

Abstract
Background
Critical limb ischemia (CLI) is the extreme manifestation of peripheral artery disease, a major unmet clinical need for which lower limb amputation is the only option for many patients. After 2 decades in development, therapeutic angiogenesis has been tested clinically via intramuscular delivery of proangiogenic proteins, genes, and stem cells. Efficacy has been modest to absent, and the largest phase 3 trial of gene therapy for CLI reported a worsening trend of plasmid fibroblast growth factor. In all clinical trials to date, gene therapy has used unregulated vectors with limited duration of expression. Only unregulated extended expression vectors such as adeno‐associated virus (AAV) and lentivirus have been tested in preclinical models.
Methods and Results
We present preclinical results of ischemia (hypoxia)‐regulated conditionally silenced (CS) AAV–human vascular endothelial growth factor (hVEGF) gene delivery that shows efficacy and safety in a setting where other strategies fail. In a BALB/c mouse model of CLI, we show that gene therapy with AAV‐CS‐hVEGF, but not unregulated AAV or plasmid, vectors conferred limb salvage, protection from necrosis, and vascular regeneration when delivered via intramuscular or intra‐arterial routes. All vector treatments conferred increased capillary density, but organized longitudinal arteries were selectively generated by AAV‐CS‐hVEGF. AAV‐CS‐hVEGF therapy reversibly activated angiogenic and vasculogenic genes, including Notch,SDF1, Angiopoietin, and Ephrin‐B2. Reoxygenation extinguished VEGF expression and inactivated the program with no apparent adverse side effects.
Conclusions
Restriction of angiogenic growth factor expression to regions of ischemia supports the safe and stable reperfusion of hindlimbs in a clinically relevant murine model of CLI.
Keywords: gene therapy, hypoxia, ischemia, peripheral vascular disease, revascularization
Subject Categories: Peripheral Vascular Disease, Gene Therapy, Angiogenesis
Introduction
Ischemia caused by occlusion of arteries is the most prevalent pathological condition that affects humans. Peripheral arterial disease affects ≈10 million people in the United States; it is often untreatable and progresses to critical limb ischemia (CLI), a condition that results in >150 000 lower limb amputations annually.1, 2 During the past 2 decades, therapeutic angiogenesis with recombinant proteins, genes, and stem cells has been tested for the treatment of myocardial ischemia and CLI.1, 3, 4, 5, 6, 7, 8, 9 Despite evidence that ischemic tissues are responsive to growth factor stimulation, clinical trials of vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF), fibroblast growth factor (FGF), constitutively active hypoxia‐inducible factor (HIF)‐1α, and developmental endothelial locus‐1 (Del‐1) have met with marginal success or failure.5, 7, 8, 10, 11, 12 Vascular regeneration after occlusion may require production of collateral vessels within and around tissues that are adjacent to the occlusion as well as regeneration of the microvasculature in the extremities downstream of the occlusion that experience the most severe ischemia (for reviews, see 13, 14, 15, 16). As part of the natural process of vascular development or regeneration, hypoxia‐inducible growth factors including VEGF are produced by ischemic tissues and diffuse out of the ischemic region to activate endothelial sprouting of adjacent vessels. Capillaries grow toward the ischemic tissue following directional cues determined at least in part by VEGF gradients. Capillaries may then remodel to form arterioles that support permanent collateral blood flow, a process that may also depend on the organization of new capillaries along gradients of hypoxia and VEGF.13, 14, 17 Similar regulation of gene expression by gradients of hypoxia and related growth factor expression may be important for angiogenic gene therapy in the setting of therapeutic angiogenesis to reproduce the natural process of microvascular regeneration in response to ischemia.
Here, we report results of hypoxia‐regulated conditionally silenced (CS) AAV vectors expressing human VEGF165 (AAV‐CS‐hVEGF) in a mouse autoamputation model of severe CLI. Gene expression, Hypoxyprobe staining, noninvasive luciferase (Luc) imaging, and hVEGF mapping revealed regions of tissue hypoxia and hVEGF expression distal and adjacent to an induced femoral artery occlusion. Physiology and laser Doppler imaging revealed increased tissue salvage and reperfusion by AAV‐CS‐hVEGF therapy compared with unregulated AAV or plasmids. Gene mapping of limb tissues by RT‐PCR during regeneration indicated reversible activation of endogenous genes associated with angiogenesis, vasculogenesis, and arteriogenesis, while DiI (1,1′‐dioctadecyl‐3,3,3′,3′‐tetramethylindocarbocyanine perchlorate) imaging and immunostaining were consistent with arteriogenesis. The results demonstrate for the first time the potential for safe and effective use of AAV, a semipermanent gene delivery vehicle for proangiogenic gene therapy.
Methods
Cell Culture, Transfections, and Hypoxia
Our methods for aerobic and hypoxic culture have been described previously.18, 19 Lipofectin (New England Biolabs) was used for all transfections. Luc was measured in cell lysates by using a Dual‐Luciferase Reporter Assay System (Promega).
Vector Constructs
Oligonucleotides containing the sequences 5′‐CTTCAGCACCGCGGACAGT‐3′ (neuronal responsive silencer elements [NRSEs]20) and 5′‐GCCTGTCACGTCCTGCACGACT‐3′ (hypoxia‐response element [HRE]) were inserted as multimers (3×) or tandem repeats of NRSE‐HRE (also 3×) into the multiple cloning site of pGL3‐PV (Promega). To create vectors with α–myosin heavy chain (α‐MHC) and phosphoglycerate kinase (PGK) promoters, the Simian virus (SV)40 promoter was excised from pGL3 and its progeny and replaced, respectively, with α‐MHC164 or PGK495. Mutations in the regulatory elements were made by replacing the core CCGCG of the NRSE element with AATCG or the core, ACGTC of the HRE with ATAGC. Plasmids containing the α‐MHC promoter were gifts from Bruce Markham (Pfizer Global Research and Development); vectors expressing wild‐type and dominant negative forms of neuronal responsive silencer factors (NRSFs) were gifts from Gail Mandel (Stony Brook University). The PGK promoter to 5′‐493 was generated by PCR by using primers designed from sequence NCBI accession number AF335420. All clones were verified through sequencing.
Generation of Recombinant AAV and In Vitro Expression
Two recombinant AAV vector plasmids were constructed by using the pAAV‐IRES‐hrGFP plasmid (Stratagene) as backbone. A 1.2‐kb Mlu/SalI containing the cytomegalovirus (CMV) promoter was replaced with the PGK or CS‐PGK promoter upstream of the hVEGF165 cDNA. After transfection into C2C12 myocytes, cultures were exposed to aerobic or hypoxic (1% O2) incubations as described previously.18, 19 Green fluorescent protein (GFP) expression was monitored with fluorescence microscopy, and secreted hVEGF165 measured in spent media was monitored with an ELISA (R&D Systems).
AAV Production
Wild‐type, single stranded (ss)AAV serotypes 2 and 9 were generated in HEK 293 cells by using adenovirus‐free pHelper plasmids to provide replication and packaging functions. Large‐scale batches of ssAAVs 9 and 2 were generated and purified at the vector core facilities of the University of Pennsylvania and University of Miami, respectively. Self‐complementary dsAAV serotype 1 was generated through site‐directed mutation of the viral long terminal repeat as described previously,21, 22 and large‐scale batches of dsAAV1‐CS‐hVEGF were generated and purified at SignaGen. Viral titers and quantification of tissue levels of AAV were determined by RT‐PCR by using primers directed to the IRES or hVEGF. For AAV infection in vitro, 30% confluent C2C12 skeletal myocytes were incubated with AAV at a density of 105 viral genomes (VG) per cell in serum‐free medium for 6 hours followed by 24 hours in the presence of 10% FBS. After an additional 24 hours, cultures were exposed to aerobic or hypoxic incubations, and hVEGF was measured in the spent media with an ELISA (R&D Systems).
Ischemic Hindlimb Model
All animal experiments were approved by the University of Miami Miller School of Medicine Institutional Animal Care and Use Committee. Our methods for implementing ischemia in BALB/c mouse hindlimbs have been described previously.23 Briefly, a skin incision is made in the middle of the left thigh longitudinally. The common femoral artery and vein are exposed proximally close to their origin at the external iliac artery and distally before their bifurcation into the popliteal and saphenous arteries. The proximal vessels, including the superficial and the deep femoral branches, and the distal portion of the saphenous artery are ligated with 7‐0 silk sutures and excised. The positions of the sutures and subsequent surgery are such that complete limb loss occurs in >80% of the untreated phosphate buffered saline (PBS) group at 2 to 3 weeks postsurgery. All arterial branches between the ligation are obliterated by excision, and the skin is closed with surgical stitches. Injections of AAV, plasmid, or PBS are made as described next.
Gene Delivery
Viral or plasmid DNAs were delivered via intramuscular injection at 4 sites into thigh and calf tissues in a total volume of 60 μL of PBS immediately after femoral artery excision or 5 days before ischemia surgery as indicated in the figure legends. Preinjection, especially of wild‐type ssAAVs 2 and 9, was found to be optimum to confer tissue salvage because of the long lag time for gene expression of wild‐type AAVs and rapid onset of necrosis and limb autoamputation in the severely ischemic BALB/c model. We found that the lag time for expression of the self‐complementary, double‐stranded (ds)AAV1 vector was significantly less, consistent with previous work.21, 22 For in vivo infection of hindlimb muscle, 1011 VGs were delivered unless indicated otherwise. Our procedures for intra‐arterial infusion of AAV were based on previously described protocols.24, 25, 26 Briefly, mice were sedated with ketamine/xylazine, and the femoral artery and vein were exposed. A 3‐0 braided silk tourniquet was placed loosely around the vessels above the site of incision. The femoral artery was punctured with a 33‐gauge needle, catheterized, and flushed with 100 μL of sterile saline. The tourniquet was applied, and 1011 VGs in 100 μL of PBS were administered with slow pressure over 1 minute followed by a dwell time of 10 minutes. Sterile saline was administered as a post flush. The catheter and the tourniquet were removed, and the wound was flushed with saline and closed.
Laser Doppler Perfusion Imaging
Laser Doppler perfusion imaging (LDI) was implemented by using an LDI2‐HR Laser Doppler Imaging System from Moor Instruments. Mice were lightly anesthetized with ketamine and placed on a heating pad to maintain temperature at 35°C to 37°C; images of both limbs were analyzed by using Moor LDI software. LDI results are presented as ratios of treatment:control contralateral limb. Amputees are scored as 0.
Immunohistology
Tissues, dissected and separated into upper thigh, lower thigh, and calf, were preserved in 10% formalin and embedded in paraffin. Standard procedures were used to deparaffinize, and fixed antigens were retrieved through incubation at 60°C with 20 μg/mL Proteinase K (Sigma‐Aldrich) for 20 minutes or immersion in boiling unmasking solution for 20 minutes. Slides were blocked in PBS with 2% serum and 0.4% Triton for 30 minutes, followed by use of the Avidin/Biotin Blocking Kit (Vector Laboratories). Primary antibody incubation was for 12 hours at 4°C with 2% serum and 0.4% Triton. Secondary antibody incubation was for 1 hour at 21°C with a biotinylated antibody in PBS/0.4% Triton. Staining was visualized by using an ABC Elite kit followed by DAB treatment (Vector Laboratories, Burlingame, CA) and hematoxylin and eosin counterstaining. Vessels were immunostained with anti‐CD31 (BD Biosciences Pharmingen) and anti–smooth muscle actin (SMA; Dako). The hVEGF antibody was from R&D Systems.
DiI Staining
DiI perfusion staining was implemented as described previously.27 After staining, the entire limb was severed and placed on a glass slide for imaging with a Zeiss LSM 510 confocal microscope. Individual images were merged by using Microsoft Paint software to create composites.
Vessel Volume
Z‐series obtained in confocal imaging were analyzed by using Volocity high‐performance imaging software.28 Vessel volume data were generated by using a uniform region of interest for each field of view. Ten fields of view from 4 separate mice were used to evaluate mean vessel density (volume of total DiI‐stained vessels) for each treatment.
Angular Deviation
Full‐size composite DiI images were imported into Volocity software,28 and a directional reference was determined manually by tracing along the path of the femoral nerve on each image. Unbranched vessels were traced by using Volocity, and the values for bearing on an XY‐axis were obtained and normalized to a 180° scale. The bearing of the reference was then subtracted from the bearing of each individual measured vessel in an image (minimum of 10 per limb), and the absolute value of each bearing was averaged to determine an overall measurement of angular deviation from the femoral nerve in collateral vessel growth for each hindlimb. Student t test was used to determine statistical significance between groups.
Magnetic Resonance Imaging
Anesthetized mice underwent magnetic resonance imaging with a 4.7‐T (200‐MHz) 40‐cm bore magnet interfaced with a Bruker Avance TM console including a gradient set with an inner diameter of 70 mm and a maximum gradient strength of 1000 mT/m. The signal was averaged 3 times, resulting in an acquisition time for each slice of ≈4 minutes. Six to 8 short‐axis slices that were 1 mm thick were used to cover the entire limb, resulting in a total scan time of 30 to 45 minutes for each mouse at each imaging session.
Hypoxyprobe
We used a Hypoxyprobe kit from Chemicon International as described by the manufacturer. Briefly, femoral artery excision was implemented in the right hindlimb as described above. Hypoxyprobe at a dose of 60 mg/kg in saline was delivered via intraperitoneal injection at intervals after ischemia; 1 hour later, mice were injected with heparin, anesthetized and perfused through the abdominal aorta with saline (5 minutes) and 2% paraformaldehyde (20 minutes). Hindlimbs were dissected, fixed in 2% paraformaldehyde for 48 hours at 4°C, rinsed, and stored in 70% ethanol. Sections were deparaffinized with xylene and prepared for immunostaining by standard procedures. Tissue peroxide was quenched with 3% H2O2 for 5 minutes and blocked with 1% BSA. Final sections were incubated with primary antibody at 4°C overnight, and secondary antibody was incubated for 1 hour at room temperature. Peroxidase chromogen DAB treatment was for 30 minutes at room temperature, and slides were counterstained with hematoxylin and eosin.
Bioluminescense Imaging
Standard cloning procedures were used to excise the hVEGF cDNA from pAAV‐CS‐hVEGF and replace it with firefly Luc cDNA. The resultant plasmid composition was confirmed by sequencing. CMV‐Ren‐Luc was from Promega Corp. Both hindlimbs each of 3 Balb/c mice per group were injected with 40 μg of pAAV‐CS‐Luc or pCMV‐Ren‐Luc in a total volume of 200 μL via 10 direct intramuscular injections along the length of the limb. After 5 days, femoral artery excision was implemented in the right hindlimb only as described earlier. At the indicated times, mice were anesthetized and injected with d‐Luciferin (150 mg/kg [PerkinElmer]) (or A‐Lume Coelenterazine [Nanolight Technology]) (CMV) in 20 μL of PBS at 6 to 8 sites along the limb. Imaging was implemented immediately using an IVIS Spectrum In Vivo Imaging System (PerkinElmer).
Statistical Analysis
Data are presented as mean±SD. Student t tests were performed to compare the means of 2 groups. P values of <0.05 were considered significant.
Results
Gene Regulation by Conditional Silencing
Gene silencers recruit HDAC to eukaryotic promoters and cause transcriptional silencing via chromatin condensation.29 Suppression of neuronal genes in non‐neuronal tissues is achieved when the ubiquitous NRSFs bind to NRSEs in the promoters of neuronal genes.20 Gene enhancers work by recruiting histone acetyltransferase, relaxing chromatin and activating transcription.29 To create a hypoxia‐regulated CS vector, we cloned alternating NRSEs and HREs in tandem upstream of a Luc reporter in a truncated SV40 promoter (called pCS‐SV). Expression of pCS‐SV in C2C12 skeletal myocytes was low when the myocytes were cultured aerobically but induced by almost 200‐fold by hypoxia. The induction was reversed through overexpression of wild‐type or dominant negative NRSF, the former as a result of retention of silencing under hypoxia and the latter as a result of an absence of aerobic silencing (Figure 1A). The effect of dnNRSF was mimicked by the pan‐HDAC inhibitor trichostatin A confirming the role of HDAC (Figure 1B). We found that CS exerted equivalent regulation of constitutively expressed promoters including SV40 and PGK and muscle‐specific promoters including α‐MHC and muscle creatine kinase in cardiac and skeletal myocytes, respectively (muscle‐specific data not shown here).
Figure 1.
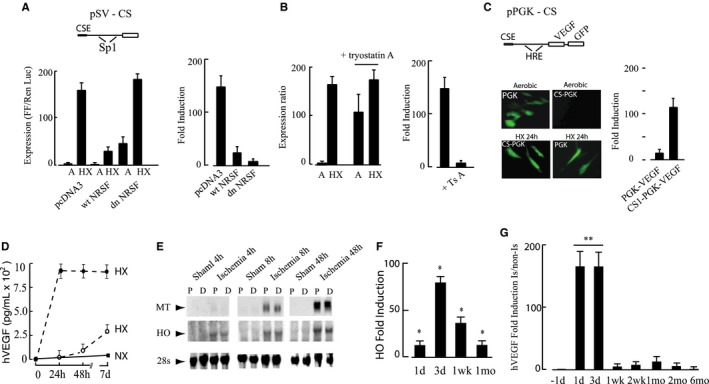
Mechanism of conditional silencing. A, C2C12 skeletal myocytes were transfected with pSV40‐CS‐Luc (pSV‐CS) (2 μg) and pcDNA, pNRSF, or pdnNRSF (2 μg), with Renilla luciferase as internal control and exposed to normoxia (21% O2) or hypoxia (0.5% O2). In the promoter diagram, CSE indicates conditional silencer element, and Sp1 depicts the positions of two Sp1 transcription factor binding sites in the SV40 promoter. B, C2C12 skeletal myocytes were transfected with pSV‐CS and subjected to aerobic or hypoxic incubation as in A; parallel plates were treated for 12 hours with trichostatin A (250 ng/mL) or vehicle. C, Adeno‐associated virus (AAV) shuttle plasmids expressing green fluorescent protein (GFP) and vascular endothelial growth factor (VEGF) phosphoglycerate kinase (PGK) directed by the PGK promoter or conditionally silenced PGK promoter (CS) were transfected into C2C12 skeletal myocyte and cultured for 48 hours under normoxia followed by 24 hours’ hypoxia. GFP was visualized by the use of fluorescence microscopy, and human VEGF (hVEGF) by the use of ELISA as described in Methods. D, C2C12 myocytes were infected with dsAAV1‐CS‐hVEGF (closed circles) or ssAAV9‐CS‐hVEGF (open circles) as described in Methods. Cultures were exposed to hypoxia (HX) or normoxia (NX) as indicated and samples of culture medium taken at the indicated times. Secreted hVEGF was measured by ELISA and values indicate rate of hVEGF production (pg/mL per 24 hours) for each condition. E, Mouse hindlimbs were made ischemic or subjected to sham operation as described in Methods. Mice were killed at 4, 8, 24, and 48 hours; the thigh muscles removed and cut into 2 longitudinal pieces proximal (P) and distal (D) to the femoral artery (FA) ligature. RNA was extracted and analyzed by Northern blot. F, Transcript levels of the HO gene in proximal adductor muscles containing the excised FA were quantified by RT‐PCR. G, Hindlimbs of BALB/c mice were injected with 1×1011 VP AAV‐CS‐VEGF, 5 days before implementing ischemia. Adductor muscle containing the injection sites was harvested and the transcript levels of hVEGF quantified by RT‐PCR. All results are mean±SEM, n=4; for F and G, *P<0.05; **P<0.01 by Student t test.
For in vivo studies, we created AAV shuttle vectors in which the hVEGF and GFP genes were driven by a CMV promoter (unregulated), PGK (partial hypoxia regulation), or PGK+NRSE/HRE (CS). The expression and regulation of PGK and PGK‐CS are shown in Figure 1C. A requirement of CS for tight regulation is again apparent from the high expression of PGK in aerobic cultures and the fold induction of PGK‐CS by hypoxia (120‐fold) compared with 6.5‐fold for PGK alone. To confirm regulated expression by whole virus, secreted hVEGF protein was measured in spent media from C2C12 myocytes infected with ssAAV9‐CS‐hVEGF or dsAAV1‐CS‐hVEGF as described in Methods. Figure 1D confirms that hVEGF secretion from cultures infected with dsAAV1‐CS‐hVEGF was rapidly induced by hypoxia, reached a maximal rate of secretion of 200±27 pg/mL per 24 hours 2 days after implementing hypoxia and remained at this level for at least 1 week. As expected, there was a lag time before expression from ssAAV9‐CS‐hVEGF was induced. Human (h)VEGF was not detected in conditioned media from uninfected normoxic or hypoxic C2C12 cells (not shown).
We used a BALB/c mouse ischemic hindlimb to model CLI and characterize gene therapy using CS‐AAV. Hindlimb muscles of BALB/c mice do not generate collateral vessels efficiently30 and more closely mimic patients with advanced peripheral artery disease relative to other mouse strains.31, 32 Severe ischemia was induced by ligation and partial excision of the femoral artery as described in Methods. This surgery results in >80% limb autoamputation in the absence of intervention (data not shown). To confirm hypoxia after surgery, we measured transcript levels of hypoxia‐inducible genes metallothionein (MT) and heme oxygenase (HO)33, 34 in anterior and posterior segments of the adductor muscles. Transcription of both genes was induced within 8 hours of ischemia and remained elevated at 72 hours (Figure 1E). PCR confirmed these results, showing peak HO expression of 80±4‐fold at 3 days that fell to 40‐ and 18‐fold at 1 and 4 weeks, respectively (Figure 1F). To determine whether the ischemic muscle–supported CS, AAV9‐CS‐hVEGF was delivered by intramuscular injection 5 days before femoral excision and hVEGF gene expression was quantified at intervals after ischemia surgery. As shown in Figure 1G, hVEGF transcripts in the ischemic limb were increased by 180‐fold 1 to 3 days after surgery, declined at 1 week but remained slightly elevated up to 1 month.
Sustained Hypoxia by FA Excision In Vivo
To confirm the presence of hypoxia in tissues downstream of FA excision, mice were injected intraperitoneally with Hypoxyprobe 7 days after surgery and muscle sections immunostained as described in Methods. Figure 2 shows representative sections of calf and lower thigh muscles from limbs with intact (left panels) and excised (right panels) FAs. Heavy brown staining was evident in the calf and thigh muscles of animals with FA excision, and myocytes with shrunken morphology were evident in the regions of intense staining, especially apparent in the calf muscle. Staining appeared more intense in the calf relative to thigh but was widespread in both muscle sets (also see Figure S1). The results confirm sustained severe hypoxia (<10 mm Hg) that is predicted to be sufficient to support hypoxia‐inducible gene expression and maintain CS‐gene vectors in a transcriptionally active state.
Figure 2.
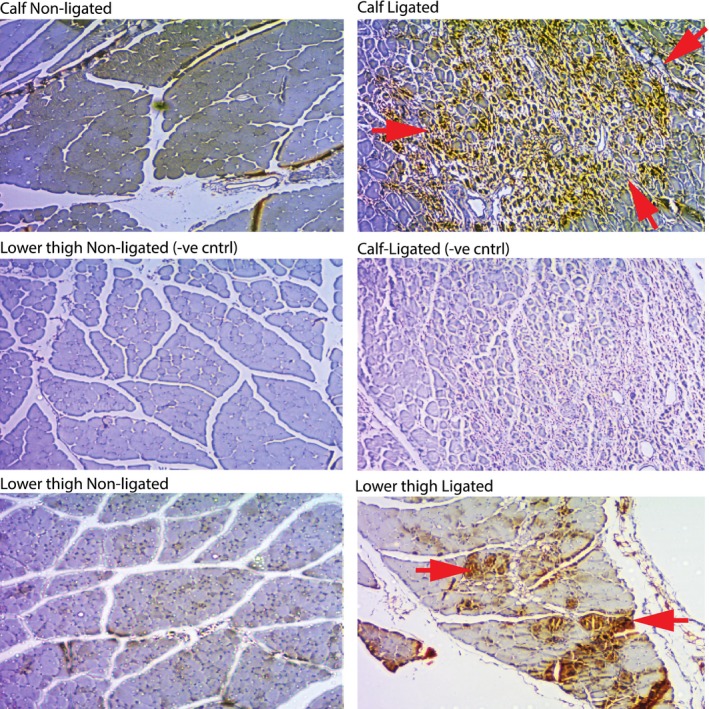
Hypoxyprobe immunostain. Representative sections of calf and lower thigh muscle stained with Hypoxyprobe as described in Methods were treated as indicated. Middle panels are without primary antibody. Red arrows denote positive staining (brown). See Methods for details of immunostaining.
Regulation of pAAV‐CS Vector In Vivo
To monitor the activity of the CS‐gene vector directly, we replaced hVEGF cDNA of pAAV‐CS‐hVEGF with Luc and used noninvasive bioluminescence imaging (IVIS) to detect expression in vivo (see Methods). Ischemia was implemented in the right limb only, and pAAV‐CS‐Luc was injected into both limbs. As shown in Figure 3, Luc expression was confined to the ischemic limb. Figure 3 (middle panels) depicts Luc expression in the ischemic limb over 13 days, during which the limb autoamputates. Luc is expressed above and below the FA excision consistent with low pO2 in both regions of tissue. By day 4, Luc was expressed in the entire limb, above and below the site of surgery but more intensely below. At days 8 to 13, the strongest expression was adjacent to the site of necrosis, predicted to be a region of severe ischemia. The lower panels in Figure 3 show expression from an unregulated pCMV‐(Ren)‐Luc vector. In this case, Luc is expressed in both limbs but repressed by ischemia in the left (surgery) limb. The results are consistent with graded hypoxia conferred by FA excision in surrounding tissues adjacent and distal to the lesion, that is sufficient to activate gene expression from a CS plasmid vector.
Figure 3.
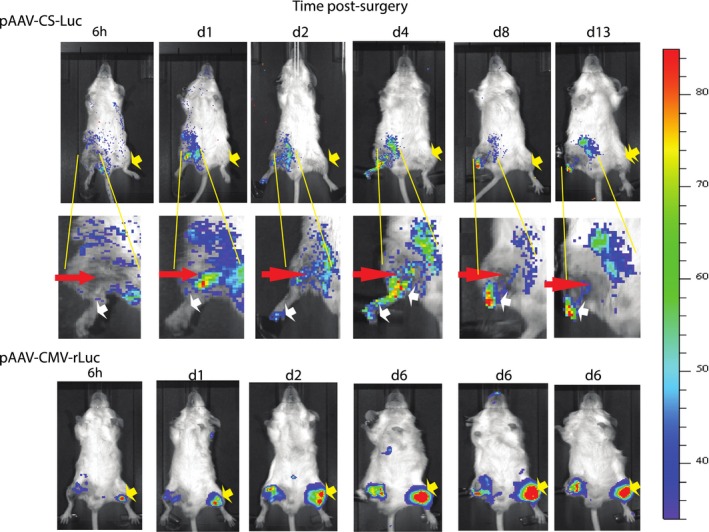
Bioluminescence imaging of pAAV‐CS expression. Mice were injected with pAAV‐CS‐Luc [conditionally silenced]‐Luc [luciferase] (top panels) or pAAV‐CMV [cytomegalovirus]‐Luc (bottom panel) in both limbs as described in Methods. After 5 days, the right hindlimb was subjected to femoral artery (FA) excision. Mice were imaged at the indicated times as described in Methods. Yellow arrows in top panels depict absence of luciferase expression in nonischemic left limbs. Red arrows in middle panels depict site of FA excision. White arrows depict graded luminescence between sutures and sites of necrosis at the extremity. Yellow arrows in bottom panels depict intense luminescence preferentially in the nonsurgery limbs by pAAV‐CMV‐Luc.
Expression of hVEGF In Vivo
To confirm expression of the hVEGF transgene in ischemic hindlimbs, sections of muscle downstream of the FA excision were immunostained with a selective hVEGF antibody. Transcripts of hypoxia‐regulated genes are rapidly induced by ischemia and global hVEGF mRNA expression by AAV9‐CS‐hVEGF peaked between 1 and 3 days after surgery (Figure 1). Immunostaining was implemented on hindlimbs from ischemia day 3, a time point when hypoxia‐induced VEGF protein production is predicted to be high. Representative sections from PBS, ssAAV9‐CS‐hVEGF, and ssAAV9‐PGK‐hVEGF treatments are shown in Figure 4. Positive (brown) heterogeneous regions of staining of both muscle and capillaries were evident in the AAV‐CS and AAV‐PGK treatments but not in PBS controls. Black arrows in Figure 4 (upper and middle panels) indicate a border between positive and negative hVEGF stain; yellow arrows indicate areas of inflammatory cell infiltration; and white arrows indicate putative endothelium. Note that endothelial and inflammatory cells in the regions of positive hVEGF stain are negative (blue) or positive (dark brown) for hVEGF stain, which may indicate hVEGF binding to receptors. Both endothelial and inflammatory cells are known to express the VEGFR2.35, 36 It is also noteworthy that capillaries appeared larger and more numerous in regions of positive hVEGF staining in both CS and PGK groups relative to the PBS group. In results not shown, we found no hVEGF staining in nonischemic muscle independent of treatment. The results are consistent with ischemia‐induced hVEGF expression from both CS and PGK promoters in vivo. The results are also consistent with previous work showing significant penetration of foreign transgene expression in hindlimb muscle by intramuscular delivery of AAV.37
Figure 4.
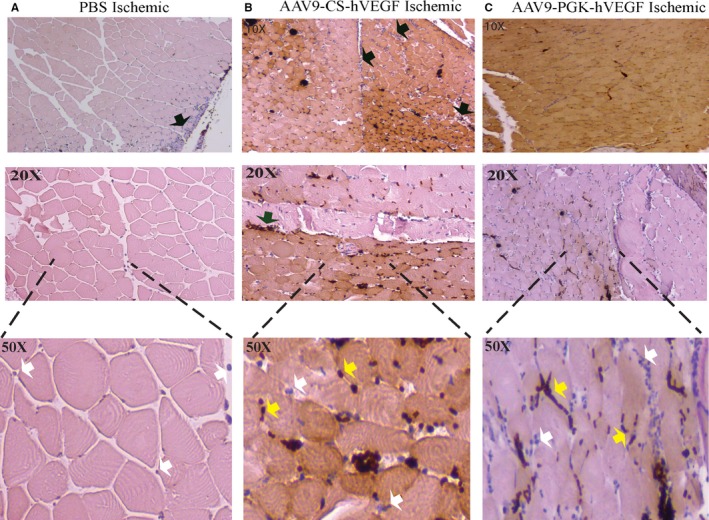
Human vascular endothelial growth factor (hVEGF) immunostain. A through C, Mouse hindlimbs were preinjected with phosphate buffered saline (PBS) or the indicated vectors (1011 VPs) 5 days before femoral artery excision as described in Methods. Lower thigh and calf muscles were harvested on day 3, dissected, fixed, and stained with anti‐hVEGF antibody, and microscopic images were obtained in a semiblinded manner as described in Methods. Black arrows indicate border of positive and negative hVEGF staining; white and yellow arrows depict capillaries and inflammatory cell infiltrates, respectively, that may be negative (blue) or positive (dark brown) for hVEGF. AAV indiactes adeno‐associated virus; CS, conditionally silenced; PGK, phosphoglycerate kinase.
Therapeutic Angiogenesis by CS hVEGF
Efficacies of CS vectors in providing hindlimb recovery after ischemia were quantified in a BALB/c mouse model of severe CLI. First, we tested plasmid vectors because these have been the most common vehicles used for CLI gene therapy clinical trials.3, 5, 7, 8, 12 We compared the effects of unregulated (CMV) and CS‐regulated plasmids at low and high doses, scaled, respectively, to be equivalent to and 5‐fold higher than the doses typically used for patient trials. At the lower dose, all limbs autoamputated within 4 weeks (Figure 5A and 5B). At high dose (Figure 5C and 5D), necrosis developed more slowly and resulted in 80% loss of limbs in the CMV group. In contrast, mice treated with high‐dose pCS‐hVEGF were protected and limbs were significantly salvaged at 20 weeks, although with low perfusion and >60% loss of toes (Figure 5C). Next, we compared regulated AAV9‐CS‐hVEGF and AAV9‐PGK‐hVEGF vectors with unregulated AAV9‐CMV‐hVEGF and PBS. Quantification of laser Doppler ratios, shown in Figure 5E, confirmed a gradual return of blood flow in AAV9‐CS‐hVEGF and AAV9‐PGK‐hVEGF groups to a maximum of 60% of the collateral limb but no such increase in either CMV or PBS groups. Examples of limbs imaged by digital photography and laser Doppler at 1 and 2 weeks respectively from mice without surgery (control) and after surgery with PBS, AAV9‐CS‐hVEGF, and AAV9‐CMV‐hVEGF treatments are shown in Figure 5F. Whereas all ischemic limbs showed evidence of necrosis, this was mostly limited to discoloration of toes in the AAV9‐CS‐hVEGF group, but there was major tissue loss in the PBS and AAV9‐CMV‐hVEGF groups. Tissue integrity correlated closely with Doppler images indicative of tissue salvage by reperfusion in CS but not CMV or PBS treatment groups (Figure 5F, lower panels). In results not shown we also found that adenovirus‐expressing CMV‐hVEGF did not support long‐term limb salvage in this model, a result that is consistent with our previous report of adenoviral‐VEGF therapy in a rabbit ischemic hindlimb model.38
Figure 5.
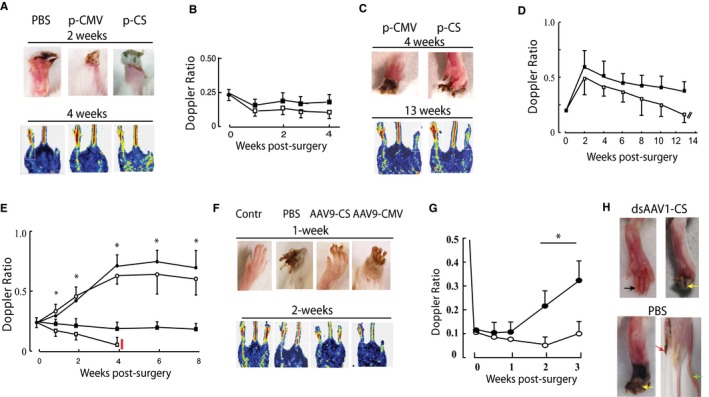
Adeno‐associated virus (AAV)‐mediated limb reperfusion and tissue salvage. A through D, Mouse hindlimbs were made ischemic and injected immediately after surgery with low‐dose (20 μg, A and B) or high‐dose (100 μg, C and D) plasmid vectors expressing human vascular endothelial growth factor (hVEGF) under the direction of cytomegalovirus (CMV) or conditionally silenced (CS) promoters as indicated. Upper and lower panels show lower limbs and Doppler images at 2 and 4 weeks and 4 and 13 weeks, respectively, for low‐ and high‐dose groups, with Doppler ratios quantified in the right panels; filled squares CS, open squares CMV (n=5). E and F, Mouse hindlimbs were injected with AAV or phosphate buffered saline (PBS) 5 days before induction of limb ischemia. E, Mean Doppler ratios quantified over 8 weeks of AAV9‐CS‐hVEGF (open circles; n=10), AAV9‐PGK‐hVEGF (filled circles, n=10), AAV9‐CMV‐hVEGF (filled squares, n=8), or PBS (open squares, n=12). *P<0.05 comparing AAV9‐CS‐hVEGF with control (PBS) at each time point by Student t test. F, Representative examples of digitally photographed limbs (top) and laser Doppler scans (lower panels) for the indicated condition and time. G and H, BALB/c mouse hindlimbs were treated as in C through F except that dsAAV1‐CS‐hVEGF (1011 VG; closed circles) or PBS (open circles) was infused through the femoral artery as described in Methods 5 days before surgery to induce ischemia. Results are mean±SEM for each time point (n=8). *P<0.05 comparing dsAAV1‐CS‐hVEGF with PBS control at weeks 2 and 3 by Student t test. Photographs in H show representative images of limbs treated as indicated at 2 weeks after ischemia. Black arrow depicts fully salvaged digits; yellow arrows depict intact feet but necrosis of toes; red arrow depicts fully amputated limb.
Previous work has shown that intra‐arterial (IA) infusion is an efficient procedure to deliver AAV to limb muscle and may be the method of choice when maximum tissue penetration is required, for example, when gene replacement is the goal.24, 25, 26 High transduction efficiency may also be an advantage for clinical application in CLI; therefore, we tested IA administration of dsAAV1‐CS‐hVEGF in the BALB/c mouse hindlimb CLI model by using procedures described in Methods. As shown in Figure 5G and 5H, IA delivery of dsAAV1‐CS‐hVEGF supported significantly enhanced perfusion (Figure 5G) and limb salvage (Figure 5H) compared with PBS. Note that the starting Doppler ratios after IA delivery (Figure 5G) were lower than those reported for IM (Figure 5E), an effect that may be a consequence of the IA procedure. Therefore, whereas the IA procedure conferred apparently lower induced Doppler scores at 2 and 3 weeks relative to intramuscular delivery, the incremental recoveries are similar. In the IA dsAAV1‐CS‐hVEGF group, 50% (4 limbs) were fully salvaged and 50% (4 limbs) were partially salvaged, whereas only full amputation and partial salvage were seen in the parallel PBS group. The IA procedure is acutely traumatic, and we experience ≈50% loss of mice in both treatment and PBS groups mostly as a result of blood clots that occluded in the FA and caused rapid limb loss; these mice were excluded from the study. Limbs that survived the procedure showed full recovery of perfusion as measured by Doppler scores at day 1 post IA delivery in all groups (data not shown). Taken together, these results confirm for the first time that gene therapy with regulated AAV or high‐dose plasmids, but not unregulated vectors, confers limb salvage in a mouse autoamputation model of severe CLI. The results further indicate that an IA route of gene delivery may be a feasible alternative to the standard intramuscular methods that are routine in most clinical gene therapy trials for CLI and that may limit efficacy.
Directional Vessel Growth and FA Regeneration
Gene therapy with AAV‐CS‐hVEGF conferred limb salvage and reperfusion to levels of 50% to 60% of control limbs. Such reperfusion would not be predicted if the therapeutic intervention were restricted to enhancement of the microvasculature at the ischemic limb extremities with no effect on upstream collateralization. Therefore, to investigate the morphology of new vessels grown in response to AAV‐CS‐hVEGF, we used in vivo DiI staining,27, 39 immunohistological examination, and MRI. Figure 6A, top panel, shows an image of the normal FA after excision of the vein, and the bottom panel shows 6 successive images (different animals) of the same region at progressive times after FA excision and gene therapy with AAV9‐CS‐hVEGF. White crosses mark the position of the sutures, and yellow arrowheads show the position of the femoral nerve. The image at day 1 after surgery confirms excision of the FA and femoral vein. One week after surgery, there is evidence of new vessel growth close to the femoral nerve. Images taken at 1, 4, and 8 months reveal vessels of large dimensions also located close to the femoral nerve. The eighth panel down shows a light micrograph of a section without staining 1 year after surgery and AAV9‐CS‐hVEGF therapy; black‐and‐white arrows depict the femoral nerve and new artery, respectively. We observed this pattern of vessel growth only in AAV9‐CS‐hVEGF groups. To quantify directional growth of vessel in response to AAV9‐CS‐hVEGF therapy, we used Volocity software to measure the average degree of deviation of DiI‐stained vessels from the orientation of the femoral nerve at time points >6 months post gene therapy (see Methods). As shown in Figure 6B, vessels generated in response to CS therapy deviated significantly less from the direction of the femoral tract than did either of the unregulated promoters, supporting the concept that CS supports directional vessel growth.
Figure 6.
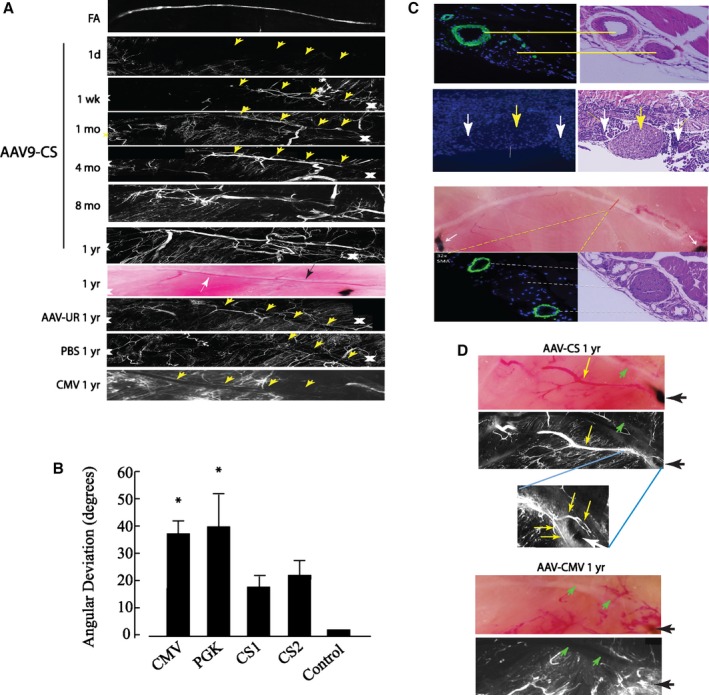
Femoral artery (FA) regeneration by conditionally silenced (CS) adeno‐associated virus (AAV)‐human vascular endothelial growth factor (hVEGF). A, Composite DiI images show vessel regeneration in AAV9‐CS‐hVEGF–treated limbs. Vessels regenerate along the femoral tract (yellow arrowheads) over the course of 1 year after FA excision. Progressive vessel regeneration is not seen in limbs treated with AAV‐CMV‐hVEGF, AAV‐PGK‐hVEGF, or phosphate buffered saline (PBS). B, Composite DiI images were analyzed by using Velocity software to measure the angular deviation of regenerated vessels from the path of the femoral nerve as described in Methods (*P<0.05; n=4 limbs×10 vessels per limb=40 vessels per group). C, Top panel, anti‐smooth muscle actin (SMA)– and DAPI‐ (left) and hematoxylin and eosin (H&E)– (right) stained sections containing the normal FA and femoral nerve; second panel, the same location immediately after hindlimb ischemia surgery; yellow arrow indicates femoral nerve; white arrows indicate region of FA excision. Bottom panels, top: light field image of hindlimb muscle with AAV9‐CS‐hVEGF treatment at 10 months after FA excision. Anti‐SMA and H&E stains confirm arterial regeneration within the femoral tract (bottom panels). D, Light field (top) and DiI image (middle) of AAV‐CS‐hVEGF treated hindlimb at 1 year after ischemia surgery; inset (bottom) shows higher magnification of vessels skirting the sutures. E, Same as (D) except treatment with AAV9‐CMV‐hVEGF. CMV indicates cytomegalovirus; PGK, phosphoglycerate kinase.
To confirm the presence of new arteries close to the femoral nerve, sections were prepared through this region from normal muscle (no surgery), immediately postsurgery, and 1 year after gene therapy and immunostained with SMA and hematoxylin and eosin. As shown in Figure 6C, in undisturbed tissue, the FA is SMA positive and runs parallel to the nerve (top panel). A similarly stained section after excision of the FA and femoral vein shows again the intact nerve but no FA. One year after FA excision and treatment with AAV9‐CS‐hVEGF, 2 SMC‐positive vessels with dimensions >0.5 mm can be seen positioned on either side of the nerve. As indicated in the light micrograph, these vessels traverse between the original sutures and parallel the original FA track. In other specimens we examined the organization of vessels around the sutures. Examples are shown in Figure 6D. In the AAV9‐CS‐hVEGF–treated limbs, small vessels skirt the suture on both sides, possibly making connections between de novo vessels and intact vessels upstream and downstream of ischemia. This was not seen in PBS‐ or AAV‐CMV–treated limbs (Figure 6D).
Angiogenesis and Arteriogenesis by CS‐Gene Therapy
Increased capillary density by gene and stem cell treatment has been confirmed in multiple models of hindlimb ischemia.15, 30, 40 We estimated endothelial growth by measuring PECAM‐1 (CD31) gene expression by RT‐PCR and capillary density by staining sections with anti‐CD31 antibody and isolectin‐B4. PECAM‐1 transcript levels (Figure 7A) increased relative to control limbs in all samples but were treatment dependent. In the PBS group, PECAM‐1 expression lagged behind the treatment groups and continued to increase over 4 weeks. In the CMV group, PECAM‐1 levels were maximal at 3 days and remained elevated at 4 weeks. In the CS groups, PECAM‐1 was elevated during the first week and fell to control after 4 weeks. Consistent with this, capillary density estimated by CD31 and isolectin‐B4 staining was consistently lower than that in control limbs in the PBS group, higher in the CMV group, and similar to untreated controls after 4 weeks in the CS group (Figure 7B). Representative sections from each condition are shown in Figure 7C and 7D. To estimate the size of SMA‐positive vessels, we used Volocity software again to generate the mean volumes of the 10 largest vessels from confocal images. The mean volume of SMA‐positive vessels of AAV9‐CS‐hVEGF–treated limbs was significantly greater than that of AAV9‐PGK–treated limbs, although neither differed significantly from normal limbs (Figure 7E). To confirm the production of large arteries in the region of the femoral vein by AAV9‐CS‐hVEGF therapy, we compared magnetic resonance images of normal limbs (no surgery) with limbs 10 months after FA excision and treatment with AAV‐CS. With this technique, the FA and femoral vein can be visualized on the anterior surface and the deep FA close to the midline of the muscle of normal limbs (Figure 7F). Vessels with slightly less intensity are visualized in the same position with the corkscrew appearance typical of regenerated collaterals.
Figure 7.
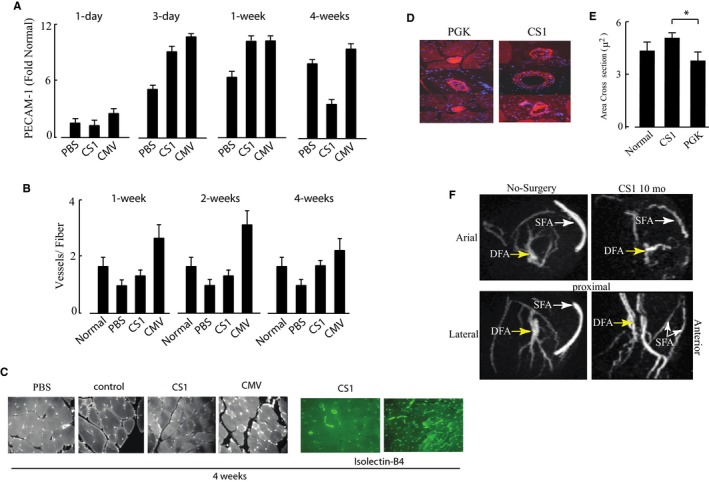
Capillary density and vessel dimensions. A, Mouse hindlimbs received intramuscular injections of AAV[adeno‐associated virus]9‐CS [conditionally silenced]‐hVEGF [human vascular endothelial growth factor] (CS), AAV9‐CMV‐hVEGF (CMV) (each 1011 viral particles (VP)), or phosphate buffered saline (PBS) prior to ischemia surgery as described in Figure 5. Following surgery, RNA was extracted from the adductor muscles within the region of gene therapy at 1, 3, and 7 days and 4 weeks as indicated and analyzed by qRT‐PCR for platelet and endothelial cell adhesion molecule 1 (PECAM‐1) expression. RNA isolated from normoxic muscle of control mice was used as a baseline for gene expression (n=3). B, Paraffin sections of hindlimb adductor muscles harvested at 1, 2, and 4 weeks after surgery and treatments were stained with anti‐CD31 or isolectin B4. Results include combined vessels identified by both staining procedures (P<0.05; n=4 limbs per group×10 vessels per limb=40 vessels per group). C, Representative fields of CD31‐ and isolectin‐stained sections with treatments indicated. D and E, Representative images and quantification of hindlimb sections stained with anti‐smooth muscle actin (SMA). The volume of SMA‐positive vessels was quantified in each group by using Volocity software. F, Magnetic resonance images of mouse hindlimbs before surgery (left panels) and 10 months after FA excision and treatment with AAV9‐CS‐hVEGF. The presence of the putative regenerated superficial FA (SFA) and deep FAs (DFA) are indicated in lateral and vertical rotational views. CMV indicates cytomegalovirus; PGK, phosphoglycerate kinase.
Reversible Activation of Angiogenic and Vasculogenic Regulatory Pathways
To further characterize the pathway of AAV9‐CS‐hVEGF–mediated vessel growth, we measured the expression of angiogenic marker genes. As shown in Figure 8, there were consistent differences in the profiles of AAV‐CS versus AAV‐CMV–treated or untreated (PBS) groups. The activities of 3 of the main effectors of Notch signaling including Dll4, Hey2, and Jag‐1 displayed the same expression trends with peak expression during the first week of AAV‐CS‐hVEGF treatment, followed by a decline to baseline after 4 weeks (Figure 8A through 8C). In contrast to this, Hey2 transcripts continued to increase over 4 weeks in both PBS and AAV‐CMV‐hVEGF groups, Dll4 was constitutively elevated in both, and Jag‐1 increased in the PBS group and remained significantly elevated at 4 weeks in both PBS and AAV‐CMV‐hVEGF groups. Hey2 is a Notch‐activated transcription factor that is required for arterial commitment41; Dll4 and Jag‐1 are also Notch‐pathway proteins that have opposing effects on endothelial sprouting; and Dll4 quenches while Jag‐1 increases tip formation.16, 42, 43 PECAM‐1 expression (shown in Figure 7) paralleled Notch signaling. The results are consistent with coordinate regulation of Notch signaling by VEGF and hypoxia only in the AAV‐CS‐hVEGF groups. The transcript levels of SDF‐1, angiopoietin‐1, and Ephrin B2 were also regulated in the CS and CMV groups but not in the PBS group (Figure 8D through 8F). These transcripts are markers, respectively, of vasculogenesis, vascular permeability, and endothelial tip extension, and the expression trends are consistent with enhanced angiogenesis in the CS and CMV groups.13, 14, 44, 45, 46 The results are consistent with coactivation and subsequent silencing of multiple genes involved in vessel growth and maturation selectively in response to CS‐gene therapy.
Figure 8.
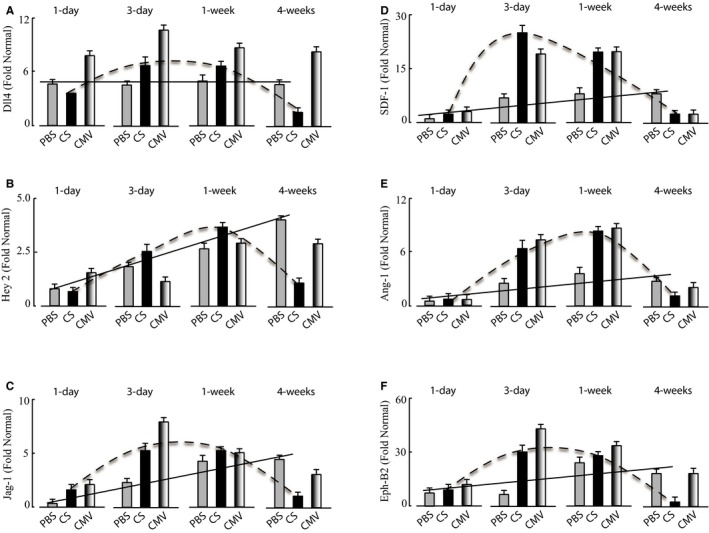
Reversible activation of angiogenic, vasculogenic, and remodeling genes. Hindlimb surgery was implemented 5 days after intramuscular gene therapy with AAV [adeno‐associated virus]9‐CS [conditionally silenced]‐hVEGF [human vascular endothelial growth factor] orAAV9‐CMV [cytomegalovirus]‐hVEGF, both 1011 viral particles (VP) per limb, or PBS as in Figure 5. Mice were killed and limbs were collected at 1, 3, 7, and 28 days after surgery. RNA was extracted from frozen muscle tissues and qRT‐PCR analysis implemented as described in Methods. Angiogenic gene transcripts in each panel were compared with the gene expression levels in naïve (normal) hindlimb tissue. A, Dll4; B, Hey2; C, Jagged‐1 (Jag‐1); D, Stromal‐derived factor‐1 (SDF‐1); E, Angiopoietin‐1; and F, Ephrin‐B2 (all n=3). Lines depict gene expression profiles in PBS (solid) and CS (dashed) treatment groups showing reversible activation of all genes by AAV‐CS‐hVEGF but not PBS.
Inhibition of Vascular Regeneration by Dibenzazepine
An essential role of Notch signaling for therapeutic angiogenesis in this model was confirmed by inhibiting Notch with dibenzazepine, a γ‐secretase inhibitor, before therapy with AAV‐CS‐hVEGF. As shown in Figure 9, such inhibition resulted in the production of disorganized, leaky vessels and rapid limb loss. This result is consistent with the known requirement of Notch for organized angiogenesis.41, 42, 43
Figure 9.
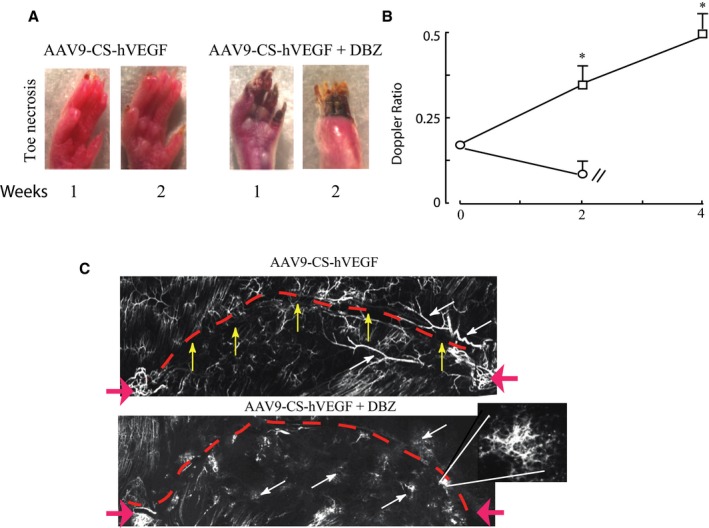
Requirement of Notch signaling for limb salvage. Gene therapy and femoral artery excision were implemented as described in Methods. Mice were treated or not with (S)‐2‐[2‐(3,5‐difluoro‐phenyl)‐acetylamino]N‐((S)‐5‐methyl‐6‐oxo‐6,7‐dihydro‐5H‐dibenzo[b,d]azepin‐7‐yl)‐propionamide (DBZ; Axon Medchem BV) delivered via intraperitoneal injection (10 μg/kg) immediately after ischemia surgery and daily thereafter for 7 days. Then, 1011 VP of adeno‐associated virus (AAV) was delivered intramuscularly to each treatment limb 5 days before surgery. A, Representative photographs of limbs 1 week after treatments with AAV9‐CS [conditionally silenced]‐hVEGF [human vascular endothelial growth factor]±DBZ. B, Doppler ratios confirm recovery of perfusion in the AAV9‐CS‐hVEGF treatment group without DBZ (squares) but not with DBZ treatment (circles). C, Composite DiI confocal images of the femoral tract reveal uncontrolled sprouting and vessel leakiness in DBZ‐treated mice. The magnified area highlights an area of vascular sprouting in a DBZ limb. Red dashed lines indicate the location of the femoral nerve; red arrows show suture locations. White and yellow arrows highlight differences in vascular morphology between treatment groups. *P<0.05; n=4 by Student t test comparing AAV treatment groups with or without DBZ at each time point. DBZ indicates dibenzazepine.
Discussion
Here, we show that gene therapy with AAV‐CS‐hVEGF confers limb salvage, regeneration of arteries, and reperfusion of limb extremities. Treated mice sustained lower degrees of necrosis, although toe loss was common in all ischemia groups and is attributed to the low tolerance of BALB/c mice to ischemia.47, 48, 49, 50 In the same model, hVEGF delivery by a constitutively active (CMV) promoter using AAV, adenovirus, or plasmid vectors did not prevent necrosis or limb loss suggesting that regulation of gene expression by hypoxia is required for limb salvage in this model. Limb salvage and moderate return of perfusion were also achieved by using a hypoxia‐regulated plasmid vector expressing hVEGF but only at a high dose that is several‐fold greater than equivalent plasmid doses that have been used in clinical trials. We propose that hypoxia regulation restricts VEGF transgene expression to ischemic tissues and recapitulates a natural process of vascular regeneration where ischemia directs the formation of growth factor gradients that guide endothelial sprouting and new vessel growth toward the regions of ischemia (reviewed in 16). In the BALB/c hindlimb model, the endogenous response to ischemia is insufficient to prevent tissue damage and limb loss, but enhanced VEGF production by AAV‐CS‐VEGF in hypoxic muscle is sufficient to counter these effects and promote limb salvage and revascularization. If this extrapolates to human CLI AAV‐CS‐VEGF may also improve amputation‐free survival in patients.
Mapping of tissue hypoxia, hypoxia‐regulated gene expression, and hVEGF immunostaining (Figures 2, 3 through 4) are consistent with the presence of hypoxia in thigh as well as calf and foot muscles after FA surgery. The widespread hypoxia and high sensitivity to ischemia of BALB/c mice relative to other mouse strains have been traced to genetic differences in a quantitative trait locus on chromosome 7 that may include defective regulation of micro‐RNAs.48, 49, 50, 51, 52, 53 The BALB/c hindlimb autoamputation phenotype more closely resembles human CLI, where gangrene and amputation are also common outcomes. In our model, the presence of widespread hypoxia is required to activate gene expression from CS vectors in muscles that are proximal (thigh) as well as distal (calf, feet) to the FA excision. Such gene activation is required for the generation of collateral vessels around the FA ligation (see Figures 6 and 7) as well as for cytoprotection and angiogenesis in tissues at the extremities. Because hypoxia may not be present in muscles surrounding occlusions of the femoral and popliteal arteries that are the most common lesions in human CLI, these tissues may need to be targeted separately to recapitulate equivalent CS silencing gene therapy in humans. Therefore, optimized human gene therapy along these lines may involve targeted delivery of factors such as HGF or FGF as plasmids to the region of occlusion combined with AAV‐CS‐VEGF delivered by IA infusion to mainly target the limb extremities. Delivery of AAV‐CS‐VEGF at the time of vascular surgery or angioplasty may improve outcomes of these surgical interventions that are often poor especially for late‐stage CLI.54, 55
Positive therapeutic angiogenesis by AAV‐CS‐VEGF may be an unexpected finding because the endogenous VEGF gene is already activated by hypoxia. Indeed, we found that mouse VEGF levels were low in normal thigh muscles but increased several‐fold after ischemia (data not shown (see references 49, 51). The muted endogenous response to ischemia and robust reversal by AAV‐CS‐VEGF therapy is paralleled by similarly depressed expression levels of SDF‐1, angiopoietin‐1, and ephrin‐B2 in placebo (PBS) mice after FA ligation but markedly augmented by AAV‐CS‐VEGF treatment (see Figure 8). This is consistent with suboptimal endogenous VEGF production by ischemic limb muscles rather than muted downstream responses to VEGF. Depressed gene activation by ischemia/hypoxia has also been reported in human CLI, especially in aged and/or diabetic subjects.56 Therefore, augmentation of appropriately regulated VEGF in muscles of human CLI patients may be a viable clinical strategy. Previous gene therapy with plasmids or adenovirus that constitutively overexpress VEGF, FGF, HGF, activated HIF‐1α, or Del‐1 have not achieved clinical efficacy despite promising results in preclinical models (reviewed in 48). Explanations for this include inadequacies of gene expression, gene persistence, and single factor approaches as well as inappropriate predictive value of preclinical models.48 Our results suggest another possibility—that clinical outcome may depend on administering separate interventions to target arteriogenic stimulators plasmid hepatocyte and/or fibroblast growth factors (pHGF, pFGF) to regions of arterial occlusions and AAV‐CS‐VEGF to the extremities to provide cytoprotection and regeneration of the microvasculature.
Our results are consistent with previous reports that VEGF is a master angiogenic switch that controls endothelial sprouting, growth, and maturation by coregulating multiple other factors.13, 16, 41, 42, 43, 44, 45, 46 Our results also support previous reports that uncontrolled VEGF expression can lead to disorganized, dysfunctional capillaries that can worsen ischemia.14, 57 A major finding of our work was the appearance of large SMA‐positive vessels close to the track of the original FA only in the AAV‐CS‐hVEGF treatment group. Our results are consistent with the following requirements for limb salvage in this model: (1) an immediate angiogenic response with coordinated, reversible activation of proangiogenic/vasculogenic and regulatory (Notch) pathways; (2) hypoxia‐regulated VEGF expression that supports directional growth of new vessels; and (3) controlled VEGF production in hypoxic but not normoxic tissue. These results are also consistent with other reports that constitutive VEGF levels must be controlled within a narrow window to avoid detrimental side effects on the vasculature58, 59 and that persistent expression is required for vessel stability.56, 60 The tight regulation conferred by CS and restriction of production to viable cells within zones of ischemia may also facilitate anastomoses with existing vessels, enhance longitudinal blood flow, and create shear stress stimuli for arterial remodeling.61, 62
It is noteworthy that in the BALB/c model described here, none of the vectors that have been used for clinical trials, including unregulated plasmids and adenovirus or unregulated AAV conferred limb salvage. Success of the AAV‐CS vectors can be attributed to the provision of growth factor gradients and cues for vessel guidance and persistent, vigilant transgene expression. CS not only confers tightly restricted gene expression required for such directional cues but also provides a stringent self‐regulating molecular switch that may be essential for the safety of growth factor gene therapy with persistent vectors such as AAV.
Sources of Funding
This work was supported by grant HL072924 from the National Institutes of Health (Dr Webster) and by a Walter G. Ross Distinguished Chair in Vascular Biology (Dr Webster).
Disclosures
Dr Webster is the founder and president of Integene LLC. The other authors have no conflicts to declare.
Supporting information
Figure S1. Representative sections of ischemic calf and lower thigh muscle stained as indicated by using a Hypoxyprobe kit as described in Methods and the legend for Figure 2. Bottom panels are negative controls lacking primary antibody.
Acknowledgments
We acknowledge the Gene Therapy Resource Program (GTRP) of the National Heart, Lung, and Blood Institute, National Institutes of Health for providing the AAV9 gene vectors used in this study. We also acknowledge Dr Lina Shehadeh for advice on statistical analyses.
(J Am Heart Assoc. 2016;5:e001815 doi: 10.1161/JAHA.115.001815)
References
- 1. Powell RJ. Update on clinical trials evaluating the effect of biologic therapy in patients with critical limb ischemia. J Vasc Surg. 2012;56:264–266. [DOI] [PubMed] [Google Scholar]
- 2. Clair D, Shah S, Weber J. Current state of diagnosis and management of critical limb ischemia. Curr Cardiol Rep. 2012;14:160–170. [DOI] [PubMed] [Google Scholar]
- 3. Gupta R, Tongers J, Losordo DW. Human studies of angiogenic gene therapy. Circ Res. 2009;105:724–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sneider EB, Nowicki PT, Messina LM. Regenerative medicine in the treatment of peripheral arterial disease. J Cell Biochem. 2009;108:753–761. [DOI] [PubMed] [Google Scholar]
- 5. Rajagopalan S, Mohler ER III, Lederman RJ, Mendelsohn FO, Saucedo JF, Goldman CK, Blebea J, Macko J, Kessler PD, Rasmussen HS, Annex BH. Regional angiogenesis with vascular endothelial growth factor in peripheral arterial disease: a phase II randomized, double‐blind, controlled study of adenoviral delivery of vascular endothelial growth factor 121 in patients with disabling intermittent claudication. Circulation. 2003;108:1933–1938. [DOI] [PubMed] [Google Scholar]
- 6. Ghosh R, Walsh SR, Tang TY, Noorani A, Hayes PD. Gene therapy as a novel therapeutic option in the treatment of peripheral vascular disease: systematic review and meta‐analysis. Int J Clin Pract. 2008;62:1383–1390. [DOI] [PubMed] [Google Scholar]
- 7. Powell RJ, Simons M, Mendelsohn FO, Daniel G, Henry TD, Koga M, Morishita R, Annex BH. Results of a double‐blind, placebo‐controlled study to assess the safety of intramuscular injection of hepatocyte growth factor plasmid to improve limb perfusion in patients with critical limb ischemia. Circulation. 2008;118:58–65. [DOI] [PubMed] [Google Scholar]
- 8. Belch J, Hiatt WR, Baumgartner I, Driver IV, Nikol S, Norgren L, Van Belle E; TAMARIS Committees and Investigators . Effect of fibroblast growth factor NV1FGF on amputation and death: a randomized placebo‐controlled trial of gene therapy in critical limb ischaemia. Lancet. 2011;377:1929–1937. [DOI] [PubMed] [Google Scholar]
- 9. Kajiguchi M, Kondo T, Izawa H, Kobayashi M, Yamamoto K, Shintani S, Numaguchi Y, Naoe T, Takamatsu J, Komori K, Murohara T. Safety and efficacy of autologous progenitor cell transplantation for therapeutic angiogenesis in patients with critical limb ischemia. Circ J. 2007;71:196–201. [DOI] [PubMed] [Google Scholar]
- 10. Creager MA, Olin JW, Belch JJ, Moneta GL, Henry TD, Rajagopalan S, Annex BH, Hiatt WR. Effect of hypoxia‐inducible factor‐1alpha gene therapy on walking performance in patients with intermittent claudication. Circulation. 2011;124:1765–1773. [DOI] [PubMed] [Google Scholar]
- 11. Rajagopalan S, Olin J, Deitcher S, Pieczek A, Laird J, Grossman PM, Goldman CK, McEllin K, Kelly R, Chronos N. Use of a constitutively active hypoxia‐inducible factor‐1alpha transgene as a therapeutic strategy in no‐option critical limb ischemia patients: phase I dose‐escalation experience. Circulation. 2007;115:1234–1243. [DOI] [PubMed] [Google Scholar]
- 12. Grossman PM, Mendelsohn F, Henry TD, Hermiller JB, Litt M, Saucedo JF, Weiss RJ, Kandzari DE, Kleiman N, Anderson RD, Gottlieb D, Karlsberg R, Snell J, Rocha‐Singh K. Results from a phase II multicenter, double‐blind placebo‐ controlled study of Del‐1 (VLTS‐ 589) for intermittent claudication in subjects with peripheral arterial disease. Am Heart J. 2007;153:874–880. [DOI] [PubMed] [Google Scholar]
- 13. Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, Capla JM, Galiano RD, Levine JP, Gurtner GC. Progenitor cell trafficking is regulated by hypoxic gradients through HIF‐1 induction of SDF‐1. Nat Med. 2004;10:858–864. [DOI] [PubMed] [Google Scholar]
- 14. Grunewald M, Avraham I, Dor Y, Bachar‐Lustig E, Itin A, Jung S, Chimenti S, Landsman L, Abramovitch R, Keshet E. VEGF‐induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell. 2006;124:175–189. [DOI] [PubMed] [Google Scholar]
- 15. Clayton JA, Chalothorn D, Faber JE. Vascular endothelial growth factor‐A specifies formation of native collaterals and regulates collateral growth in ischemia. Circ Res. 2008;103:1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Herbert SP, Stainier DY. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol. 2011;12:551–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Patel‐Hett S, D'Amore PA. Signal transduction in vasculogenesis and developmental angiogenesis. Int J Dev Biol. 2011;55:353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Webster KA, Discher DJ, Kaiser S, Hernandez O, Sato B. Hypoxia‐activated apoptosis of cardiac myocytes requires reoxygenation or a pH shift and is independent of p53. J Clin Invest. 1999;104:239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kubasiak L, Webster KA. Hypoxia and acidosis activate cardiac myocyte death through the Bcl‐2 family protein BNIP3. Proc Natl Acad Sci USA. 2002;99:12825–12830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Naruse Y, Aoki T, Kojima T, Mori N. Neural restrictive silencer factor recruits mSin3 and histone deacetylase complex to repress neuron‐specific target genes. Proc Natl Acad Sci USA. 1999;96:13691–13696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McCarty DM, Monahan PE, Samulski RJ. Self‐complementary recombinant adeno‐associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001;8:1248–1254. [DOI] [PubMed] [Google Scholar]
- 22. Wang Z, Ma HI, Li J, Sun L, Zhang J, Xiao X. Rapid and highly efficient transduction by double‐stranded adeno‐associated virus vectors in vitro and in vivo. Gene Ther. 2003;10:2105–2111. [DOI] [PubMed] [Google Scholar]
- 23. Layman H, Spiga MG, Brooks T, Pham S, Webster KA, Andreopoulos FM. The effect of the controlled release of basic fibroblast growth factor from ionic gelatin‐based hydrogels on angiogenesis in a murine critical limb ischemic model. Biomaterials. 2007;16:2646–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gonin P, Arandel L, Van Wittenberghe L, Marais T, Perez N, Danos O. Femoral intra‐arterial injection: a tool to deliver and assess recombinant AAV constructs in rodents whole hind limb. J Gene Med. 2005;7:782–791. [DOI] [PubMed] [Google Scholar]
- 25. Fougerousse F, Bartoli M, Poupiot J, Arandel L, Durand M, Guerchet N, Gicquel E, Danos O, Richard I. Phenotypic correction of alpha‐sarcoglycan deficiency by intra‐arterial injection of a muscle‐specific serotype 1 rAAV vector. Mol Ther. 2007;15:53–61. [DOI] [PubMed] [Google Scholar]
- 26. Chicoine LG, Montgomery CL, Bremer WG, Shontz KM, Griffin DA, Heller KN, Lewis S, Malik V, Grose WE, Shilling CJ, Campbell KJ, Preston TJ, Coley BD, Martin PT, Walker CM, Clark KR, Sahenk Z, Mendell JR, Rodino‐Klapac LR. Plasmapheresis eliminates the negative impact of AAV antibodies on microdystrophin gene expression following vascular delivery. Mol Ther. 2014;22:338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boden J, Wei J, McNamara G, Layman H, Abdulreda M, Andreopoulos F, Webster KA. Whole‐mount imaging of the mouse hindlimb vasculature using the lipophilic carbocyanine dye DiI. Biotechniques. 2012;53:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Abdulreda MH, Faleo G, Molano RD, Lopez‐Cabezas M, Molina J, Tan Y, Echeverria OA, Zahr‐Akrawi E, Rodriguez‐Diaz R, Edlund PK, Leibiger I, Bayer AL, Perez V, Ricordi C, Caicedo A, Pileggi A, Berggren PO. High‐resolution, noninvasive longitudinal live imaging of immune responses. Proc Natl Acad Sci USA. 2011;108:12863–12868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peserico A, Simone C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J Biomed Biotechnol. 2011;2011:371832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chalothorn D, Clayton JA, Zhang H, Pomp D, Faber JE. Collateral density, remodeling, and VEGF‐A expression differ widely between mouse strains. Physiol Genomics. 2007;30:179–191. [DOI] [PubMed] [Google Scholar]
- 31. Schultz A, Lavie L, Hochberg I, Beyar R, Stone T, Skorecki K, Lavie P, Roguin A, Levy AP. Interindividual heterogeneity in the hypoxic regulation of VEGF: significance for the development of the coronary artery collateral circulation. Circulation. 1999;100:547–552. [DOI] [PubMed] [Google Scholar]
- 32. Marfella R, Esposito K, Nappo F, Siniscalchi M, Sasso FC, Portoghese M, Di Marino MP, Baldi A, Cuzzocrea S, Di Filippo C, Barboso G, Baldi F, Rossi F, D'Amico M, Giugliano D. Expression of angiogenic factors during acute coronary syndromes in human type 2 diabetes. Diabetes. 2004;53:2383–2391. [DOI] [PubMed] [Google Scholar]
- 33. Murphy BJ, Andrews GK, Bittel D, Discher DJ, McCue J, Green CJ, Yanovsky M, Giaccia A, Sutherland RM, Laderoute KR, Webster KA. Activation of metallothionine gene expression by hypoxia involves metal response elements and metal transcription factor‐1. Cancer Res. 1999;59:1315–1322. [PubMed] [Google Scholar]
- 34. Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J, Semenza GL, Choi AM. Hypoxia‐inducible factor‐1 mediates transcriptional activation of the heme oxygenase‐1 gene in response to hypoxia. J Biol Chem. 1997;272:5375–5381. [PubMed] [Google Scholar]
- 35. David Dong ZM, Aplin AC, Nicosia RF. Regulation of angiogenesis by macrophages, dendritic cells, and circulating myelomonocytic cells. Curr Pharm Des. 2009;15:365–379. [DOI] [PubMed] [Google Scholar]
- 36. Dineen SP, Lynn KD, Holloway SE, Miller AF, Sullivan JP, Shames DS, Beck AW, Barnett CC, Fleming JB, Brekken RA. Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res. 2008;68:4340–4346. [DOI] [PubMed] [Google Scholar]
- 37. Pacak CA, Walter GA, Gaidosh G, Bryant N, Lewis MA, Germain S, Mah CS, Campbell KP, Byrne BJ. Long‐term skeletal muscle protection after gene transfer in a mouse model of LGMD‐2D. Mol Ther. 2007;15:1775–1781. [DOI] [PubMed] [Google Scholar]
- 38. Gounis MJ, Spiga MG, Graham RM, Wilson A, Haliko S, Lieber BB, Wakhloo AK, Webster KA. Capillary growth is confined to the transient period of VEGF expression that follows adenoviral gene delivery to ischemic muscle. Gene Ther. 2005;12:762–771. [DOI] [PubMed] [Google Scholar]
- 39. Li Y, Song Y, Zhao L, Gaidosh G, Laties AM, Wen R. Direct labeling and visualization of blood vessels with lipophilic carbocyanine dye DiI. Nat Protoc. 2008;3:1703–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Abarbanell AM, Herrmann JL, Weil BR, Wang Y, Tan J, Moberly SP, Fiege JW, Meldrum DR. Animal models of myocardial and vascular injury. J Surg Res. 2010;162:239–249. [DOI] [PubMed] [Google Scholar]
- 41. Fischer A, Schumacher N, Maier M, Sendtner M, Gessler M. The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes Dev. 2004;18:901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hellström M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, Alva J, Nilsson AK, Karlsson L, Gaiano N, Yoon K, Rossant J, Iruela‐Arispe ML, Kalén M, Gerhardt H, Betsholtz C. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445:776–780. [DOI] [PubMed] [Google Scholar]
- 43. Noguera‐Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, Lin HC, Yancopoulos GD, Thurston G. Blockade of Dll4 inhibits tumour growth by promoting non‐productive angiogenesis. Nature. 2006;444:1032–1037. [DOI] [PubMed] [Google Scholar]
- 44. Liekens S, Schols D, Hatse S. CXCL12‐CXCR4 axis in angiogenesis, metastasis and stem cell mobilization. Curr Pharm Des. 2010;16:3903–3920. [DOI] [PubMed] [Google Scholar]
- 45. Gavard J, Patel V, Gutkind JS. Angiopoietin‐1 prevents VEGF‐induced endothelial permeability by sequestering Src through mDia. Dev Cell. 2008;14:25–36. [DOI] [PubMed] [Google Scholar]
- 46. Sawamiphak S, Seidel S, Essmann CL, Wilkinson GA, Pitulescu ME, Acker T, Acker‐Palmer A. Ephrin‐B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature. 2010;465:487–491. [DOI] [PubMed] [Google Scholar]
- 47. Lotfi S, Patel AS, Mattock K, Egginton S, Smith A, Modarai B. Towards a more relevant hind limb model of muscle ischaemia. Atherosclerosis. 2013;227:1–8. [DOI] [PubMed] [Google Scholar]
- 48. Cooke JP, Losordo DW. Modulating the vascular response to limb ischemia: angiogenic and cell therapies. Circ Res. 2015;116:1561–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Helisch A, Wagner S, Khan N, Drinane M, Wolfram S, Heil M, Ziegelhoeffer T, Brandt U, Pearlman JD, Swartz HM, Schaper W. Impact of mouse strain differences in innate hindlimb collateral vasculature. Arterioscler Thromb Vasc Biol. 2006;26:520–526. [DOI] [PubMed] [Google Scholar]
- 50. Dokun AO, Keum S, Hazarika S, Li Y, Lamonte GM, Wheeler F, Marchuk DA, Annex BH. A quantitative trait locus (LSq‐1) on mouse chromosome 7 is linked to the absence of tissue loss after surgical hindlimb ischemia. Circulation. 2008;117:1207–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Onimaru M, Yonemitsu Y, Tanii M, Nakagawa K, Masaki I, Okano S, Ishibashi H, Shirasuna K, Hasegawa M, Sueishi K. Fibroblast growth factor‐2 gene transfer can stimulate hepatocyte growth factor expression irrespective of hypoxia‐mediated downregulation in ischemic limbs. Circ Res. 2002;91:923–930. [DOI] [PubMed] [Google Scholar]
- 52. McClung JM, McCord TJ, Keum S, Johnson S, Annex BH, Marchuk DA, Kontos CD. Skeletal muscle‐specific genetic determinants contribute to the differential strain‐dependent effects of hindlimb ischemia in mice. Am J Pathol. 2012;180:2156–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hazarika S, Farber CR, Dokun AO, Pitsillides AN, Wang T, Lye RJ, Annex BH. MicroRNA‐93 controls perfusion recovery after hindlimb ischemia by modulating expression of multiple genes in the cell cycle pathway. Circulation. 2013;127:1818–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sottiurai V, White JV. Extensive revascularization or primary amputation: which patients with critical limb ischemia should not be revascularized? Semin Vasc Surg. 2007. Mar;20:68–72. [DOI] [PubMed] [Google Scholar]
- 55. Bosma J, Vahl A, Wisselink W. Systematic review on health‐related quality of life after revascularization and primary amputation in patients with critical limb ischemia. Ann Vasc Surg. 2013;27:1105–1114. [DOI] [PubMed] [Google Scholar]
- 56. Semenza GL. Regulation of vascularization by hypoxia‐inducible factor 1. Ann N Y Acad Sci. 2009;1177:2–8. [DOI] [PubMed] [Google Scholar]
- 57. Dor Y, Djonov V, Abramovitch R, Itin A, Fishman GI, Carmeliet P, Goelman G, Keshet E. Conditional switching of VEGF provides new insights into adult neovascularization and pro‐angiogenic therapy. EMBO J. 2002;21:1939–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ozawa CR, Banfi A, Glazer NL, Thurston G, Springer ML, Kraft PE, McDonald DM, Blau HM. Microenvironmental VEGF concentration, not total dose, determines a threshold between normal and aberrant angiogenesis. J Clin Invest. 2004;113:516–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Von Degenfeld G, Banfi A, Springer ML, Wagner RA, Jacobi J, Ozawa CR, Merchant MJ, Cooke JP, Blau HM. Microenvironmental VEGF distribution is critical for stable and functional vessel growth in ischemia. FASEB J. 2006;20:2657–2659. [DOI] [PubMed] [Google Scholar]
- 60. Tafuro S, Ayuso E, Zacchigna S, Zentilin L, Moimas S, Dore F, Giacca M. Inducible adeno‐associated virus vectors promote functional angiogenesis in adult organisms via regulated vascular endothelial growth factor expression. Cardiovasc Res. 2009;83:663–671. [DOI] [PubMed] [Google Scholar]
- 61. Schierling W, Troidl K, Troidl C, Schmitz‐Rixen T, Schaper W, Eitenmüller IK. The role of angiogenic growth factors in arteriogenesis. J Vasc Res. 2009;46:365–374. [DOI] [PubMed] [Google Scholar]
- 62. Heil M, Schaper W. Insights into pathways of arteriogenesis. Curr Pharm Biotechnol. 2007;8:35–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Representative sections of ischemic calf and lower thigh muscle stained as indicated by using a Hypoxyprobe kit as described in Methods and the legend for Figure 2. Bottom panels are negative controls lacking primary antibody.