

Abstract
Summary: Analysis of the composition of heterogeneous tissue has been greatly enabled by recent developments in single-cell transcriptomics. We present SCell, an integrated software tool for quality filtering, normalization, feature selection, iterative dimensionality reduction, clustering and the estimation of gene-expression gradients from large ensembles of single-cell RNA-seq datasets. SCell is open source, and implemented with an intuitive graphical interface. Scripts and protocols for the high-throughput pre-processing of large ensembles of single-cell, RNA-seq datasets are provided as an additional resource.
Availability and Implementation: Binary executables for Windows, MacOS and Linux are available at http://sourceforge.net/projects/scell, source code and pre-processing scripts are available from https://github.com/diazlab/SCell.
Supplementary information: Supplementary data are available at Bioinformatics online.
Contact: aaron.diaz@ucsf.edu
1 Introduction
Single-cell sequencing enables heterogeneity assessments at unprecedented resolution. At a cost comparable to sequencing a sample in bulk, hundreds of single-cell datasets can instead be generated. We present SCell, a software tool to perform outlier filtering, to normalize cell-cycle effects, to select genes for dimensionality reduction and to estimate inter-sample expression gradients. Several groups have proposed reconstructing the gene-expression kinetics of developmental processes from transcriptomics data, by summarizing gene expression along the backbone of a spanning graph of the samples’ PCA coordinates (Bendall et al., 2014; Magwene et al, 2003; Trapnell et al, 2014). As single-cell sequencing becomes more widely adopted, the large number of available samples makes direct regression of gene expression on PCA coordinates an attractive alternative. SCell can regress/interpolate gene expression on PCA space, visualize expression gradients, and estimate expression kinetics along minimum spanning trees and minimum weight paths. These tools are accessible through an interactive, graphical interface.
2 Results
2.1 Quality control and pre-processing
To identify outlier libraries, we developed a strategy to estimate genes expressed at background levels in a given sample. We then filter samples whose background fraction is significantly larger than average. Our approach builds on previous methods for sequencing data based on order-statistics (Diaz et al., 2012; Xu et al., 2010) (Supplementary Materials M1, Supplementary Figure S1A). In our tests, samples that had a small q-value for our Lorenz-statistic had low complexity, as measured by Gini-Simpson index, and/or they had low coverage, as estimated by the Good-Turing statistic (Supplementary Figure S1B). Moreover, in our data the Lorenz-statistic correlated with the results of live-dead staining (Pearson-correlation 0.74). SCell displays these quality metrics, Gini-Simpson index and user’s metadata, including the number of mapped reads and the results of live-dead staining, in an interactive expression profiler (Supplementary Figure S1C). SCell also reports library coverage and marginal-return estimates based on PRESEQ (Daley and Smith, 2014) (Supplementary Materials M1).
2.2 Normalization and feature selection
We extend remove-unwanted-variation using control genes (RUVg) (Risso et al., 2014) to normalize single-cell data for dimensionality reduction and clustering. RUVg utilizes ordinary-least-squares regression to produce normalized counts, we implement a robust variant: iteratively reweighted least squares with a bisquare weight function (Supplementary methods M2). SCell can produce counts normalized by any combination of: (i) Cyclins and cyclin-dependent kinase (CDK) expression, which corresponds to cell-cycle state, and (ii) a user supplied count matrix. Additionally, SCell utilizes canonical-correlation analysis to correlate cell-cycle and gene expression (Supplementary Materials M2). This estimates the percentage of genome-wide variance explained by cyclin/CDKs, the specific cyclins/CDKs explaining the most variance and genes correlating with cyclin/CDs (Supplementary Figure S2A). By default, SCell normalizes samples by sequencing depth.
SCell provides statistics for feature selection. We use a score statistic, from a generalized-Poisson model, to test for gene-wise zero-inflation and identify technical dropouts (Supplementary Materials M2). We use a power function based on the index-of-dispersion to prioritize genes by variability (Supplementary Materials M2). SCell implements an interactive viewer to visualize gene variance versuss sampling (Supplementary Fig S3), and to select genes based on these statistics, and their false discovery rates.
2.3 Dimensionality reduction, clustering and expression kinetics
SCell implements PCA, and optionally varimax-rotation. Two interactive windows allow the user to explore samples in PCA space, with gene-level and sample-level metadata displayed upon mouse-over (Fig. S4). SCell offers several methods for clustering: k-means, Minkowski-weighted k-means (de Amorim, 2012), Gaussian mixture model, the clustering ‘with scatter’ algorithm DBSCAN and user-defined clusters. Genes and samples can be added by cluster or individually to user-defined gene and sample lists. A PCA can be recomputed at any time from the user’s sample list, enabling ‘iterative’ PCA learning of population sub-structure. SCell implements minimum-spanning-tree (MST) and Gabriel graph minimum-cost paths, for semi-supervised estimation of cell-state transitions (Supplementary Materials M3). SCell automatically fits loess/lowess regressions, as well as several interpolation types (linear, cubic spline, biharmonic and thin-plate spline) on gene expression in PCA space. This allows users to visualize expression gradients, and evaluate gene-expression kinetics along MST and minimum-cost paths. To illustrate, we performed quality filtering, feature selection and clustering on 96 cells sequenced from gestational-week 10 human fetal neocortex, 258 cells from gestational-weeks 16–18, human fetal neocortex (Supplementary Materials M4, dbGaP phs000989.v3.p1), as well as 393 cells previously published from gestational-week 16-18 human fetal neocortex (Supplementary Figure S4B, dbGaP phs000989.v1.p1). We identified 501 cells which passed quality control at a Lorenz-statistic q-value cutoff of q = 0.05. 2169 genes were chosen for dimensionality reduction, based on a zero-inflation q-value of 0.1 (to control for under-sampling) and an index-or-dispersion power of 90% (to enrich for variable genes). We identified clusters expressing markers of interneurons and these could be distinguished from cells of the excitatory-neuronal lineage. Iterative PCA analysis of the interneurons identified sub-clusters expressing markers of different classes of inhibitory neurons (Supplementary Figure S4B and C). Lowess regression, along a Gabriel-graph shortest path of the excitatory-neuronal lineage, predicts a rapid decline in markers of neural stem cells, a gradual increase in neuronal markers and a transient spike in markers of intermediate-progenitors along pseudo-time (Fig. 1).
Fig. 1.
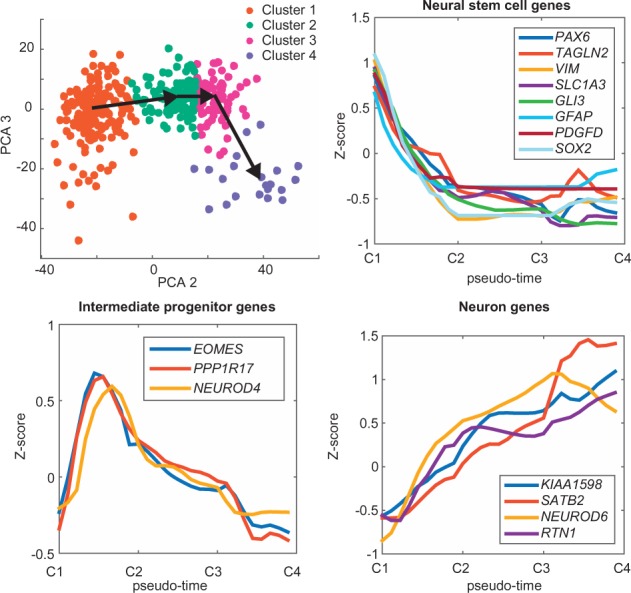
747 cells sequenced from human fetal neocortex. Lineage reconstruction, via a Gabriel-graph shortest distance path and LOWESS regression, models the kinetics of gene expression during commitment to the excitatory-neuronal lineage (Color version of this figure is available at Bioinformatics online.)
Funding
This work has been supported by UCSF-CTSI UL1 TR000004 and a Shurl and Kay Curci Foundation Research grant (to A.D.), a Damon Runyon Cancer Research Foundation postdoctoral fellowship (DRG-2166-13) (to AAP), a VA award 5I01 BX000252 (to D.A.L.) and by NIH awards U01 MH105989 and R01NS075998 to (A.R.K.).
Conflict of Interest: none declared.
Supplementary Material
References
- Bendall S.C. et al. (2014) Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell, 157, 714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley T., Smith A.D. (2014) Modeling genome coverage in single cell sequencing. Bioinformatics, 30, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Amorim R.C. (2012) Constrained clustering with Minkowski Weighted K-Means. In: 2012 IEEE 13th International Symposium of Computing Intelligence of Informatics, pp. 13–17. [Google Scholar]
- Diaz A. et al. (2012) Normalization, bias correction, and peak calling for ChIP-seq. Stat. Appl. Genet. Mol. Biol., 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magwene P.M. et al. (2003) Reconstructing the temporal ordering of biological samples using microarray data. Bioinformatics, 19, 842–850. [DOI] [PubMed] [Google Scholar]
- Risso D. et al. (2014) Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol., 32, 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C. et al. (2014) The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol., 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H. et al. (2010) A signal-noise model for significance analysis of ChIP-seq with negative control. Bioinformatics, 26, 1199–1204. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.