

Abstract
Recent studies have revealed that bile acids (BAs) are not only facilitators of dietary lipid absorption but also important signaling molecules exerting multiple physiological functions. Some major signaling pathways involving the nuclear BAs receptor farnesoid X receptor and the G protein-coupled BAs receptor TGR5/M-BAR have been identified to be the targets of BAs. BAs regulate their own homeostasis via signaling pathways. BAs also affect diverse metabolic pathways including glucose metabolism, lipid metabolism and energy expenditure. This paper suggests the mechanism of controlling metabolism via BA signaling and demonstrates that BA signaling is an attractive therapeutic target of the metabolic syndrome.
Keywords: Bile acids, TGR5/M-BAR, Farnesoid X receptor, Glucose metabolism, Energy metabolism, Lipid metabolism, Bariatric surgery, Microbiota, Incretin, Bile acid binding resin
Core tip: Bile acids (BAs) are important molecules that participate in various metabolic pathways. BA signaling mechanisms are attractive therapeutic targets of the metabolic syndrome. In this review, we show the mechanisms of controlling glucose, lipid and energy metabolism via BA signaling. Furthermore, the authors also describe how those basic scientific studies have been applied to the clinical setting. Particularly, bile acid binding resin (BABR) originally used to treat hypercholesterolemia also stimulates incretin secretion and improves glucose metabolism. In addition to BABR, the clinical application of farnesoid X receptor and TGR5/M-BAR agonists are ongoing for the treatment of metabolic syndrome. The effects of bariatric surgery on glycemic control are also associated with BA metabolism.
INTRODUCTION
Bile acids (BAs) are the main constituents of bile and amphipathic molecules, containing both hydrophilic and hydrophobic regions. BAs are synthesized from cholesterol in the liver, stored in the gall bladder, and flow into the small intestine after meal ingestion. Intestinal BAs facilitate digestion and absorption of lipids and fat-soluble vitamins[1].
Recent reports suggest that BAs are responsible not only for the absorption of lipids but also for signal transduction. Some major signaling mechanisms have been identified, including the MAPK pathways, nuclear hormone receptor farnesoid X receptor (FXR)-mediated pathway and G protein-coupled receptor TGR5/M-BAR (also named GPR131)-mediated pathway[2-5]. BAs have been demonstrated to be natural ligands of FXR. The main role of the FXR signaling pathway is regulating both enterohepatic circulation and BA biosynthesis to maintain the homeostasis of BA[6]. In addition, FXR signaling has been known to regulate lipogenesis gene expression and improve hepatic steatosis[7]. Moreover, recent studies have shown that BAs and FXR signaling are associated with the beneficial glycemic effects of bariatric surgery and regulation of autophagy[8-10]. BAs also activate TGR5/M-BAR. The TGR5/M-BAR signaling pathway stimulates energy expenditure in both brown adipose tissue (BAT) as well as skeletal muscle[11]. Furthermore, TGR5/M-BAR plays a role in hepatic microcirculation as well as cytokine release from macrophages[12]. Taken together, BAs not only participate in the digestion and absorption of lipids but also in various metabolic pathways. BA signaling participates in various diseases such as cancer, immune disorders, and metabolic syndrome[13-15]. In this review, we summarize the current knowledge of the metabolic regulation mechanisms of BAs and propose BA signaling pathways as a therapeutic target of the metabolic syndrome.
BILE ACIDS METABOLISM
The majority of synthesized BAs are secreted into the bile and kept in the gallbladder. When food enters the gastrointestinal tract, bile flows into the small intestine, and are efficiently absorbed by active transport and passive diffusion in the terminal ileum. BAs are then transported again to the liver through the portal vein and re-uptaken at the sinusoidal membranes of hepatocytes. These BAs are then secreted into the bile again; each BA molecule can complete 4-12 cycles of circulation per day[16].
BA synthesis has two differential pathways: The “classic (or neutral) pathway” and the “alternative (or acidic) pathway”. In the classic pathway, the enzyme cholesterol-7α-hydroxylase (CYP7A1) hydroxylates the C7α position during the first step. In the alternate pathway, the enzyme sterol-27α-hydroxylase (CYP27A1) first hydroxylates the C27 position. The classic pathway seems more important than the alternative pathway because the classic pathway is responsible for maintaining cholesterol homeostasis by controlling BA synthesis[17]. The rate-limiting enzyme CYP7A1 converts cholesterol to 7α-hydroxycholesterol, and other enzymes including sterol-12α-hydroxylase (CYP8B1), 25-hydroxycholesterol-7α-hydroxylase (CYP7B1) and CYP27A1 convert 7α-hydroxycholesterol to primary BAs, including cholic acid (CA) and chenodeoxycholic acid (CDCA)[18]. CYP8B1 controls the production of CA, and CA regulates the CA/CDCA ratio in humans or the CA/MCA ratio in mice by mediating feedback regulation[19]. Regulation of this ratio is important because previous studies demonstrated that the ratio of CA/CDCA is associated with liver diseases in humans[20]. For example, this ratio is decreased in patients with liver cirrhosis and hepatic cancer but is increased in cholestasis. Most of the BAs are conjugated to glycine or taurine, and the ratio of BAs conjugated to taurine and glycine differ depending on the animal species. In humans, the ratio of BAs conjugated to taurine and glycine are approximately 1:2, and most BAs are conjugated with taurine in mice. BAs inhibit the expression of CYP7A1 and CYP8B1 in liver through several pathways, which are mainly FXR-dependent. BAs activate FXR, leading to the upregulation of a small heterodimer partner (SHP; NR0B2), which suppresses the activity of hepatocyte nuclear factor-4α (HNF-4α; NR2A1), liver X receptor (LXR; Nr1h3) and liver receptor homolog-1 (LRH-1; NR5A2), which are both required for transcriptional induction of BA synthesis enzymes via binding to BA-response elements in promoters[21-23]. Additionally, the intestinal activation of FXR by BAs causes an increased expression of fibroblast growth factor (FGF)-15 in rodents and FGF-19 in humans. BAs absorbed in the terminal ileum activate intestinal FXR and induce enterocytic production of FGF-15/19. This FGF-15/19 is passed from the portal vein to the hepatocytes and couples with a receptor, FGF receptor 4 (FGFR4). These signaling pathways via FGF-15/19 and FGFR4 induce receptor dimerization, autophosphorylation, and c-Jun N-terminal kinase pathway activation resulting in the repression of CYP7A1 transcription (Figure 1)[24,25]. A second BA receptor, TGR5/M-BAR, also contributes to regulation of BA homeostasis. TGR5/M-BAR knockout mice present with a decrease in the BA pool size and the impaired suppression of CYP7A1 expression upon BA administration[26,27]. Vitamin D also regulates BAs synthesis. Vitamin D receptor activation induces the expression of FGF-15/19, and BA synthesis is decreased by reducing CYP7A1 expression[28,29]. BAs regulate BA homeostasis via FXR, TGR5/M-BAR and other signaling pathways primarily by maintaining gene expression of the rate-limiting enzymes CYP7A1 and CYP8B1.
Figure 1.
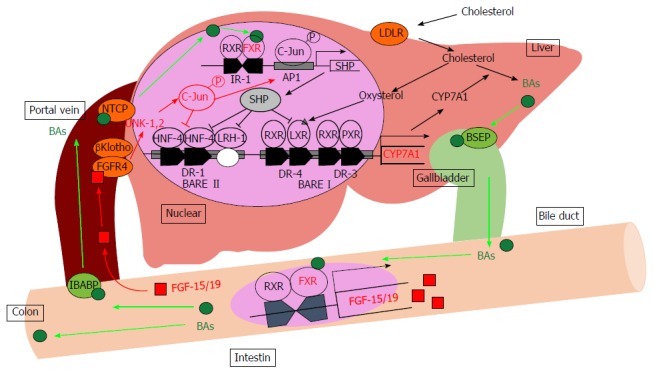
Bile acid metabolism in the liver. BAs induce the FXR-SHP-mediated pathway and repress BA synthesis enzyme gene expression such as CYP7A1 and CYP8B1. Synthesized BAs increase the expression of FGF-15/19 in the small intestine. FGF-15/19 signaling induces JNK pathway activation resulting in the repression of CYP7A1 transcription. AP1: Activator protein 1; BAs: Bile acids; BARE: Bile acid response element; BSEP: Bile salt export pump; CYP7A1: Cholesterol-7α-hydroxylase; DR: Direct repeat element; FGF-15/19: Fibroblast growth factor-15/19; FGFR4: Fibroblast growth factor receptor 4; FXR: Farnesoid X receptor; HNF-4: Hepatocyte nuclear factor; IBABP: Intestinal bile acid-binding protein; IR-1: Inverted repeat element-1; JNK: Jun-N-terminal kinase; LDLR: Low-density lipoprotein receptor; LRH-1: Liver receptor homolog-1; LXR: Liver X receptor; NTCP: Sodium-taurocholate cotransporting polypeptide; PXR: Pregnane X receptor; RXR: Retinoid X receptor; SHP: Small heterodimer partner.
BILE ACIDS IN GLUCOSE METABOLISM
Previous studies have clarified that BAs affect glucose metabolism. Glucose induces the expression of FXR and CYP7A1, and insulin reduces their expression in vitro[30]. Further studies have shown that BAs seem to regulate gluconeogenesis, but the mechanisms remain poorly understood. Some studies have indicated that the expression of phosphoenolpyruvate carboxykinase (PEPCK), which is the rate-limiting enzyme of gluconeogenesis, is suppressed by BAs in human liver cancer cells (HepG2 cells) and the mouse liver[31-33]. Additionally, enzymes such as glucose 6-phosphatase and fructose 1,6-bisphosphatase 1 which also participate in gluconeogenesis are repressed by BAs[31]. These effects are decreased in FXR and SHP knockout mice, which supports the idea that BAs suppress gluconeogenesis in a FXR-SHP-dependent manner[33]; however, others have reported that FXR-dependent signaling induces PEPCK expression and increases gluconeogenesis in primary hepatocytes and rat hepatoma cell lines[34]. In terms of glycogen synthesis, BAs increase hepatic glycogen synthesis and storage, resulting in decreased blood glucose levels in an FXR-dependent manner (Figure 2B)[35]. A previous study demonstrated that long-term FXR activation (3 mo) with a synthetic FXR agonist, GW4064, worsened glucose intolerance and insulin resistance in high-fat fed C57BL/6J mice[33,36]. The mechanism behind the effects of GW4064 is lowering the BA pool size following FXR activation. Some reports have suggested that short-term (10 d) FXR activation by the synthetic FXR agonist GW4064 reduced glycolytic gene expression and improved insulin resistance in ob/ob or db/db mice[35,37]. In contrast, the difference of the GW4064 administration period may lead to the opposite result. Long-term administration of BAs, the endogenous natural ligands of FXR, did not decrease the BA pool size and subsequently improved glucose intolerance and insulin resistance[36].
Figure 2.
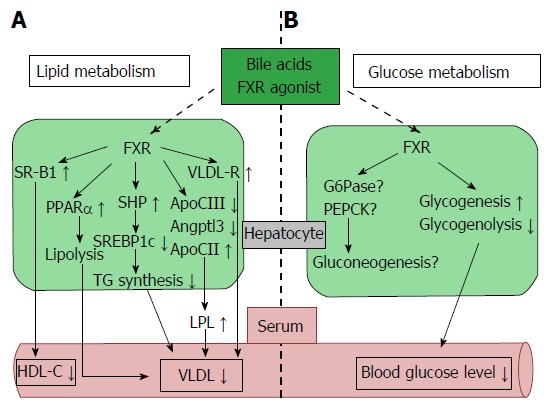
Farnesoid X receptor-dependent metabolic regulation in the liver. Hepatic FXR signaling regulates lipid and glucose metabolism. A: FXR signaling reduces lipogenesis (SREBP1c) and induces fatty acid β oxidation (PPARα) and plasma TG clearance (LPL and VLDL-R), resulting in decreased plasma VLDL levels. Plasma HDL-C uptake is also increased by FXR and SRB1 activity; B: FXR signaling up-regulates glycogenesis, down-regulates glycogenolysis, and reduces blood glucose levels. Hepatic FXR signaling is also associated with gluconeogenesis, but the controlling mechanism is still unclear. Angptl3: Angiopoietin-like protein 3; ApoCII/CIII: Apolipoprotein-CII/CIII; FXR: Farnesoid X receptor; G6Pase: Glucose-6-phosphatase; HDL-C: High density lipoprotein-cholesterol; LPL: Lipoprotein lipase; PEPCK: Phosphoenolpyruvate carboxykinase; PPARα: Peroxisome proliferator-activated receptor α; SR-B1: Scavenger receptor-B1; SREBP1c: Sterol regulatory element-binding protein 1c; TG: Triglyceride; VLDL-R: Very low density lipoprotein-receptor.
BA administration improved metabolism including glucose tolerance and insulin resistance. The beneficial effects of BAs, such as a decrease in gluconeogenesis and increase in glycogen synthesis, seem to occur not only through FXR signaling but also through a number of other signaling molecules, such as TGR5/M-BAR. BAs stimulate incretins, such as glucagon-like peptide-1 (GLP-1; Figure 3). GLP-1 is secreted by dietary stimulation from enteric L cells and promotes insulin secretion by binding to the GLP-1 receptor in the pancreatic β cell. Further, GLP-1 maintains pancreatic function, and GLP-1 receptor agonists have been developed for the treatment of diabetes[38]. TGR5/M-BAR signaling causes GLP-1 secretion in mouse enteroendocrine STC-1 cells[39]. Moreover, 6-ethyl-23(S)-methylcholic acid (6EMCA or INT-777[40]), a semisynthetic TGR5/M-BAR agonist, stimulates the secretion of GLP-1 in both mouse and human enteroendocrine cells. In the present study, knockdown of TGR5/M-BAR by shRNA decreased 6EMCA-induced secretion of GLP-1 in STC-1 cells[41]. The natural TGR5/M-BAR agonist oleanolic acid also improves the metabolism of glucose[42]. This evidence indicates the importance of TGR5/M-BAR in GLP-1 secretion. An in vivo study with TGR5/M-BAR transgenic and TGR5/M-BAR knockout mice strongly supports the relationship between TGR5/M-BAR and GLP-1 secretion[43]. Considering the current mechanism, TGR5/M-BAR activation increases cAMP levels and the ATP/ADP ratio, which then leads to depolarization of the plasma membrane as well as Ca2+ mobilization, resulting in increased GLP-1 release[41]. Additionally, a human genetic study revealed an association between a single nucleotide polymorphism, rs3731859, of the TGR5/M-BAR gene and various metabolic indexes including BMI, waist circumference, intramyocellular lipid, and fasting serum GLP-1 levels[44]. Hence, these findings suggested that GLP-1 secretion was stimulated by TGR5/M-BAR signaling in vivo. BAs and TGR5/M-BAR could become therapeutic targets of diabetes.
Figure 3.
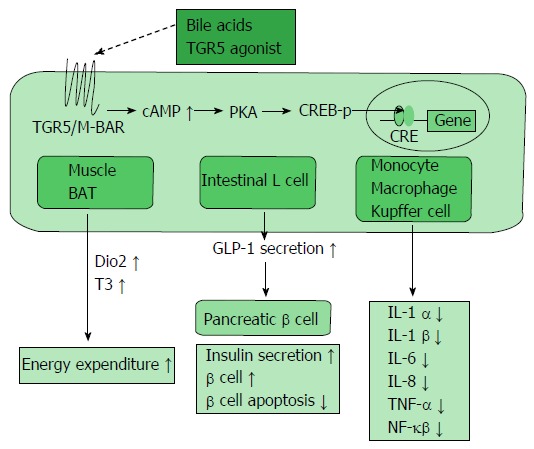
TGR5/M-BAR-dependent metabolic regulation. TGR5/M-BAR activation leads to increased intracellular cAMP levels, the activation of PKA and induction of CREB phosphorylation. This series of signaling activity induces the expression of genes bearing CRE and exists in various tissues. TGR5/M-BAR signaling induces energy expenditure in the muscle and BAT, increases GLP-1 secretion in the intestinal L cell, and reduces inflammatory cytokine release in immune cells. CREB-p: cAMP response element-binding protein phosphorylation; Dio2: Deiodinase iodothyronine type II; T3: Tri-iodothyronine; BAT: Brown adipose tissue; GLP-1: Glucagon-like peptide-1.
BILE ACIDS IN LIPID METABOLISM
BAs are important in regulating triglyceride (TG) metabolism as well as cholesterol metabolism. The relationship between BAs and TG was first reported in the treatment of gallstones with CDCA. CDCA treatment decreased the serum TG level in patients with gallstones[45]. In fact, BAs or a synthetic FXR agonist affected TG metabolism via several mechanisms including the FXR-mediated pathway. The target of FXR, SHP, suppressed up-regulation of sterol regulatory element-binding protein-1c (SREBP-1c), the master regulator of fatty acid and TG synthesis, to reduce the expression of the lipogenic genes such as acetyl CoA synthetase, acetyl CoA carboxylase, stearoyl CoA desaturase 1, and fatty acid synthase[7,46]. In addition, the TG-lowering effects were attenuated in SHP knockout mice, indicating that lipogenesis mediated by SREBP-1c is suppressed in an FXR-SHP-dependent manner[7]. Additionally, FXR activation by BAs increases expression of apolipoprotein (Apo) CII. Apo CII activates lipoprotein lipase, which in turn stimulates TG hydrolysis in very low density lipoprotein (VLDL) and chylomicrons, and also facilitates the clearance of TG from the serum[47]. The expression of ApoCIII and angiopoietin-like protein 3, which inhibits the activity of lipoprotein lipase, were repressed by FXR stimulation with BAs[48-50]. In addition, FXR induces the expression of the VLDL receptor, which acts to clear plasma TG (Figure 2A)[51].
BAs also represses the expression of microsomal triglyceride transfer protein (MTP) and ApoB in an FXR-independent manner to suppress the formation of VLDL and chylomicrons[52]. Not only VLDL but also high density lipoprotein (HDL) clearance are suggested to be subject to modulation by BAs. Expression of scavenger receptor B1 (SRB1), a molecule in charge of hepatic uptake of HDL, is decreased, and HDL-C (HDL-cholesterol) is elevated in FXR knockout mice[53]. In addition, the administration of an FXR ligand increases hepatic SRB1 expression and decreases HDL-C levels (Figure 2A)[54].
BAs control other major regulators of lipid metabolism such as proliferator-activated receptor α (PPARα) and pyruvate dehydrogenase kinase-4 (PDK4). The nuclear receptor PPARα, which is activated by free fatty acids (FFA), decreases serum TG levels and exerts an important role for controlling enzymes participating in fatty acid β oxidation (Figure 2A)[55]. A study suggested that BAs directly regulate PPARα through FXR in humans, but not in mice[56]. PDK4 is also up regulated by BAs in an FXR-dependent manner, resulting in inactivation of pyruvate dehydrogenase, decreased glycolysis and increased oxidation of fatty acid β[57]. BAs are also associated with atherosclerosis[58,59]. Treatment with TGR5/M-BAR agonist INT-777 represses the activation of inflammatory cytokines such as NF-κB and inhibits foam cell formation and subsequent atherosclerotic plaques. In addition, INT-777 does not inhibit atherosclerosis in TGR5/M-BAR knockout mice, supporting the anti-atherosclerotic effect of TGR5/M-BAR (Figure 3)[58].
BILE ACIDS IN ENERGY METABOLISM
BAs have been reported to stimulate adaptive thermogenesis and energy expenditure via TGR5/M-BAR (Figure 3)[11]. TGR5/M-BAR activation leads to increased intracellular cAMP levels, activation of PKA and induction of CREB phosphorylation. This series of signaling activity induces the expression of genes bearing a cAMP responsive element and exists in various tissues[60,61].
In the BAT, TGR5/M-BAR stimulation increases the intracellular cAMP level and induces cAMP-dependent iodothyronine deiodinase type 2 (Dio2) expression, which converts inactive thyroxine (T4) to active 3,5,3’-triiodothyronine (T3) to evoke increased energy expenditure[11]. Dio2 increases the nuclear T3 level without various unwanted side effects caused by increased blood T3 levels. Only 20% of nuclear T3 is produced and secreted from the human thyroid gland, and the remaining nuclear T3 is supplemented from other tissues. Dio2 supplies approximately 50% of the T3 in the nucleus including the BAT[62]. The BAT is one of the most important targets of BAs to increase energy expenditure. Although BAT had been regarded as a tissue only in newborn infants, recent studies with FDG-PET revealed the existence of BAT in the shoulders and neck in adult humans, especially with brief cold exposure[63-65]. Furthermore, several groups have shown the importance of BAT in adult humans. In healthy patients, the amount of BAT is large and its activity is high but are reduced in obese patients[66-68]. In addition, TGR5/M-BAR and Dio2 are co-expressed in skeletal muscle in humans, which suggests the presence of a thermogenic mechanism in humans[11]. Moreover, a recent study found another type of adipocyte termed “beige” cells which are derived from white adipose tissue. These adipocytes also respond to cyclic AMP stimulation with high uncoupling protein (UCP) 1 expression and respiration rates similar to BAT cells[69,70]. These accumulating findings suggest a therapeutic approach to improve obesity and metabolic syndrome by increasing energy expenditure through TGR5/M-BAR stimulation.
BILE ACIDS IN AUTOPHAGY
Autophagy is an evolutionarily conserved catabolic system that maintains energy homeostasis by recycling nutrients in the fasted state. Recent studies have revealed that FXR stimulation suppresses autophagy in the liver. FXR and peroxisome PPARα competitively bind to the promoter regions of autophagic genes, and these receptors show conflicting effects on transcription[8]. In the liver, PPARα activation under fasted conditions promotes autophagic lipolysis, while FXR activation under fed conditions suppresses autophagy. That is, PPARα and FXR competitively regulate autophagy based on the nutritional condition (Figure 4A). Another study also revealed that FXR and cAMP response element-binding protein (CREB), which is a transcriptional activator under starvation, competitively regulate autophagy in the liver[9]. In the fasted condition, CREB binding to its coactivator CREB regulated the transcription coactivator 2 (CRTC2) to induce CRTC2 activity and subsequent autophagic-related gene expression. Additionally, FXR stimulation caused by feeding disrupts the functional CREB–CRTC2 complex and downregulates autophagy (Figure 4B). In any case, there is no doubt that FXR acts as a suppresser of autophagy.
Figure 4.
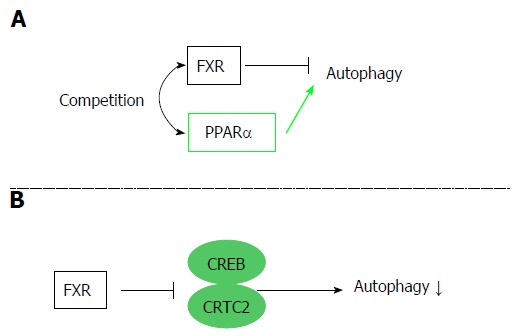
Autophagy regulation by the farnesoid X receptor. FXR is associated with regulation of autophagy. Two different mechanisms are reported. A: FXR and PPARα competitively bind to the promoter regions of autophagic genes, and FXR activation suppresses autophagy; B: FXR stimulation disrupts the functional CREB–CRTC2 complex and suppresses autophagy. FXR: Farnesoid X receptor; PPARα: Peroxisome proliferator-activated receptor α; CREB: cAMP response element-binding protein; CRTC2: CREB regulated transcription coactivator 2.
ROLES OF BILE ACIDS IN THE GASTROINTESTINAL TRACT
Intestinal FXR has been recently identified as a possible target for improving metabolic syndrome. Intestinal FXR activation induces the expression of FGF-15/19, and several studies have demonstrated that FGF-15/19 affects glucose and energy homeostasis. FGF-19 transgenic mice showed increased hepatic β oxidation, reduced adipose tissue weight, and improved glucose tolerance and insulin sensitivity[71]. In mice, hepatic acetyl-CoA carboxylase 2 (ACC2) mRNA was decreased, and the mass of the BAT was increased. ACC2 exists at the mitochondrial membrane and converts acetyl-CoA to malonyl-CoA. ACC2 activation results in an elevation of malonyl-CoA levels, which inhibit carnitine palmitoyl transferase-1 (CPT-1) activation[72]. CPT-1 transfers FFA from the cytoplasm to the mitochondria and induces fatty acid β oxidation. Thus, the overexpression of FGF-19 suppresses ACC2 mRNA levels, decreases malonyl-CoA levels, activates CPT-1, and thereby increases β-oxidation in the liver. In addition, hyperglycemia is improved upon administration of FGF-19 protein in obese mice[73]. Furthermore, activation of intestinal FXR by administration of fexaramine, an FXR agonist, improved obesity and insulin resistance by inducing FGF-15, changing the serum BA composition and stimulating systemic TGR5/M-BAR[74]. These results suggest the possibility that metabolic disease is improved through the intestinal FXR-FGF-15/19 signaling pathway (Figure 5B).
Figure 5.
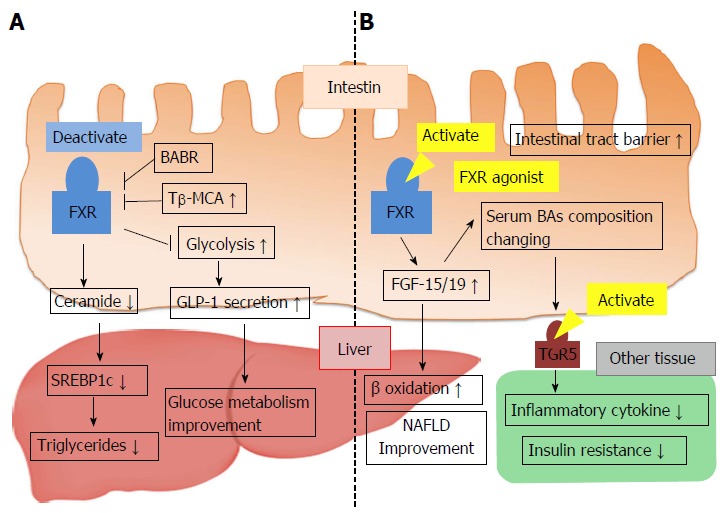
Conflicting mechanisms of metabolic regulation via intestinal farnesoid X receptor activity. A: FXR activation decreases hepatic TG levels and improves glucose metabolism; B: Intestinal FXR activation of FXR agonist leads to FGF-15/19 production and improves nonalcoholic fatty liver disease. Synthesized FGF-15/19 changes BA metabolism and serum BA composition, which causes TGR5/M-BAR activation, reduced inflammatory cytokine release, and improved insulin resistance. BABR: Bile acid binding resin; FGF-15/19: Fibroblast growth factor-15/19; FXR: Farnesoid X receptor; NAFLD: Nonalcoholic fatty liver disease; SREBP1c: Sterol regulatory element-binding protein 1c; Tβ-MCA: Tauro-β-muricholic acid; GLP-1: Glucagon-like peptide-1; TG: Triglyceride.
The primary BAs excreted into the intestine become deconjugated BAs and are converted into various secondary BAs by microbial enzymes[75]. In germ-free (GF) mice, a decrease in the gut microbiota that facilitate BA deconjugation leads to increased tauro-beta-muricholic acid (T-β-MCA). In comparison to conventionally raised mice, FXR-dependent BA synthesis is reduced in GF mice. Therefore, T-β-MCA is an FXR antagonist, and the microbiota affect bile acid homeostasis via the inhibition of intestinal FXR signaling by change in the BA composition[76]. In contrast to previous reports, recent studies have noted that alteration of the BA composition by microbiota and inhibition of intestinal FXR activity improved lipid and glucose metabolism. Increased T-β-MCA reduced intestinal FXR activation and decreased serum ceramide levels through repression of ceramide synthesis. Decreased ceramide downregulated expression of hepatic SREBP-1c and resulted in an improvement of obesity and nonalcoholic fatty liver disease (NAFLD)[77-79]. Additionally, intestinal FXR deactivation may also improve glucose metabolism as well as lipid metabolism. FXR activation in L cells decreased glycolysis, proglucagon expression and cAMP levels. Thus, GLP-1 production and secretion were inhibited (Figure 5A)[80]. Conflicting opinions suggest that microbiota regulation of BA homeostasis and intestinal FXR activation are involved in controlling hepatic lipid accumulation and glucose metabolism. Further studies are needed to clarify the roles of intestinal FXR signaling for improving metabolic diseases.
Bariatric surgery provides another clue to identifying the link between BAs and glucose homeostasis. Bariatric surgery, particularly gastric bypass surgery, is an established modality for obesity and type 2 diabetes mellitus, albeit that the mechanism of its effectiveness remains unclear. Interestingly, an improvement in glycemic control is seen soon after the surgery, when the body weight remains unchanged. Therefore, some of the anti-metabolic syndrome effects of this surgical intervention appear to be independent of body weight reduction. One recent study suggested that BAs might participate in this immediate effect of bariatric surgery. Following gastric bypass, the bile flow is changed, which leads to an increase in plasma BA level and incretin secretion[81]. Hormonal factors and the gut microbiota might also be involved in the effects of this surgery. The gut microbiota is responsible for the enteral BA metabolism, and the normal spectrum of gut microbiota is impacted by gastrointestinal surgery. As one example, the predominant presence of Firmicutes was reportedly diminished, and other species, such as methanogens and Prevotellacaea, were also inhibited after bariatric surgery[82]. In addition to these studies, recent research has revealed that FXR is associated with the effect of bariatric surgery[83]. Interestingly, in FXR knockout mice, metabolic improvements such as weight loss and improved glucose tolerance were reduced after bariatric surgery. Furthermore, the surgery changed the gut microbial communities differently between wild type and FXR knockout mice. This study suggested that BAs may affect glucose homeostasis via FXR signaling and alterations of the gut microbiota after bariatric surgery. Further investigations are expected.
Bile acid binding resin (BABR) is an effective drug for the treatment of hypercholesterolemia by lowering LDL-cholesterol. BABR absorbs BAs in the intestine, thereby preventing their uptake in the ileum, interrupting their enterohepatic circulation, and facilitating their excretion in the feces. The inhibition of the enterohepatic circulation leads to a reduction of the BA pool size, repression of FXR-SHP and FGF-15/19 signaling, and induction of CYP7A1 expression and synthesis of BAs from the cholesterol to maintain the BA pool size. A decrease in intrahepatic cholesterol levels activates SREBP-2, which induces the expression of the LDL receptor (LDLR) to enhance cholesterol uptake, reducing serum cholesterol levels. In addition to lowering the serum cholesterol effect, there is interaction between BABR and glucose metabolism[84]. In a diet-induced obesity rat model, BABR decreased serum glucose and improved glucose tolerance[85,86]. In a clinical trial, cholestyramine, a first generation BABR, improved glycemia by 13% in patients with type 2 diabetes[87]. In addition, a second generation BABR also improved glucose clearance and increased serum GIP and GLP-1 levels in patients with type 2 diabetes mellitus[88]. These studies clarified that BABR is not absorbed in the body and there are few unwanted side effects. Furthermore, BABR can decrease blood glucose levels only in high glucose situations. As a result, in January 2008, this drug was approved as a therapeutic drug for diabetes by the Food and Drug Administration (FDA) in the United States[87,89-92].
Although how BABR improves diabetes remains unknown, several possible mechanisms have been proposed. BABR-mediated improvement of hepatic insulin sensitivity depends on downregulating the hepatic cholesterol-LXR-IRS2 pathway[93]. In addition, BABR induces GLP-1 secretion via the activation of TGR5/M-BAR or GPR40, each being activated by BAs binding with BABR or unabsorbed long-chain fatty acids[39,94,95]. Further, BABR affects the make-up of the BA pool and peripheral BAs, which results in the induction of peripheral energy expenditure and improved glucose tolerance[84]. The BABR effects of improving diabetes may be explained by the inhibition of intestinal FXR as well as TGR5/M-BAR signaling[80]. BABR colesevelam inhibits intestinal FXR activation and improves glucose metabolism by increasing proglucagon gene expression and inducing GLP-1 secretion in ob/ob mice[80]. These findings suggest that inhibiting FXR in the L cell via BABR could be a new target for diabetes.
CLINICAL APPLICATION IN BA SIGNALING
Currently, BABR has been approved by the FDA and has been clinically used as a diabetes treatment drug. The association between bariatric surgery and BA homeostasis was confirmed. In addition to BABR and bariatric surgery, other clinical applications based on the mechanism of metabolic control via BA signaling are ongoing. For instance, INT-747 (also named 6-ethyl-CDCA), which is a synthetic FXR agonist, exerts a hepatoprotective effect in patients of primary biliary cirrhosis (PBC)[96-98], and a phase III clinical study has already been completed and confirmed the effect of PBC. In addition to medicine, INT-747 has also entered into a study for NAFLD treatment. A phase II clinical trial for NAFLD has been completed, and an improvement was observed in type 2 diabetes mellitus patients with NAFLD. Clinical trials with TGR5 agonists, such as INT-777, are ongoing, and future studies are expected[40,41,99].
Altogether, these clinical applications will elucidate the BA signaling mechanisms that will lead to the improvement of metabolic disorders including obesity and diabetes.
CONCLUSION
Today, BAs have become important molecules to control metabolic homeostasis. In this review we discussed the relationship between BA metabolism and signal transmission, such as the FXR and TGR5/M-BAR pathways and the possibility that BAs may improve metabolic diseases. Current evidence shows that BAs regulate lipid, glucose, and energy metabolism via FXR or TGR/M-BAR-mediated pathways. Furthermore, the clinical application of FXR and TGR/M-BAR agonists are ongoing.
Recent studies have focused on intestinal FXR signaling; however, conflicting data have been reported regarding the metabolic regulation of intestinal FXR activity. Further studies are necessary to determine intestinal FXR signaling taking into consideration various factors such as microbiota regulation, BA pool size, and BA composition.
Footnotes
Conflict-of-interest statement: No potential conflicts of interest. No financial support.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Manuscript source: Invited manuscript
Peer-review started: October 9, 2015
First decision: November 6, 2015
Article in press: May 27, 2016
P- Reviewer: Das UN, Maroni L, Mikov MM S- Editor: Wang JL L- Editor: A E- Editor: Li D
References
- 1.Hofmann AF, Borgstroem B. The Intraluminal Phase of Fat Digestion In Man: The Lipid Content OF The Micellar And Oil Phases of Intestinal Content Obtained During Fat Digestion and Absorption. J Clin Invest. 1964;43:247–257. doi: 10.1172/JCI104909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Urizar NL, Dowhan DH, Moore DD. The farnesoid X-activated receptor mediates bile acid activation of phospholipid transfer protein gene expression. J Biol Chem. 2000;275:39313–39317. doi: 10.1074/jbc.M007998200. [DOI] [PubMed] [Google Scholar]
- 3.Maruyama T, Miyamoto Y, Nakamura T, Tamai Y, Okada H, Sugiyama E, Nakamura T, Itadani H, Tanaka K. Identification of membrane-type receptor for bile acids (M-BAR) Biochem Biophys Res Commun. 2002;298:714–719. doi: 10.1016/s0006-291x(02)02550-0. [DOI] [PubMed] [Google Scholar]
- 4.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 5.Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 6.Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T, Noonan DJ, Burka LT, Mcmorris T, Lamph WW, et al. Identification of a Nuclear Receptor That Is Activated by Farnesol Metabolites. Cell. 1995;81:687–693. doi: 10.1016/0092-8674(95)90530-8. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, Auwerx J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113:1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, Moore DD. Nutrient-sensing nuclear receptors coordinate autophagy. Nature. 2014;516:112–115. doi: 10.1038/nature13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar S, Sun X, Yoon G, Kang Y, Zhong W, et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature. 2014;516:108–111. doi: 10.1038/nature13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Settembre C, Ballabio A. Cell metabolism: autophagy transcribed. Nature. 2014;516:40–41. doi: 10.1038/nature13939. [DOI] [PubMed] [Google Scholar]
- 11.Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 12.Keitel V, Reinehr R, Gatsios P, Rupprecht C, Görg B, Selbach O, Häussinger D, Kubitz R. The G-protein coupled bile salt receptor TGR5 is expressed in liver sinusoidal endothelial cells. Hepatology. 2007;45:695–704. doi: 10.1002/hep.21458. [DOI] [PubMed] [Google Scholar]
- 13.Ichikawa R, Takayama T, Yoneno K, Kamada N, Kitazume MT, Higuchi H, Matsuoka K, Watanabe M, Itoh H, Kanai T, et al. Bile acids induce monocyte differentiation toward interleukin-12 hypo-producing dendritic cells via a TGR5-dependent pathway. Immunology. 2012;136:153–162. doi: 10.1111/j.1365-2567.2012.03554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsuzaki J, Suzuki H, Tsugawa H, Watanabe M, Hossain S, Arai E, Saito Y, Sekine S, Akaike T, Kanai Y, et al. Bile acids increase levels of microRNAs 221 and 222, leading to degradation of CDX2 during esophageal carcinogenesis. Gastroenterology. 2013;145:1300–1311. doi: 10.1053/j.gastro.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 15.Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 2007;67:863–867. doi: 10.1158/0008-5472.CAN-06-1078. [DOI] [PubMed] [Google Scholar]
- 16.Houten SM, Watanabe M, Auwerx J. Endocrine functions of bile acids. EMBO J. 2006;25:1419–1425. doi: 10.1038/sj.emboj.7601049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwarz M, Russell DW, Dietschy JM, Turley SD. Alternate pathways of bile acid synthesis in the cholesterol 7alpha-hydroxylase knockout mouse are not upregulated by either cholesterol or cholestyramine feeding. J Lipid Res. 2001;42:1594–1603. [PubMed] [Google Scholar]
- 18.Russell DW, Setchell KD. Bile acid biosynthesis. Biochemistry. 1992;31:4737–4749. doi: 10.1021/bi00135a001. [DOI] [PubMed] [Google Scholar]
- 19.Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 20.Amuro Y, Endo T, Higashino K, Uchida K, Yamamura Y. Serum, fecal and urinary bile acids in patients with mild and advanced liver cirrhosis. Gastroenterol Jpn. 1981;16:506–513. doi: 10.1007/BF02774522. [DOI] [PubMed] [Google Scholar]
- 21.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 22.Brendel C, Schoonjans K, Botrugno OA, Treuter E, Auwerx J. The small heterodimer partner interacts with the liver X receptor alpha and represses its transcriptional activity. Mol Endocrinol. 2002;16:2065–2076. doi: 10.1210/me.2001-0194. [DOI] [PubMed] [Google Scholar]
- 23.Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, Mangelsdorf DJ. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell. 2000;6:507–515. doi: 10.1016/s1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- 24.Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, Donahee M, Wang DY, Mansfield TA, Kliewer SA, et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003;17:1581–1591. doi: 10.1101/gad.1083503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 26.Maruyama T, Tanaka K, Suzuki J, Miyoshi H, Harada N, Nakamura T, Miyamoto Y, Kanatani A, Tamai Y. Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J Endocrinol. 2006;191:197–205. doi: 10.1677/joe.1.06546. [DOI] [PubMed] [Google Scholar]
- 27.Vassileva G, Golovko A, Markowitz L, Abbondanzo SJ, Zeng M, Yang S, Hoos L, Tetzloff G, Levitan D, Murgolo NJ, et al. Targeted deletion of Gpbar1 protects mice from cholesterol gallstone formation. Biochem J. 2006;398:423–430. doi: 10.1042/BJ20060537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bouillon R, Carmeliet G, Lieben L, Watanabe M, Perino A, Auwerx J, Schoonjans K, Verstuyf A. Vitamin D and energy homeostasis: of mice and men. Nat Rev Endocrinol. 2014;10:79–87. doi: 10.1038/nrendo.2013.226. [DOI] [PubMed] [Google Scholar]
- 29.Han S, Li T, Ellis E, Strom S, Chiang JY. A novel bile acid-activated vitamin D receptor signaling in human hepatocytes. Mol Endocrinol. 2010;24:1151–1164. doi: 10.1210/me.2009-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duran-Sandoval D, Mautino G, Martin G, Percevault F, Barbier O, Fruchart JC, Kuipers F, Staels B. Glucose regulates the expression of the farnesoid X receptor in liver. Diabetes. 2004;53:890–898. doi: 10.2337/diabetes.53.4.890. [DOI] [PubMed] [Google Scholar]
- 31.Yamagata K, Daitoku H, Shimamoto Y, Matsuzaki H, Hirota K, Ishida J, Fukamizu A. Bile acids regulate gluconeogenic gene expression via small heterodimer partner-mediated repression of hepatocyte nuclear factor 4 and Foxo1. J Biol Chem. 2004;279:23158–23165. doi: 10.1074/jbc.M314322200. [DOI] [PubMed] [Google Scholar]
- 32.Yamagata K, Yoshimochi K, Daitoku H, Hirota K, Fukamizu A. Bile acid represses the peroxisome proliferator-activated receptor-gamma coactivator-1 promoter activity in a small heterodimer partner-dependent manner. Int J Mol Med. 2007;19:751–756. [PubMed] [Google Scholar]
- 33.Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. 2006;116:1102–1109. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stayrook KR, Bramlett KS, Savkur RS, Ficorilli J, Cook T, Christe ME, Michael LF, Burris TP. Regulation of carbohydrate metabolism by the farnesoid X receptor. Endocrinology. 2005;146:984–991. doi: 10.1210/en.2004-0965. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y, Lee FY, Barrera G, Lee H, Vales C, Gonzalez FJ, Willson TM, Edwards PA. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci USA. 2006;103:1006–1011. doi: 10.1073/pnas.0506982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watanabe M, Horai Y, Houten SM, Morimoto K, Sugizaki T, Arita E, Mataki C, Sato H, Tanigawara Y, Schoonjans K, et al. Lowering bile acid pool size with a synthetic farnesoid X receptor (FXR) agonist induces obesity and diabetes through reduced energy expenditure. J Biol Chem. 2011;286:26913–26920. doi: 10.1074/jbc.M111.248203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cariou B, van Harmelen K, Duran-Sandoval D, van Dijk TH, Grefhorst A, Abdelkarim M, Caron S, Torpier G, Fruchart JC, Gonzalez FJ, et al. The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J Biol Chem. 2006;281:11039–11049. doi: 10.1074/jbc.M510258200. [DOI] [PubMed] [Google Scholar]
- 38.Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87:1409–1439. doi: 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- 39.Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Biophys Res Commun. 2005;329:386–390. doi: 10.1016/j.bbrc.2005.01.139. [DOI] [PubMed] [Google Scholar]
- 40.Pellicciari R, Gioiello A, Macchiarulo A, Thomas C, Rosatelli E, Natalini B, Sardella R, Pruzanski M, Roda A, Pastorini E, et al. Discovery of 6alpha-ethyl-23(S)-methylcholic acid (S-EMCA, INT-777) as a potent and selective agonist for the TGR5 receptor, a novel target for diabesity. J Med Chem. 2009;52:7958–7961. doi: 10.1021/jm901390p. [DOI] [PubMed] [Google Scholar]
- 41.Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sato H, Genet C, Strehle A, Thomas C, Lobstein A, Wagner A, Mioskowski C, Auwerx J, Saladin R. Anti-hyperglycemic activity of a TGR5 agonist isolated from Olea europaea. Biochem Biophys Res Commun. 2007;362:793–798. doi: 10.1016/j.bbrc.2007.06.130. [DOI] [PubMed] [Google Scholar]
- 43.Thomas C, Auwerx J, Schoonjans K. Bile acids and the membrane bile acid receptor TGR5--connecting nutrition and metabolism. Thyroid. 2008;18:167–174. doi: 10.1089/thy.2007.0255. [DOI] [PubMed] [Google Scholar]
- 44.Müssig K, Staiger H, Machicao F, Machann J, Schick F, Schäfer SA, Claussen CD, Holst JJ, Gallwitz B, Stefan N, et al. Preliminary report: genetic variation within the GPBAR1 gene is not associated with metabolic traits in white subjects at an increased risk for type 2 diabetes mellitus. Metabolism. 2009;58:1809–1811. doi: 10.1016/j.metabol.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 45.Fromm H, Eschler A, Töllner D, Canzler H, Schmidt FW. [In vivo dissolving of gall-stones: the effect of chenodeoxycholic acid. (author’s transl)] Dtsch Med Wochenschr. 1975;100:1619–1624. doi: 10.1055/s-0028-1106432. [DOI] [PubMed] [Google Scholar]
- 46.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kast HR, Nguyen CM, Sinal CJ, Jones SA, Laffitte BA, Reue K, Gonzalez FJ, Willson TM, Edwards PA. Farnesoid X-activated receptor induces apolipoprotein C-II transcription: a molecular mechanism linking plasma triglyceride levels to bile acids. Mol Endocrinol. 2001;15:1720–1728. doi: 10.1210/mend.15.10.0712. [DOI] [PubMed] [Google Scholar]
- 48.Ginsberg HN, Le NA, Goldberg IJ, Gibson JC, Rubinstein A, Wang-Iverson P, Norum R, Brown WV. Apolipoprotein B metabolism in subjects with deficiency of apolipoproteins CIII and AI. Evidence that apolipoprotein CIII inhibits catabolism of triglyceride-rich lipoproteins by lipoprotein lipase in vivo. J Clin Invest. 1986;78:1287–1295. doi: 10.1172/JCI112713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Claudel T, Inoue Y, Barbier O, Duran-Sandoval D, Kosykh V, Fruchart J, Fruchart JC, Gonzalez FJ, Staels B. Farnesoid X receptor agonists suppress hepatic apolipoprotein CIII expression. Gastroenterology. 2003;125:544–555. doi: 10.1016/s0016-5085(03)00896-5. [DOI] [PubMed] [Google Scholar]
- 50.Inaba T, Matsuda M, Shimamura M, Takei N, Terasaka N, Ando Y, Yasumo H, Koishi R, Makishima M, Shimomura I. Angiopoietin-like protein 3 mediates hypertriglyceridemia induced by the liver X receptor. J Biol Chem. 2003;278:21344–21351. doi: 10.1074/jbc.M213202200. [DOI] [PubMed] [Google Scholar]
- 51.Sirvent A, Claudel T, Martin G, Brozek J, Kosykh V, Darteil R, Hum DW, Fruchart JC, Staels B. The farnesoid X receptor induces very low density lipoprotein receptor gene expression. FEBS Lett. 2004;566:173–177. doi: 10.1016/j.febslet.2004.04.026. [DOI] [PubMed] [Google Scholar]
- 52.Hirokane H, Nakahara M, Tachibana S, Shimizu M, Sato R. Bile acid reduces the secretion of very low density lipoprotein by repressing microsomal triglyceride transfer protein gene expression mediated by hepatocyte nuclear factor-4. J Biol Chem. 2004;279:45685–45692. doi: 10.1074/jbc.M404255200. [DOI] [PubMed] [Google Scholar]
- 53.Sinal CJ, Yoon M, Gonzalez FJ. Antagonism of the actions of peroxisome proliferator-activated receptor-alpha by bile acids. J Biol Chem. 2001;276:47154–47162. doi: 10.1074/jbc.M107000200. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Y, Yin L, Anderson J, Ma H, Gonzalez FJ, Willson TM, Edwards PA. Identification of novel pathways that control farnesoid X receptor-mediated hypocholesterolemia. J Biol Chem. 2010;285:3035–3043. doi: 10.1074/jbc.M109.083899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 56.Pineda Torra I, Claudel T, Duval C, Kosykh V, Fruchart JC, Staels B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol Endocrinol. 2003;17:259–272. doi: 10.1210/me.2002-0120. [DOI] [PubMed] [Google Scholar]
- 57.Savkur RS, Bramlett KS, Michael LF, Burris TP. Regulation of pyruvate dehydrogenase kinase expression by the farnesoid X receptor. Biochem Biophys Res Commun. 2005;329:391–396. doi: 10.1016/j.bbrc.2005.01.141. [DOI] [PubMed] [Google Scholar]
- 58.Pols TW, Nomura M, Harach T, Lo Sasso G, Oosterveer MH, Thomas C, Rizzo G, Gioiello A, Adorini L, Pellicciari R, et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011;14:747–757. doi: 10.1016/j.cmet.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang YD, Chen WD, Yu D, Forman BM, Huang W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor κ light-chain enhancer of activated B cells (NF-κB) in mice. Hepatology. 2011;54:1421–1432. doi: 10.1002/hep.24525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 61.Sato H, Macchiarulo A, Thomas C, Gioiello A, Une M, Hofmann AF, Saladin R, Schoonjans K, Pellicciari R, Auwerx J. Novel potent and selective bile acid derivatives as TGR5 agonists: biological screening, structure-activity relationships, and molecular modeling studies. J Med Chem. 2008;51:1831–1841. doi: 10.1021/jm7015864. [DOI] [PubMed] [Google Scholar]
- 62.Bianco AC, Silva JE. Nuclear 3,5,3’-triiodothyronine (T3) in brown adipose tissue: receptor occupancy and sources of T3 as determined by in vivo techniques. Endocrinology. 1987;120:55–62. doi: 10.1210/endo-120-1-55. [DOI] [PubMed] [Google Scholar]
- 63.Cohade C, Mourtzikos KA, Wahl RL. “USA-Fat”: prevalence is related to ambient outdoor temperature-evaluation with 18F-FDG PET/CT. J Nucl Med. 2003;44:1267–1270. [PubMed] [Google Scholar]
- 64.Hany TF, Gharehpapagh E, Kamel EM, Buck A, Himms-Hagen J, von Schulthess GK. Brown adipose tissue: a factor to consider in symmetrical tracer uptake in the neck and upper chest region. Eur J Nucl Med Mol Imaging. 2002;29:1393–1398. doi: 10.1007/s00259-002-0902-6. [DOI] [PubMed] [Google Scholar]
- 65.Saito M. Brown adipose tissue as a regulator of energy expenditure and body fat in humans. Diabetes Metab J. 2013;37:22–29. doi: 10.4093/dmj.2013.37.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ. Cold-activated brown adipose tissue in healthy men. N Engl J Med. 2009;360:1500–1508. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 67.Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–1517. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, Taittonen M, Laine J, Savisto NJ, Enerbäck S, et al. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360:1518–1525. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- 69.Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scimè A, Devarakonda S, Conroe HM, Erdjument-Bromage H, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu J, Boström P, Sparks LM, Ye L, Choi JH, Giang AH, Khandekar M, Virtanen KA, Nuutila P, Schaart G, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150:366–376. doi: 10.1016/j.cell.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tomlinson E, Fu L, John L, Hultgren B, Huang X, Renz M, Stephan JP, Tsai SP, Powell-Braxton L, French D, et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology. 2002;143:1741–1747. doi: 10.1210/endo.143.5.8850. [DOI] [PubMed] [Google Scholar]
- 72.Abu-Elheiga L, Matzuk MM, Kordari P, Oh W, Shaikenov T, Gu Z, Wakil SJ. Mutant mice lacking acetyl-CoA carboxylase 1 are embryonically lethal. Proc Natl Acad Sci USA. 2005;102:12011–12016. doi: 10.1073/pnas.0505714102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fu L, John LM, Adams SH, Yu XX, Tomlinson E, Renz M, Williams PM, Soriano R, Corpuz R, Moffat B, et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology. 2004;145:2594–2603. doi: 10.1210/en.2003-1671. [DOI] [PubMed] [Google Scholar]
- 74.Fang S, Suh JM, Reilly SM, Yu E, Osborn O, Lackey D, Yoshihara E, Perino A, Jacinto S, Lukasheva Y, et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat Med. 2015;21:159–165. doi: 10.1038/nm.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47:241–259. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 76.Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall HU, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013;17:225–235. doi: 10.1016/j.cmet.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 77.Li F, Jiang C, Krausz KW, Li Y, Albert I, Hao H, Fabre KM, Mitchell JB, Patterson AD, Gonzalez FJ. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat Commun. 2013;4:2384. doi: 10.1038/ncomms3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Qi Y, Jiang C, Cheng J, Krausz KW, Li T, Ferrell JM, Gonzalez FJ, Chiang JY. Bile acid signaling in lipid metabolism: metabolomic and lipidomic analysis of lipid and bile acid markers linked to anti-obesity and anti-diabetes in mice. Biochim Biophys Acta. 2015;1851:19–29. doi: 10.1016/j.bbalip.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jiang C, Xie C, Li F, Zhang L, Nichols RG, Krausz KW, Cai J, Qi Y, Fang ZZ, Takahashi S, et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest. 2015;125:386–402. doi: 10.1172/JCI76738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Trabelsi MS, Daoudi M, Prawitt J, Ducastel S, Touche V, Sayin SI, Perino A, Brighton CA, Sebti Y, Kluza J, et al. Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nat Commun. 2015;6:7629. doi: 10.1038/ncomms8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nakatani H, Kasama K, Oshiro T, Watanabe M, Hirose H, Itoh H. Serum bile acid along with plasma incretins and serum high-molecular weight adiponectin levels are increased after bariatric surgery. Metabolism. 2009;58:1400–1407. doi: 10.1016/j.metabol.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 82.Zhang H, DiBaise JK, Zuccolo A, Kudrna D, Braidotti M, Yu Y, Parameswaran P, Crowell MD, Wing R, Rittmann BE, et al. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci USA. 2009;106:2365–2370. doi: 10.1073/pnas.0812600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ryan KK, Tremaroli V, Clemmensen C, Kovatcheva-Datchary P, Myronovych A, Karns R, Wilson-Pérez HE, Sandoval DA, Kohli R, Bäckhed F, et al. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature. 2014;509:183–188. doi: 10.1038/nature13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Watanabe M, Morimoto K, Houten SM, Kaneko-Iwasaki N, Sugizaki T, Horai Y, Mataki C, Sato H, Murahashi K, Arita E, et al. Bile acid binding resin improves metabolic control through the induction of energy expenditure. PLoS One. 2012;7:e38286. doi: 10.1371/journal.pone.0038286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shang Q, Saumoy M, Holst JJ, Salen G, Xu G. Colesevelam improves insulin resistance in a diet-induced obesity (F-DIO) rat model by increasing the release of GLP-1. Am J Physiol Gastrointest Liver Physiol. 2010;298:G419–G424. doi: 10.1152/ajpgi.00362.2009. [DOI] [PubMed] [Google Scholar]
- 86.Chen L, McNulty J, Anderson D, Liu Y, Nystrom C, Bullard S, Collins J, Handlon AL, Klein R, Grimes A, et al. Cholestyramine reverses hyperglycemia and enhances glucose-stimulated glucagon-like peptide 1 release in Zucker diabetic fatty rats. J Pharmacol Exp Ther. 2010;334:164–170. doi: 10.1124/jpet.110.166892. [DOI] [PubMed] [Google Scholar]
- 87.Garg A, Grundy SM. Cholestyramine therapy for dyslipidemia in non-insulin-dependent diabetes mellitus. A short-term, double-blind, crossover trial. Ann Intern Med. 1994;121:416–422. doi: 10.7326/0003-4819-121-6-199409150-00004. [DOI] [PubMed] [Google Scholar]
- 88.Suzuki T, Oba K, Igari Y, Matsumura N, Watanabe K, Futami-Suda S, Yasuoka H, Ouchi M, Suzuki K, Kigawa Y, et al. Colestimide lowers plasma glucose levels and increases plasma glucagon-like PEPTIDE-1 (7-36) levels in patients with type 2 diabetes mellitus complicated by hypercholesterolemia. J Nippon Med Sch. 2007;74:338–343. doi: 10.1272/jnms.74.338. [DOI] [PubMed] [Google Scholar]
- 89.Zieve FJ, Kalin MF, Schwartz SL, Jones MR, Bailey WL. Results of the glucose-lowering effect of WelChol study (GLOWS): a randomized, double-blind, placebo-controlled pilot study evaluating the effect of colesevelam hydrochloride on glycemic control in subjects with type 2 diabetes. Clin Ther. 2007;29:74–83. doi: 10.1016/j.clinthera.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 90.Bays HE, Goldberg RB, Truitt KE, Jones MR. Colesevelam hydrochloride therapy in patients with type 2 diabetes mellitus treated with metformin: glucose and lipid effects. Arch Intern Med. 2008;168:1975–1983. doi: 10.1001/archinte.168.18.1975. [DOI] [PubMed] [Google Scholar]
- 91.Fonseca VA, Rosenstock J, Wang AC, Truitt KE, Jones MR. Colesevelam HCl improves glycemic control and reduces LDL cholesterol in patients with inadequately controlled type 2 diabetes on sulfonylurea-based therapy. Diabetes Care. 2008;31:1479–1484. doi: 10.2337/dc08-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Goldberg RB, Fonseca VA, Truitt KE, Jones MR. Efficacy and safety of colesevelam in patients with type 2 diabetes mellitus and inadequate glycemic control receiving insulin-based therapy. Arch Intern Med. 2008;168:1531–1540. doi: 10.1001/archinte.168.14.1531. [DOI] [PubMed] [Google Scholar]
- 93.Tagawa H, Irie J, Itoh A, Kusumoto Y, Kato M, Kobayashi N, Tanaka K, Morinaga R, Fujita M, Nakajima Y, et al. Bile acid binding resin improves hepatic insulin sensitivity by reducing cholesterol but not triglyceride levels in the liver. Diabetes Res Clin Pract. 2015;109:85–94. doi: 10.1016/j.diabres.2015.04.025. [DOI] [PubMed] [Google Scholar]
- 94.Henry RR, Aroda VR, Mudaliar S, Garvey WT, Chou HS, Jones MR. Effects of colesevelam on glucose absorption and hepatic/peripheral insulin sensitivity in patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2012;14:40–46. doi: 10.1111/j.1463-1326.2011.01486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Porez G, Prawitt J, Gross B, Staels B. Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease. J Lipid Res. 2012;53:1723–1737. doi: 10.1194/jlr.R024794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, Morelli A, Parks DJ, Willson TM. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002;45:3569–3572. doi: 10.1021/jm025529g. [DOI] [PubMed] [Google Scholar]
- 97.Fiorucci S, Antonelli E, Rizzo G, Renga B, Mencarelli A, Riccardi L, Orlandi S, Pellicciari R, Morelli A. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology. 2004;127:1497–1512. doi: 10.1053/j.gastro.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 98.Fiorucci S, Clerici C, Antonelli E, Orlandi S, Goodwin B, Sadeghpour BM, Sabatino G, Russo G, Castellani D, Willson TM, et al. Protective effects of 6-ethyl chenodeoxycholic acid, a farnesoid X receptor ligand, in estrogen-induced cholestasis. J Pharmacol Exp Ther. 2005;313:604–612. doi: 10.1124/jpet.104.079665. [DOI] [PubMed] [Google Scholar]
- 99.Li T, Holmstrom SR, Kir S, Umetani M, Schmidt DR, Kliewer SA, Mangelsdorf DJ. The G protein-coupled bile acid receptor, TGR5, stimulates gallbladder filling. Mol Endocrinol. 2011;25:1066–1071. doi: 10.1210/me.2010-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]