in mice serum.
Figure 1.
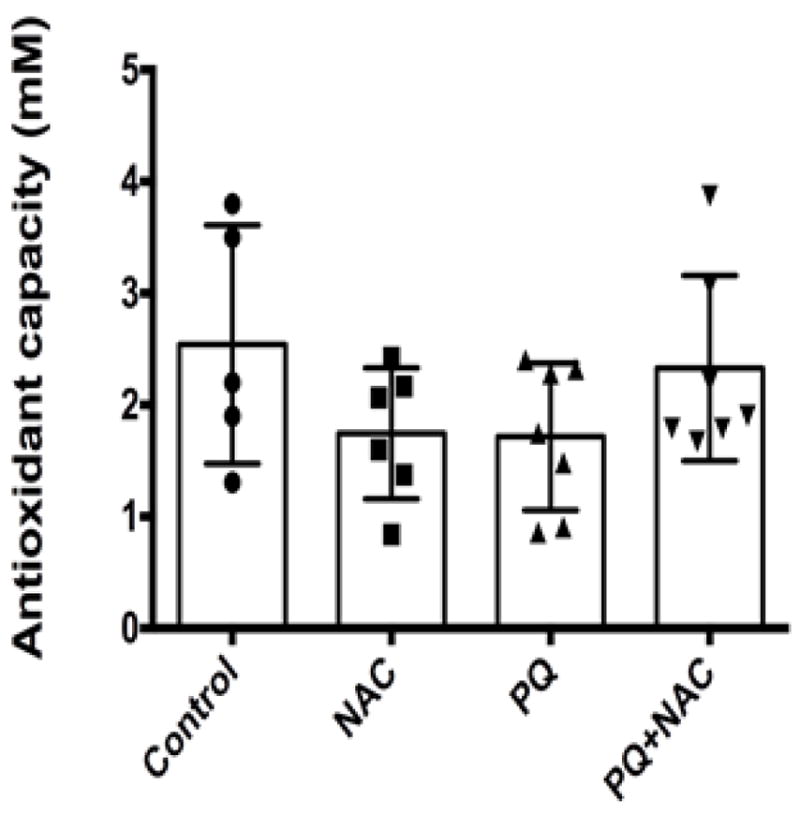
Antioxidant capacity in term of Trolox (mM) equivalent units in mice serum. Bars represent mean ±1.96 (SD) with distribution of samples per group in scatter blot column. Interval of confidence at 95% were as follow: Control: 2.54+/−1.96 (0.40), NAC: 1.74 +/− 1.96 (0.31), PQ: 1.71 +/− 1.96 (0.30), and PQ+NAC: 2.32 +/− 1.96 (0.34). The sample size of the experimental groups was as follow: Control (N=5), NAC (N=6), PQ (N=7), and PQ+NAC (N=7).
NAC is a precursor of one of the most versatile antioxidant, glutathione. GSH levels were analyzed to determine to what extent ROS due to oxidative stress induced by PQ interferes with GSH levels and the effect of NAC on GSH levels. Results are depicted in Fig. 2(A, B, C). As shown in Fig. 2A, One-way ANOVA analysis was performed showing strongly significant differences among the experimental groups (**p=0.001). Multiple comparison analysis by Tukey test (post-hoc) showed statistical significance among the experimental groups (Control vs. PQ, NAC vs. PQ). As expected in liver tissue at Fig. 2A, PQ induced oxidative stress reduced the amount of GSH in comparison with the control group. NAC appears to provide a partial protective effect when administered simultaneously to PQ. The effect of NAC administered to PQ and PQ+NAC mice, did not reach statistical significance, but nonetheless showed an expected tendency. GSH levels in brain tissue of treated mice are depicted in Fig. 2B. One-way ANOVA analysis was performed showing significant differences among the experimental groups (**p=0.04). Multiple comparison analysis by Tukey test (post-hoc) showed statistical significance among the experimental groups (NAC vs. PQ). PQ treatment reduced total GSH levels while NAC supplementation increased total GSH levels in comparison with the Control group. When both drugs were together (PQ+NAC) they showed an intermediate profile indicating that NAC was capable of offsetting the oxidative stress damage induced by PQ. Likewise, the GSH levels in lung tissue are depicted in Fig. 2C. Given the variable nature and composition of lung tissue our data showed a non significant trend. One-way ANOVA analysis showed no significant differences (p=0.14) among the experimental groups. Each tissue displayed a unique profile of GSH levels, but the three tissues confirmed that NAC alone and even in presence of PQ partially restored the GSH synthesis. However, these observations were more prominent in liver and lung tissues due to the fact that both tissues were involved in the main principal site of action of NAC and PQ.
Figure 2.

Glutathione (GSH) levels (μM) of (A) liver, (B) brain and (C) lung tissue of mice. Bars represent mean ±1.96 (SD) with distribution of samples per group in scatter blot column. (A) Interval of confidence at 95% were as follow: Control: 5.40+/−1.96 (0.42), NAC: 4.66 +/− 1.96 (0.43), PQ: 1.82 +/− 1.96 (0.40), and PQ+NAC: 3.34 +/− 1.96 (0.55). The sample size of the experimental groups was as follow: Control (N=6), NAC (N=7), PQ (N=5), and PQ+NAC (N=6). (B) Interval of confidence at 95% were as follow: Control: 22.50 +/−1.96 (0.80), NAC: 35.03 +/− 1.96 (1.35), PQ: 10.86 +/− 1.96 (1.24), and PQ+NAC: 23.01 +/− 1.96 (1.52). The sample size of the experimental groups was as follow: Control (N=4), NAC (N=5), PQ (N=6), and PQ+NAC (N=5). (C) Interval of confidence at 95% were as follow: Control: 0.09 +/−1.96 (0.19), NAC: 1.60 +/− 1.96 (0.30), PQ: 0 +/− 1.96 (0) and PQ+NAC: 2.92 +/− 1.96 (0.56). The sample size of the experimental groups was as follow: Control (N=6), NAC (N=7), PQ (N=7), and PQ+NAC (N=7).
Lipid peroxidation is a well-established oxidative stress biomarker. We determined if PQ-induced oxidative stress increases the MDA levels and how the antioxidant effects of NAC would affect lipid peroxidation. The MDA levels are depicted in Fig. 3 (A, B, C). One-way ANOVA analysis showed significant differences among the experimental groups (*p<0.02). Multiple comparison analysis by Tukey test (post-hoc) showed statistical significance among the experimental groups (Control vs. PQ, PQ vs. PQ+NAC). As depicted in Fig. 3A, PQ-induced oxidative stress by a significant increase in the MDA levels compared to the control group. One-way ANOVA analysis showed strongly significant differences among the experimental groups (**p=0.001). Multiple comparison analysis by Tukey test (post-hoc) showed statistical significance among the experimental groups (Control vs. NAC, Control vs. PQ, and Control vs. PQ+NAC). In Fig. 3B, a significant increase in MDA levels were observed in comparison with control group. We observed a nominal increase in MDA levels in NAC group. The MDA levels in lung tissue as depicted in Fig. 3C, shown no significant differences among the experimental groups (p=0.90) by One-way ANOVA analysis.
Figure 3.

Lipid peroxidation (MDA) levels of (A) liver, (B) brain and (C) lung tissue of mice. Bars represent mean ±1.96 (SD) with distribution of samples per group in scatter blot column. (A) Interval of confidence at 95% were as follow: Control: 11.02+/−1.96 (0.47), NAC: 12.92 +/− 1.96 (0.66), PQ: 16.77 +/− 1.96 (0.64), and PQ+NAC: 14.69 +/− 1.96 (0.78). The sample size of the experimental groups was as follow: Control (N=6), NAC (N=7), PQ (N=7), and PQ+NAC (N=7). (B) Interval of confidence at 95% were as follow: Control: 6.33+/−1.96 (0.33), NAC: 13.28 +/− 1.96 (0.71), PQ: 11.58 +/− 1.96 (0.59), and PQ+NAC: 14.47 +/− 1.96 (0.84). The sample size of the experimental groups was as follow: Control (N=6), NAC (N=7), PQ (N=7), and PQ+NAC (N=6). (C) Interval of confidence at 95% were as follow: Control: 23.31+/−1.96 (1.18), NAC: 22.93 +/− 1.96 (1.18), PQ: 21.46 +/− 1.96 (1.01), and PQ+NAC: 22.42 +/− 1.96 (0.91). The sample size of the experimental groups was as follow: Control (N=6), NAC (N=7), PQ (N=7), and PQ+NAC (N=7).
One of the principal causes of mtDNA damage is the excess ROS generation in the inner membrane of the mitochondria due to the ETC. Given the proximity of the mtDNA to the ETC, it is more prone to DNA damage in comparison with the nDNA. We examined the mtDNA integrity to determine if NAC reduces the PQ-induced oxidative damage of mtDNA. ROS induce a number of different lesions in the DNA, but under our experimental conditions we aimed to amplify the overall damage contained in two specific mtDNA amplicons: small amplicon (117 bp) and long amplicon (10 Kb). The small amplicon served as an internal control to normalize the mtDNA copy number due to the low sensitivity to DNA damage, while the long amplicon has a higher probability of being attacked by ROS resulting in a higher sensitivity for DNA damage. This method involved the isolation of DNA from tissues treated under the identical conditions of drug treatments followed by the screening of the relative amplification levels and frequency of lesions in liver and brain tissue by PCR. We measured the relative amplification of the mtDNA, which is inversely proportional to the DNA damage. Also, we analyzed the frequency of lesions with the Poisson equation analysis (Damage=-Ln Abs damage/Abs no damage).
As depicted in Fig. 4A, our data showed that mice with no treatment (control) showed the highest level of relative amplification (1.0), suggesting that mtDNA genome did not carried any lesion that delays the progression of the DNA polymerase activity. Also, One-way ANOVA analysis was performed showing strongly significant differences among the experimental groups (****p<0.0001) in liver tissue. Multiple comparison analysis by Tukey test (post-hoc) showed statistical significance among the experimental groups (Control vs. NAC, Control vs. PQ, PQ vs. PQ+NAC, and NAC vs. PQ). Our results showed that PQ induced oxidative stress decreases the relative amplification levels of mtDNA and NAC reestablished these levels even comparable to the basal system. Also, as depicted in Fig. 4A, One-way ANOVA analysis showed strongly significant differences among the experimental groups (****p<0.0001) in brain tissues. Multiple comparison analysis by Tukey test (post-hoc) showed statistical significance among the experimental groups (Control vs. PQ, NAC vs. PQ, NAC vs. PQ+NAC). Both relative amplification levels in liver and brain tissue showed statistically significant differences.
Figure 4.

Mitochondrial DNA damage (A) Relative amplification of mtDNA and (B) Poisson analysis for detection of lesions frequency in mtDNA both of liver and brain tissues of mice. Bars represent mean ±1.96 (SD) with distribution of samples per group in scatter blot column. (A) Relative amplification for liver tissue-Interval of confidence at 95% were as follow: Control: 1+/−1.96(0), NAC: 0.92 +/− 1.96 (0.10), PQ: 0.71 +/− 1.96 (0.07), and PQ+NAC: 0.90 +/− 1.96 (0.09). The sample size of the experimental groups was as follow: Control (N=5), NAC (N=5), PQ (N=4), and PQ+NAC (N=5). Relative amplification for brain tissue-Interval of confidence at 95% were as follow: Control: 1+/−1.96(0), NAC: 0.95 +/− 1.96 (0.18), PQ: 0.63 +/− 1.96 (0.16), and PQ+NAC: 0.69 +/− 1.96 (0.15). The sample size of the experimental groups was as follow: Control (N=5), NAC (N=4), PQ (N=5), and PQ+NAC (N=5). (B) Poisson analysis for liver tissue-Interval of confidence at 95% were as follow: NAC: 0.07 +/− 1.96 (0.10), PQ: 0.33 +/− 1.96 (0.08), and PQ+NAC: 0.07 +/− 1.96 (0.10). The sample size of the experimental groups was as follow: NAC (N=5), PQ (N=4), and PQ+NAC (N=5). Poisson analysis for brain tissue-Interval of confidence at 95% were as follow: NAC: 0.11 +/− 1.96 (0.14), PQ: 0.47 +/− 1.96 (0.20), and PQ+NAC: 0.30 +/− 1.96 (0.16). The sample size of the experimental groups was as follow: NAC (N=4), PQ (N=5), and PQ+NAC (N=4).
As shown in Fig. 4B, we confirmed that frequency of lesions was inversely proportional of the DNA damage in liver and brain tissue. Specifically, One-way ANOVA analysis showed strongly significant differences among the experimental groups (****p<0.001) in liver tissue. Multiple comparison analysis by Tukey test (post-hoc) showed statistical significance among the experimental groups (NAC vs. PQ and PQ vs. PQ+NAC). In brain tissue, One-way ANOVA showed strongly significant differences among the experimental groups (**p<0.02). Multiple comparison analysis by Tukey test showed statistical significance among the experimental groups (NAC vs. PQ). The results depicted in Fig. 4B were consistent and representative with the relative amplification levels obtained in Fig. 4A. Also, as depicted in Fig. 4A, an internal parameter (50%) was used to confirm that our results were in the exponential phase (40% – 60%). In both tissues this parameter was achieved, as expressed in liver tissue with 60% and in brain tissue with a 52%.
The current paradigm states that ROS and oxidative stress cause mutations in DNA. We wanted to test this paradigm by quantifying the levels of 8-OH-dG in DNA extracted from liver and brain tissue from our experimental groups. 8-OH-dG is the oxidized form of deoxyguanosine found in DNA and is a well-established indicator of DNA damage due to oxidative stress. Levels of 8-OH-dG are an important biomarker for the activity of ROS. As depicted in Fig. 5A, One-way ANOVA analysis showed strongly significant differences among the experimental groups (*p<0.003). Multiple comparison analysis by Tukey test (post-hoc) showed statistical significance among the experimental groups (PQ vs. PQ+NAC). Our results showed that PQ induced oxidative stress by increasing the 8-OH-dG levels in liver tissue. NAC treatment might replenish GSH levels and maintains the background level of 8-OH-dG as depicted in the control group. Besides, the effect of NAC in presence of PQ reduces statistically significant the 8-OH-dG levels in liver tissue even to lower levels of the basal system. In addition, as depicted in Fig. 5B, One-way ANOVA analysis showed significant differences among the experimental groups (p= 0.20). In brain tissue, NAC did not decrease 8-OH-dG levels. For instance, a similar distribution of 8-OH-dG levels was observed among the four experimental groups.
Figure 5.

The levels of oxidized DNA base 8-oxodeoxyguanosine (8-OHdG) in (A) liver and (B) brain tissues of mice. Bars represent mean ±1.96 (SD) with distribution of samples per group in scatter blot column. (A) Interval of confidence at 95% was as follow: Control: 1.15+/−1.96 (0.30), NAC: 1.02 +/− 1.96 (0.19), PQ: 1.46 +/− 1.96 (0.19), and PQ+NAC: 0.85 +/− 1.96 (0.22). The sample size of the experimental groups was as follow: Control (N=5), NAC (N=5), PQ (N=5), and PQ+NAC (N=4). (B) Interval of confidence at 95% were as follow: Control: 1.50+/−1.96 (0.31), NAC: 1.33 +/− 1.96 (0.20), PQ: 1.82 +/− 1.96 (0.34), and PQ+NAC: 1.37 +/− 1.96 (0.25). The sample size of the experimental groups was as follow: Control (N=4), NAC (N=5), PQ (N=4), and PQ+NAC (N=4).
4. Conclusion
Present study investigated the PQ-induced oxidative stress in vital body tissues and the counteractive effect of NAC by evaluating the serum antioxidant capacity, lipid peroxidation, antioxidant GSH levels, and DNA integrity and damage in mice. The authors in [3, 36, 57] noticed that the homeostatic control of antioxidant capacity in blood and other tissues has been extensively studied including how this capacity is modified during the oxidative stress process. Our data suggest that under the experimental conditions neither PQ nor NAC modulated the mice serum antioxidant capacity. We speculated that both PQ and NAC displayed a limited potential in serum due to several reasons involving short half-lives, fast metabolic degradation, and the pharmacokinetics of the drugs. For example, NAC is metabolized very fast and has a low toxicity profile by oral administration with a half-life of 5.0 – 6.25 hrs [2]. As the results in [32, 68] indicated that NAC has <5% bioavailability after passing through N-deacelytation in the intestinal mucosa followed by the first pass metabolism in the liver tissue. Moreover NAC does not accumulate in blood or tissues and it is rapidly excreted by the renal system (30%) and by non-renal system (70%) [72]. On the other hand, PQ shows a high toxicity profile whether by oral ingestion or by intraperitoneal administration but it is not well absorbed from the gastrointestinal tract [30]. PQ is mainly distributed to lung, liver, kidneys and lastly plasma [11, 76]. Most importantly, PQ is poorly metabolized (<5%) and 64% is excreted in 1 hour as the parental compound in feces and urine.
Based on the pharmacokinetics, we speculated that short half lives of ROS induced by PQ were scavenged quickly in the serum exerted a limited oxidant effects, which in turn makes a weak signal to be target by antioxidant defense system. We also believe that the transitory effects exerted by PQ and NAC during the 3-weeks treatment needs to be optimized so as to determine the window for sample collection [39, 78]. Another possible reason is that our rodent model (Balb/C mice) has neither genetic modifications nor oxidative stress enzymes deficiencies. Our data suggest that present rodent model has a robust REDOX response and was able to overcome oxidative stress response induced by PQ without supplementation of the exogenous antioxidant NAC. This is in agreement with the data reported in rodent models by authors in [8, 45, 81] which evidenced that the baseline levels for antioxidant capacity fluctuate from 0.7 – 2.50 mM equivalents of Trolox. Our data are in agreement with these finding because our control mice showed an antioxidant capacity of 2.54 mM units equivalents of Trolox. With these findings, we predict that mice with lower antioxidant capacity levels would be unlikely to overcome the oxidative stress response and mice with a higher antioxidant capacity would be likely to overcome these responses [42].
In line with these data, we might correlate the antioxidant capacity with the response of the GSH metabolism in mice exposed to PQ and NAC. The authors in [49, 50] showed that the distribution of GSH levels might vary depending on the specific requirements and physiological functions in the specific organs or tissues of mice. Our data showed significant differences of GSH levels in liver and brain tissues, but not in lung tissue. For example, in liver tissue we observed as expected the GSH depletion induced by PQ and the boost of GSH induced by NAC. GSH enriched levels in liver tissue explained the expected result of total GSH levels, specifically in NAC group. This is because GSH concentration in cytosol ranges from 1–10 mM, but in hepatocytes the concentration is high as 10 mM due to hepatocytes active transport of GSH to the rest of the blood and tissues [29, 53]. In concordance with our data, author in [14] showed that both PQ and Diquat subcutaneous dose at 130 mg/kg between 0.5 hrs and 4 hrs interferes with GSH recycling mechanisms inducing subsequently GSH depletion in liver tissue and biliary duct of male CD-1 mice. Indeed, we did not measure the oxidized glutathione (GSSG)/reduced glutathione (GSH) ratio, we predict that this balance will provide more insight of the potential indicators of hepatic oxidative stress caused by PQ.
Our data further showed that mice exposed to only NAC presented the highest GSH levels in comparison with the other groups, suggesting that NAC reached the brain tissue. This is in agreement with previous studies conducted by authors in [4, 25] in the central nervous system. As the result in [70], NAC impact the modulation of the antioxidant defenses against oxidative damage as a result of ROS scavenge in the dopaminergic system of neuronal cells. As expected, GSH depletion by PQ exposure is a direct consequence of repeated PQ doses administrations that get accumulate into the brain tissue eliciting neurotoxicity as reported by authors in [30, 52]. PQ modulates REDOX regimen showing a reduction of GSH levels and increase in the oxidized glutathione GSSG [39]. There were no significant differences of GSH levels among the experimental groups in lung tissue, which is the main site action by PQ. However a trend in the effects induced by either NAC or PQ was observed in the lung tissue of mice. The authors in [1, 64] have demonstrated that GSH content in lung decreases in the presence of thiol depleter as PQ due to the destruction of alveolar type II cells. Also, supplementation of antioxidants such as NAC over prolonged period resulted in an elevation of intracellular GSH pool and decreased the susceptibility of tissue to oxidant-induced tissue injury and delayed the lung tissue inflammation [20]. Depletion of GSH levels prone the membrane lipids to severe oxidative damage. The correlation of the former conditions with lipid peroxidation (MDA levels) was an indicator of the ongoing oxidative stress process in the tissues that have been under studied. Present data showed significant differences in the MDA levels among the experimental groups in liver and brain tissues, but not in lung tissue.
Furthermore liver tissue was more likely to undergo oxidative lipid peroxidation than the other tissues due to the high amount of lipid composition and rates of detoxification [60]. In agreement with study reported by author in [59], it was showed that mice hepatocytes that were treated with PQ showed higher cytotoxicity and MDA levels. Hepatocytes are sensitive to quinones as PQ treatment due to their low glutathione content, which in turn makes them more prone to lipid peroxidation. Hepatocytes are the major inducer of GSH synthesis as observed in mice undergoing NAC supplementation in comparison with the rest of the experimental groups. Data further showed significant differences of MDA levels among the experimental groups in brain tissue. However, our results showed a tendency of NAC as a pro-oxidant agent not prominently noted in the literature. There are few studies that reported the exerting action of NAC as a pro-oxidant. Our findings are in agreement with the proposed mechanism of thyl radicals as a consequence of the interaction of PQ with steady state levels of superoxide. The basic mechanism of action of thiols is to scavenge the hydroxyl and superoxide radicals. When thiol groups interact with the former radicals they form a potent thyl radical [13]. Thyl radical reacts with free fatty acids, as those found in brain tissues, and forms potent peroxyl radicals [69]. Therefore, it is suggested that thyl radicals act as a strong oxidant that interferes with GHS metabolism and at the same time increasing the peroxidation of neuronal biological membranes [77]. We observed that PQ induced less lipid peroxidation than NAC, yet PQ in the presence of NAC showed similar rates of lipid peroxidation as NAC group, confirming the effect of NAC as a pro-oxidant. It is supported by our data on GSH levels that NAC group showed the highest concentration of GSH levels compared to other experimental groups. On the other hand, we expected to detect higher MDA levels in mice lung tissue after PQ exposure since its toxicity have been often used by authors in [38, 80] to create models for acute lung injury and pulmonary fibrosis. However, our data showed no significant differences in the MDA levels among the experimental and control group. The possible reason is that lung tissue contained high levels of oxidative stress background due to the high composition of lipids that surround the alveolar cells accustomed to an oxidant rich environment. Earlier study has also reported that prolonged PQ treatment resulted in a continual slow rate of lipid peroxidation and a slow rate of antioxidant defenses replenishment in lungs [24]. Based on the fact that even though lipid peroxidation predicts the levels of oxidative membrane damage, MDA levels are not an absolute indicator of lipid peroxidation because of its rapid degradation in vivo models [7].
Our study further investigated the extent of oxidative genomic damage in mice exposed to PQ and amelioration by NAC measured by PCR and ELISA assays which are highly sensitive and specific for detecting genomic damage in mtDNA and nuclear DNA (nDNA, 8-OH-dG). The authors in [22, 37, 48, 74, 82] have reported that mtDNA is a sensitive biomarker for oxidative damage due to lacks of DNA protecting histones, has limited DNA repairing mechanisms, and its close proximity to the electron transport chain (ETC). Studies reported by authors in [16, 82] showed for the first time the incorporation of PCR with the purpose of screening the effects of a variety of genotoxicant agents in the relative amplification and frequency of lesions in the mtDNA of mice, rats, human, rhesus monkeys, and yeast models. However, we are the first one to report the effects of PQ and NAC in mtDNA amplification and frequency of lesions in a rodent model by this technique. Both liver and brain tissues showed strong significant differences (p <0.0001) in relation to amplification levels and frequency of lesions among the experimental groups due of the high susceptibility of mtDNA to oxidative damage. One of the earlier study conducted by author in [26] have shown that PQ exacerbates the generation of ROS in the mitochondria, subsequently inhibiting the normal function of complex I and IV, leading to the reduction of ATP synthesis, genome instability and ultimately cell death. Most reports showed a decrease in the complex 1 and to a lesser extent in complex IV, while complex II and III are mostly unaffected [21]. Due to the fact that complex I and IV are encoded by mtDNA whereas complex II and III are encoded by nDNA, it was expected to detect a greater presence of mtDNA levels in liver and brain tissues [66]. Subsequently, the amount of mtDNA damage represents an early indicator of liver and brain mitochondrial dysfunction which was ameliorated by thiols that increased the flow of the ETC by substituting reducing equivalents whom may be absent in the ETC [16, 44, 47, 48, 55]. Therefore, we speculate the effects of NAC in restoring the amplification levels of mtDNA as reducing equivalents improved the energy status, restored the genome stability because it acts as a scavenger of ROS over the rate of ROS generation induce by PQ exposure.
As the results indicated in [41, 46], guanine is the main target of ROS and its oxidized form, 8-OH-dG levels, is the most frequent base lesion in nDNA which make it an ubiquitous biomarker to provide information of ongoing oxidative stress in the system. As expected, our data showed significant differences among the experimental groups in liver tissue. PQ significantly induced oxidative damage to nDNA, while according to previous studies NAC exerted its effects through a non-enzymatic mechanism in the regulation of cellular redox homeostasis, enhancing intracellular biosynthesis of GSH, and ROS scavenging activity [56, 83]. These findings were confirmed by an earlier study conducted by author in [65], which showed how NAC decreased 8-OH-dG levels as close as in the wild type in ATM deficient rodent model (C57bl6J/p). The modulating effects of NAC can be seen in a rodent model lacking genes to cope with oxidative stress and in our Balb/C rodent model that also have no genetic modification. The levels of 8-OH-dG in brain tissue did not show significant differences among the experimental groups. Our data showed a similar distribution of 8-OH-dG levels suggesting that nDNA has a greater capacity of DNA repair mechanism to detect 8-OH-dG lesions from the system more efficiently. Our results are in agreement with a previous report [40]. that brain tissue has intracellular antioxidant defenses, which are highly competent. Usually, the terminals of the neuronal cells need high levels of ascorbic acid, in which GSH is required for their modulation and for maintaining ascorbic acid in it reduced form. We speculated that mice undergoing NAC treatment were more likely to modulate the levels of GSH, which subsequently modulates the ascorbic acid for a better antioxidant response. Therefore, our results suggested that PQ effect was more evident in liver tissue by inducing 8-oxo lesions showing more susceptibility to nuclear DNA damage rather than in brain tissue, which showed less susceptibility to nDNA damage. In conclusion, taking together these results demonstrated that Paraquat induced oxidative stress response by inducing hepatic membrane lipid peroxidation, hepatic oxidative nDNA damage, liver and brain mtDNA damage as well as by depleting liver, brain and lung GSH levels in mice. Treatment with antioxidant (NAC) ameliorated oxidative stress response in the liver of mice. These results have important implications in the future as in the use of a non-invasive and cost effective therapy as a reliable reducing agent of oxidative stress effects mediated by PQ.
We considered the following future steps in order to extend this project into a new research phase in order to overcome few limitation of the study. In the antioxidant capacity test, due to the multivariable nature of redox status, we are considering to conduct a battery of tests in mice serum that include dose-dependent studies in order to determine the pharmacological trend and the peak value of PQ and NAC. Also, the total GSH levels that were measured in our study included the amount of reduced GSH and oxidized GSSG levels per tissue. We could not explore the individual levels of GSSG and establish a ratio in respect of GSH levels. Therefore, the further steps would be to proceed with the GSSG derivatization. In regards to the lipid peroxidation analysis, we would complement this analysis with the glutathione peroxidase (GP) studies in order to compare the reduced hydroperoxides to the corresponding alcohols and to reduce free hydrogen peroxide to water with MDA levels. DNA integrity analysis conducted by PCR was one of the most sensitive analyses that we perform in this research study. Even though the results obtained were the expected, it will be interestingly to extend this assay by exploring the possible effect of the genotoxicity of PQ and antioxidant activity of NAC but including the nDNA. The selection of specific primers in nDNA of the analogous segments that were used in the mtDNA can be used to compare the degree of damage or repair in nDNA vs. mtDNA in the same biological sample. In this way, we would be able to compare the rates of damage in mtDNA and nDNA under the same experimental conditions. Lastly, we would suggest to include in a future experimental design another tissue or biological source to screen for 8-oxo levels, as for example mice urine. The major route of excretion of ubiquitous oxidative stress lesions, such as 8-oxo, is by body fluid excretion.
Acknowledgments
This project was granted by the Research Initiative for Scientific Enhancement (RISE) Program of the University of Puerto Rico in Ponce, Campus by the National Institute of General Medical Sciences/National Institute of Health (R25-GM096955), the Ponce Health Sciences Education Foundation (Endowment Fund) of the Ponce Health Sciences University Institutional Funds, National Institute of Health of the Minority Biomedical Research Support (GM088087), and the Molecular and Genomic Core (NIMHD/MD007579). In addition, authors wanted to thanks to the National Institute on Minority Health and Health Disparities (G12MD007579) for the financial assistance to defray publication fee of this research article. The authors wanted to express their genuine gratitude to undergraduate students from the UPR-Ponce: Michael Manoharan, Rafael Rivera, Jamie Rosado, Melanie Ortiz, and Ralphdy Vergne for their excellent technical support. Thanks to Mrs. Lissette Laboy (supervisor of animal house) for her valuable assistance regarding the mice handling and drug administration training at the Animal House of Ponce Health Sciences University (PHSU). In addition, the authors wanted to provide a special acknowledgement to Ms. Maria Acosta and Mr. Eliezer Cruz in terms of technical assistance, mice handling, mice surgery and the blood and tissue collection for the oxidative stress analysis. Ms. Nelly and Dr. Jonathan Flores for sharing their expertise and knowledge in the PCR and molecular analysis. Mrs. Myrella Cruz for helping in the experiments regarding antioxidant capacity and 8-OHdG tests. Dr. Diego Zavala and Dr. Luisa Morales, faculty members, for reviewing and statistical analysis.
Contributor Information
Maricelly Santiago Ortiz, Email: maricelly.santiago@gmail.com.
Kevin Muñoz Forti, Email: kevinmunoz2@upr.edu.
Edu B. Suárez Martinez, Email: edu.suarez@upr.edu.
Lenin Godoy Muñoz, Email: lgodoy@psm.edu.
Kazim Husain, Email: kazimhusain@hotmail.com.
Wilfredo Hernández Muñiz, Email: whernandez@psm.edu.
References
- 1.Anderson ME, Luo JL. Glutathione therapy: from prodrugs to genes. Semin Liver Dis. 1998;18:415–424. doi: 10.1055/s-2007-1007174. [DOI] [PubMed] [Google Scholar]
- 2.Atkuri KR, Mantovani JJ, Herzenberg LA, Herzenberg LA. N-Acetylcysteine a safe antidote for cysteine/glutathione deficiency. Curr Opin Pharmacol. 2007;7:355–359. doi: 10.1016/j.coph.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balcerczyk A, Bartosz G. Thiols are main determinants of total antioxidant capacity of cellular homogenates. Free Radical Research. 2003;37:537–541. doi: 10.1080/1071576031000083189. [DOI] [PubMed] [Google Scholar]
- 4.Ballatori N, Krance SM, Marchan R, Hammond CL. Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology. Mol Aspects Med. 2009;1:13–28. doi: 10.1016/j.mam.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banaclocha MM, Martinez N. N-acetylcysteine elicited increase in cytochrome c oxidase activity in mice synaptic mitochondria. Brain Res. 1999;842:249–251. doi: 10.1016/s0006-8993(99)01819-3. [DOI] [PubMed] [Google Scholar]
- 6.Banaclocha MM. N-acetylcysteine elicited increase in complex 1 activity in synaptic mitochondria from aged mice: implications for treatment of Parkinson’s disease. Brain Res. 2000;859:173–175. doi: 10.1016/s0006-8993(00)02005-9. [DOI] [PubMed] [Google Scholar]
- 7.Barber A, Bernheim F. Lipid peroxidation: Its measurement, occurrence, and significance in animal tissues. Advance in Geronotological research. 1967;2:355–356. [PubMed] [Google Scholar]
- 8.Bauer A, Banek C, Needham K, Gillham H, Capoccia S, Regal J, et al. Pravastatin attenuates hypertension, oxidative stress, and angiogenic imbalance in rat model of placental ischemia-induced hypertension. Hypertension. 2013;61:1103–1110. doi: 10.1161/HYPERTENSIONAHA.111.00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berck M, Malhi GS, Gray LJ, Dean O. The promise of N-acetylcysteine in neuropsychiatry. Trends Pharmacol Science. 2013;34:167–177. doi: 10.1016/j.tips.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Bindu M, Guha I, Mallika T, Behera P, Chakrabarti S. Age-related oxidative decline of mitochondrial functions in rat brain is prevented by long term oral antioxidant supplementation. Biogerontology. 2011;12:119–131. doi: 10.1007/s10522-010-9301-8. [DOI] [PubMed] [Google Scholar]
- 11.Blanco T, Anderica C, Pedraza J. New insights into antioxidant strategy against Paraquat toxicity. Free Radical Research. 2015;48:623–639. doi: 10.3109/10715762.2014.899694. [DOI] [PubMed] [Google Scholar]
- 12.Bus JS, Gibson JE. Paraquat: model for oxidant-initiated toxicity. Environ Health Perspect. 1984;55:37–46. doi: 10.1289/ehp.845537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calabrese V, Ragusa N, Antico A, Mangiameli S, Rizza V. Cysteine induced enhancement of lipid peroxidation in susbtantia nigra: Comparative effect with exogenous administration of reduced glutathione. Drugs under Exp Clin Res. 1997;23:25–31. [PubMed] [Google Scholar]
- 14.Cao L, Waldon D. Ratios of biliary glutathione disulfide (GSSG) to glutathione (GSH): a potential index to screen drug-induced hepatic oxidative stress in rat and mice. Anal Bioanalytical Chem. 2013;405:2635–2642. doi: 10.1007/s00216-012-6661-8. [DOI] [PubMed] [Google Scholar]
- 15.Castello P, Drechsel D, Patel M. Mitochondria are a major source of Paraquat-induced reactive oxygen species in the brain. The journal of biological chemistry. 2007;282:14186–14193. doi: 10.1074/jbc.M700827200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Castro M, Suarez E, Kraiselburd E, Isidro A, Paz J, Ferder L, et al. Aging increase mitochondrial DNA damage and oxidative stress in liver of rhesus monkeys. Exp Gerontology. 2012;47:29–37. doi: 10.1016/j.exger.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chanyachukul T, Yoovathaworn K, Thongsaard W, Chongthammakun S, Navasumrit P, Satayavivad J. Attenuation of paraquat-induced motor behavior and neurochemical disturbances by L-valine in vivo. Toxicology. 2004;150:259–69. doi: 10.1016/j.toxlet.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 18.Chen Q, Niu Y, Zhang R, Guo H, Gao Y, Li Y, et al. The toxic influence of paraquat on hippocampus of mice: involvement of oxidative stress. Neurotoxicology. 2010;31:310–316. doi: 10.1016/j.neuro.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 19.Coco T, Sgobbo P, Clemente M, Lopriore B, Grattagliano I, Paola M, et al. Tissue specific changes of mitochondria function in aged rats: effect of a long term dietary treatment with N-acetylcysteine. Free Radic Biol Med. 2005;38:796–805. doi: 10.1016/j.freeradbiomed.2004.11.034. [DOI] [PubMed] [Google Scholar]
- 20.Deneke SM. Thiol-based antioxidants. Curr Top Cell Regulatory. 2000;36:151–180. doi: 10.1016/s0070-2137(01)80007-8. [DOI] [PubMed] [Google Scholar]
- 21.Desai VG, Feurers RJ, Hart RW, Ali SF. MPP+ induced neurotoxicity in mouse is age dependant. Evidence by selective inhibition of complexes of electron transporter. Brain Res. 1996;715:1–8. doi: 10.1016/0006-8993(95)01255-9. [DOI] [PubMed] [Google Scholar]
- 22.Desler C, Marcker ML, Singh KK, Rasmussen LJ. The importance of mitochondrial DNA in aging and cancer. J Aging Res. 2011;11:407536. doi: 10.4061/2011/407536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Devasagayam T, Tilak JC, Boloor KK, Sane Ketaki S, Ghaskadbi Saroj S, Lele RD. Free Radicals and Antioxidants in Human Health: Current Status and Future Prospects. Journal of Association of Physicians of India (JAPI) 2004;52:796–797. [PubMed] [Google Scholar]
- 24.Di Luzio NR. Antioxidants, lipid peroxidation, and chemical induced liver injury. Fed Proc. 1975;32:1875–1881. [PubMed] [Google Scholar]
- 25.Dringen R. Glutathione metabolism and oxidative stress in neurodegeneration. Eur J Biochem. 2000;16:4903–4904. doi: 10.1046/j.1432-1327.2000.01651.x. [DOI] [PubMed] [Google Scholar]
- 26.Dunette SB, Bjorklund A. Prospects for new restorative and neuro-protective treatments in Parkinson disease. Nature. 1999;99:A32–A39. doi: 10.1038/399a032. [DOI] [PubMed] [Google Scholar]
- 27.Eakin K, Baratz-Goldstein R, Pick C, Zindel O, Balaban C, Hoffer M, et al. Efficacy of N-acetylcysteine in traumatic brain injury. Plos ONE. 2014;9:1–7. doi: 10.1371/journal.pone.0090617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farr S, Poon H, Dogrukol D, Drake J, Banks W, Eyerman E, et al. The antioxidants α-lipoic acid and NAC reverse memory impairment and brain oxidative stress in aged SAMP8 mice. Journal of Neurochemistry. 2003;84:1173–1183. doi: 10.1046/j.1471-4159.2003.01580.x. [DOI] [PubMed] [Google Scholar]
- 29.Forman H, Zhang Hongqiao, Rinna Alessandra. Glutathione: overview of its protective roles, measurement, and biosynthesis. Molecular Aspects of Medicine. 2009;30:1–12. doi: 10.1016/j.mam.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Franco R, Li S, Rodriguez-Rocha H, Burns M, Panayiotidis MI. Molecular mechanisms of pesticide-induced neurotoxicity: Relevance to Parkinson’s disease. Chem Biol Interact. 2010;188:289–300. doi: 10.1016/j.cbi.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gillissen A, Nowak D. Characterization of N-acetylcysteine and ambroxol in anti-oxidant therapy. Respir Med. 1998;92:609–23. doi: 10.1016/s0954-6111(98)90506-6. [DOI] [PubMed] [Google Scholar]
- 32.Giustarini D, Milzani A, Dalle-Donne I, Tsikas D, Rossi R. N-Acetylcysteine ethyl ester (NACET): a novel lipophilic cell-permeable cysteine derivative with an unusual pharmacokinetic feature and remarkable antioxidant potential. Biochem Pharmacol. 2012;84:1522–1533. doi: 10.1016/j.bcp.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 33.Gonzalez R, Ferrin G, Hidalgo AB, Ranchal I, Lopez-Cirello, Santos M, et al. N-acetylcysteine, coenzyme Q (10) and superoxide dismutase mimetic prevent mitochondrial cell dysfunction and cell death induced by D-galactosamine in primary culture of human hepatocytes. Chem Biol Interact. 2009;181:95–106. doi: 10.1016/j.cbi.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 34.Harman D. Aging: a theory based on free radical and radiation chemistry. Journal of Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 35.Harman D. The biologic clock: the mitochondria? J Am Geriatr. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 36.Sae-Young Hong, Jong-Oh Yang, Eun-Young Lee, See-Won Lee. Effects of NAC and glutathione on antioxidant status of human serum and 3T3 fibroblasts. J Korean Med Science. 2003;18:649–654. doi: 10.3346/jkms.2003.18.5.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hunter S, Jung D, Giulio R, Meyer J. The QPCR assay for analysis of mitochondrial DNA damage repair, and relative copy number. Methods. 2010;51:444–451. doi: 10.1016/j.ymeth.2010.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jenkinson SG. Free radical effects on lung metabolism. Clin Chest Med. 1989;10:37–47. [PubMed] [Google Scholar]
- 39.Kang MJ, Gil SJ, Koh HC. Paraquat induces alternation of the dopamine catabolic pathways and glutathione levels in the substantia nigra of mice. Toxicol Lett. 2009;188:148–52. doi: 10.1016/j.toxlet.2009.03.026. [DOI] [PubMed] [Google Scholar]
- 40.Kang MJ, Gil SJ, Lee JE, Koh HC. Selective vulnerability of the striatal subregions of C57BL/6 mice to paraquat. Toxicol Lett. 2010;195:127–34. doi: 10.1016/j.toxlet.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 41.Kino K, Sugiyama H. GC>CH transversion mutation might be caused by 8 oxoguanine oxidation product. Nucleic Acid Symp. 2000;44:139–140. doi: 10.1093/nass/44.1.139. [DOI] [PubMed] [Google Scholar]
- 42.Koracevic D, Koracevic G, Djordjevic V, Andrejevic S, Cosic V. Method for the measurement of antioxidant activity in human fluids. Journal of Clinical Pathology. 2001;54:356–361. doi: 10.1136/jcp.54.5.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kowaltoski AJ, Castilho RF, Vercesi AE. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 2001;495:12–15. doi: 10.1016/s0014-5793(01)02316-x. [DOI] [PubMed] [Google Scholar]
- 44.Kumaran S, Subathra M, Bahu M, Panneerselvan C. Supplementacion of L-carnitine improves mitochondria enzymes in heart and skeletal muscle of aged rats. Exp Aging Res. 2005;31:55–67. doi: 10.1080/03610730590882846. [DOI] [PubMed] [Google Scholar]
- 45.Shaoqing Lei, Lei S, Liu Y, Liu H, Yu H, Wang H, Xia Z. Effects of N-acetylcysteine on nicotinamide dinucleotide phosphate oxidase activation and antioxidant status in heart, lung, liver and kidney in streptozotocin-induced diabetic rats. Yonsei Med J. 2012;53:294–303. doi: 10.3349/ymj.2012.53.2.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lindahl T, Barnes DE. Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol. 2000;65:127–133. doi: 10.1101/sqb.2000.65.127. [DOI] [PubMed] [Google Scholar]
- 47.Mandavilli BS, Ali SF, Van Houten B. DNA damage in brain mitochondria caused by aging and MPTP treatment. Brain Res. 2000;885:45–52. doi: 10.1016/s0006-8993(00)02926-7. [DOI] [PubMed] [Google Scholar]
- 48.Mandavilli BS, Santos JH, Van Houten B. Mitochondrial DNA repair and aging. Mutat Res. 2002;509:127–51. doi: 10.1016/s0027-5107(02)00220-8. [DOI] [PubMed] [Google Scholar]
- 49.Mari M, Morales A, Colell A, Garcia C, Fernandez JC. Mitochondrial glutathione, a key survival antioxidant. Antioxidant Redox Signal. 2009;11:2685–2700. doi: 10.1089/ars.2009.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mari M, Morales A, Colell A, Garcia C, Kaplowitz N, Fernandez J. Mitochondrial glutathione: features, regulation and role in disease. Biochimica et Biophysica Act. 2013;1830:3317–3328. doi: 10.1016/j.bbagen.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martin G, Loeb L. Mice and mitochondria. Nature. 2004;429:357–359. doi: 10.1038/429357a. [DOI] [PubMed] [Google Scholar]
- 52.McCormack AL, Thiruchelvam M, Manning-Bog AB, Thiffault C, Langston JW, Cory-Slechta DA, et al. Environmental risk factors and Parkinson’s disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol Dis. 2002;2:119–27. doi: 10.1006/nbdi.2002.0507. [DOI] [PubMed] [Google Scholar]
- 53.Meister A. Glutathione metabolism and its selective modification. J Biol Chem. 1988;263:17205–17208. [PubMed] [Google Scholar]
- 54.Miquel J, Ferrandiz ML, Dejuan E, Sevila I, Martinez M. N-acetylcysteine protects against age-related decline of oxidative phosphorylation in liver mitochondria. Eur J Pharmacology. 1995;292:333–335. doi: 10.1016/0926-6917(95)90041-1. [DOI] [PubMed] [Google Scholar]
- 55.Navarro A, Boveris A. Rat brain and liver mitochondria develop oxidative stress and lose enzymatic activities on aging. Am J Physiol Regul Integr Comp Physiol. 2004;287:1244–1249. doi: 10.1152/ajpregu.00226.2004. [DOI] [PubMed] [Google Scholar]
- 56.Neal R, Matthews RH, Lutz P, Ercal N. Antioxidant role of N-acetylcyseine isomers following high dose of irradiation. Free Radic Biol Med. 2003;34:689–695. doi: 10.1016/s0891-5849(02)01372-2. [DOI] [PubMed] [Google Scholar]
- 57.Nikki E. Assessment of antioxidant capacity in vitro and in vivo. Free Radical Biology Med. 2010;49:503–515. doi: 10.1016/j.freeradbiomed.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 58.Noszal B, Visky D, Kranzni M. Population, acid-base, and redox properties of N-acetylcysteine conformers. J Med Chem. 2000;43:2176–2182. doi: 10.1021/jm9909600. [DOI] [PubMed] [Google Scholar]
- 59.Orrenius S, Bellomo G. Altered thiol and calcium homeostasis in oxidative hepatocellular injury. Hepatology. 1985;5:876–882. doi: 10.1002/hep.1840050529. [DOI] [PubMed] [Google Scholar]
- 60.Poli G, Albano E, Dianzani MU. The role of lipid peroxidation in liver damage. Chem Phys Lipids. 1987;45:117–142. doi: 10.1016/0009-3084(87)90063-6. [DOI] [PubMed] [Google Scholar]
- 61.Prasad K, Tarasewicz E, Mathew J, Strickland PA, Buckley B, Richardson JR, et al. Toxicokinetics and toxicodynamics of paraquat accumulation in mouse brain. Exp Neurol. 2009;215:358–367. doi: 10.1016/j.expneurol.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Prasad K, Winnik B, Thiruchelvam MJ, Buckley B, Mirochnitchenko O, Richfield EK. Prolonged toxicokinetics and toxicodynamics of paraquat in mouse brain. Environ Health Perspect. 2007;115:1448–1453. doi: 10.1289/ehp.9932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Radomska-Lesniewska DM, Skopinski P. N-acetylcysteine as an antioxidant and anti-inflamatory drug and its some clinical applications. Cent Eur J Immun. 2012;37:57–66. [Google Scholar]
- 64.Rahman Q, Abidi P, Afaq F, Schiffmann D, Mossman BT, Kamp DW, et al. Glutathione redox system in oxidative lung injury. Crit Rev Toxicol. 1999;6:543–68. doi: 10.1080/10408449991349276. [DOI] [PubMed] [Google Scholar]
- 65.Reliene R, Fischer E, Schiestl R. Effects of N-Acetylcysteine on oxidative DNA damage and the frequency of DNA deletions in Atm-deficient mice. Cancer research. 2012;64:5148–5153. doi: 10.1158/0008-5472.CAN-04-0442. [DOI] [PubMed] [Google Scholar]
- 66.Richter C, Park JW, Ames B. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Science. 1995;85:6465–6467. doi: 10.1073/pnas.85.17.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Samuni Y, Goldstein S, Dean O, Berck M. The chemistry and the biological activities of N-acetylcysteine. Biochimica et Biophysica Acta. 2013;1830:4117–4129. doi: 10.1016/j.bbagen.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 68.Santangelo F. Intracellular thiol concentration modulating inflammatory response: influence on the regulation of cell functions through cysteine prodrug approach. Curr Med Chem. 2003;23:2599–2610. doi: 10.2174/0929867033456567. [DOI] [PubMed] [Google Scholar]
- 69.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radical Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 70.Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur J Biochem. 2000;16:4904–11. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- 71.Mauro Serafani, Daniele Del Rio. Understanding the association between dietary antioxidants, redox status and disease: is the total antioxidant capacity is the right tool? Redox Report. 2004;9:145–152. doi: 10.1179/135100004225004814. [DOI] [PubMed] [Google Scholar]
- 72.Shahripour B, Harrigan MR, Alexandrov AV. N-acetylcysteine (NAC) in neurological disorders: mechanisms of action and therapeutic opportunities. Brain Behav. 2014;2:108–122. doi: 10.1002/brb3.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sheffner AL, Medler EM, Bailey KR, Gallo DG, Mueller AJ, Sarett HP. Metabolic studies with acetylcysteine. Biochem Pharmacol. 1966;15:1523–1535. doi: 10.1016/0006-2952(66)90197-3. [DOI] [PubMed] [Google Scholar]
- 74.Shinega M, Gagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sunitha K, Hemshekhar M, Thushara RM, Sebastin M, Yariswamy M, Kemparaju K, et al. N-acetylcysteine amide: a derivative to fulfill the promises of N-acetylcysteine. Free Radical Research. 2013;47:357–367. doi: 10.3109/10715762.2013.781595. [DOI] [PubMed] [Google Scholar]
- 76.Suntres Z. Role of antioxidant in Paraquat toxicity. Toxicology. 2002;180:65–77. doi: 10.1016/s0300-483x(02)00382-7. [DOI] [PubMed] [Google Scholar]
- 77.Tamba M, Torregiani A, Tubertini O. Thiyl and thyl-peroxyl radical produced from the irradiation of antioxidant thiol compounds. Rad Phys Chem. 1995;46:569–574. [Google Scholar]
- 78.Thiruchelvam M, McCormack A, Richfield EK, Baggs RB, Tank AW, Di Monte DA, et al. Age-related irreversible progressive nigrostriatal dopaminergic neurotoxicity in the paraquat and maneb model of the Parkinson’s disease phenotype. Eur J Neurosci. 2003;3:589–600. doi: 10.1046/j.1460-9568.2003.02781.x. [DOI] [PubMed] [Google Scholar]
- 79.Tudek B, Winczura A, Janik J, Siomek A, Foksinski M, Olinski R. Involvement of oxidatively damaged DNA and repair in cancer development and aging. Am J Transl Res. 2010;2:254–284. [PMC free article] [PubMed] [Google Scholar]
- 80.Weiss SJ. Oxygen, ischemia and inflammation. Acta Physiol Scand. 1986;548:9–37. [PubMed] [Google Scholar]
- 81.Wold L, Ying Z, Hutchinson KR, Velten M, Gorr MW, Velten C, et al. Cardiovascular remodeling in response to long-term exposure to fine particulate matter air pollution. Circ Heart Fail. 2012;4:452–61. doi: 10.1161/CIRCHEARTFAILURE.112.966580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci. 2003;60:6–20. doi: 10.1007/s000180300001. [DOI] [PMC free article] [PubMed] [Google Scholar]