

Abstract
Background
Estimates of long‐term survival are required to adequately assess the variety of health and social services required by those with congenital heart disease (CHD) throughout their lives.
Methods and Results
Medline, Embase, and Scopus were searched from inception to June 2015 using MeSH headings and keywords. Population‐based studies that ascertained all persons born with CHD within a predefined area and reported survival estimates at ≥5 years were included. Unadjusted survival estimates for each CHD subtype at ages 1 year, 5 years, 10 years, and so forth were extracted. Pooled survival estimates for each age were calculated using meta‐analyses. Metaregression was performed to examine the impact of study period on survival. Of 7840 identified articles, 16 met the inclusion criteria. Among those with CHD, pooled 1‐year survival was 87.0% (95% CI 82.1–91.2), pooled 5‐year survival was 85.4% (95% CI 79.4–90.5), and pooled 10‐year survival was 81.4% (95% CI 73.8–87.9). There was significant heterogeneity of survival estimates among articles (P<0.001 for 1‐, 5‐, and 10‐year survival). A more recent study period was significantly associated with greater survival at ages 1 year (P=0.047), 5 years (P=0.013), and 10 years (P=0.046). Survival varied by CHD subtype, with 5‐year survival being greatest for those with ventricular septal defect (96.3%, 95% CI 93.7–98.2) and lowest for those with hypoplastic left heart (12.5%, 95% CI 0.0–41.4).
Conclusions
Among persons with CHD, the mortality rate is greatest during the first year of life; however, this systematic review and meta‐analysis showed that survival decreases gradually after infancy and into adulthood.
Keywords: congenital, heart defects, survival
Subject Categories: Meta Analysis, Mortality/Survival, Epidemiology, Congenital Heart Disease, Pediatrics
Introduction
Congenital heart disease (CHD) composes the largest group of congenital anomalies and affects ≈1% of births in the United States and Europe.1, 2, 3 CHD is a leading cause of stillbirth and infant death and accounts for 4.2% of neonatal deaths in the United States.4 Babies with severe CHD subtypes require complex surgeries for survival. With advances in medical, surgical, and intensive care interventions, an estimated 83% of babies with CHD now survive infancy in the United States.5 Although 1‐year survival estimates have been described,3, 6, 7, 8, 9, 10, 11 long‐term survival estimates are not well researched, and survival may continue to decrease into adulthood.
A previous systematic review of the long‐term prognosis of CHD included only hospital‐based studies that ascertained cases postsurgically or in adulthood; estimates were not representative of all persons with CHD.12 We conducted a systematic review and meta‐analysis of population‐based studies reporting long‐term survival of persons born with CHD. The aim was to assess and quantify long‐term survival to inform health services planning and decision making.
Methods
Search Strategy
We conducted comprehensive literature searches of Medline, Embase, and Scopus from inception (1946, 1974, and 1996, respectively) to June 18, 2015. MeSH terms and keywords were entered systematically into the databases. The keywords included congenital and heart or cardiac or cardiovascular and subject heading searches such as “exp Heart Defects, Congenital/ep, mo” but varied according to database. The list of search terms is available from the authors.
After systematic searches of each database, the citations were extracted, and titles and abstracts were screened according to the inclusion criteria. Full articles were retrieved for all relevant citations. Reference lists of included articles were scanned and examined, and key journals were searched using keywords.
Inclusion Criteria
Population‐based original studies were included if they (1) ascertained all persons born with CHD within a predefined geopolitical area; (2) reported survival estimates (or the number of patients born and the number or proportion alive) at age ≥5 years; (3) reported survival estimates for all CHD combined or a single CHD subtype including ventricular septal defect, pulmonary valve stenosis, atrial septal defect, aortic valve atresia or stenosis, atrioventricular septal defect, coarctation of aorta, common arterial truncus, pulmonary valve atresia (with ventricular septal defect or with intact ventricular septum), tetralogy of Fallot, total anomalous pulmonary venous return, transposition of great vessels, tricuspid atresia, single ventricle, hypoplastic left heart, and Ebstein's anomaly; (4) were available from the British Library or the Internet and were written in the English language.
Exclusion Criteria
Articles were excluded if patients were not followed from birth (eg, follow‐up began in adulthood or after surgical correction); patients were not born in well‐defined regions (ie, hospital‐based); survival was not estimated as a proportion of those born with CHD (eg, age‐specific population mortality rates); survival was reported only for certain subtype groups (eg, “severe” CHD). For multiple articles reported on the same data set, the largest study or the study with the most recent study period was included. Both articles were included if they reported survival for different CHD subtypes or ages.
Data Extraction
K.E.B. performed the literature searches, screened citations, and reviewed 40 full papers. J.R. screened 10% of the titles and all abstracts to confirm decisions about inclusion, and extracted data from all included papers. There were no discrepancies between reviewers regarding article inclusion.
Study characteristics including study design, quality, data sources, prevalence estimates, and the percentage of cases with extracardiac anomalies (ie, cases of CHD occurring with another congenital anomaly not of the cardiovascular system, such as Down syndrome or cleft lip) were extracted from each article. If it was unclear whether cases with extracardiac anomalies were included, the authors were contacted.
Kaplan–Meier survival estimates and corresponding 95% CIs were obtained from each included study at ages 1 year, 5 years, 10 years, and so forth. If 95% CIs were not reported, the authors were contacted. If this was unsuccessful, the number of patients born and the proportion that survived were used to estimate binomial 95% CIs, assuming no cases were censored. Survival estimates for all CHD subtypes combined and for each CHD subtype were extracted. If survival estimates were presented only graphically, the authors were contacted for survival estimates. If this was unsuccessful, survival estimates were extracted using Plot Digitizer software.13, 14
Statistical Analysis
If there were at least 3 studies reporting survival, pooled estimates of survival were calculated using a meta‐analysis with random effects. Weighting for each article was allocated using the inverse of the variance. If the number of studies is small, the estimation of between‐study variance is thought to be imprecise in random‐effects models.15 Consequently, if there were only 3 studies reporting survival, the pooled survival was also estimated using fixed‐effects meta‐analysis to allow comparison. To stabilize the variance and adjust the study weights, a simplified double‐arcsine transformation was performed on the survival estimates and 95% CIs.16 The Cochrane Q test and the I2 statistic were used to test for heterogeneity in survival estimates between articles, with I2>50% indicating substantial heterogeneity.17 Random‐effects metaregression was performed for all CHD subtypes combined to assess year of delivery as a source of heterogeneity. In this analysis, the year in which the study commenced was used as an explanatory variable. The adjusted R 2 value was used to estimate the proportion of between‐article variation accounted for by the year of study commencement. A bubble plot was used to present the fitted metaregression model. In this analysis, bubbles represent each article, with sizes dependent on the precision of the survival estimates. Publication bias was assessed with the Egger test.18
Analysis was performed in Stata 13 (StataCorp), and P<0.05 was considered statistically significant.
Quality Appraisal
Quality appraisal was based on 4 of the 6 domains developed by Hayden et al to assess potential bias in systematic reviews of prognostic studies.19 The domains used were study ascertainment, study attrition, outcome ascertainment, and analysis. The domains relating to confounding and prognostic factors were not relevant to this review because the primary aim was to investigate unadjusted survival estimates.
Results
Figure 1 shows a Preferred Reporting Items for Systematic Reviews and Meta‐Analyses diagram for the flow of articles through the review. Of 7840 identified articles, 16 met the inclusion criteria.20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35
Figure 1.
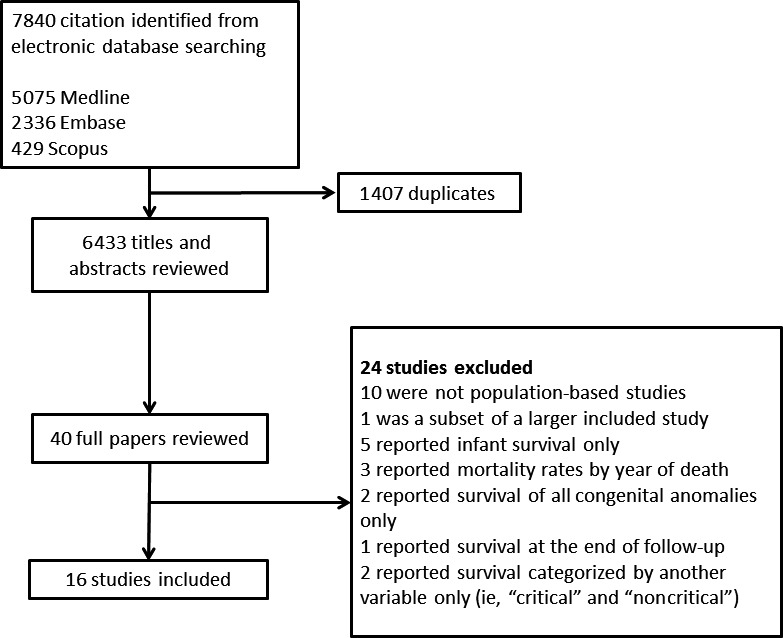
Preferred Reporting Items for Systematic Reviews and Meta‐Analyses diagram for the flow of articles through the review.
Study Characteristics
All included studies were conducted in high‐income Western populations, with 10 in Europe and 6 in the United States (Table 1). Although several articles reported survival of subsets of the same population, all were included because survival was reported for different CHD subtypes or at different ages. The oldest article included patients born between 1973 and 1997,22 and the most recent article included patients born between 1991 and 2007.35 Of the 16 included articles, 9 included cases with extracardiac anomalies, with ≈20% of cases occurring with other congenital anomalies in each article.23, 24, 25, 26, 28, 30, 33, 34, 35 Four articles excluded patients with trisomy 13 (Patau syndrome) and 18 (Edward syndrome) only.21, 22, 27, 29 Two articles excluded cases of CHD with any extracardiac anomalies,20, 32 and 1 did not state whether cases with extracardiac anomalies were included.31 Prevalence estimates were reported by most studies and ranged from 3.730 to 10.226 per 1000 live births when considering all CHD as a composite group.
Table 1.
Descriptions of the Included Articles
| Study | Included Birth Years | Study Location | Included CHD Subtypes (ICD Codes) | Inclusion of ECAs | Age Limit for Diagnosis | Source of Cases | Source of Death Information | Percentage of Traced Cases | Prevalence per 1000 Live Births |
|---|---|---|---|---|---|---|---|---|---|
| Dastgiri et al20 | 1980–1997 | Glasgow, Scotland | All CHD subtypes (ICD 10: Q20–26) | Author's response: excluded | No age limit | Glasgow register of Congenital Anomalies | Registrar General for Scotland | 97% (all congenital anomalies) | Not stated |
| Fixler et al21 | 1996–2003 | Texas, USA | SV physiology: HLH (ICD 9: 746.7), PVA‐IVS (746.0), SV (745.3), TA (746.1), d‐TGV (745.1) | Cases with trisomy 13 or 18 were excluded; 14.1% of HLH, 21.0% of SV, 15.3% of PVA‐IVS, 17.9% of TA, 9.3% of d‐TGV had ECAs | 1 year | Texas Birth Defects Registry | Medical records, death certificates, national death index | N/A, nontraced cases considered alive | Not stated |
| Frid et al22 | 1973–1997 | Sweden | AVSD (ICD 9: 745G, ICD 10: 21.2) | Cases with trisomy 13 or 18 were excluded; 68.9% had trisomy 21 | None stated | Register of Congenital Malformations, Register of Congenital Heart Malformations, and the Medical Birth Register; local registries at 4 pediatric cardiology centers were also searched for the beginning of the study period | National population database and medical records | 98.7% of all cases with AVSD | 0.3 |
| Garne23 | 1986–1998 | Funen County, Denmark | All CHD subtypes (EUROCAT criteria ie, ICD 10: Q20–26) | Cases with ECAs were included, 21% of cases | 5 years and diagnosed before 2002 | EUROCAT Registry of Congenital Malformations for Funen County | National registration system | 99.6% | 7.9 |
| Idorn et al24 | 1977–2009 | Denmark, Europe | HLH (ICD 10: Q234), PVA‐IVS (Q220), TA (Q224) | Cases with ECAs were included, 10% of cases | All ages | Danish register of congenital heart disease, local surgical registries, medical records, local fetal ultrasound registries | Civil registration system | Not stated | 0.4 |
| Jackson et al25 | 1979–1988 | Merseyside, England | All CHD subtypes (ICD 9: 745.00–747.49) | Cases with ECAs were included, percentage not stated | No age limit | Liverpool Registry of Congenital Malformations | Liverpool Registry of Congenital Malformations and hospital records | Not stated | 7.6 |
| Meberg et al26 | 1982–1996 | Vestfold, Norway, Europe | All CHD subtypes (no ICD codes stated) | Cases with ECAs were included, 20% of cases | None stated | Vestfold County Central Hospital, regional cardiology services, Child Health Centers and pediatric departments of the hospitals in neighboring counties | Hospital records | 100% | 10.2 |
| Miller et al27 | 1979–2003 | Metropolitan Atlanta, GA, USA | AVSD (ICD 9: 745.000–747.999) | Cases with trisomy 13 or 18 were excluded, 52.4% had trisomy 21 | None stated | Metropolitan Atlanta Congenital Defects Program | Hospital records and vital records from the state of Georgia, National Death Index | Not stated but number of untraced “assumed to be small” | Not stated |
| Moons et al28 | 2002 | Belgium | All CHD subtypes (no ICD codes specified) | Author response: cases with ECAs were included, percentage not stated | 5 years | Pediatric cardiology database covering 7 tertiary care centers in Belgium | Medical records | Not stated | 8.3 |
| Nembhard et al29 | 1996–2003 | Texas, USA | ICD 9 (746–747) | Cases with trisomy 13 or 18 were excluded, 20.7% of cases had ECAs | 1 year | Texas birth defects register | Death certificates linked to the Texas birth defects register | Not stated | 8.7 |
| Olsen et al30 | 1977–2006 | Denmark | All CHD subtypes: ICD 8: 746 to 747 (except 746.7 and 747.5–747.9) and ICD‐10: Q20–Q26 (except Q26.5–Q26.6) | Cases with ECAs were included, 20.0% of cases | 1 year | Danish National Registry of Patients | Civil registration system | 100% | 3.7 |
| Samanek and Voriskova31 | 1980–1990 | Bohemia, Czech Republic | All CHD subtypes (no ICD codes specified) | Not stated | None stated | Hospital records | Autopsy reports | Not stated | 6.2 |
| Tennant et al32 | 1985–2003 | Northeast England | All CHD subtypes (ICD 10: Q20–26) | Cases with ECAs were excluded, percentage not stated | 16 years of age (1985–2001) or, from 2001, to age 12 years | Northern Congenital Abnormality Survey | Office for National Statistics death registrations | 99% (of all congenital anomalies) | 6.8 |
| Wang et al (2011)33 | 1983–2006 | New York State, USA | TGV (ICD 9: 745.10–745.12, 745.19), ToF (745.2), HLH (746.7), AVA/S (746.3), CAT (745.0), AVSD (745.6), CoA (747.10) | Cases with ECAs were included, percentage not stated | None stated | Congenital Malformations Registry | Death certificates files maintained by the New York State Department of Health | 97% (of all congenital anomalies) | 9.5 |
| Wang et al (2013)34 | 1983–2006 | New York State, USA | TGV (ICD 9: 745.10–745.12, 745.19), ToF (745.2), HLH (746.7), CoA (747.10) | Cases with ECAs were included, percentage not stated | 2 years | Congenital Malformations Registry | Death certificate files maintained by the New York State Department of Health | Not stated | Not stated |
| Wang et al (2015)35 | 1991–2007 | Arizona, Colorado, Florida, Georgia (5 counties of Metropolitan Atlanta), Illinois, Massachusetts, Michigan, Nebraska, New Jersey, New York (excluding New York City), North Carolina, Texas | TGV (ICD 9: 745.10–745.12, 745.19), ToF (745.2), HLH (746.7), AVA/S (746.3), CAT (745.0), AVSD (745.6), CoA (747.10) | Cases with ECAs included, percentage not stated | None stated | Arizona Birth Defects Monitoring Program, Metropolitan Atlanta Congenital Defects Program, Colorado Responds to Children with Special Needs, Florida Birth Defects Registry, Illinois Adverse Pregnancy Outcomes Reporting System, Massachusetts Birth Defects Monitoring Program, Michigan Birth Defects Registry, Nebraska Birth Defects Registry, New Jersey Special Child Health Services Registry, New York State Congenital Malformations Registry, North Carolina Birth Defects Monitoring Program, and Texas Birth Defects Epidemiology, and Surveillance Branch | Death certificates, hospital discharge files (Arizona, Texas), medical records (Arizona, Texas), and the National Death Index (Georgia, Michigan) | Not stated | 2.1 |
AVA/S, aortic valve atresia or stenosis; AVSD, atrioventricular septal defect; CAT, common arterial truncus; CHD, congenital heart disease; CoA, coarctation of aorta; d‐TGV, dextro‐TGV; ECA, extracardiac anomaly; HLH, hypoplastic left heart; ICD, International Classification of Disease; IVS, intact ventricular septum; N/A, not available; PVA, pulmonary valve atresia (with ventricular septal defect or IVS); SV, single ventricle; TA, tricuspid atresia; TGV, transposition of great vessels; ToF, tetralogy of Fallot.
Survival Estimates
Survival was reported to age 5 years in 5 articles,20, 21, 23, 28, 29 to age 8 years in 1 article,35 to age 10 years in 3 articles,25, 26, 27 to age 15 years in 2 articles,22, 31 to age 20 years in 1 article,32 to age 25 years in 3 articles,30, 33, 34 and to age 30 years in 1 article.24
For all CHD (as a composite group), pooled 1‐year survival from 6 articles was 87.0% (95% CI 82.1–91.2), pooled 5‐year survival from 8 articles was 85.4% (95% CI 79.4–90.5), and pooled 10‐year survival from 4 articles was 81.4% (95% CI 73.8–87.9) (Figure 2). It was not possible to pool estimates beyond 10 years because there were too few articles; however, Figure 3 shows the survival estimates plotted over increasing age, up to age 25 years. The fitted metaregression showed that survival decreases very gradually with increasing age over 25 years. There was no evidence of publication bias according to Egger tests (P=0.748 for 1 year, P=0.237 for 5 years, and P=0.601 for 10 years). There was significant heterogeneity between articles for survival at 1 year (I2=99.0%, P<0.001), 5 years (I2=99.6%, P<0.001), and 10 years (I2=99.5%, P<0.001). Metaregression showed that a more recent study period was significantly associated with increased 1‐, 5‐, and 10‐year survival (P=0.047, P=0.013, and P=0.046, respectively) (Figure 4). According to the adjusted R 2 values, study period accounted for 50.9%, 62.8%, and 87.0% of the between‐article variance for 1‐, 5‐, and 10‐year survival. After adjustment for study period, however, substantial residual heterogeneity remained that was attributable to between‐study heterogeneity (I2=98.2% at age 1 year, I2=98.4% for survival at age 5 years, and I2=93.7% for survival at age 10 years).
Figure 2.
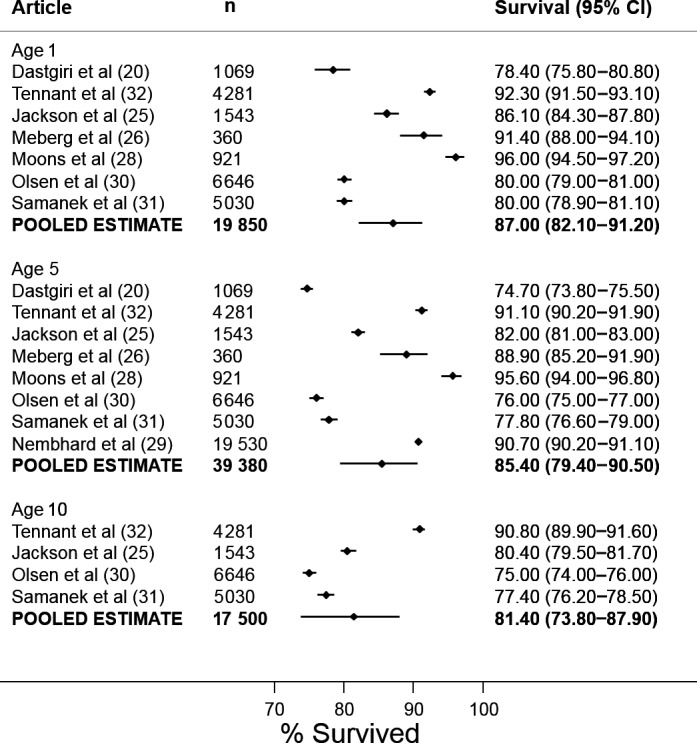
Forest plot for all congenital heart disease at ages 1, 5, and 10 years.
Figure 3.
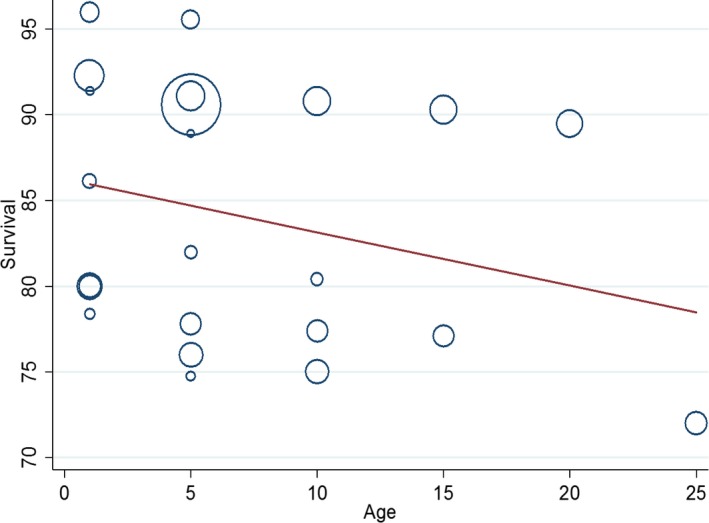
Bubble plot of survival estimates for all congenital heart disease at ages 1 to 25 years.
Figure 4.
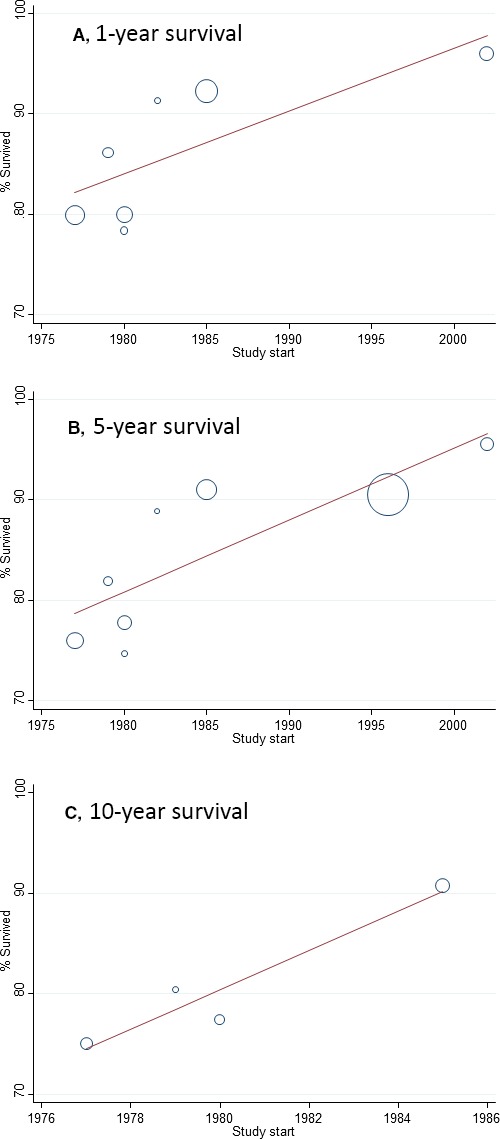
Bubble plots showing the association between study period and survival for all congenital heart disease. A, 1‐year survival. B, 5‐year survival. C, 10‐year survival.
Table 2 shows the survival estimates and pooled survival estimates for persons with CHD by subtype. Pooled 1‐year survival was lowest for those with hypoplastic left heart (17.4%, 95% CI 0.0–54.5) and greatest for those with ventricular septal defect (95.5%, 95% CI 89.0–99.2). There was significant heterogeneity of survival estimates among articles for all CHD subtypes, with the exception of tetralogy of Fallot (I2=0%, P=0.169). Heterogeneity of estimates for single ventricle was of borderline statistical significance (I2=65.0%, P=0.057). Pooled 5‐year survival varied by subtype, with survival for hypoplastic left heart at 12.5% (95% CI 0.0–41.4) and survival for ventricular septal defect at 97.7% (95% CI 93.5–99.8). With the exception of tetralogy of Fallot (I2=0.0%, P=0.957) and single ventricle (I2=26.9%, P=0.250), there was significant heterogeneity of survival estimates among articles (Table 2). It was possible to calculate pooled 15‐year survival estimates for aortic valve atresia or stenosis, atrioventricular septal defect, common arterial truncus, and coarctation of aorta but not for any other CHD subtypes. There were too few studies to calculate pooled survival beyond age 15 years, although in the few studies that reported survival into adulthood, survival was still gradually declining.
Table 2.
Survival Estimates at Age One to 25
| Subtype | Article | N | Survival Estimates (95% CI) | |||||
|---|---|---|---|---|---|---|---|---|
| 1 year | 5 years | 10 years | 15 years | 20 years | 25 years | |||
| All congenital heart disease | Dastgiri et al20 | 1069 | 78.4 (75.8–80.8)a | 74.7 (73.8–75.5)b | ||||
| Jackson et al25 | 1543 | 86.1 (84.3–87.8)a | 82.0 (81.0–83.0) | 80.4 (79.5–81.7)b | ||||
| Meberg et al26 | 360 | 91.4 (88.0–94.1)a | 88.9 (85.2–91.9)a | |||||
| Moons et al28 | 921 | 96.0 (94.5–97.2)a | 95.6 (94.0–96.8)a | |||||
| Nembhard et al29 | 19 530 | 90.7 (90.2–91.1)a | ||||||
| Olsen et al30 | 6646 | 80 (79–81) | 76 (75–77)a | 75 (74–76) | 72 (70–73) | |||
| Samanek et al31 | 5030 | 80.0 (78.9–81.1) | 77.8 (76.6–79.0) | 77.4 (76.2–78.5) | 77.1 (75.9–78.3) | |||
| Tennant et al32 | 4281 | 92.3 (91.5–93.1) | 91.1 (90.2–91.9) | 90.8 (89.9–91.6) | 90.3 (89.3–91.2) | 89.5 (88.4–90.6) | ||
| Pooled estimate (95% CI) | 87.0 (82.1–91.2) | 85.4 (79.4–90.5) | 81.4 (73.8–87.9) | |||||
| Heterogeneity I2 & P–value | 99.0%, P<0.001 | 99.6%, P<0.001 | 99.5%, P<0.001 | |||||
| Ventricular septal defect | Garne23 | 195 | 96.9 (93.4, 98.9)a | |||||
| Moons et al28 | 303 | 99.3 (97.6–99.9)a | ||||||
| Nembhard et al29 | 10 382 | 93.9 (93.5–94.4)a | ||||||
| Olsen et al30 | 1559 | 94 (93–95) | 90 (89–91.7) | |||||
| Samanek et al31 | 2092 | 91.1 (89.8–92.3)a | 89.4 (88.0–90.7) | |||||
| Tennant et al32 | 1805 | 99.2 (98.7–99.5) | 99.1 (98.6–99.5) | 99.1 (98.5–99.4) | 99.1 (98.5–99.4) | 98.3 (96.6–99.1) | ||
| Pooled estimate (95% CI) | 95.5 (89.0–99.2) | 97.7 (93.5–99.8) | ||||||
| 95.5 (95.0–96.0) | ||||||||
| Heterogeneity I2 & P‐value | 99.0%, P<0.001 | 98.1%, P<0.001 | ||||||
| Pulmonary valve stenosis | Garne 23 | 33 | 97.0 (84.2–99.9)a | |||||
| Nembhard et al29 | 1170 | 91.6 (89.9–93.1)a | ||||||
| Samanek et al31 | 292 | 96.2 (94.0–98.5) | 95.6 (93.1–98.0) | 95.6 (93.1–98.0) | 95.6 (93.1–98.0) | |||
| Tennant et al32 | 382 | 98.7 (96.8–99.5) | 98.1 (96.1–99.1) | 98.1 (96.1–99.1) | 98.1 (96.1–99.1) | 98.1 (96.1–99.1) | ||
| Pooled estimate (95% CI) | 95.6 (91.1–98.6) | |||||||
| Heterogeneity I2 & P‐value | 89.6%, P<0.001 | |||||||
| Atrial septal defect | Garne23 | 78 | 98.7 (93.1, 100.0)a | |||||
| Moons et al28 | 162 | 99.4 (96.6–100.0)a | ||||||
| Nembhard et al29 | 9164 | 89.9 (89.3–90.5)a | ||||||
| Olsen et al30 | 361 | 93 (90–95.3) | 91 (88–95.6) | 84 (72–91) | ||||
| Samanek et al31 | 436 | 94.0 (92.4–96.3) | 92.9 (90.1–95.1)a | 92.9 (90.1–95.1)a | ||||
| Tennant et al32 | 365 | 97.3 (95.0–98.5) | 97.0 (94.6–98.3) | 97.0 (94.6–98.3) | 96.3 (93.3–98.0) | 96.3 (93.3–98.0) | ||
| Pooled estimate (95% CI) | 94.9 (92–97.2) | 96.8 (90.8–99.7) | 94.0 (89.9–97.1) | |||||
| 94.8 (93.5–96.0) | 95.4%, P<0.001 | 94.3 (92.7–95.6) | ||||||
| Heterogeneity I2 & P‐value | 77.4%, P<0.001 | 81.6%, P=0.004 | ||||||
| Aortic valve atresia/stenosis | Garne23 | 24 | 87.5 (67.6, 97.3)a | |||||
| Moons et al28 | 36 | 100.0 (90.3–100.0)a | ||||||
| Samanek31 | 391 | 90.3 (87.3–93.3) | 88.4 (85.1–91.7) | |||||
| Tennant et al32 | 171 | 92.4 (87.3–95.5) | 91.2 (85.9–94.6) | 91.2 (85.9–94.6) | 89.3 (83.2–3.3) | 89.3 (83.2–3.3) | ||
| Wang et al33 | 877 | 74.1 (71.0–77.0) | 73.4 (70.1–76.4) | |||||
| Wang et al35 | 2646 | 83.6 (82.1–84.9) | 81.5 (79.7–83.2) | |||||
| Pooled estimate (95% CI) | 88.7 (82.4–93.8) | 92.1 (81.3–98.4) | 84.4 (73.1–93.1) | |||||
| 85.0 (83.7–86.2) | 92.7%, P<0.001 | 82.2 (80.3–84.0) | ||||||
| Heterogeneity I2 & P‐value | 91.3%, P<0.001 | 96.8%, P<0.001 | ||||||
| Atrioventricular septal defect | Frid et al22 | 502 | 77.1 (73.2–80.7)a | 66.5 (62.2–70.7)a | 64.3 (59.9–68.5)a | 63.1 (58.8–67.4)a | ||
| Garne23 | 20 | 50 (27.2–72.8)a | ||||||
| Miller et al27 | 338 | 57.9 (49.7–65.3) | ||||||
| Moons et al28 | 37 | 91.9 (78.1–98.3)a | ||||||
| Olsen et al30 | 354 | 75 (70–79) | 65 (59–70) | 59 (51–65) | ||||
| Samanek et al31 | 201 | 62.2 (55.4–69.0) | 54.7 (47.7–61.8) | 54.2 (47.1–61.2) | 54.2 (47.1–61.2) | |||
| Tennant et al32 | 94 | 84.0 (74.9–90.1) | 80.9 (71.3–87.5) | 79.7 (70.1–86.6) | 79.7 (70.1–86.6) | 79.7 (70.1–86.6) | ||
| Wang et al33 | 1004 | 59.5 (56.3–62.6) | 58.1 (56.5–61.4) | 56.6 (52.8–60.2) | ||||
| Wang et al34 | 4884 | 80.1 (79.0–81.2) | 76.7 (75.3–78.1) | |||||
| Pooled estimate (95% CI) | 75.9 (70.5–81.0) | 71.2 (61.9–79.6) | 64.0 (57.2–70.5) | 63.4 (56.3–70.3) | ||||
| Heterogeneity I2 & P‐value | 89.0%, P<0.001 | 92.7%, P<0.001 | 81.4%, P<0.001 | 85.9%, P<0.001 | ||||
| Coarctation of aorta | Garne23 | 12 | 58.3 (27.7–84.8)a | |||||
| Moons et al28 | 46 | 91.3 (79.2–97.6)a | ||||||
| Nembhard et al29 | 1145 | 78.6 (76.1–80.9) | ||||||
| Olsen et al30 | 334 | 84 (79–87) | 82 (77–85) | 78 (61–82) | ||||
| Samanek et al31 | 266 | 68.0 (62.3–73.8) | 65.4 (59.6–71.3) | 65.0 (59.2–70.9) | 65.0 (59.2–70.8) | |||
| Tennant et al32 | 189 | 91.5 (86.6–94.7) | 91.5 (86.6–94.7) | 90.9 (85.8–94.3) | 90.9 (85.8–94.3) | 89.6 (83.7–93.5) | ||
| Wang et al33 | 2529 | 79.4 (77.8–81.0) | 77.0 (75.4–78.6) | 76.0 (74.3–77.7) | 75.2 (73.3–77.0) | |||
| Wang et al34 | 6365 | 84.5 (83.6–85.4) | 81.9 (80.7–83.0) | |||||
| Pooled estimate (95% CI) | 82.7 (75.4–89.0) | 81.0 (70.7–89.4) | 80.3 (65.0–92.0) | 78.2 (65.9–88.4) | ||||
| 79.5 (76.6–82.2) | 76.2 (74.6–77.7) | |||||||
| Heterogeneity I2 & P‐value | 93.7%, P<0.001 | 93.0%, P<0.001 | 87.3%, P<0.001 | 95.6%, P<0.001 | ||||
| Common arterial trunk | Moons et al28 | 7 | 85.7 (42.1–99.6)a | |||||
| Olsen et al30 | 78 | 45 (34–55) | 45 (34–55) | 45 (34–55) | 45 (34–55) | 45 (34–55) | 45(34–55) | |
| Samanek et al31 | 55 | 12.7 (3.7–21.7) | 10.5 (4.1–22.2)a | 7.3 (0–15.4) | 7.3 (0–15.4) | |||
| Tennant et al32 | 36 | 36.1 (21.0–51.4) | 36.1 (21.0–51.4) | 36.1 (21.0–51.4) | ||||
| Wang et al33 | 460 | 59.2 (54.4–63.6) | 55.2 (49.5–60.5) | |||||
| Wang et al35 | 956 | 75.1 (72.7–77.7) | ||||||
| Pooled estimate (95% CI) | 41.8 (14.1–72.6) | 47.4 (21.8–73.8) | 28.9 (16.3–43.3) | 36.5 (14.6–62) | ||||
| 35.4 (30.0–41.0) | 54.4(50.2–58.6) | |||||||
| Heterogeneity I2 & P‐value | 97.6%, P<0.001 | 96.3%, P<0.001 | 87.3%, P<0.001 | 94.5%, P<0.001 | ||||
| Pulmonary valve atresia (with IVS) | Fixler et al21 | 118 | 59.3 (49.9–67.6) | 55.7 (45.8–64.4) | ||||
| Idorn et al24 | 75 | 41.7 (30.1–53.3)a | 37.5 (26.4–49.2)a | 35.3 (24.0–46.5)a | 37.5 (26.4–49.2)a | 35.3 (24.0–46.5)a | 37.5 (26.4–49.2)a | |
| Moons et al28 | 6 | 83.3 (36.5–99.1)a | ||||||
| Samanek et al31 | 53 | 18.9 (8.1–29.6) | 7.6 (0.3–14.8) | 7.6 (0.3–14.8) | 7.6 (0.3–14.8) | |||
| Pooled estimate (95% CI) | 39.7 (18.5–63.3) | 41.1 (17.2–67.6) | ||||||
| 45.5 (39.2–52.0) | ||||||||
| Heterogeneity I2 & P‐value | 92.1%, P<0.001 | 92.0%, P<0.001 | ||||||
| Pulmonary atresia | Garne et al23 | 5 | 60.0 (14.7–94.7)a | |||||
| Pulmonary valve atresia (with VSD) | Moons et al28 | 6 | 67 (19–96)a | 50 (11.8–88.2)a | ||||
| Samanek et al31 | 55 | 61.8 (48.7–74.9) | 54.5 (41.1–68.0) | 45.2 (30.8–59.6) | 45.2 (30.8–59.6) | |||
| Tetralogy of Fallot | Garne23 | 7 | 82.6 (61.2–95.0)a | |||||
| Moons et al28 | 52 | 83 (70–92)a | 82.7 (69.7–91.8)a | |||||
| Olsen et al30 | 381 | 83 (79–87) | 70 (65–74) | 67 (58–74) | ||||
| Samanek et al31 | 169 | 84.6 (79.0–90.2) | 76.6 (70.1–83.2) | 76.6 (70.1–83.2) | ||||
| Tennant et al32 | 190 | 90.5 (85.4–93.9) | 83.7 (77.6–88.2) | 83.1 (76.9–87.7) | 83.1 (76.9–87.7) | 80.8 (72.8–86.6) | ||
| Wang et al34 | 5208 | 87.1 (86.1–87.9) | 84.7 (83.5–85.8) | |||||
| Wang et al34 | 1739 | 86.9 (85.3–88.4) | ||||||
| Pooled estimate (95% CI) | 86.3 (83.7–88.6) | 84.6 (83.5–85.7) | 81.4 (77.5–85) | |||||
| 81.6 (78.6–84.4) | ||||||||
| Heterogeneity I2 & P‐value | 0.0%, P=0.097 | 0.0%, P=0.957 | 36.1%, P=0.209 | |||||
| Total anomalous pulmonary venous return | Garne23 | 5 | 20 (0.5–71.6)a | |||||
| Samanek et al31 | 40 | 52.5 (36.7–8.23) | 50.0 (34.2–65.8) | 50.0 (34.2–65.8) | 50.0 (34.2–65.8) | |||
| Tennant et al32 | 54 | 72.2 (58.2–82.2) | 72.2 (58.2–82.2) | 72.2 (58.2–82.2) | 72.2 (58.2–82.2) | 72.2 (58.2–82.2) | ||
| Pooled estimate (95% CI) | 53.7 (30–76.6) | |||||||
| 61.2 (51.2–70.6) | ||||||||
| Heterogeneity I2 & P‐value | 76.6%, P=0.014 | |||||||
| Transposition of the great vessels | Garne23 | 21 | 76.2 (52.8, 91.8)a | |||||
| Moons et al28 | 29 | 100.0 (88.1–100.0)a | ||||||
| Olsen et al30 | 461 | 74 (70–78) | 62 (38–67) | 50 (41–59) | ||||
| Samanek et al31 | 271 | 61.6 (56.7–67.5) | 56.5 (50.3–62.4)a | 53.9 (46.8–60.9) | 53.9 (46.8–60.9) | |||
| Tennant et al32 | 189 | 82.5 (76.3–87.3) | 81.0 (74.6–85.9) | 80.3 (73.8–85.3) | 78.4 (71.6–83.9) | 74.1 (64.4–81.5) | ||
| Wang et al34 | 1840 | 74.5 (72.4–76.4) | ||||||
| Wang et al34 | 4330 | 83.7 (82.6–84.4) | 81.1 (79.7–82.4) | |||||
| Pooled estimate (95% CI) | 76.0 (65.5–85.1) | 81.9 (68.9–91.9) | 66.1 (46.0–83.5) | |||||
| 67.1 (62.5–71.5) | ||||||||
| Heterogeneity I2 & P‐value | 96.9%, P<0.001 | 95.9%, P<0.001 | 93.6%, P<0.001 | |||||
| Tricuspid atresia | Fixler et al21 | 67 | 76.1 (64.0–84.6) | 74.6 (62.4–83.4) | ||||
| Idorn et al24 | 106 | 68.0 (58.2–76.7)a | 61.7 (51.4–70.6)a | 60.5 (50.4–69.7)a | 57.4 (47.6–67.1)a | 57.4 (47.6–67.1)a | 57.4 (47.6–67.1)a | |
| Moons et al28 | 4 | 100 (39.8–100.0)a | 100 (39.8–100.0)a | |||||
| Samanek et al31 | 39 | 46.2 (30.2–62.1) | 35.9 (20.5–51.3) | 35.9 (20.5–51.3) | ||||
| Tennant et al32 | 24 | 83.3 (61.5–93.4) | 66.7 (44.3–81.7) | 62.5 (40.3–78.4) | 62.5 (40.3–78.4) | |||
| Pooled estimate (95% CI) | 71.4 (57.2–83.7) | 53.7 (30.0–76.6) | 53.1 (36.5–69.2) | 53.3 (37.2–69.1) | ||||
| 55.1 (47.3–62.9) | 56.2 (49.1–63.1) | |||||||
| Heterogeneity I2 & P‐value | 74.4%, P=0.004 | 93.9%, P<0.001 | 72.4%, P=0.027 | 72.9%, P=0.025 | ||||
| Hypoplastic left heart | Garne | 22 | 4.5 (0.1–22.8)a | |||||
| Idorn et al24 | 252 | 12.5 (8.9–17.5)a | 10.4 (6.9–14.8)a | 10.4 (6.9–14.8)a | 8.8 (5.6–12.9)a | |||
| Moons et al28 | 10 | 50 (18.7–81.3)a | 40.0 (12.2–73.8)a | |||||
| Samanek et al31 | 172 | 0 (0.0–2.1)a | 0 (0.0–2.1)a | 0 (0.0–2.1)a | 0 (0.0–2.1)a | |||
| Tennant et al32 | 73 | 4.1 (1.1–10.5) | 2.9 (0.5–8.9) | |||||
| Wang et al34 | 33.1 (30.6–35.7) | |||||||
| Wang et al34 | 2976 | 55.2 (53.4–56.9) | 50.6 (48.4–52.7) | |||||
| Pooled estimate (95% CI) | 17.4 (0.0–54.5) | 12.5 (0.0–41.4) | ||||||
| Heterogeneity I2 & P‐value | 99.5%, P<0.001 | 99.1%, P=0.036 | ||||||
| Single ventricle | Fixler et al21 | 286 | 64.7 (58.8–69.9) | 56.1 (49.9–61.7) | ||||
| Garne23 | 16 | 56.3 (29.9– 80.2)a | ||||||
| Moons et al28 | 9 | 56 (21–86)a | 55.6 (21.2–86.3)a | |||||
| Tennant et al32 | 31 | 83.9 (65.5–93.0) | 74.2 (55.0–86.2) | 74.2 (55.0–86.2) | 64.5 (43.1–80.0) | |||
| Pooled estimate (95% CI) | 70.4 (54.1–84.4) | 59.8 (50.4–68.8) | ||||||
| 69.5 (63.3–75.3) | ||||||||
| Heterogeneity I2 & P‐value | 65.0%, P=0.057 | 26.9%, P=0.250 | ||||||
| Ebstein's anomaly | Garne23 | 5 | 60.0 (14.7–94.7)a | |||||
| Moons et al28 | 3 | 100 (29.2–100.0)a | ||||||
| Nembhard et al29 | 160 | 68.8 (61.0–75.8)a | ||||||
| Samanek et al31 | 22 | 67.9 (50.2–86.5) | 64.3 (46.2–82.4) | 64.3(46.2–82.4) | 64.3(46.2–82.4) | |||
| Tennant et al32 | 55 | 67.3 (53.2–78.0) | 58.0 (43.8–69.7) | 58.0 (43.8–69.7) | 54.6 (39.7–67.2) | 54.6 (39.7–67.2) | ||
| Pooled estimate (95% CI) | 65.6 (57.5–73.2) | |||||||
| Heterogeneity I2 & P‐value | 18.0%, P=0.300 | |||||||
Pooled estimated are calculated using random effects meta‐analysis. But where there are ≤3 studies, pooled estimates are also calculated using fixed effects meta‐analysis with these results being shown in italics. AVA/S in Wang et al's studies refers to aortic valve stenosis only. IVS indicates intact ventricular septum; VSD, ventricular septal defect.
Indicates that 95% CIs were not reported in the study, but 95% binomial exact 95% CIs were calculated by the authors.
95% CIs obtained from author
For subtypes for which just 3 studies reported survival, pooled estimates were also calculated using fixed‐effect meta‐analysis (Table 2). Pooled survival estimates were generally similar for the random‐ and fixed‐effects models, with the exception of the 10‐ and 15‐year pooled estimates for common arterial trunk (28.9% versus 35.4% and 36.5% versus 54.4%, respectively).
Quality Appraisal
Quality appraisal is shown in Table 3. All articles satisfied the study ascertainment domain because, by definition, population‐based studies are representative of the population. The attrition domain was satisfied by 31% of articles because of studies failing to report the proportion of untraced cases; however, many of the studies classed unmatched cases as alive, so it is possible that all cases were traced. The outcome ascertainment domain was satisfied by 94% of studies, and the analysis domain was satisfied by 81%. Studies that did not satisfy the analysis domain were those that did not perform survival analysis and instead reported the proportion alive, which does not account for case censorship. This may have slightly inflated survival in these studies.
Table 3.
Quality Appraisal of Included Articles
| Domain | Quality Items, Potential Bias | Yes | No | Not Stated | Number of Studies, % |
|---|---|---|---|---|---|
| Study ascertainment | The study population is adequately described for key characteristics (ie, CHD subtype frequency, sex distribution, ethnicity) | 21, 22, 23, 25, 27, 29, 30, 33, 34 | 20, 24, 26, 27, 28, 31, 32, 35 | 9 (56%) | |
| Ascertainment is adequately described, including method of ascertainment included birth years, study location | 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35 | 16 (100%) | |||
| Inclusion and exclusion criteria are adequately described (ie, ICD codes stated and inclusion of extracardiac anomalies | 21, 22, 23, 24, 25, 26, 27, 29, 30, 32, 33, 34, 35 | 20, 27, 28, 31 | 13 (81%) | ||
| There is adequate ascertainment | 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35 | 16 (100%) | |||
| POTENTIAL BIAS: The study sample represents the population of interest on key characteristics sufficient to limit potential bias to the results | 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35 | 16 (100%) | |||
| Study attrition | The proportion of traced cases is stated and adequate | 20, 22, 23, 29, 32 | 21, 24, 25, 26, 27, 28, 30, 31, 33, 34, 35 | 5 (31%) | |
| Reasons for untraced cases are provided | 20, 23, 29, 32 | 22 | 21, 24, 25, 26, 27, 28, 30, 31, 33, 34, 35 | 4 (25%) | |
| Untraced cases are adequately described for key characteristics (ie, CHD subtype) | 20, 22, 23, 29, 32 | 21, 24, 25, 26, 27, 28, 30, 31, 33, 34, 35 | 5 (31%) | ||
| There are no important differences between key characteristics and outcomes in participants who were traced and untraced | 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35 | 0 (0%) | |||
| POTENTIAL BIAS: Untraced cases are not associated with key characteristics (ie, the study data adequately represent the sample), sufficient to limit potential bias | 20, 22, 23, 29, 32 | 21, 24, 25, 26, 27, 28, 30, 31, 33, 34, 35 | 5 (31%) | ||
| Outcome ascertainment | Frequency of outcome is recorded | 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 32, 33, 34, 35 | 30, 31 | 14 (88%) | |
| The method of ascertainment of deaths is valid and reliable to limit misclassification bias | 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35 | 25 | 15 (94%) | ||
| POTENTIAL BIAS: The outcome of interest is adequately measured in study participants to sufficiently limit potential bias | 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35 | 25 | 15 (94%) | ||
| Analysis | There is sufficient presentation of results (ie, number of cases and 95% CIs) | 21, 24, 25, 27, 29, 30, 31, 32, 33, 34, 35 | 20, 22, 23, 26, 27, 28 | 11 (69%) | |
| The analysis is adequate for the design of the study | 20, 21, 24, 25, 27, 28, 29, 30, 31, 32, 33, 34, 35 | 22, 23, 26 | 13 (81%) | ||
| Results are not selectively reported | 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35 | 16 (100%) | |||
| POTENTIAL BIAS: The statistical analysis is appropriate for the design of the study, limiting potential for presentation of invalid results | 20, 21, 24, 25, 27, 28, 29, 30, 31, 32, 33, 34, 35 | 22, 23, 26 | 13 (81%) |
CHD indicates congenital heart disease; ICD, International Classification of Disease.
Discussion
In this systematic review and meta‐analysis, we found that 87.0% of individuals born with CHD survived to age 1 year, 85.4% survived to age 5 years, and 81.4% survived to age 10 years. Few studies reported survival beyond age 10 years, but survival appeared to continue to gradually decrease into adulthood. There was substantial variation in survival estimates among articles, some of which was accounted for by study period, which positively affected survival.
The main strength of this systematic review is its restriction to population‐based studies. Although including hospital‐based studies would have increased the amount of data available, such studies underascertain milder CHD subtypes that do not require major medical intervention. In addition, children with severe CHD may travel to centers with specialist expertise; therefore, the survival estimates reported by hospital‐based studies can be unrepresentative of the general population of individuals with CHD. The robustness of the individual rates of bias was examined using a quality assessment with previously published domains and items.19 Although each study failed to satisfy at least 1 quality item because of the population‐based study designs, the potential for bias in each domain remained low. Moreover, for all CHD, we did not identify any significant publication bias according to the Egger test.
A further strength is the comprehensive nature of our search strategy. Three databases were searched for relevant citations, along with key journals and reference lists; therefore, the likelihood of missing key studies was limited. Full articles were reviewed by both authors to ensure that they fully met the inclusion criteria and that data were extracted correctly. A further strength is that we reported pooled estimates calculated from fixed‐ and random‐effects meta‐analyses if there were just 3 studies reporting survival. Random‐effects meta‐analysis may calculate pooled estimates using an imprecise between‐study variance if the number of studies is low.15 The pooled estimates from the fixed‐effect meta‐analyses were broadly similar to those from the random‐effects meta‐analyses but with smaller confidence intervals.
There were also several limitations. The maximum follow‐up was just 30 years, with 5 of the included studies reporting survival to just 5 years. The greatest risk of death occurred in infancy, but survival continued to decrease over follow‐up, although at a much lesser rate. A study of CHD‐related mortality rates between 1999 and 2006 in the United States showed a high mortality rate of 41.5 per 100 000 in infancy, which decreased to 1.38 between ages 1 and 4 years and stabilized at ≈0.55 between the ages of 5 and 65 years. After age 65 years, the mortality rate doubled to 1.10 per 100 000.36
A further limitation is that longer term survival estimates may not be representative of children born with CHD today. Even in the most recent studies, 25‐year survival rates related to persons born in the 1990s; in our metaregression of 1‐, 5‐, and 10‐year survival, we showed that survival estimates improved over time.
All included studies were performed in high‐income Western populations. Evidence suggests that infant mortality rates associated with congenital anomalies are greater in low‐income countries.37 Consequently, the survival estimates in this review are not likely to be globally representative. Although we included only articles written in the English language, we did not identify any relevant articles written in other languages.
Most of the included articles included cases with extracardiac anomalies20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 33, 34; therefore, it is difficult to assess how much of the mortality was accounted for by CHD as opposed to the co‐occurring congenital anomalies. Nevertheless, cases with extracardiac anomalies accounted for only 20% of cases, and some extracardiac anomalies were not likely to be life threatening; therefore, the impact on survival is likely to be low. All articles used all‐cause mortality, meaning that deaths may not have been directly related to the CHD diagnosis.
Although this review provides insight into long‐term mortality associated with CHD, we did not account for morbidity. Research suggests that quality of life is lower in those with CHD and that those who live with CHD can have morbidities such as endocarditis, cerebrovascular accidents, myocardial infarctions, and arrhythmias.38, 39, 40 The American Heart Association has also reported that children with CHD are at increased risk of developmental disorders.41 Research suggests that children with CHD are more likely to require special education services, regardless of CHD severity.42
In our metaregression, we found that a more recent study period positively affected survival estimates; however, even after adjustment for study period, there was still a high degree of heterogeneity. Although we adjusted for study period using the year of study commencement, the lengths of the study periods varied by article; therefore, our adjustment for the year of study commencement is not likely to have fully accounted for the changes in survival over time. Further heterogeneity is likely attributable to a variety of sources. Case ascertainment is likely a major cause. Olsen et al reported lower survival estimates even after accounting for study period, but their prevalence of CHD was almost half that of other studies. Given that they included only cases diagnosed before age 1 year, it is likely that they underascertained cases with milder CHD subtypes, such as ventricular septal defect.30 The data sources used may also have contributed to variation in ascertainment, with articles using hospital records as opposed to congenital anomaly registers (which use multiple sources for ascertainment) contributing to lower survival estimates, likely due to the milder cases being underascertained.31
Variation in study periods is arguably the greatest source of heterogeneity for survival estimates. Survival has improved over time because of advances in surgical correction. The Fontan operation, for example, for repair of single ventricle, hypoplastic left heart, and tricuspid atresia and the conduit repair for cases of common arterial trunk were introduced in the late 1970s and developed throughout the 1980s and 1990s.43, 44 The arterial switch operation for treatment of transposition of the great vessels was introduced in 197545 and fully replaced the atrial switch operations in the early 1990s, resulting in improved long‐term survival.46 Survival is also likely to have improved over time because of advances in prenatal diagnosis. Greater prenatal diagnosis rates may have led to an increase in rates of termination (for fetal anomaly). If cases with the more severe subtypes were terminated, this would have resulted in better survival. Prenatal diagnosis also allows quicker intervention at birth or even in utero, which may also improve survival.47 In addition, survival is likely to have improved because of the introduction of prostaglandin, which underwent trials in neonates with cyanotic CHD in the 1970s,48, 49 although it was not frequently administered until the 1980s.
The improvement in survival rates over time has led to an emerging population of adolescents and adults with CHD. These patients require long‐term follow‐up, sometimes leading to reinvestigation and reoperation. Consequently, population‐based surveillance of CHD is crucial to adequately assess the variety of health and social services required by those with CHD throughout their lives.
Sources of Funding
Best is funded by the British Heart Foundation (FS/12/23/29511).
Disclosures
None.
Acknowledgments
Thanks to Harper Gilmour, Saeed Dastgiri, Wendy Nembhard, Philip Moons and Ying Wang for providing further information on their studies. We thank Dr Svetlana Glinianaia, Prof Fiona Matthews and Dr Angela McBrien for their helpful comments on this manuscript.
(J Am Heart Assoc. 2016;5:e002846 doi: 10.1161/JAHA.115.002846)
References
- 1. Hoffman JIE, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890–1900. [DOI] [PubMed] [Google Scholar]
- 2. Reller MD, Strickland MJ, Riehle‐Colarusso T, Mahle WT, Correa A. Prevalence of congenital heart defects in metropolitan Atlanta, 1998–2005. J Pediatr. 2008;153:807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dolk H, Loane M, Garne E. Congenital heart defects in Europe: prevalence and perinatal mortality, 2000 to 2005. Circulation. 2011;123:841–849. [DOI] [PubMed] [Google Scholar]
- 4. Centers for Disease Control Prevention . Racial differences by gestational age in neonatal deaths attributable to congenital heart defects—United States, 2003–2006. MMWR Morb Mortal Wkly Rep. 2010;59:1208. [PubMed] [Google Scholar]
- 5. Oster ME, Lee KA, Honein MA, Riehle‐Colarusso T, Shin M, Correa A. Temporal trends in survival among infants with critical congenital heart defects. Pediatrics. 2013;131:e1502–e1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bourdial H, Jamal‐Bey K, Edmar A, Caillet D, Wuillai F, Bernede‐Bauduin C, Boumahni B, Robillard PY, Kauffmann E, Laffitte A, Touret Y, Cuillier F, Fourmaintraux A, Alessandri JL, Gérardin P, Randrianaivo H. Congenital heart defects in La Réunion Island: a 6‐year survey within a EUROCAT‐affiliated congenital anomalies registry. Cardiol Young. 2012;22:547–557. [DOI] [PubMed] [Google Scholar]
- 7. Dadvand P, Rankin J, Shirley MDF, Rushton S, Pless‐Mulloli T. Descriptive epidemiology of congenital heart disease in Northern England. Paediatr Perinat Epidemiol. 2009;23:58–65. [DOI] [PubMed] [Google Scholar]
- 8. Dilber D, Malcic I. Spectrum of congenital heart defects in Croatia. Eur J Pediatr. 2010;169:543–550. [DOI] [PubMed] [Google Scholar]
- 9. Cleves MA, Ghaffar S, Zhao W, Mosley BS, Hobbs CA. First‐year survival of infants born with congenital heart defects in Arkansas (1993–1998): a survival analysis using registry data. Birth Defects Res A Clin Mol Teratol. 2003;67:662–668. [DOI] [PubMed] [Google Scholar]
- 10. Lee K, Khoshnood B, Chen L, Wall SN, Cromie WJ, Mittendorf RL. Infant mortality from congenital malformations in the United States, 1970–1997. Obstet Gynecol. 2001;98:620–627. [DOI] [PubMed] [Google Scholar]
- 11. Knowles RL, Bull C, Wren C, Wade A, Goldstein H, Dezateux C. Modelling survival and mortality risk to 15 years of age for a national cohort of children with serious congenital heart defects diagnosed in infancy. PLoS One. 2014;9:e106806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Verheugt CL, Uiterwaal CSPM, Grobbee DE, Mulder BJM. Long‐term prognosis of congenital heart defects: a systematic review. Int J Cardiol. 2008;131:25–32. [DOI] [PubMed] [Google Scholar]
- 13. Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta‐analyses of the published literature for survival endpoints. Stat Med. 1998;17:2815–2834. [DOI] [PubMed] [Google Scholar]
- 14. Plot Digitizer . 2015. Available at: http://plotdigitizer.sourceforge.net/. Accessed June 2015.
- 15. Borenstein M, Hedges L, Rothstein H. Introduction to meta‐analysis. 2007.
- 16. Barendregt JJ, Doi SA, Lee YY, Norman RE, Vos T. Meta‐analysis of prevalence. J Epidemiol Community Health. 2013;67:974–978. [DOI] [PubMed] [Google Scholar]
- 17. Higgins J, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ. 2003;327:557–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Egger M, Smith GD, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ. 1997;315:629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hayden JA, Côté P, Bombardier C. Evaluation of the quality of prognosis studies in systematic reviews. Ann Intern Med. 2006;144:427–437. [DOI] [PubMed] [Google Scholar]
- 20. Dastgiri S, Gilmour WH, Stone DH. Survival of children born with congenital anomalies. Arch Dis Child. 2003;88:391–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fixler DE, Nembhard WN, Salemi JL, Ethen MK, Canfield MA. Mortality in first 5 years in infants with functional single ventricle born in Texas, 1996 to 2003. Circulation. 2010;121:644–650. [DOI] [PubMed] [Google Scholar]
- 22. Frid C, Bjorkhem G, Jonzon A, Sunnegardh J, Anneren G, Lundell B. Long‐term survival in children with atrioventricular septal defect and common atrioventricular valvar orifice in Sweden. Cardiol Young. 2004;14:24–31. [DOI] [PubMed] [Google Scholar]
- 23. Garne E. Congenital heart defects—occurrence, surgery and prognosis in a Danish County. Scand Cardiovasc J. 2004;38:357–362. [DOI] [PubMed] [Google Scholar]
- 24. Idorn L, Olsen M, Jensen AS, Juul K, Reimers JI, Sorensen K, Johnsen SP, Sondergaard L. Univentricular hearts in Denmark 1977 to 2009: incidence and survival. Int J Cardiol. 2013;167:1311–1316. [DOI] [PubMed] [Google Scholar]
- 25. Jackson M, Walsh KP, Peart I, Arnold R. Epidemiology of congenital heart disease in Merseyside—1979 to 1988. Cardiol Young. 1996;6:272–280. [Google Scholar]
- 26. Meberg A, Otterstad JE, Froland G, Lindberg H, Sorland SJ. Outcome of congenital heart defects–a population‐based study. Acta Paediatr. 2000;89:1344–1351. [DOI] [PubMed] [Google Scholar]
- 27. Miller A, Siffel C, Lu C, Riehle‐Colarusso T, Frías JL, Correa A. Long‐term survival of infants with atrioventricular septal defects. J Pediatr. 2010;156:994–1000. [DOI] [PubMed] [Google Scholar]
- 28. Moons P, Sluysmans T, De Wolf D, Massin M, Suys B, Benatar A, Gewillig M. Congenital heart disease in 111 225 births in Belgium: birth prevalence, treatment and survival in the 21st century. Acta Paediatr. 2009;98:472–477. [DOI] [PubMed] [Google Scholar]
- 29. Nembhard WN, Salemi JL, Ethen MK, Fixler DE, Dimaggio A, Canfield MA. Racial/Ethnic disparities in risk of early childhood mortality among children with congenital heart defects. Pediatrics. 2011;127:e1128–e1138. [DOI] [PubMed] [Google Scholar]
- 30. Olsen M, Christensen TD, Pedersen L, Johnsen SP, Hjortdal VE. Late mortality among Danish patients with congenital heart defect. Am J Cardiol. 2010;106:1322–1326. [DOI] [PubMed] [Google Scholar]
- 31. Samanek M, Voriskova M. Congenital heart disease among 815,569 children born between 1980 and 1990 and their 15‐year survival: a prospective Bohemia survival study. Pediatr Cardiol. 1999;20:411–417. [DOI] [PubMed] [Google Scholar]
- 32. Tennant PW, Pearce MS, Bythell M, Rankin J. 20‐year survival of children born with congenital anomalies: a population‐based study. Lancet. 2010;375:649–656. [DOI] [PubMed] [Google Scholar]
- 33. Wang Y, Hu J, Druschel CM, Kirby RS. Twenty‐five‐year survival of children with birth defects in New York State: a population‐based study. Birth Defects Res Part A Clin Mol Teratol. 2011;91:995–1003. [DOI] [PubMed] [Google Scholar]
- 34. Wang Y, Liu G, Druschel CM, Kirby RS. Maternal race/ethnicity and survival experience of children with congenital heart disease. J Pediatr. 2013;163:1437–1442.e1431–1432. [DOI] [PubMed] [Google Scholar]
- 35. Wang Y, Liu G, Canfield MA, Mai CT, Gilboa SM, Meyer RE, Anderka M, Copeland GE, Kucik JE, Nembhard WN, Kirby RS. Racial/ethnic differences in survival of United States children with birth defects: a population‐based study. J Pediatr. 2015;166:819–826.e812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gilboa SM, Salemi JL, Nembhard WN, Fixler DE, Correa A. Mortality resulting from congenital heart disease among children and adults in the United States, 1999 to 2006. Clinical perspective. Circulation. 2010;122:2254–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rosano A, Botto LD, Botting B, Mastroiacovo P. Infant mortality and congenital anomalies from 1950 to 1994: an international perspective. J Epidemiol Community Health. 2000;54:660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Knowles RL, Day T, Wade A, Bull C, Wren C, Dezateux C; Defects UKCSoCH . Patient‐reported quality of life outcomes for children with serious congenital heart defects. Arch Dis Child. 2014;99:413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Engelfriet P, Boersma E, Oechslin E, Tijssen J, Gatzoulis MA, Thilén U, Kaemmerer H, Moons P, Meijboom F, Popelová J. The spectrum of adult congenital heart disease in Europe: morbidity and mortality in a 5 year follow‐up period the Euro Heart Survey on adult congenital heart disease. Eur Heart J. 2005;26:2325–2333. [DOI] [PubMed] [Google Scholar]
- 40. Warnes CA. The adult with congenital heart diseaseborn to be bad? J Am Coll Cardiol. 2005;46:1–8. [DOI] [PubMed] [Google Scholar]
- 41. Marino BS, Lipkin PH, Newburger JW, Peacock G, Gerdes M, Gaynor JW, Mussatto KA, Uzark K, Goldberg CS, Johnson WH. Neurodevelopmental outcomes in children with congenital heart disease: evaluation and management a scientific statement from the American Heart Association. Circulation. 2012;126:1143–1172. [DOI] [PubMed] [Google Scholar]
- 42. Riehle‐Colarusso T, Autry A, Razzaghi H, Boyle CA, Mahle WT, Braun KVN, Correa A. Congenital heart defects and receipt of special education services. Pediatrics. 2015;136:496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Girinath MR. Case presentation: truncus arteriosus: repair with homograft reconstruction in infancy In: Barratt‐Boyes BG, Neutze JM, Harris EA, eds. Heart Disease in Infancy: Diagnosis and Surgical Treatment. Edinburgh: Churchill Livingstone; 1973:234. [Google Scholar]
- 44. Sanders JH Jr. Gibbon's surgery of the chest. Arch Surg. 1976;111:1411. [Google Scholar]
- 45. Jatene AD, Fontes VF, Paulista PP, de Souza LC, Neger F, Galantier M, Souza JE. Successful anatomic correction of transposition of the great vessels. A preliminary report. Arq Bras Cardiol. 1975;28:461–464. [PubMed] [Google Scholar]
- 46. Hörer J, Schreiber C, Cleuziou J, Vogt M, Prodan Z, Busch R, Holper K, Lange R. Improvement in long‐term survival after hospital discharge but not in freedom from reoperation after the change from atrial to arterial switch for transposition of the great arteries. J Thorac Cardiovasc Surg. 2009;137:347–354. [DOI] [PubMed] [Google Scholar]
- 47. Freud LR, McElhinney DB, Marshall AC, Marx GR, Friedman KG, Pedro J, Emani SM, Lafranchi T, Silva V, Wilkins‐Haug LE. Fetal aortic valvuloplasty for evolving hypoplastic left heart syndrome: postnatal outcomes of the first 100 patients. Circulation. 2014;130:638–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Elliott RB, Starling MB, Neutze JM. Medical manipulation of the ductus arteriosus. Lancet. 1975;305:140–142. [DOI] [PubMed] [Google Scholar]
- 49. Olley PM, Coceani F, Bodach E. E‐type prostaglandins: a new emergency therapy for certain cyanotic congenital heart malformations. Circulation. 1976;53:728–731. [DOI] [PubMed] [Google Scholar]