

Abstract
Mitotic kinesin Eg5 is an attractive anti-cancer drug target. Discovery of Eg5 inhibitors has been focused on targeting the “monastrol-binding site”. However, acquired drug resistance has been reported for such inhibitors. Therefore, identifying new Eg5 inhibitors which function through a different mechanism(s) could complement current drug candidates and improve drug efficacy. In this study, we explored a novel allosteric site of Eg5 and identified new Eg5 inhibitors through structure-based virtual screening. Experiments with the Saturation-Transfer Difference (STD) NMR demonstrated that the identified Eg5 inhibitor SRI35566 binds directly to Eg5 without involving microtubules. Moreover, SRI35566 and its two analogs significantly induced monopolar spindle formation in colorectal cancer HCT116 cells and suppressed cancer cell viability and colony formation. Together, our findings reveal a new allosteric regulation mechanism of Eg5 and a novel drug targeting site for cancer therapy.
Introduction
Kinesins represent a family of cytoskeletal motor proteins that utilize the energy from ATP hydrolysis to perform mechanical work along microtubules (MT) and mediate cellular processes such as cargo transport, spindle assembly and chromosome movement.1 Mitotic kinesins are required for various aspects of mitosis, including bipolar spindle assembly, chromosome alignment, chromosome segregation and cytokinesis.2 Mitotic kinesin Eg5, a plus-end directed member of the kinesin-5 subfamily, is an attractive anti-cancer drug target. Inhibition of Eg5 function blocks centrosome migration and leads to cell cycle arrest, and eventually apoptotic cell death.3 Since Eg5 is exclusively involved in mitotic spindle of proliferating cells, Eg5 specific inhibitors exhibit improved safety profiles compared to other traditional anti-mitotic drugs which target the multi-function relevant MT. Discovery of small molecule inhibitors of Eg5 has attracted much attentions in the past decade and several Eg5 inhibitors have advanced into clinical trials.4-18
Monastrol is the first Eg5 inhibitor that was identified over a decade ago.19 Since then, drug discovery studies targeting Eg5 have been mainly focused on an allosteric site where monastrol binds. However, mutagenesis studies have demonstrated that subtle changes at this “monastrol-binding site” conferred resistance to ligand binding without affecting the enzymatic function of Eg5, indicating acquired drug resistance of current Eg5 inhibitors.20, 21 In addition, mutations at the monastrol-binding site were found in Eg5 inhibitor (ispinesib) resistant cancer cells.22 Therefore, identification of new inhibitors that interact at a different binding site(s) of Eg5 could be a unique and important strategy to complement current Eg5 drug candidates.
Experimental studies have demonstrated the existence of multiple allosteric sites of Eg5.23-25 However, the exact locations of these allosteric sites are not clear. We have previously conducted molecular dynamics (MD) simulation studies of Eg5 and identified several novel allosteric sites (Figure 1) by using correlation-analysis and binding site mapping based on the simulation-generated structural and dynamics results.26 The identified S1 and S2 sites have been predicted as suitable for tight binding of small molecules, and therefore are potential targeting sites for the discovery of novel Eg5 inhibitors. A recently published crystal structure of an Eg5-inhibitor complex27 confirmed the existence of the predicted S2 site and validated our computational strategy for identifying novel allosteric sites.

The other identified allosteric S1 site locates nearby the functionally important kinesin neck linker region.28 It has long been recognized that conformational changes of kinesin neck linker are correlated with structural relaxation of kinesin α4–helix, and such a correlation is crucial for the force-generating behavior of kinesin proteins.29-31 A ligand that binds at the S1 site would block the interactions between the neck linker and the kinesin core structure, interrupt the force generating conformational changes, and consequently inhibit the kinesin functions. So far, no published studies have targeted this allosteric S1 site. In the present study, we explored the S1 site of Eg5 and identified several novel inhibitors through structure-based virtual screening (SBVS). These Eg5 inhibitors significantly induced monopolar spindle formation and suppressed cell viability of colorectal cancer cells.
Methods and Materials
Preparation of Eg5 motor protein
Protein expression and purification were based on published protocols.32-34 Eg5 motor domain (amino acids 15-368) was cloned into pEt24a and expressed in Escherichia coli BL21 (DE3) cells for 12 h at 30 °C. Protein purification was carried out in two steps with His-tag affinity and gel filtration chromatography. Specifically, cell pellet was resuspended in the lysis buffer (75 mM Tris-HCI pH 8.0, 300 mM NaCl, 5% glycerol, 20 mM imidazole, 0.1% Triton X-100, 0.5 mM TCEP) supplemented with a protease-inhibitor cocktail and 1μg/ml Benzonase nuclease, and was lysed by two passages through a French press. Cell debris was removed by centrifugation and the clear supernatant was passed through a Nickel-chelating Sepharose column (Amersham Bioscience) equilibrated with the lysis buffer. The column was then washed extensively with washing buffer (20 mM Tris-HCI pH 8.0, 300 mM NaCI, 5% glycerol, 20 mM imidazole and 0.5 mM TCEP) to remove non-specific proteins. The bound proteins were eluted using a 300 ml linear gradient of 50-400 mM imidazole in an elution buffer (20 mM Tris-HCI pH 8.0, 300 mM NaCI, 5% glycerol, 400 mM imidazole and 0.5 mM TCEP). Eluted fractions were analyzed by SDS-PAGE, and fractions containing Eg5 were pooled. Further purification was performed with a gel-filtration column (Superdex 200 26/60 Amersham Biosciences) (buffer: 50 mM phosphate pH 7.4, 100 mM NaCI and 1 mM DTT). Purified protein fractions were pooled together and concentrated to 10 mg/ml, frozen quickly in liquid nitrogen, and stored at −20°C.
Virtual compound library
For the purpose of applying structure-based virtual screening, we assembled a compound library consisting of approximately 500,000 structurally diverse compounds selected from different commercial sources. Specifically, structures of a total amount of approximately eight million compounds were downloaded directly from the websites of ten vendors (Asinex, Chembridge, ChemDiv, Enamine, FCH group, InterBioScreen, Life chemicals, TIMTEC, SPECS and Vitas-M). From the eight million commercially available compounds, we first identified the most diverse set of 100,000 structurally representative compounds using the clustering and diversity analysis protocols of Pipeline Pilot;35 then for each of the 100,000 compounds, four structurally most similar analogs were selected based on the Tanimoto coefficients calculated from the 2D structural fingerprints. Such an assembled library covers a large portion of chemical space, contains diverse structures, and could provide initial structure-activity relationship (SAR) information.
Molecular modeling
Structural model generation and molecular docking studies were conducted using the programs of the Schrödinger Suite 2014 (Schrödinger, LLC, New York, NY, 2014). The Eg5 model was generated based on the crystal structure of ADP-bound Eg5 complex (PDB ID: 1II6) using the protein preparation wizard of the maestro program.36 The SiteMap program25 was used to identify potential binding pocket(s) on Eg5 by mapping the surface of the structural models. The 3D structures of ligands were prepared using the LigPrep program.37 The Glide program38 was used for docking studies with the default parameters. Specifically, the Induced-Fit-Docking (IFD) protocol of Glide,39 which is capable of sampling dramatic side-chain conformational changes as well as minor changes in protein backbone structure, was applied to explore the binding mode of the identified active compounds. Residues within 5 Å of the docked ligands were allowed to be flexible and the docked results were scored using the extra-precision (XP) mode of Glide.
Virtual screening
Structure-based virtual screening was performed using the virtual screening workflow of maestro, which uses a three-step docking/scoring protocol implemented in Glide. 3D conformations of the 500,000 assembled compounds were prepared using LigPrep, and a total of 923,408 conformers were generated. All of the 923,408 conformers were first docked into the S1 site of Eg5 using the high-throughput virtual screening (HTVS) mode; the top 5% best-scored conformers were then re-docked and scored using the standard-precision (SP) mode; the top 10% of the SP docking resulted conformers were further docked and scored using the extra-precision (XP) mode. Finally, the top 25% of the best XP-scored conformers were outputted for visual examination.
Enzyme inhibition assay
The ADP Hunter™ Plus assay from DiscoveRx, which detects the MT-stimulated enzymatic ATPase activity of Eg5, was used to evaluate compound activity. Compounds were first tested in triplicates at 100 μM; hit compounds were further tested with a 10-point two-fold serial dilution to confirm their activity and determine their IC50 values. S-Trityl-L-cysteine (STLC), a selective Eg5 inhibitor,40, 41 was used as the control compound. Specifically, 20 μl of 15 mM PIPES (pH 7.0) containing 1 mM MgCl2, 50 nM MT, 20 μM Paclitaxel, 200 μM ATP, 5% DMSO, 60 nM Eg5 proteins and 1:2 serial dilutions of each individual compound starting from 1000 μM were added to each well of a 96-well plate. The plate was incubated at room temperature for 0.5 h and then the ADP Hunter™ Plus reagents were added. The plate was further incubated for 0.5 h and then read for fluorescence (ex.530/em.590) on Synergy 4 (BioTek).
NMR spectroscopy
STD-NMR data were collected following established protocols42, 43 using a Bruker DRX500. Samples containing SRI35566 and Eg5 protein at a concentration ratio of 20:1 were prepared in D2O. STD-NMR spectra were recorded with a total of 32 K points, 80 scans, and selective saturation of protein resonances at 0, 0.65, 1.67, and 7.61 ppm (−8.18 ppm for the reference spectra), using a series of SEDUCE pulses (1000 points, 50 ms), for a total saturation time of 10 s (SEDUCE-1 pulse is similar to a Gaussian pulse, and has been used by other laboratories.44 Reference experiments using the free ligands themselves (i.e. without Eg5) were performed under the same experimental conditions to verify true ligand binding.
Cell culture
Colorectal cancer cell line HCT116 was obtained from ATCC, and cultured in RPMI1640 medium containing 10% fetal bovine serum, 2 mM of L-glutamine, 100 units/ml of penicillin, and 100 μg/ml of streptomycin. Cells were grown under standard cell culture conditions at 37 °C in a humidified atmosphere with 5% CO2.
Confocal microscopy
HCT116 cells were grown on glass coverslips. After overnight incubation, the cells were treated with each individual compound for 16 h. The cells were fixed with 4% paraformaldehyde in phosphate-buffered saline for 20 min, and permeabilized with 0.2% Triton X-100 for 10 min at room temperature. The cells were then incubated with anti-α-tubulin-FITC antibody (Sigma) for 45 min, and nuclei were stained with NucRed647 for 10 min. The cells were then examined by a laser-scan confocal microscope (Leica DMI 4000 B). All images were captured with an HCX PL Apo 63x oil immersion objective. Images were processed and analyzed using Leica's LAS Image Analysis software. The percentage of mitotic cells with monopolar spindles was calculated out of a total number of a minimum of 20 mitotic cells per coverslip that were detected in different and randomly chosen microscopic fields.
Cell viability assay
Cells were seeded into 96-well tissue culture treated microtiter plates at a density of 4000 cells/well. After overnight incubation, the cells were treated with compounds for 96 h. Cell viability was measured by the CellTiter-Glo Assay (Promega).
Colony formation assay
HCT116 cells were seeded at a density of 500 cells/well into 6-well plates. After overnight culture, the cells were treated with compounds at 80 μM, and media were replenished every 3 days. After being incubated for 14 days, colonies were fixed with 4% formaldehyde, stained with 0.5 mg/mL crystal violet, and imaged on a FluorChem HD2 Imager System (Alpha Innotech).
Results
Structural characterization of the allosteric S1 site
Our previous study based on MD simulations demonstrated that the residues at the S1 site of Eg5 correlate dynamically with the active site (nucleotide-binding site) residues, suggesting compounds that bind to the S1 site could allosterically affect the function of Eg5.26 We analyzed the Eg5 crystal structure (PDB ID: 1II6) using the SiteMap program, which was designed for mapping and scoring potential binding site(s) based on properties such as binding pocket size, exposure/enclosure, contact, hydrophobic/hydrophilic balance, donor/acceptor character, etc. A SiteScore value of 0.80 has been shown to accurately distinguish a drug-binding site from non-drug-binding sites.25 The predicted SiteScore value of the Eg5 S1 site is 0.98, suggesting an excellent site for tight binding of small molecule drugs. As shown in Figure 2, the S1 site locates at the opposite side of the active site and consists of residues from the short α5-helix and the surrounding beta-sheets. It is an open pocket formed mainly by hydrophobic residues, including Leu160, Leu161, Ile163, Ile196, Leu199, Val238, Val264, Ile319 and Leu320, with several polar (Ser159, Ser240, Asn262 and Ser323) and charged (Asp322, Lys260) residues at the entrance area.

Identification of an Eg5 inhibitor through structure-based virtual screening
To explore whether the S1 pocket is a real binding site that can be targeted to modulate Eg5 function, we conducted SBVS to identify compounds that can potentially bind to the S1 site. We assembled 500,000 structurally diverse compounds from approximately eight million commercially available compounds. Using the Eg5 crystal structure as the receptor template, we screened these 500,000 compounds through a three-stage docking process. From the top-scored SBVS results, we selected 50 compounds that showed sufficient structural complementarity to the S1 site based on visual examination of the docked Eg5-compound complex models. Among the 50 selected compounds, 37 commercially available compounds were finally purchased and tested in the Eg5 ATPase assays. One compound, SRI35566 (Figure 3A), was confirmed by the serial dilution assay as an Eg5 inhibitor with an IC50 value of 65 μM.

SRI35566 binds directly to Eg5 without involving microtubule
Since the ADP Hunter™ plus assay detects the MT-stimulated Eg5 ATPase activities, there is a chance that the identified active compounds may inhibit Eg5 function through a MT related mechanism, which is unwanted due to the side effects of interfering MT.45 For instance, compounds that bind to MT (such as taxanes46) or bind to kinesin-MT complex (such as AZ8247) will be detected as actives in this assay. To examine whether the inhibitory effect of SRI35566 is related to MT, we applied STD-NMR to test the Eg5-SRI35566 binding in the absence of MT. STD-NMR is a technique that not only detects transient binding, but can also provide information regarding which part(s) of a ligand interacts directly with a receptor.42, 44, 48 In the case that the ligand does not bind to the protein in a ligand-protein mixture sample, no STD-NMR signal will be detected. The observed STD-NMR spectrum of the SRI35566/Eg5 mixture sample proved that SRI35566 bound directly to Eg5 (Figure 4). As a control, no STD signals were present in the free ligand sample (data not shown), which confirmed that the observed STD signals in the presence of Eg5 were due to a true saturation transfer from the protein.
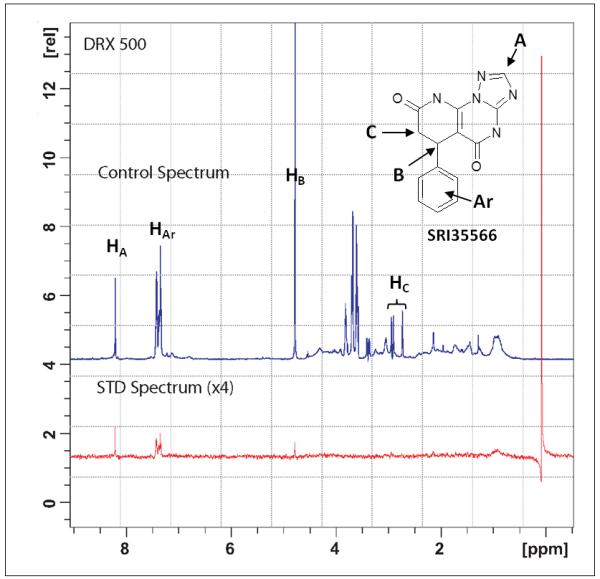
To further explore the structural insight of Eg5-SRI35566 interactions, we re-docked the compound using an Induce-Fit Docking (IFD) protocol to take into consideration structural flexibility of the binding site residues. The predicted Eg5-SRI35566 complex model (Figure 5A) shows that SRI35566 fits well into the S1 pocket: the phenyl ring of SRI35566 is buried deeply into the hydrophobic core; and the heterocyclic fragment of SRI35566 further stabilizes its binding by forming multiple hydrogen bonds with the sidechain and/or mainchain atoms of the residues near the entrance. Interestingly, these hydrogen atoms that showed clear STD-NMR signals (Figure 4) are in close proximity to the Eg5 residues in the docked model (Figure 5A). The distances of the closest protein-ligand atom pairs for HAr, HA and HB are 2.24, 2.68 and 2.22 Å, respectively. As a comparison, the closest distance for the HC pairs, which did not show observable STD-NMR signal, is 4.21 Å. The consistency between the docked model and the STD-NMR results supports the predicted binding mode at the S1 site. Taken together, these results suggest that SRI35566 inhibits Eg5 through a direct binding at the allosteric S1 site.

Analog exploration
A hit expansion effort based on structural similarity and substructure searches was further devoted to identifying commercial analogs of SRI35566. About 5000 compounds were selected using different search strategies. Those compounds were then docked into the S1 site of Eg5 and visually examined for their structural complementary to the binding site. Thirty-five SRI35566 “analogs” that docked well into the S1 site were purchased and tested in the Eg5 ATPase assay. Two compounds, SRI35565 and SRI35564, were confirmed as Eg5 inhibitors with IC50 values of 78.9 and 118.3 μM, respectively (Figure 3B & 3C). For both compounds, docking studies using the IFD protocol resulted in similar binding modes as SRI35566, with strong hydrophobic interactions at the main pocket and additional polar interactions at the gate region (Figure 5B & 5C).
Identified Eg5 inhibitors trigger monopolar spindle formation in cancer cells
Eg5 is a highly conserved plus-end-directed MT motor that plays a critical role in bipolar spindle assembly, and loss of Eg5 induces monopolar spindle formation.40, 49-51 Therefore, we performed spindle formation assay in colorectal cancer HCT116 cells. As shown in Figure 6, SRI35566, SRI35565 and SRI35564 at 80 μM significantly enhanced the percentage of HCT116 cells containing monopolar spindles.

Identified Eg5 inhibitors suppress cancer cell viability
Given that our identified Eg5 inhibitors can induce monopolar spindle formation in colorectal HCT116 cells, we then examined the effect of the Eg5 inhibitors on HCT116 cell viability. As shown in Figure 7, SRI35566, SRI35565 and SRI35564 inhibited HCT116 cancer cell viability at a concentration shown to suppress Eg5 activity and induce monopolar spindle formation in colorectal cancer cells. Moreover, we performed colony formation assays, and found that SRI35566, SRI35565 and SRI35564 at 80 μM significantly suppressed colony formation in HCT116 cells.

Discussion
Allostery is one of the most common and powerful means to regulate protein function.52, 53 Residues at the allosteric site(s) of a protein are evolutionarily less conserved compared to its active site residues. Allosteric ligands thus have better chances to bind specifically to the target protein and modulate its functions. While targeting allosteric site has been proven a promising strategy for the discovery and development of target-selective ligands, identifying allosteric site remains a challenging task. Most known allosteric sites, including the monastrol-site of Eg5, were found after the discovery of allosteric ligands which were identified either by serendipity or through a high-throughput screening campaign.54 Currently, there are no established experimental strategies for the discovery of novel allosteric sites.
Protein flexibility and conformational dynamics are the key elements of allosteric regulations. To this end, MD simulation is a very powerful tool for studying protein dynamics as well as the related intra-molecular interactions,55-63 and simulation-based computational methods have already been utilized to understand how a known allosteric ligand(s) regulates protein functions.60, 61, 64-66 In our previous study, we utilized such computational approaches to predict unknown allosteric sites and identified several Eg5 allosteric sites as potential drug targets.26 However, such theoretically predicted allosteric sites need to be experimentally validated. Notably, the crystal structure of Eg5-BI8 complex (PDB ID:3ZCW),27 which was solved about two years after our predictions, confirmed the existence of the allosteric S2 site of Eg5, and demonstrated the usefulness of such computational predictions.
The validation of the predicted S2 site of Eg5 is a rare case in that a ligand (BI8) that binds to the S2 site is available (although its binding site was unknown). In most cases, to confirm a novel allosteric site, a ligand that binds to this site needs to be identified first. In the current study, we applied SBVS to identify inhibitors that bind to the S1 site of Eg5. While a compound may bind to a certain site(s) of a protein without affecting its function, our results demonstrated that the identified compounds bound directly to the Eg5 protein, induced monopolar spindle formation, a phenotype of Eg5 inhibition, in colorectal cancer cells, and suppressed cancer cell viability and colony formation. These results confirmed the existence of the S1 site and its feasibility as a potential drug targeting site for modulating Eg5 functions. The identified Eg5 inhibitors provide a useful starting point for the development of lead compounds.
To the best of our knowledge, this is the first experimental demonstration of the allosteric S1 site. So far, three allosteric sites of Eg5 have been identified, including the monastrol-site, the S2 site and this S1 site. Unlike the monastrol-site, which is Eg5-specific due to a unique long loop-5 that forms part of the site, both S1 and S2 sites locate at the structurally conserved regions of kinesins, thus may be ubiquitous for the kinesin family. Several kinesin proteins have been demonstrated as promising drug targets for cancer treatment in recent years.67-69 The identified allosteric S1 and S2 sites thus provide an opportunity to develop selective kinesin inhibitors for drug development. We conducted multiple sequence alignment using ClustalX program,70 and compared the S1 site residues of several representative human kinesins, including KHC, the conventional N-terminal kinesin,1 KIFC1, a C-terminal kinesin,71 CENP-E, which possesses a long, kinesin-7 family-specific neck region,1, 72 and Eg5. As shown in Supplemental Table 1, while residues at the core and the gate regions of the S1 site keep their hydrophobic or polar properties, only a few of them are conserved, other binding site residues are also not conserved among these kinesins, indicating that compounds could be designed to selectively inhibit a specific kinesin by targeting the S1 site. By saying so, the existence of S1 and S2 on other kinesins needs to be further validated.
While the present study demonstrated the existence of the allosteric S1 site, whether the identified compounds selectively inhibit Eg5 needs to be further evaluated. Among the three Eg5 inhibitors identified in this study, SRI35564 displayed the highest cytotoxic effect on colorectal cancer HCT116, but has a relatively weaker potency on Eg5 ATPase inhibition, indicating a potential off-target effect of this compounds. Further experimental studies, such as mutagenesis and crystallographic studies, could provide additional structural and biological details of the interactions between Eg5 and its inhibitors and help to develop selective inhibitors of Eg5 for drug discovery purposes.
Supplementary Material
Conclusions Acknowledgments
We thank Dr. N. Rama Krishna and Dr. Ronald Shin of the UAB High Field NMR Facility for their supports on the collection of NMR spectrum. This work was partially supported by grants from Southern Research Institute, the National Institutes of Health R21CA182056 and Alabama Innovation Fund.
Footnotes
Conflict of Interest:
The authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.Miki H, Okada Y, Hirokawa N. Analysis of the kinesin superfamily: insights into structure and function. Trends in Cell Biology. 2005;15:467–476. doi: 10.1016/j.tcb.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 2.Huszar D, Theoclitou ME, Skolnik J, Herbst R. Kinesin motor proteins as targets for cancer therapy. Cancer and Metastasis Reviews. 2009;28:197–208. doi: 10.1007/s10555-009-9185-8. [DOI] [PubMed] [Google Scholar]
- 3.Tao WK, et al. Induction of apoptosis by an inhibitor of the mitotic kinesin KSP requires both activation of the spindle assembly checkpoint and mitotic slippage. Cancer Cell. 2005;8:49–59. doi: 10.1016/j.ccr.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 4.Rickert KW, et al. Discovery and biochemical characterization of selective ATP competitive inhibitors of the human mitotic kinesin KSP. Arch Biochem Biophys. 2008;469:220–31. doi: 10.1016/j.abb.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 5.Luo LS, et al. Conformation-dependent ligand regulation of ATP hydrolysis by human KSP: Activation of basal hydrolysis and inhibition of microtubule-stimulated hydrolysis by a single, small molecule modulator. Journal of the American Chemical Society. 2008;130:7584–7591. doi: 10.1021/ja710889h. [DOI] [PubMed] [Google Scholar]
- 6.Cox CD, et al. Kinesin spindle protein (KSP) inhibitors. 9. Discovery of (2S)-4-(2,5-difluorophenyl)-N-[(3R,4S)-3-fluoro-1-methylpiperidin-4-yl]- 2-(hydroxymethyl)-N-methyl-2-phenyl-2,5-dihydro-1H-pyrrole-1-carboxamide (MK-0731) for the treatment of taxane-refractory cancer. Journal of Medicinal Chemistry. 2008;51:4239–4252. doi: 10.1021/jm800386y. [DOI] [PubMed] [Google Scholar]
- 7.Pinkerton AB, Lee TT, Hoffman TZ, Wang Y, Kahraman M. Synthesis and SAR of thiophene containing kinesin spindle protein (KSP) inhibitors. Bioorganic & Medicinal Chemistry Letters. 2007;17:3562–3569. doi: 10.1016/j.bmcl.2007.04.076. [DOI] [PubMed] [Google Scholar]
- 8.Parrish CA, et al. Novel ATP-competitive kinesin spindle protein inhibitors. Journal of Medicinal Chemistry. 2007;50:4939–4952. doi: 10.1021/jm070435y. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Saez I, et al. Structure of human Eg5 in complex with a new monastrol-based inhibitor bound in the R configuration. J Biol Chem. 2007;282:9740–7. doi: 10.1074/jbc.M608883200. [DOI] [PubMed] [Google Scholar]
- 10.Maliga Z, et al. A pathway of structural changes produced by monastrol binding to eg5. Journal of Biological Chemistry. 2006;281:7977–7982. doi: 10.1074/jbc.M511955200. [DOI] [PubMed] [Google Scholar]
- 11.Kim KS, et al. Synthesis and SAR of pyrrolotriazine-4-one based Eg5 inhibitors. Bioorg Med Chem Lett. 2006;16:3937–42. doi: 10.1016/j.bmcl.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 12.Zhang YJ, Xu WF. Progress on kinesin spindle protein inhibitors as anti-cancer agents. Anti-Cancer Agents in Medicinal Chemistry. 2008;8:698–704. [PubMed] [Google Scholar]
- 13.El-Nassan HB. Advances in the discovery of kinesin spindle protein (Eg5) inhibitors as antitumor agents. Eur J Med Chem. 2013;62:614–31. doi: 10.1016/j.ejmech.2013.01.031. [DOI] [PubMed] [Google Scholar]
- 14.Knight SD, Parrish CA. Recent progress in the identification and clinical evaluation of inhibitors of the mitotic kinesin KSP. Current Topics in Medicinal Chemistry. 2008;8:888–904. doi: 10.2174/156802608784911626. [DOI] [PubMed] [Google Scholar]
- 15.Jiang C, You QD, Li ZY, Guo QL. Kinesin spindle protein inhibitors as anticancer agents. Expert Opinion on Therapeutic Patents. 2006;16:1517–1532. [Google Scholar]
- 16.Coleman PJ, Fraley ME. Inhibitors of the mitotic kinesin spindle protein. Expert Opinion on Therapeutic Patents. 2004;14:1659–1667. [Google Scholar]
- 17.Xing ND, et al. A potent chemotherapeutic strategy in prostate cancer: S-(methoxytrityl)-L-cysteine, a novel Eg5 inhibitor. Asian Journal of Andrology. 2011;13:236–241. doi: 10.1038/aja.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barsanti PA, et al. The discovery of tetrahydro-beta-carbolines as inhibitors of the kinesin Eg5. Bioorg Med Chem Lett. 2010;20:157–60. doi: 10.1016/j.bmcl.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 19.Mayer TU, et al. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science. 1999;286:971–974. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]
- 20.Brier S, Lemaire D, DeBonis S, Forest E, Kozielski F. Molecular dissection of the inhibitor binding pocket of mitotic kinesin Eg5 reveals mutants that confer resistance to antimitotic agents. J Mol Biol. 2006;360:360–76. doi: 10.1016/j.jmb.2006.04.062. [DOI] [PubMed] [Google Scholar]
- 21.Maliga Z, Mitchison TJ. Small-molecule and mutational analysis of allosteric Eg5 inhibition by monastrol. BMC Chem Biol. 2006;6:2. doi: 10.1186/1472-6769-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo L, et al. ATP-competitive inhibitors of the mitotic kinesin KSP that function via an allosteric mechanism. Nature Chemical Biology. 2007;3:722–726. doi: 10.1038/nchembio.2007.34. [DOI] [PubMed] [Google Scholar]
- 23.Matsuno K, Sawada J, Sugimoto M, Ogo N, Asai A. Bis(hetero)aryl derivatives as unique kinesin spindle protein inhibitors. Bioorganic & Medicinal Chemistry Letters. 2009;19:1058–1061. doi: 10.1016/j.bmcl.2009.01.018. [DOI] [PubMed] [Google Scholar]
- 24.Learman SS, et al. NSC 622124 Inhibits Human Eg5 and Other Kinesins via Interaction with the Conserved Microtubule-Binding Site. Biochemistry. 2009;48:1754–1762. doi: 10.1021/bi801291q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheth PR, et al. Novel Benzimidazole inhibitors Bind to a Unique Site in the Kinesin Spindle Protein Motor Domain. Biochemistry. 2010;49:8350–8358. doi: 10.1021/bi1005283. [DOI] [PubMed] [Google Scholar]
- 26.Zhang W. Exploring the intermediate states of ADP-ATP exchange: a simulation study on Eg5. J Phys Chem B. 2011;115:784–95. doi: 10.1021/jp107255t. [DOI] [PubMed] [Google Scholar]
- 27.Ulaganathan V, et al. Structural insights into a unique inhibitor binding pocket in kinesin spindle protein. J Am Chem Soc. 2013;135:2263–72. doi: 10.1021/ja310377d. [DOI] [PubMed] [Google Scholar]
- 28.Turner J, et al. Crystal structure of the mitotic spindle kinesin Eg5 reveals a novel conformation of the neck-linker. J Biol Chem. 2001;276:25496–502. doi: 10.1074/jbc.M100395200. [DOI] [PubMed] [Google Scholar]
- 29.Sindelar CV, Downing KH. An atomic-level mechanism for activation of the kinesin molecular motors. Proc Natl Acad Sci U S A. 2010;107:4111–6. doi: 10.1073/pnas.0911208107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenfeld SS, Xing J, Jefferson GM, King PH. Docking and rolling, a model of how the mitotic motor Eg5 works. J Biol Chem. 2005;280:35684–95. doi: 10.1074/jbc.M506561200. [DOI] [PubMed] [Google Scholar]
- 31.Goulet A, et al. The structural basis of force generation by the mitotic motor kinesin-5. J Biol Chem. 2012;287:44654–66. doi: 10.1074/jbc.M112.404228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cochran JC, Gatial JE, Kapoor TM, Gilbert SP. Monastrol inhibition of the mitotic kinesin Eg5. Journal of Biological Chemistry. 2005;280:12658–12667. doi: 10.1074/jbc.M413140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan YW, et al. Inhibition of a mitotic motor protein: Where, how, and conformational consequences. Journal of Molecular Biology. 2004;335:547–554. doi: 10.1016/j.jmb.2003.10.074. [DOI] [PubMed] [Google Scholar]
- 34.DeBonis S, et al. Interaction of the mitotic inhibitor monastrol with human kinesin Eg5. Biochemistry. 2003;42:338–49. doi: 10.1021/bi026716j. [DOI] [PubMed] [Google Scholar]
- 35.Baurin N, et al. Drug-like annotation and duplicate analysis of a 23-supplier chemical database totalling 2.7 million compounds. Journal of Chemical Information and Computer Sciences. 2004;44:643–651. doi: 10.1021/ci034260m. [DOI] [PubMed] [Google Scholar]
- 36.Maestro. version 9.8 Schrödinger, LLC; New York, NY: 2014. [Google Scholar]
- 37.Behnke-Parks WM, et al. Loop L5 Acts as a Conformational Latch in the Mitotic Kinesin Eg5. Journal of Biological Chemistry. 2011;286:5242–5253. doi: 10.1074/jbc.M110.192930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glide. version 6.3 Schrödinger, LLC; New York, NY: 2014. [Google Scholar]
- 39.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. Journal of Medicinal Chemistry. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 40.Skoufias DA, et al. S-trityl-L-cysteine is a reversible, tight binding inhibitor of the human kinesin Eg5 that specifically blocks mitotic progression. J Biol Chem. 2006;281:17559–69. doi: 10.1074/jbc.M511735200. [DOI] [PubMed] [Google Scholar]
- 41.DeBonis S, et al. In vitro screening for inhibitors of the human mitotic kinesin Eg5 with antimitotic and antitumor activities. Mol Cancer Ther. 2004;3:1079–90. [PubMed] [Google Scholar]
- 42.Jayalakshmi V, Krishna NR. CORCEMA refinement of the bound ligand conformation within the protein binding pocket in reversibly forming weak complexes using STD-NMR intensities. Journal of Magnetic Resonance. 2004;168:36–45. doi: 10.1016/j.jmr.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 43.Krishna NR, Jayalakshmi V. Complete relaxation and conformational exchange matrix analysis of STD-NMR spectra of ligand-receptor complexes. Progress in Nuclear Magnetic Resonance Spectroscopy. 2006;49:1–25. [Google Scholar]
- 44.Peng JW, Lepre CA, Fejzo J, Abdul-Manan N, Moore JM. Nuclear magnetic resonance-based approaches for lead generation in drug discovery. Methods Enzymol. 2001;338:202–30. doi: 10.1016/s0076-6879(02)38221-1. [DOI] [PubMed] [Google Scholar]
- 45.Quasthoff S, Hartung HP. Chemotherapy-induced peripheral neuropathy. J Neurol. 2002;249:9–17. doi: 10.1007/pl00007853. [DOI] [PubMed] [Google Scholar]
- 46.Hagiwara H, Sunada Y. Mechanism of taxane neurotoxicity. Breast Cancer. 2004;11:82–5. doi: 10.1007/BF02968008. [DOI] [PubMed] [Google Scholar]
- 47.Wu J, et al. Discovery and mechanistic study of a small molecule inhibitor for motor protein KIFC1. ACS Chem Biol. 2013;8:2201–8. doi: 10.1021/cb400186w. [DOI] [PubMed] [Google Scholar]
- 48.Jayalakshmi V, Krishna NR. Complete relaxation and conformational exchange matrix (CORCEMA) analysis of intermolecular saturation transfer effects in reversibly forming ligand-receptor complexes. Journal of Magnetic Resonance. 2002;155:106–118. doi: 10.1006/jmre.2001.2499. [DOI] [PubMed] [Google Scholar]
- 49.Kapitein LC, et al. The bipolar mitotic kinesin Eg5 moves on both microtubules that it crosslinks. Nature. 2005;435:114–8. doi: 10.1038/nature03503. [DOI] [PubMed] [Google Scholar]
- 50.Kapoor TM, Mayer TU, Coughlin ML, Mitchison TJ. Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J Cell Biol. 2000;150:975–88. doi: 10.1083/jcb.150.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blangy A, et al. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell. 1995;83:1159–69. doi: 10.1016/0092-8674(95)90142-6. [DOI] [PubMed] [Google Scholar]
- 52.MOUND J. Chance and Necessity: Essay on the Natural Philosophy of Modern Biology. Penguin Books; 1977. [Google Scholar]
- 53.Fenton AW. Allostery: an illustrated definition for the 'second secret of life'. Trends Biochem Sci. 2008;33:420–5. doi: 10.1016/j.tibs.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lindsley JE, Rutter J. Whence cometh the allosterome? Proceedings of the National Academy of Sciences of the United States of America. 2006;103:10533–10535. doi: 10.1073/pnas.0604452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grant BJ, et al. Novel allosteric sites on Ras for lead generation. PLoS One. 2011;6:e25711. doi: 10.1371/journal.pone.0025711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dixit A, Verkhivker GM. Computational modeling of allosteric communication reveals organizing principles of mutation-induced signaling in ABL and EGFR kinases. PLoS Comput Biol. 2011;7:e1002179. doi: 10.1371/journal.pcbi.1002179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhuravlev PI, Papoian GA. Protein functional landscapes, dynamics, allostery : a tortuous path towards a universal theoretical framework. Quarterly Reviews of Biophysics. 2010;43:295–332. doi: 10.1017/S0033583510000119. [DOI] [PubMed] [Google Scholar]
- 58.Ivetac A, McCammon JA. Mapping the druggable allosteric space of G-protein coupled receptors: a fragment-based molecular dynamics approach. Chem Biol Drug Des. 2010;76:201–17. doi: 10.1111/j.1747-0285.2010.01012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smock RG, Gierasch LM. Sending signals dynamically. Science. 2009;324:198–203. doi: 10.1126/science.1169377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kong Y, Karplus M. Signaling pathways of PDZ2 domain: a molecular dynamics interaction correlation analysis. Proteins. 2009;74:145–54. doi: 10.1002/prot.22139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bradley MJ, Chivers PT, Baker NA. Molecular dynamics simulation of the Escherichia coli NikR protein: Equilibrium conformational fluctuations reveal interdomain allosteric communication pathways. Journal of Molecular Biology. 2008;378:1155–1173. doi: 10.1016/j.jmb.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Silvestre-Ryan J, Lin YC, Chu JW. “Fluctuograms” Reveal the Intermittent Intra-Protein Communication in Subtilisin Carlsberg and Correlate Mechanical Coupling with Co-Evolution. Plos Computational Biology. 2011;7 doi: 10.1371/journal.pcbi.1002023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lange OF, Grubmuller H. Full correlation analysis of conformational protein dynamics. Proteins-Structure Function and Bioinformatics. 2008;70:1294–1312. doi: 10.1002/prot.21618. [DOI] [PubMed] [Google Scholar]
- 64.Ma BY, Tsai CJ, Haliloglu T, Nussinov R. Dynamic Allostery: Linkers Are Not Merely Flexible. Structure. 2011;19:907–917. doi: 10.1016/j.str.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shen A. Allosteric regulation of protease activity by small molecules. Molecular Biosystems. 2010;6:1431–1443. doi: 10.1039/c003913f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang J, et al. Conformational transition pathway in the allosteric process of human glucokinase. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:13368–13373. doi: 10.1073/pnas.0605738103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Y, et al. KIFC1 is a novel potential therapeutic target for breast cancer. Cancer Biol Ther. 2015;16:1316–22. doi: 10.1080/15384047.2015.1070980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Barakat MT, Humke EW, Scott MP. Kif3a is necessary for initiation and maintenance of medulloblastoma. Carcinogenesis. 2013;34:1382–92. doi: 10.1093/carcin/bgt041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wood KW, et al. Antitumor activity of an allosteric inhibitor of centromere-associated protein-E. Proc Natl Acad Sci U S A. 2010;107:5839–44. doi: 10.1073/pnas.0915068107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research. 1997:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mountain V, et al. The kinesin-related protein, HSET, opposes the activity of Eg5 and cross-links microtubules in the mammalian mitotic spindle. J Cell Biol. 1999;147:351–66. doi: 10.1083/jcb.147.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Garcia-Saez I, Yen T, Wade RH, Kozielski F. Crystal structure of the motor domain of the human kinetochore protein CENP-E. J Mol Biol. 2004;340:1107–16. doi: 10.1016/j.jmb.2004.05.053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.