

Abstract
We investigate the relationship between inherent secondary structure and aggregation propensity of peptides containing chameleon sequences (i.e., sequences that can adopt either α or β structure depending on context) using a combination of replica exchange molecular dynamics simulations, ion-mobility mass spectrometry, circular dichroism and transmission electron microscopy. We focus on an eight-residue long chameleon sequence that can adopt an α-helical structure in the context of the iron-binding protein from Bacillus anthracis (PDB id 1JIG) and a β-strand in the context of the baculovirus P35 protein (PDB id 1P35). We show that the isolated chameleon sequence is intrinsically disordered, interconverting between α-helical and β-rich conformations. The inherent conformational plasticity of the sequence can be constrained by addition of flanking residues with a given secondary structure propensity. Intriguingly, we show that the chameleon sequence with helical flanking residues aggregates rapidly into fibrils, whereas the chameleon sequence with flanking residues that favor β-conformations has weak aggregation propensity. This work sheds new insights into the possible role of α-helical intermediates in fibril formation.
Keywords: chameleon peptides, α-helix, β-hairpin, aggregation cascade, molecular dynamics, ion-mobility mass spectrometry, replica exchange molecular dynamics
INTRODUCTION
The secondary structure propensity of amino acids in a given sequence plays a central role in dictating the overall topology of the protein. Among the most important secondary structure elements are the α-helix and the β-sheet. These two types of structures are often mutually exclusive; a peptide sequence that favors an α-helix is unlikely to adopt β-sheet structure (either β-strand or β-hairpin) under the same experimental condition. However, there are some exceptions. The term “chameleon sequence” was first defined by Kabsch and Sander for sequences that can fold into either an α-helical structure or a β-sheet/strand structure.1 They identified an eleven-residue long sequence that adopts an α-helix when placed in one position of the IgG-binding domain of a protein and a β-sheet when it is in another position.2 Secondary structure conversion between an α-helix and a β-structure in chameleon sequences have been induced by sequence mutations,3–4 ligand binding5 and pH changes.6 Subsequent studies based on an analysis of structures deposited in the Protein Data Bank (http://www.rcsb.org/) uncovered additional six-residue, seven-residue and eight-residue long sequences.7–10 These can adopt an α-helix structure in one protein and β-structure (strand, sheet or hairpin) in the context of other proteins.
The chameleon sequences are valuable for investigating the relationship between inherent α-helix and β-strand propensity and protein self-assembly, as previous research has suggested that α-helix/β-strand “discordant” sequences are essential for amyloid fibril formation.11 The process of protein aggregation into amyloid fibrils is strongly correlated with diseases, including Alzheimer’s, Parkinson’s, Huntington’s, and type II diabetes.12–16 Early work established that sequences with high β-sheet propensity can nucleate amyloid fibrils,17–19 but that only specific types of β-sheets can grow into amyloid fibrils.20–24 For example, Hoyer et al. showed that constraining the monomeric Alzheimer’s amyloid β (Aβ) peptides into β-hairpin structure inhibited fibril formation.21 Larini et al.20 and Bleiholder et al.25 further emphasized the need for a dynamic β-hairpin structure in their studies and showed that the aggregation of toxic Aβ(25–35) fragments is initiated by a conformational transition from β-hairpin monomers to β-strand oligomers. In addition, recent studies point to a role of α-helix-rich intermediates in the early stages of the aggregation.26–39 Because many aggregating peptides are amphipathic and intrinsically disordered, it is very challenging to elucidate the role of specific secondary structure in promoting fibril assembly. Here, we use chameleon sequence containing peptides to study the fibril formation propensities of β-hairpin and α-helix structures.
The paper is structured as followed. We first investigate the intrinsic conformational properties of an isolated chameleon sequence, VLYVKLHN, using replica exchange molecular dynamics (REMD) simulations. We then consider this sequence in the context of a protein in which it forms α-helical structure (residues 8–31 of PDB 1JIG40) or β-strand structure (residues 263–286 of PDB 1P3541). We refer to the original 8 amino-acid long chameleon sequence as HS (helix/strand), and the two larger 24-residue constructs as HS-α and HS-β. All sequences and their structures in their cognate proteins are shown in Figure 1. The structure and aggregation of these peptides are studied computationally, as well as through ion-mobility mass spectrometry (IM-MS), transmission electron microscopy (TEM) and circular dichroism. Finally, we perform point mutations in the flanking sequences of HS-α and HS-β to further investigate computationally the effect of environment in modulating the secondary structure of the chameleon sequence.
Figure 1.
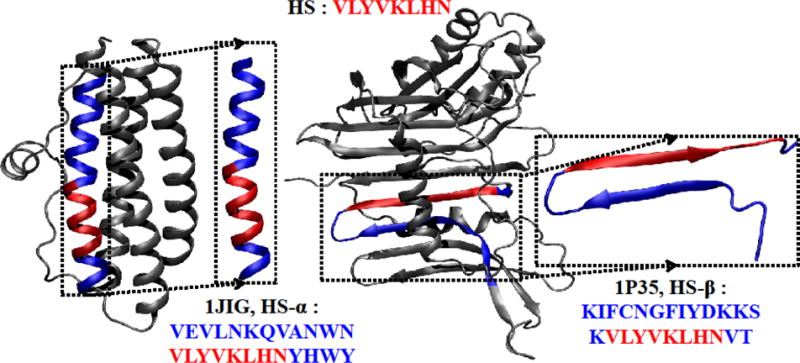
A chameleon-HS sequence VLYVKLHN (red) in 1JIG (α-helix) and in 1P35 (β-strand). The sequence fragments extracted from the original PDB structure are shown in the dotted boxes: the HS-α sequence (chameleon sequence with α-helix conformation) and the HS-β sequence (chameleon sequence with β-strand conformation) are highlighted in red. The flanking residues are shown in blue, with the original chameleon sequence in red. The figure was prepared by VMD.42
MATERIALS and METHODS
Models
The HS sequence (chameleon-HS sequence as an isolated fragment) is VLYVKLHN. We further considered the two sequences, HS-α and HS-β shown in Figure 1, to investigate how flanking residues affect folding tendencies. The length of residues added to the HS sequence is long enough to include at least one secondary structure unit, i.e., an α-helix, and two complementary β-strands or β-sheet/hairpin (see Figure 1). In addition, a second chameleon sequence, the seven-residue long chameleon-HE (Helix and shEet) sequence, RVQDNIV was studied as described in the Supporting Information).
Molecular Modeling
To enhance sampling in the molecular dynamics simulations, the REMD method43 is used. It is performed by running a set of independent simulations at different temperatures and exchanges determined by the Metropolis criterion in each period of time. REMD has been known to be an efficient sampling method44 and has been applied successfully to α-helical peptides,34, 45–46 β-sheet peptides,47 β-hairpin peptides48, small proteins49 and amyloid assembly.20, 50–54 The temperature-based REMD method, which allows enhanced conformational sampling, is a suitable tool for studying the structural plasticity of the isolated chameleon sequence HS and the factors that induce chameleon sequences to fold into an α-helix conformation in HS-α and a β-hairpin in HS-β. Details about simulation parameters and settings can be found in Supporting Information section S1.
Ion-mobility mass spectrometry
Ion-mobility based mass spectrometry (IM-MS) was utilized to measure the collision cross sections of monomers of HS-α and HS-β peptides. The experimental values were compared to theoretical cross sections of structures obtained from simulations. Here, the ions are generated by nano-electrospray, captured by a capillary, transferred to an ion funnel, gently desolvated, trapped and pulsed into a drift cell filled with helium gas. The ion packet is pulled through the cell by a weak electric field, exits the cell, is mass-selected and detected. The mass filter can be set to select a certain value of m/z and an arrival time distribution measured, with t = 0 set by the pulse into the cell and t = tA when the ions arrive at the detector. By varying the voltage across the cell at constant cell pressure very accurate values of the reduced K0, can be obtained. These values can be transformed into collision cross sections using a well established kinetic theory relationship.55 If mass spectra are desired, the ions are continuously transmitted from the ion funnel to the drift cell. In the experiments described here two separated drift cell IM-MS were used; one with a short (5 cm) drift cell56 and one with a much longer cell (2 m).57 The latter instrument has much higher resolution and because of the design of its source it very gently transfers the ions from the nano-electrospray droplets to the drift cell. This is important in order to retain solution-like structures for cross section determinantion.58–62
Transmission Electron Microscopy and Circular Dichroism
Transmission electron microscopy (TEM) images were taken at t = 0 and t = 480 h using a JEOL 1200 EX transmission electron microscope with an accelerating voltage of 80 kV. Circular dichroism spectra were taken using an Aviv circular dichroism spectrometer model 202 (Instruments Inc.) and a J-810 spectropolarimeter (JASCO, Tokyo, Japan) for time-course studies. Detailed experimental procedures are described in details in Supporting Information sections S1.3 and S1.4.
RESULTS and DISCUSSION
The chameleon sequence samples both α-helical and β-conformations
The Potential Mean Force (PMF) of the isolated chameleon-HS sequence fragment is shown in Figure 2 as a function of the RMSD from the reference helical structure and the reference strand structure, as described in the figure captions. From this plot, it is apparent that the peptide samples helical, strand and coil conformations. From the α-helix region to the β-strand region, the potential well depth (estimated from the PMF) varied by only about 0.5 kcal/mol. Hence the conversion between α and β secondary structures, through the random coil, can occur quite readily. An α-helix structure with six residues participating in helix formation, as calculated by DSSP63, was populated in 33.3% of the total structures (see Figure 2 (a)) with an RMSD from the reference α-helix structure of 0.201 Å. A single-turn α-helix comprising four residues was populated only in 6.8% of the total structures (see Figure 2 (d)). The coil-like structure (b) in Figure 2 had no helical content and had an occurrence frequency of 14.2%. Finally, an extended β-strand structure with occurrence frequency of 14.0% was observed that had an RMSD from the reference β structure of 0.734 Å. The higher propensity for helical structure over strand structure can be attributed to the relative ease of forming an isolated helix (as opposed to a strand that is stabilized in the context of the protein by a complementary strand). The chameleon-HS sequence has no charged residues other than one lysine so it cannot have an intermediate structure having a salt bridge53 in transit between α-helices (Figure 2(a)), random-coils (Figure 2(b)) and fully stretched structures (Figure 2(c)). It is interesting to note that statistical analyses of structures in the PDB,8–10 show that Val, Leu, Ile and Ala (amino acids with either strong helical or β-propensities) occur frequently in chameleon sequences, while Cys, His, Met, Pro and Trp amino acids (helix breakers, disulfide bond formers, or amino acids with “neutral” secondary structure propensities) are less frequent. For our particular peptide (VLYVKLHN), the two Val residues present in the sequence lend α-helix propensity while the two Leu residues favor a β-fold, the combination of both leading to the “frustrated” nature of this sequence, and its ability to populate both helical and strand structures.
Figure 2.
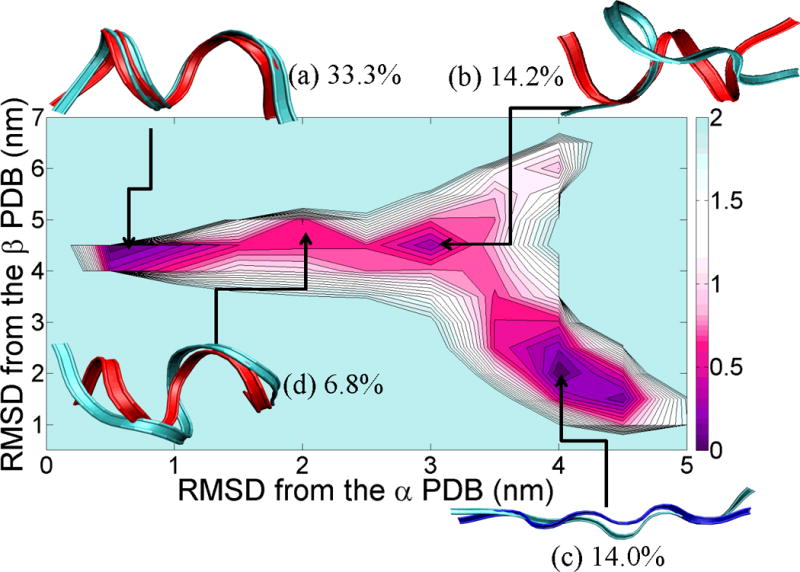
PMF landscape of the chameleon-HS sequence fragment along with the result of cluster analysis. X-axis of the PMF graph is the RMSD (nm) from the reference α-helix fragment of the protein (shown in red color), PDB id = 1JIG and Y-axis of the PMF graph is the RMSD (nm) from the reference β-strand fragment of the protein (shown in blue), PDB id = 1P35. Each of the (a), (b), (c) and (d) structures reflected the lowest RMSD structure from each centroid and ordered by population (indicated as a percentage next to the letter label).
Flanking residues induce chameleon sequences to favor one type of secondary structures
Having established computationally that the isolated HS fragment can populate both helical and β-conformations, we now turn to the effect of flanking sequences on conformation. We first consider the flanking residues that are present in the context of the full length proteins 1JIG (in which the HS sequence adopts an α-helix) and 1P35 (in which the peptide adopts a β-strand). The HS-α sequence was seen to fold predominantly into an α-helix (population 89.7%), as shown in the PMF in Figure 3A. Helical content (i.e., the number of residues in an α-helix) calculated by the DSSP program63 ranged from 20 to 22 (56% for 22, 12% for 21, and 32% for 20). Reference structure helical content from the PDB is 24. The overlay of the simulation and PDB structures is shown in (a) in Figure 3A. The lowest RMSD from the reference α-helix PDB structure to the structures in region (a) in Figure 3A is 2.31 Å. The discrepancy arises from fraying in the terminal regions of HS-α. These ends are stabilized in the context of the full protein. No other significantly populated basin emerges from the PMF or clustering analysis.
Figure 3.
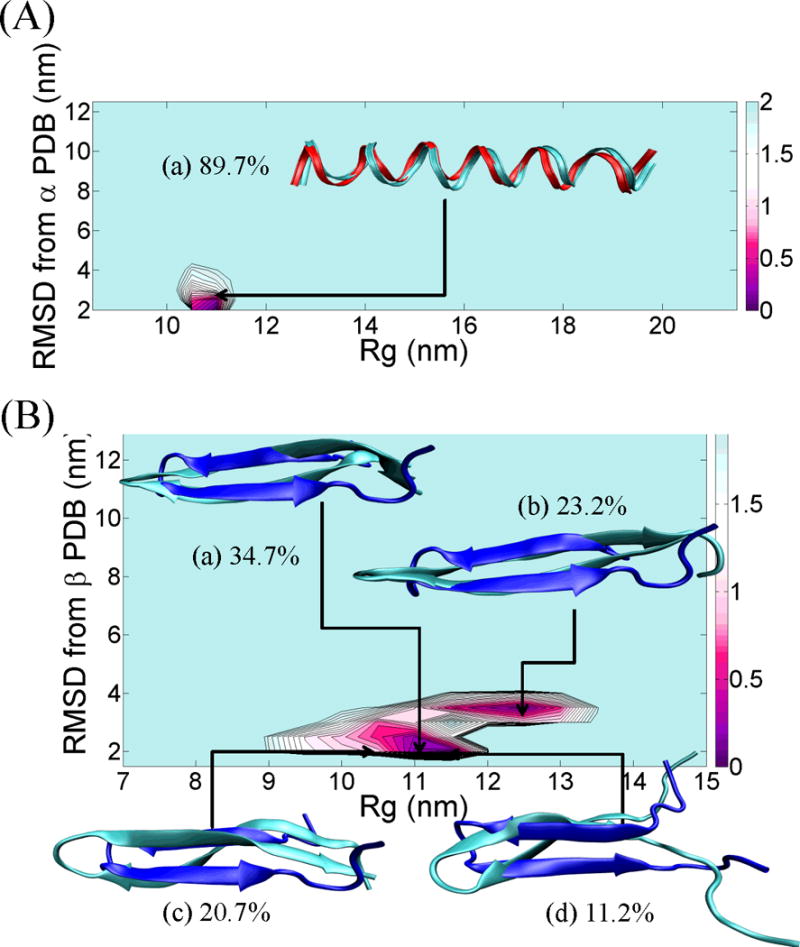
(A) PMF landscape of the chameleon- HS-α sequence fragment along with the result of cluster analysis. X-axis of the PMF graph is the radius of gyration, Rg (nm) and Y-axis of the PMF graph is the RMSD (nm) from the reference α-helix fragment of the protein, PDB id = 1JIG. The structure (a) reflected the lowest RMSD structure from each centroid and ordered by population. The red ribbon structure indicates the reference α-helix structure. (B) PMF landscape of the chameleon- HS-β sequence fragment along with the result of cluster analysis. X-axis of the PMF graph is the radius of gyration, Rg (nm) and Y-axis of the PMF graph is the RMSD (nm) from the reference β-strand fragment of the protein, PDB id = 1P35. Each of the (a), (b), (c) and (d) structures reflected the lowest RMSD structure from each centroid and ordered by population. The blue ribbon structure indicates the reference β-strand structure. All ribbon structures were prepared using VMD.42 The energy unit on this PMF landscape is kcal/mol.
The HS-β sequence, in turn, folded into β-strand secondary structure as shown in the PMF in Figure 3B. All significantly populated basins consisted of β-hairpin structures. The most populated structure (34.7%) in Figure 3B is centroid (a), which comprises β-strand content (i.e., the number of residues in β-strand) ranging from 14 to 20. Reference structure β-strand content from the PDB is 18. The second most populated centroid (23.2%) exhibits a β-strand content of 8~14 (centroid (b)). Two other centroids were populated at levels of 20.7% (Figure 3B (c)) and 11.2% (Figure 3B (d)), respectively. The structures in each of the basins in the PMF had the same overall β-fold, but the radii of gyration differed because of fraying in the N- and C-terminal regions. The lowest RMSD from the reference β-strand PDB structure to the structures in region (a) in Figure 3B is 2.37 Å. No structures with helical content were observed during the last 50 ns of the REMD simulations.
Although each peptide sequence possesses an instrinsic secondary structure propensity, adding flanking residues to this sequence may introduce allosteric effects (due to changes in intra-chain hydrogen bonding networks, hydrophobicity, etc.), affecting the overall secondary structure. Here, we find that adding residues from the reference α-helix structure to the head and tail of the HS chameleon sequence dramatically increased the frequency of α-helix conformations (Figure 3A). Similarly, adding to the tail of the chameleon-HS the flanking residues found in the PDB structure in which the segment has β-content restricts the folding pathway to predominantly β-strand conformations (Figure 3B).
As shown in the supplemental section S2.2, a second sequence studied, chameleon-HE sequence which has a weaker helix propensity (Arg and Gln replace Ala and Leu residues) also behaves similarly to the chameleon-HS sequence. It forms only α-helix conformations upon addition of capping residues stemming from the original PDB structure that led to helix formation in the context of the protein (i.e., the HE-α sequence; see Figure S4A). The chameleon-HE has stronger β-sheet propensity than the chameleon-HS sequence due to the presence of three residues with strong β-sheet propensity. It formed β-sheet and hairpin structures when β-favoring flanking residues were added (see Figure S4B). Results obtained from REMD simulations demonstrate strong competition among secondary structures in these peptides and the crucial roles of residue contexts in altering the secondary structure equilibria. Figure 4 illustrates the competition between α-helix and β-sheet in HS, HS-α and HS- β peptides.
Figure 4.
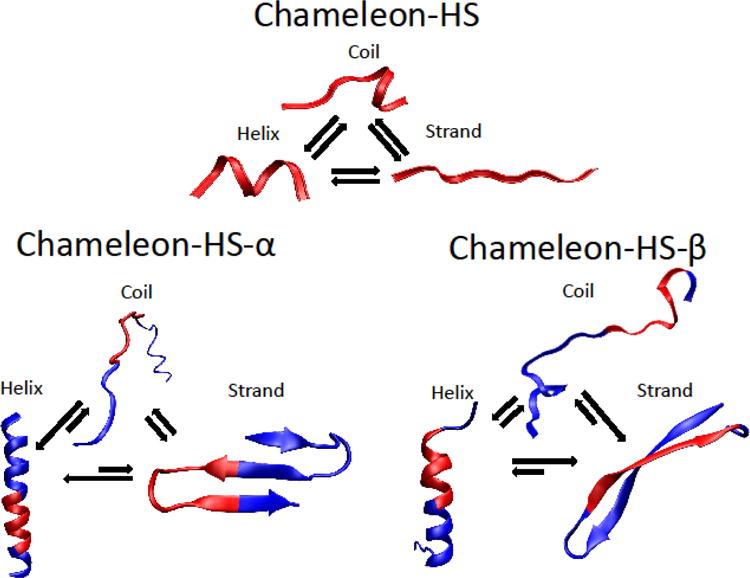
A schematic of secondary structure equilibria in chameleon-HS, HS-α and HS-β obtained from REMD simulations showing the strong competition among α-helix, random coil and β-sheet structures.
To further probe the dependence of secondary structure propensity on residue contexts, we performed point mutations on HS-α and HS-β to alter the secondary structure preference (see Figure 5A). The data for mutations performed on the HE-α and HE-β sequences are shown, in the Supporting Information.
Figure 5.
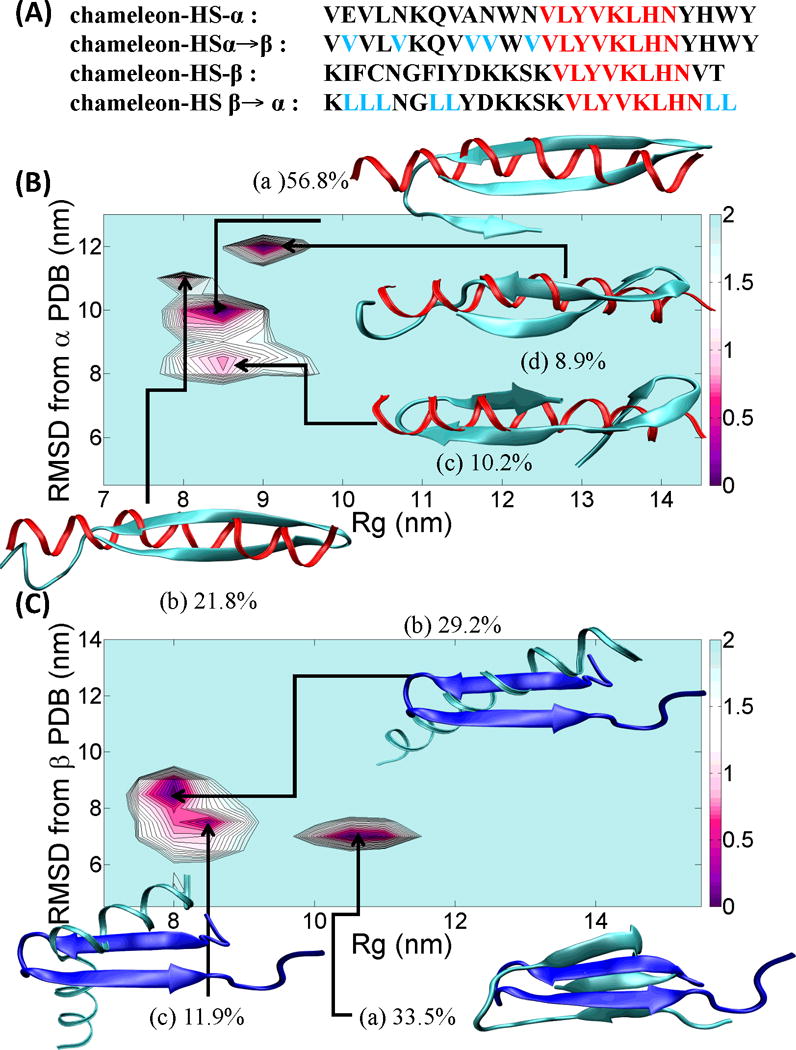
(A) four chameleon-HS sequence fragments, chameleon-HS-α, chameleon-HSα→β, chameleon-HS-β and chameleon-HSβ→α are shown with black (flanking residues), red (chameleon) and cyan (mutation) colors. (B) PMF landscape of the chameleon-HSα→β sequence fragment along with the result of cluster analysis. X-axis of the PMF graph is the radius of gyration, Rg (nm) and Y-axis of the PMF graph is the RMSD (nm) from the reference α-helix fragment of the protein, PDB id = 1JIG. Each of the (a), (b), (c) and (d) structures reflected the highest populations indicated above. The red ribbon structure indicates the reference α-helix structure. (C) PMF landscape of the chameleon-HSβ→α sequence fragment along with the result of cluster analysis. X-axis of the PMF graph is the radius of gyration, Rg and Y-axis of the PMF graph is the RMSD from the reference β-strand fragment of the protein, PDB id = 1P35. Each of the (a), (b) and (c) structures reflected the highest populations indicated above. The blue ribbon structure indicates the reference β-strand structure. All ribbon structures were prepared using VMD.42 The energy unit on this PMF landscape is kcal/mol.
We construct the HSα→β peptide (Helix-Sheet chameleon sequence with Helical context mutated to β) by mutating the second, fifth, ninth, tenth and twelfth residues (i.e., residues with weak β-sheet propensity) from HS-α into Val residues (a residue with high β-sheet propensity). The potential of mean force is shown in Figure 5B. The HSα→β’s helical structures were negligible (<4%) in the last 50 ns of the 250 ns REMD simulation and structures primarily adopted either two strands (centroid (b) with 21.8% population and centroid (d) with 8.9% population in Figure 5B) or three strands (centroids (a) 56.8% (most populated) and (c) 10.2% populated). β-strand contents calculated by the DSSP program ranged from 14 to 22.
Similarly, we constructed the HSβ→α (Helix-Sheet chameleon sequence and Sheet context mutated to Helix) sequence by mutating the second, third, fourth, seventh, eighth, twenty third and twenty fourth residues HS-β into Leu residues. The population of HSβ→α’s β-strand structures was reduced to 33.5% vs. 89.8% from the original HSβ sequence. The single β-rich centroid found had three strands (Figure 5C (a)). Two new helical clusters induced by the point mutations consisted of either one α-helix (centroid (b) with population 29.2%) or two α-helices (centroid (c) with 11.9% population, in Figure 5C). α-helix contents ranged from 18 to 22.
HS-α and HS-β aggregate through different pathways
We also investigated the aggregation propensity of HS-β and HS-α peptides using TEM to examine the structures present initially, and after incubation for 3–4 weeks. Interestingly, HS-α forms fibrils at t = 0 while HS-β forms curved worm-like aggregates, but no straight fibrils (see Figure 6). By the end of the aggregation time-course, we did not observe fibrils for HS-α, likely due to precipitation or adhesion to the cuvette wall. The structures formed by HS-β remained essentially unchanged throughout the time course.
Figure 6.
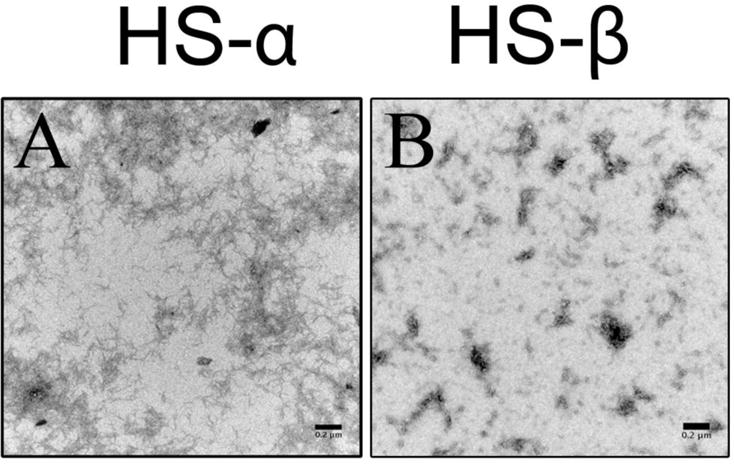
Representative TEM images of 150 μM (A) HS-α and (B) HS-β in 20 mM ammonium acetate buffer (pH = 7) obtained at t = 0. HS-α forms predominantly fibrils whereas HS-β forms curved worm-like aggregates. Scale bars are 0.2 μm.
Time-course CD studies suggest that HS-β (Figure 7, left panel) is initially unstructured (minimum at 198 nm). The peptide monomer and oligomers display increasing regular β-structure over the course of 20 days (480 hours), as evidenced by the broad minimum ranging between 200 nm and 214 nm. HS-α monomer and oligomers (Figure 7, right panel), on the other hand, adopt mainly α-helical conformations over a 20-day time course. HS-α starts partially unstructured, as indicated by molar ellipticity minima at 200 nm and 217 nm. During the course of aggregation, non-coil secondary structure elements increase in frequency, as shown by a rise in molar ellipticity at 200 nm (i.e., more α-helical) and a concomitant decrease in molar ellipticity at 206 nm and 216 nm, indicating the presence of both α and β-rich oligomer structures after 20 days.
Figure 7.
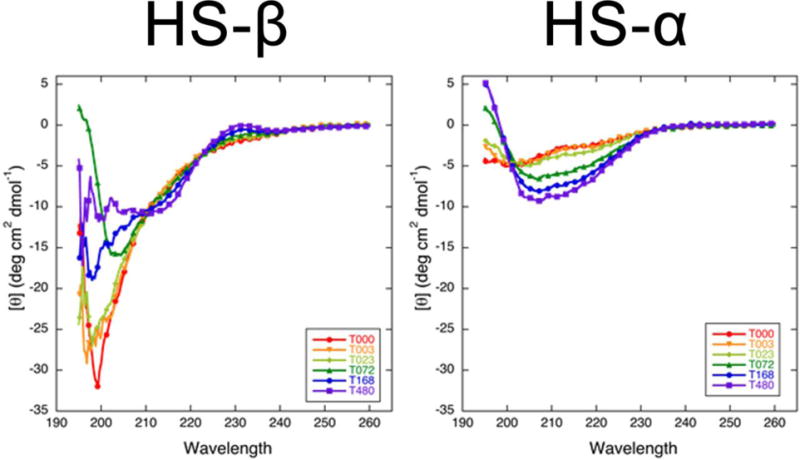
Time-course CD spectra of HS-β and HS-α in 20 mM ammonium acetate buffer. Different colors are used to annotate the spectra collected at different time points (e.g., T072 is the spectrum collected at t = 72 hours).
Additional secondary structure analysis using KD2 method on the 48-hour incubated peptides were also performed. In HS-β, the β-structure population is about 50% of the total, whereas the α-helix population is only half of that (Figure S6, top panel). For HS-α peptides, the α-helices are 60% of the total population, and their β-hairpin counterparts are only 8–12% (Figure S6 and Table S1).
Ion-mobility experiments
We performed IM experiments to: (1) determine collision cross sections of HS-α and HS-β monomers (for comparison with cross sections obtained from the REMD calculations of these peptides); and (2) evaluate the time-dependent aggregation of both peptides (for comparison with TEM results).
The mass spectra are simple for both HS-α and HS-β with results in acetate buffer shown in Figure 8, panels A and B from instrument 1. The z = +3 charge state is dominant for both species. The ATDs for the z = +3 charge states are given in panels C and D of Figure 8 with the peptide cross sections noted on the features. Both ATDs appear bimodal. The longer time feature is due to the solution (dehydrated) structure and the shorter time feature is due to the gas phase structure. This biomodality is often observed for peptides in this size range since solution-to-gas phase structural transition barriers are low enough for both structures to be observed.64
Figure 8.
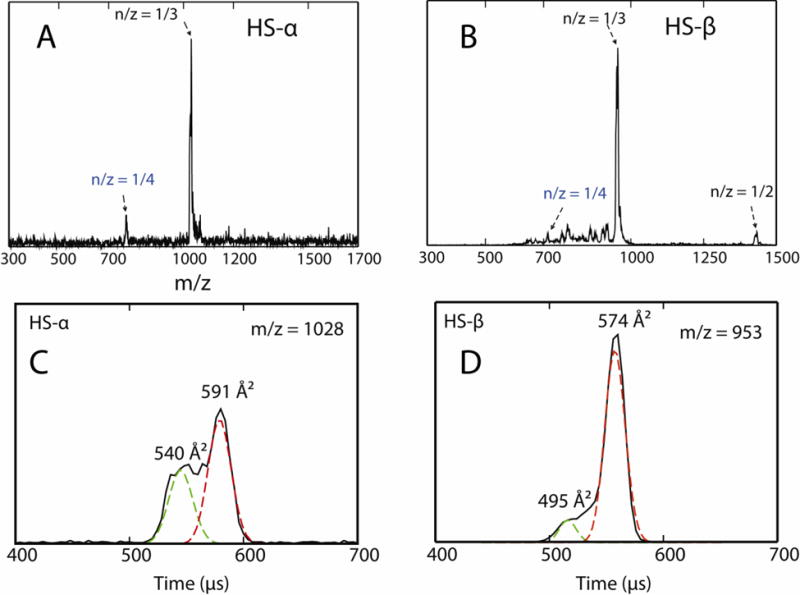
(A, B) ESI-quadrupole mass spectra of 200 μM HS-α and HS-β in 20 mM ammonium acetate buffer. (C, D) Representative ATDs of n/z = 1/3 mass spectral peaks where n is the oligomer size and z is the charge. The experimental cross sections σ are shown.
The data for averaged cross sections of charge states z = +3 and +4 (Supporting Information Figures S7 and S9) are tabulated in Table 1 and compared with cross sections calculated from the modeling studies discussed earlier. The model structures were first “dehydrated”64 before cross sections were determined using the trajectory65–66 and PSA algorithms.67–68 In general, the agreement between experiment and theory is good, with computationally derived cross sections ~3–5% larger than those determined experimentally. There is also good agreement in the relative values as the HS-α experimental cross sections are about 2% greater than the experimental HS-β cross sections while for theory HS-α is about 1% greater than HS-β. These systematic agreements provide important support for the theoretical structures presented in Figure 3.
Table 1.
Comparison of experimental and theoretical cross sections for HS-α and HS-β. All units are in Å2.
The cross sections are averages of results from instruments 1 and 2 and have uncertainties of less than 1%.
S results are average cross sections from the smaller radius of gyration distribution.
L results are average cross sections from the larger radius of gyration distribution. For HS-α essentially all structures occupy the same potential well (see Figure 3A).
Mass spectra taken using a prototype Q-TOF instrument69 at t = 0 and t = 2 weeks are given in Figure 9 for both HS-α and HS-β. In HS-α the presence of unresolved high mass aggregates is obvious even at earliest times. The spectrum changes little after two weeks indicating a quasi steady state has been reached between the aggregates and monomer species. The situation is much different for HS-β. At early times similar monomer peaks are observed as for HS-α (i.e., charge state z = +3 is dominant along with z = +4 and +2 charge states being observed). However, after two weeks no high mass aggregates are observed, only the presence of smaller oligomers (n/z = 2/3, 2/5, 4/5, 4/7, etc.). Hence while some amyloid fibrils may have been formed, as suggested by the TEM data in Figure 6, their rate of formation and overall propensity is much less than observed for HS-α.
Figure 9.
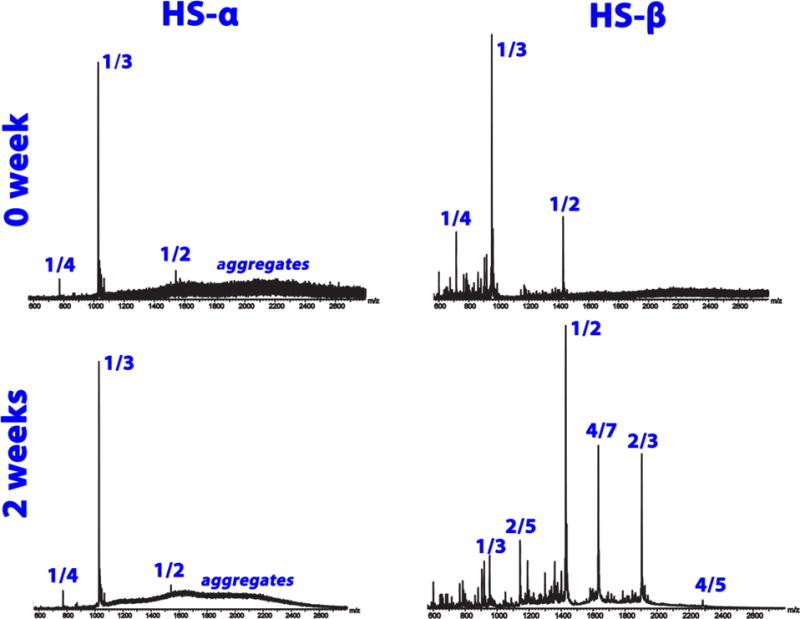
Time course ESI-TOF mass spectra of 200 μM HS-α (left) and HS-β (right) in 20 mM ammonium acetate buffer obtained at t = 0 and 2 weeks. The mass spectral peaks are annotated by their n/z ratios where n is the oligomer number and z is the charge.
It is interesting to note that HS-α aggregates more readily than HS-β. While this may appear surprising at first glance, there are several reports in the literature, both experimental27, 29–30, 70–71 and theoretical,36, 53, 72 of accumulation of helical intermediates en route to aggregation. The presence of such intermediates has been observed in a number of intrinsically disordered peptides implicated in amyloid diseases, including the Aβ peptide,33, 73 IAPP,34, 74 insulin59 and Tau,29 as well as in the aggregation of functional β-rich aggregates, such as silk.75 Formation of helical rich intermediates may be a means of accumulating a critical concentration of peptides that can then rearrange into β-sheet structure to form fibrils.76 This process could be further facilitated in the cell by the presence of surfaces such as membranes.34, 77–78 Helical-promoting solvents such as HFIP are routinely used in vitro aggregation experiments and have been shown to promote aggregation when present at moderate concentrations.79 Osmolytes such as TMAO can also promote helical structure and accelerate fibril formation, as was seen for a fragment of Tau and Aβ.53, 80 Of note, fibrils that form through helical intermediates do not necessarily result in β-rich amyloid fibrils, but can form helical fibrils, as is the case for the aggregation of the Apolipoprotein A-1 mimetic peptide.27, 32 The CD data collected here show that the HS-α peptide gains helical structure as aggregation proceeds. It is difficult to unambiguously state whether the fibrils seen in experiment are helical or sheet, or a mixture of both.
Aggregation via helical intermediates is just one possible means by which β-rich aggregates can form. Another viable pathway would involve population of an aggregate prone β-rich conformer that would catalyze the formation of β-sheets. This mechanism has been supported by computational20, 81 and experimental studies.18, 82 Formation of amyloid fibrils from peptides that adopt β-hairpins as monomer (such as HS-β) requires a certainly structural plasticity of the monomer, as overly stable hairpins will inhibit amyloid fibril formation. This was nicely shown in experiments on the Aβ peptide in which an artificial stabilized β-hairpin structure lead to off-pathway oliogmers.21 Our simulations on the Aβ(25–35) peptide, a strongly aggregating fragment of Aβ, confirmed this picture by showing that a transition from β-hairpin to extended strand is required in order for fibrilization to proceed.20 Hence, while β-hairpins are possible candidates for fibril formation, they must possess the ability to readily extend in order to form fibrils. The CD data (Figure 7) shows that the monomer of HS-β is already enriched in β-content, and that β-sheet content increases over the 20-day window in which aggregation was monitored. The resulting wormlike fibrils are therefore likely β-sheet rich amyloid-like fibrils (or protofibrils). We do not see significant evidence of helical intermediates. The transition from β-hairpin to a more extended aggregation prone structure does not appear to proceed readily for HS-β, hence its more limited ability to form fibrils.
SUMMARY and CONCLUSIONS
In this study, we analyzed the structural flexibilities of HS- and HE-type chameleon sequences. Chameleon sequences can produce α-helix or a β-sheet conformers, but no one conformer is formed predominately. The chameleon peptides thus behave in a manner akin to intrinsically disordered proteins, which populate relative large volumes of conformational space but can form stable secondary structure elements under the right conditions. These conditions include the presence of flanking residues with strong α-helix or β-sheet propensities. The neutral folding tendencies of a chameleon sequence allows the allosteric effects of flanking residues to dictate the structural evolution and the overall folding pathway of protein.
We show by IM-MS and microscopy imaging that HS-α has a strong propensity to form higher order aggregates and fibrils, suggesting α-helices can facilitate (nucleate) aggregation. Interestingly, the β-hairpin HS-β does not favor fibril formation. The fibrils observed after a two-week incubation may be due to the very small population of HS-β α-helix that initiates aggregation, or to a slower secondary aggregation pathway involving the β-hairpins. One might naively have expected that the β-favoring sequences would be more aggregation prone than the α-helical favoring sequences, but this is not the case, highlighting the challenges associated with attempting to predict aggregation propensities based on secondary structure propensity scales alone. We have identified in this work a novel aggregating sequence, shown that helical structure can induce formation of fibrillar aggregates11 and that judiciously chosen single point mutations in flanking sequences can bias secondary structure formation and thereby aggregation properties.
Supplementary Material
Acknowledgments
We gratefully acknowledge Ms. Margaret Condron at UCLA for synthesizing the HS-β peptide. This work was supported by the National Science Foundation MCB-1158577 (J.-E.S), CHE-1301032 (M.T.B), the David and Lucile Packard Foundation (J.-E.S), and National Institutes of Health grants NS038328 and AG041295 (D.B.T.). The authors acknowledge the Texas Advanced Computing Center (TACC) at University of Texas at Austin and the National Institute for Computational Sciences at Oak Ridge National Laboratory for providing HPC resources through the XSEDE (grant no. TG-MCA05S027), and the Center for Scientific Computing at the CNSI and MRL: an NSF MRSEC (DMR-1121053) and NSF CNS-0960316. A prototype Synapt instrument was graciously provided by the Waters Corporation.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Kabsch W, Sander C. On the use of sequence homologies to predict protein structure – identical pentapeptides can have completely different conformations. Proc Natl Acad Sci U S A. 1984;81:1075–1078. doi: 10.1073/pnas.81.4.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Minor DL, Kim PS. Context-dependent secondary structure formation of a designed protein sequence. Nature. 1996;380:730–734. doi: 10.1038/380730a0. [DOI] [PubMed] [Google Scholar]
- 3.Tidow H, Lauber T, Vitzithum K, Sommerhoff CP, Rosch P, Marx UC. The solution structure of a chimeric lekti domain reveals a chameleon sequence. Biochemistry. 2004;43:11238–11247. doi: 10.1021/bi0492399. [DOI] [PubMed] [Google Scholar]
- 4.Yang WZ, Ko TP, Corselli L, Johnson RC, Yuan HS. Conversion of a beta-strand to an alpha-helix induced by a single-site mutation observed in the crystal structure of fis mutant Pro(26)Ala. Protein Sci. 1998;7:1875–1883. doi: 10.1002/pro.5560070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abel K, Yoder MD, Hilgenfeld R, Jurnak F. An α to β conformational switch in EF-TU. Structure. 1996;4:1153–1159. doi: 10.1016/s0969-2126(96)00123-2. [DOI] [PubMed] [Google Scholar]
- 6.Carr CM, Kim PS. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell. 1993;73:823–832. doi: 10.1016/0092-8674(93)90260-w. [DOI] [PubMed] [Google Scholar]
- 7.Cohen BI, Presnell SR, Cohen FE. Origins of structural diversity within sequentially identical hexapeptides. Protein Sci. 1993;2:2134–2145. doi: 10.1002/pro.5560021213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mezei M. Chameleon sequences in the PDB. Protein Eng. 1998;11:411–414. doi: 10.1093/protein/11.6.411. [DOI] [PubMed] [Google Scholar]
- 9.Zhou XH, Alber F, Folkers G, Gonnet GH, Chelvanayagam G. An analysis of the helix-to-strand transition between peptides with identical sequence. Proteins. 2000;41:248–256. doi: 10.1002/1097-0134(20001101)41:2<248::aid-prot90>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 10.Guo JT, Jaromczyk JW, Xu Y. Analysis of chameleon sequences and their implications in biological processes. Proteins. 2007;67:548–558. doi: 10.1002/prot.21285. [DOI] [PubMed] [Google Scholar]
- 11.Kallberg Y, Gustafsson M, Persson B, Thyberg J, Johansson J. Prediction of amyloid fibril-forming proteins. J Biol Chem. 2001;276:12945–12950. doi: 10.1074/jbc.M010402200. [DOI] [PubMed] [Google Scholar]
- 12.Kelly JW. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr Opin Struc Biol. 1998;8:101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 13.Eisenberg D, Nelson R, Sawaya MR, Balbirnie M, Sambashivan S, Ivanova MI, Madsen AO, Riekel C. The structural biology of protein aggregation diseases: Fundamental questions and some answers. Acc Chem Res. 2006;39:568–575. doi: 10.1021/ar0500618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thirumalai D, Klimov DK, Dima RI. Emerging ideas on the molecular basis of protein and peptide aggregation. Curr Opin Struc Biol. 2003;13:146–159. doi: 10.1016/s0959-440x(03)00032-0. [DOI] [PubMed] [Google Scholar]
- 15.Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo JS, Taddei N, Ramponi G, Dobson CM, Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 16.Kelly JW. Alternative conformations of amyloidogenic proteins govern their behavior. Curr Opin Struc Biol. 1996;6:11–17. doi: 10.1016/s0959-440x(96)80089-3. [DOI] [PubMed] [Google Scholar]
- 17.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJW, McFarlane HT, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 19.Groveman BR, Dolan MA, Taubner LM, Kraus A, Wickner RB, Caughey B. Parallel in-register intermolecular beta-sheet architectures for prion-seeded prion protein (prp) amyloids. J Biol Chem. 2014;289:24129–24142. doi: 10.1074/jbc.M114.578344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larini L, Shea JE. Role of β-hairpin formation in aggregation: The self-assembly of the amyloid-β(25–35) peptide. Biophys J. 2012;103:576–586. doi: 10.1016/j.bpj.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoyer W, Gronwall C, Jonsson A, Stahl S, Hard T. Stabilization of a beta-hairpin in monomeric alzheimer’s amyloid-beta peptide inhibits amyloid formation. Proc Natl Acad Sci U S A. 2008;105:5099–5104. doi: 10.1073/pnas.0711731105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kajava AV, Baxa U, Steven AC. Beta arcades: Recurring motifs in naturally occurring and disease-related amyloid fibrils. FASEB J. 2010;24:1311–1319. doi: 10.1096/fj.09-145979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phelps EM, Hall CK. Structural transitions and oligomerization along polyalanine fibril formation pathways from computer simulations. Proteins. 2012;80:1582–1597. doi: 10.1002/prot.24052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheon M, Hall CK, Chang I. Structural conversion of Aβ17–42 peptides from disordered oligomers to u-shape protofilaments via multiple kinetic pathways. PLOS Comput Biol. 2015;11:e1004258. doi: 10.1371/journal.pcbi.1004258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bleiholder C, Do TD, Wu C, Economou NJ, Bernstein SS, Buratto SK, Shea J-E, Bowers MT. Ion mobility spectrometry reveals the mechanism of amyloid formation of Aβ(25–35) and its modulation by inhibitors at the molecular level: Epigallocatechin gallate and scyllo-inositol. J Am Chem Soc. 2013;135:16926–16937. doi: 10.1021/ja406197f. [DOI] [PubMed] [Google Scholar]
- 26.Pertinhez TA, Bouchard M, Tomlinson EJ, Wain R, Ferguson SJ, Dobson CM, Smith LJ. Amyloid fibril formation by a helical cytochrome. FEBS Lett. 2001;495:184–186. doi: 10.1016/s0014-5793(01)02384-5. [DOI] [PubMed] [Google Scholar]
- 27.Lazar KL, Miller-Auer H, Getz GS, Orgel JPRO, Meredith SC. Helix-turn-helix peptides that form alpha-helical fibrils: Turn sequences drive fibril structure. Biochemistry. 2005;44:12681–12689. doi: 10.1021/bi0509705. [DOI] [PubMed] [Google Scholar]
- 28.Kunjithapatham R, Oliva FY, Doshi U, Perez M, Avila J, Munoz V. Role for the α-helix in aberrant protein aggregation. Biochemistry. 2005;44:149–156. doi: 10.1021/bi048564t. [DOI] [PubMed] [Google Scholar]
- 29.Sadqi M, Hernandez F, Pan UM, Perez M, Schaeberle MD, Avila J, Munoz V. Alpha-helix structure in alzheimer’s disease aggregates of tau-protein. Biochemistry. 2002;41:7150–7155. doi: 10.1021/bi025777e. [DOI] [PubMed] [Google Scholar]
- 30.Anderson VL, Ramlall TF, Rospigliosi CC, Webb WW, Eliezer D. Identification of a helical intermediate in trifluoroethanol-induced alpha-synuclein aggregation. Proc Natl Acad Sci U S A. 2010;107:18850–18855. doi: 10.1073/pnas.1012336107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pannuzzo M, Raudino A, Milardi D, La Rosa C, Karttunen M. Alpha-helical structures drive early stages of self-assembly of amyloidogenic amyloid polypeptide aggregate formation in membranes. Sci Rep. 2013;3:2781. doi: 10.1038/srep02781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Do TD, Chamas A, Zheng X, Barnes A, Chang D, Veldstra T, Takhar H, Dressler N, Trapp B, Miller K, et al. Elucidation of the aggregation pathways of helix-turn-helix peptides: Stabilization at the turn region is critical for fibril formation. Biochemistry. 2015;54:4050–4062. doi: 10.1021/acs.biochem.5b00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirkitadze MD, Condron MM, Teplow DB. Identification and characterization of key kinetic intermediates in amyloid β-protein fibrillogenesis. J Mol Biol. 2001;312:1103–1119. doi: 10.1006/jmbi.2001.4970. [DOI] [PubMed] [Google Scholar]
- 34.Guo C, Cote S, Mousseau N, Wei G. Distinct helix propensities and membrane interactions of human and rat IAPP(1–19) monomers in anionic lipid bilayers. J Phys Chem B. 2015;119:3366–3376. doi: 10.1021/jp5111357. [DOI] [PubMed] [Google Scholar]
- 35.Nguyen HD, Hall CK. Spontaneous fibril formation by polyalanines; discontinuous molecular dynamics simulations. J Am Chem Soc. 2006;128:1890–1901. doi: 10.1021/ja0539140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tarus B, Straub JE, Thirumalai D. Probing the initial stage of aggregation of the abeta(10–35)-protein: Assessing the propensity for peptide dimerization. J Mol Biol. 2005;345:1141–1156. doi: 10.1016/j.jmb.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 37.Xu Y, Shen J, Luo X, Zhu W, Chen K, Ma J, Jiang H. Conformational transition of amyloid β-peptide. Proc Natl Acad Sci U S A. 2005;102:5403–5407. doi: 10.1073/pnas.0501218102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding F, Borreguero JM, Buldyrey SV, Stanley HE, Dokholyan NV. Mechanism for the α-helix to β-hairpin transition. Proteins. 2003;53:220–228. doi: 10.1002/prot.10468. [DOI] [PubMed] [Google Scholar]
- 39.Daidone I, Simona F, Roccatano D, Broglia RA, Tiana G, Colombo G, Di Nola A. β-hairpin conformation of fibrillogenic peptides: Structure and alpha-beta transition mechanism revealed by molecular dynamics simulations. Proteins. 2004;57:198–204. doi: 10.1002/prot.20178. [DOI] [PubMed] [Google Scholar]
- 40.Papinutto E, Dundon WG, Pitulis N, Battistutta R, Montecucco C, Zanotti G. Structure of two iron-binding proteins from bacillus anthracis. J Biol Chem. 2002;277:15093–15098. doi: 10.1074/jbc.M112378200. [DOI] [PubMed] [Google Scholar]
- 41.Fisher AJ, Cruz W, Zoog SJ, Schneider CL, Friesen PD. Crystal structure of baculovirus p35: Role of a novel reactive site loop in apoptotic caspase inhibition. EMBO J. 1999;18:2031–2039. doi: 10.1093/emboj/18.8.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Humphrey W, Dalke A, Schulten K. Vmd: Visual molecular dynamics. J Mol Graph Model. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 43.Sugita Y, Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett. 1999;314:141–151. [Google Scholar]
- 44.Trebst S, Troyer M, Hansmann UHE. Optimized parallel tempering simulations of proteins. J Chem Phys. 2006;124 doi: 10.1063/1.2186639. [DOI] [PubMed] [Google Scholar]
- 45.Zhang W, Wu C, Duan Y. Convergence of replica exchange molecular dynamics. J Chem Phys. 2005;123 doi: 10.1063/1.2056540. [DOI] [PubMed] [Google Scholar]
- 46.Lei H, Wu C, Wang ZX, Zhou Y, Duan Y. Folding processes of the b domain of protein a to the native state observed in all-atom ab initio folding simulations. J Chem Phys. 2008;128:235105. doi: 10.1063/1.2937135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roe DR, Hornak V, Simmerling C. Folding cooperativity in a three-stranded β-sheet model. J Mol Biol. 2005;352:370–381. doi: 10.1016/j.jmb.2005.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou RH, Berne BJ. Can a continuum solvent model reproduce the free energy landscape of a beta-hairpin folding in water? Proc Natl Acad Sci U S A. 2002;99:12777–12782. doi: 10.1073/pnas.142430099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pitera JW, Swope W. Understanding folding and design: Replica-exchange simulations of “trp-cage” miniproteins. Proc Natl Acad Sci U S A. 2003;100:7587–7592. doi: 10.1073/pnas.1330954100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qi RX, Luo Y, Wei GH, Nussinov R, Ma BY. A β “stretching-and-packing” cross-seeding mechanism can trigger tau protein aggregation. J Phys Chem Lett. 2015;6:3276–3282. [Google Scholar]
- 51.Laghaei R, Mousseau N, Wei G. Structure and thermodynamics of amylin dimer studied by hamiltonian-temperature replica exchange molecular dynamics simulations. J Phys Chem B. 2011;115:3146–3154. doi: 10.1021/jp108870q. [DOI] [PubMed] [Google Scholar]
- 52.Miyashita N, Straub JE, Thirumalai D, Sugita Y. Transmembrane structures of amyloid precursor protein dimer predicted by replica-exchange molecular dynamics simulations. J Am Chem Soc. 2009;131:3438–3439. doi: 10.1021/ja809227c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levine ZA, Larini L, LaPointe NE, Feinstein SC, Shea JE. Regulation and aggregation of intrinsically disordered peptides. Proc Natl Acad Sci U S A. 2015;112:2758–2763. doi: 10.1073/pnas.1418155112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ganguly P, Do TD, Larini L, LaPointe NE, Sercel AJ, Shade MF, Feinstein SC, Bowers MT, Shea JE. Tau assembly: The dominant role of PHF6 (VQIVYK) in microtubule binding region repeat R3. J Phys Chem B. 2015;119:4582–4593. doi: 10.1021/acs.jpcb.5b00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mason EA. Transport properties of ions in gases. 99. John Wiley & Sons; 1988. [Google Scholar]
- 56.Wyttenbach T, Kemper PR, Bowers MT. Design of a new electrospray ion mobility mass spectrometer. Int J Mass Spectrom. 2001;212:13–23. [Google Scholar]
- 57.Kemper PR, Dupuis NF, Bowers MT. A new, higher resolution, ion mobility mass spectrometer. Int J Mass Spectrom. 2009;287:46–57. [Google Scholar]
- 58.Silveira JA, Fort KL, Kim D, Servage KA, Pierson NA, Clemmer DE, Russell DH. From solution to the gas phase: Stepwise dehydration and kinetic trapping of substance p reveals the origin of peptide conformations. J Am Chem Soc. 2013;135:19147–19153. doi: 10.1021/ja4114193. [DOI] [PubMed] [Google Scholar]
- 59.Susa AC, Wu C, Bernstein SL, Dupuis NF, Wang H, Raleigh DP, Shea JE, Bowers MT. Defining the molecular basis of amyloid inhibitors: Human islet amyloid polypeptide-insulin interactions. J Am Chem Soc. 2014;136:12912–12919. doi: 10.1021/ja504031d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen S-H, Russell DH. How closely related are conformations of protein ions sampled by im-ms to native solution structures? J Am Soc Mass Spectrom. 2015;26:1433–1443. doi: 10.1007/s13361-015-1191-1. [DOI] [PubMed] [Google Scholar]
- 61.Benesch JLP, Ruotolo BT. Mass spectrometry: Come of age for structural and dynamical biology. Curr Opin Struc Biol. 2011;21:641–649. doi: 10.1016/j.sbi.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dickinson ER, Jurneczko E, Pacholarz KJ, Clarke DJ, Reeves M, Ball KL, Hupp T, Campopiano D, Nikolova PV, Barran PE. Insights into the conformations of three structurally diverse proteins: Cytochrome c, P53, and MDM2, provided by variable-temperature ion mobility mass spectrometry. Anal Chem. 2015;87:3231–3238. doi: 10.1021/ac503720v. [DOI] [PubMed] [Google Scholar]
- 63.Kabsch W, Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 64.Baumketner A, Bernstein SL, Wyttenbach T, Bitan G, Teplow DB, Bowers MT, Shea JE. Amyloid beta-protein monomer structure: A computational and experimental study. Protein Sci. 2006;15:420–428. doi: 10.1110/ps.051762406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shvartsburg AA, Jarrold MF. An exact hard-spheres scattering model for the mobilities of polyatomic ions. Chem Phys Lett. 1996;261:86–91. [Google Scholar]
- 66.Mesleh MF, Hunter JM, Shvartsburg AA, Schatz GC, Jarrold MF. Structural information from ion mobility measurements: Effects of the long range potential. J Phys Chem A. 1996;100:16082–16086. [Google Scholar]
- 67.Bleiholder C, Wyttenbach T, Bowers MT. A novel projection approximation algorithm for the fast and accurate computation of molecular collision cross sections (I). Method. Int J Mass Spectrom. 2011;308:1–10. [Google Scholar]
- 68.Bleiholder C, Contreras S, Do TD, Bowers MT. A novel projection approximation algorithm for the fast and accurate computation of molecular collision cross sections (II). Parameterization and application to biomolecules. Int J Mass Spectrom. 2013:345–347. [Google Scholar]
- 69.Pringle SD, Giles K, Wildgoose JL, Williams JP, Slade SE, Thalassinos K, Bateman RH, Bowers MT, Scrivens JH. An investigation of the mobility separation of some peptide and protein ions using a new hybrid quadrupole/travelling wave IMS/OA-TOF instrument. Int J Mass Spectrom. 2007;261:1–12. [Google Scholar]
- 70.Fezoui Y, Hartley DM, Walsh DM, Selkoe DJ, Osterhout JJ, Teplow DB. A de novo designed helix-turn-helix peptide forms nontoxic amyloid fibrils. Nat Struc Biol. 2000;7:1095–1099. doi: 10.1038/81937. [DOI] [PubMed] [Google Scholar]
- 71.Ghosh D, Singh PK, Sahay S, Jha NN, Jacob RS, Sen S, Kumar A, Riek R, Maji SK. Structure based aggregation studies reveal the presence of helix-rich intermediate during α-synuclein aggregation. Sci Rep. 2015;5:9228. doi: 10.1038/srep09228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rojas AV, Liwo A, Scheraga HA. A study of the alpha-helical intermediate preceding the aggregation of the amino-terminal fragment of the β amyloid peptide (Aβ(1–28)) J Phys Chem B. 2011;115:12978–12983. doi: 10.1021/jp2050993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vivekanandan S, Brender JR, Lee SY, Ramamoorthy A. A partially folded structure of amyloid-β(1–40) in an aqueous environment. Biochem Biophys Res Commun. 2011;411:312–316. doi: 10.1016/j.bbrc.2011.06.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Qi R, Luo Y, Ma B, Nussinov R, Wei G. Conformational distribution and alpha-helix to beta-sheet transition of human amylin fragment dimer. Biomacromolecules. 2014;15:122–131. doi: 10.1021/bm401406e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dicko C, Kenney JM, Vollrath F. β-silks: Enhancing and controlling aggregation. Adv Protein Chem. 2006;73:17–53. doi: 10.1016/S0065-3233(06)73002-9. [DOI] [PubMed] [Google Scholar]
- 76.Hauser CA, Deng R, Mishra A, Loo Y, Khoe U, Zhuang F, Cheong DW, Accardo A, Sullivan MB, Riekel C, et al. Natural tri- to hexapeptides self-assemble in water to amyloid beta-type fiber aggregates by unexpected α-helical intermediate structures. Proc Natl Acad Sci U S A. 2011;108:1361–1366. doi: 10.1073/pnas.1014796108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Apostolidou M, Jayasinghe SA, Langen R. Structure of alpha-helical membrane-bound human islet amyloid polypeptide and its implications for membrane-mediated misfolding. J Biol Chem. 2008;283:17205–17210. doi: 10.1074/jbc.M801383200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cao P, Abedini A, Wang H, Tu LH, Zhang X, Schmidt AM, Raleigh DP. Islet amyloid polypeptide toxicity and membrane interactions. Proc Natl Acad Sci U S A. 2013;110:19279–19284. doi: 10.1073/pnas.1305517110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yanagi K, Ashizaki M, Yagi H, Sakurai K, Lee YH, Goto Y. Hexafluoroisopropanol induces amyloid fibrils of islet amyloid polypeptide by enhancing both hydrophobic and electrostatic interactions. J Biol Chem. 2011;286:23959–23966. doi: 10.1074/jbc.M111.226688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cho SS, Reddy G, Straub JE, Thirumalai D. Entropic stabilization of proteins by tmao. J Phys Chem B. 2011;115:13401–13407. doi: 10.1021/jp207289b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Reddy G, Straub JE, Thirumalai D. Influence of preformed Asp23-Lys28 salt bridge on the conformational fluctuations of monomers and dimers of abeta peptides with implications for rates of fibril formation. J Phys Chem B. 2009;113:1162–1172. doi: 10.1021/jp808914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chimon S, Shaibat MA, Jones CR, Calero DC, Aizezi B, Ishii Y. Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of alzheimer’s β-amyloid. Nat Struc Mol Biol. 2007;14:1157–1164. doi: 10.1038/nsmb1345. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.