

Abstract
Background
The concept of innate immunity is well recognized within the spectrum of atherosclerosis, which is primarily dictated by macrophages. Although current insights to this process are largely based on murine models, there are fundamental differences in the atherosclerotic microenvironment and associated inflammatory response relative to humans. In this light, we characterized the cellular aspects of innate immune response in normal, nonprogressive, and progressive human atherosclerotic plaques.
Methods and Results
A systematic analysis of innate immune response was performed on 110 well‐characterized human perirenal aortic plaques with immunostaining for specific macrophage subtypes (M1 and M2 lineage) and their activation markers, neopterin and human leukocyte antigen–antigen D related (HLA‐DR), together with dendritic cells (DCs), natural killer (NK) cells, mast cells, neutrophils, and eosinophils. Normal aortae were devoid of low‐density lipoprotein, macrophages, DCs, NK cells, mast cells, eosinophils, and neutrophils. Early, atherosclerotic lesions exhibited heterogeneous populations of (CD68+) macrophages, whereby 25% were double positive “M1” (CD68+/ inducible nitric oxide synthase [iNOS]+/CD163−), 13% “M2” double positive (CD68+/iNOS −/CD163+), and 17% triple positive for (M1) iNOS (M2)/CD163 and CD68, with the remaining (≈40%) only stained for CD68. Progressive fibroatheromatous lesions, including vulnerable plaques, showed increasing numbers of NK cells and fascin‐positive cells mainly localized to the media and adventitia whereas the M1/M2 ratio and level of macrophage activation (HLA‐DR and neopterin) remained unchanged. On the contrary, stabilized (fibrotic) plaques showed a marked reduction in macrophages and cell activation with a concomitant decrease in NK cells, DCs, and neutrophils.
Conclusions
Macrophage “M1” and “M2” subsets, together with fascin‐positive DCs, are strongly associated with progressive and vulnerable atherosclerotic disease of human aorta. The observations here support a more complex theory of macrophage heterogeneity than the existing paradigm predicated on murine data and further indicate the involvement of (poorly defined) macrophage subtypes or greater dynamic range of macrophage plasticity than previously considered.
Keywords: aorta, atherosclerosis, immune system, inflammation, macrophage
Subject Categories: Atherosclerosis, Inflammation
Introduction
Macrophages and, possibly, other cellular components of the innate immune system are paramount in the initiation, progression, and complications of atherosclerosis.1, 2, 3 By the same token, data from animal studies show that these cells orchestrate atherosclerosis regression and plaque stabilization, underlining the central role of these cell types in the atherosclerotic process.4, 5, 6
It is now recognized that macrophages constitute a highly heterogeneous and dynamic cell population that were initially labeled as proinflammatory M1 macrophages and tissue regenerative M2 macrophages, although more‐elaborative classifications have been brought forward and some even pointed out that macrophage differentiation is a continuum.7 Experimental studies mainly involving mice imply a major shift in macrophage identity during the atherosclerotic process with proinflammatory processes (classically activated M1 macrophages) involved in the initiation and progression of the disease, and alternatively (activated M2 macrophages) linked to resolution and repair.8 Much of the theory in this area has been driven by in vitro studies exploring gene/protein expression patterns and functional attributes of monocytes or macrophages subjected to various treatments.9, 10
Knowledge on the innate immune system in the atherosclerotic process essentially relies on observations from rodent models.11 Yet, there are fundamental immunological and inflammatory differences between rodents and humans.12, 13, 14 Moreover, lesions in established murine atherosclerosis models fail to progress to advanced vulnerable plaques complicated by rupture, hence inflammatory responses in advanced stages of the disease cannot be characterized in these models.15 Consequently, it is becoming recognized that observations from animal models may not necessarily translate to the human atherosclerotic process, particularly when considering the inflammatory milieu.16, 17, 18
In this regard, we considered a systematic evaluation of the cellular components of the innate immune system (macrophages and their subtypes, dendritic cells [DCs], mast cells, natural killer [NK] cells, neutrophils, and eosinophils) throughout the process of human atherosclerosis, particularly in relation to complicated plaques, relevant to symptomatic disease. To that end, we performed a systematic histological evaluation of these components using a unique collection of biobanked human arterial tissues that covers the full spectrum of lesion progression.
Results from this study confirm an extensive and dynamic inflammatory process involving specific cellular components of the innate immune system occurring throughout disease progression. Furthermore, it was concluded that a simple dichotomous classification system for macrophage differentiation falls short in the biological context.
Material and Methods
Patients and Tissue Sampling
Tissue sections were selected from a large tissue bank containing over 400 individual abdominal aortic wall patches (AAWPs) that were obtained during liver, kidney, or pancreas transplantation (viz all material was from cadaveric donors). Details of this bank have been described previously by van Dijk et al.19 All patches were harvested from grafts that were eligible for transplantation (ie, all donors met the criteria set by The Eurotransplant Foundation) and because of national regulations, only transplantation‐relevant data for donation are available. The study did not need approval by an institutional review committee, and no informed consent was needed from the subjects. Sample collection and handling was performed in accord with the guidelines of the Medical and Ethical Committee in Leiden, The Netherlands, and the code of conduct of the Dutch Federation of Biomedical Scientific Societies (https://www.federa.org/sites/default/files/digital_version_first_part_code_of_conduct_in_uk_2011_12092012.pdf).
Histological sections of AAWPs were stained by hematoxylin and eosin (H&E) and Movat pentachrome for classification of the lesions (in accord with the modified American Heart Association classification as proposed by Virmani et al.20, 21). Classification was performed by two independent observers without knowledge of the character of the aortic tissue.19, 20 In all cases, the aortic tissue block showing the most advanced plaque from each patient was used for further analysis. The lesion types included in this classification are described below. In order to obtain a balanced and representative study group, 10 to 12 samples were randomly selected from representative lesion morphologies. A total of 110 samples were used for further immunohistochemistry staining and analysis.
Characterization of the Lesions and Histological Definitions
A detailed description of plaque characterization and morphological analysis is provided in van Dijk et al.19 Plaque morphologies included adaptive intimal thickening (AIT), intimal xanthoma (IX), pathological intimal thickening (PIT), early (EFA) and late fibroatheroma (LFA), thin‐cap fibroatheroma (TCFA), acute plaque rupture (PR), healed plaque rupture (HR), and fibrotic calcific plaque (FCP).
Immunohistochemistry
Single immunostaining for visualizing macrophages, DCs, mast cells, and neutrophils
Histological sections of formalin‐fixed, paraffin‐embedded aortic tissues decalcified in Kristensen fluid were immunostained using antibodies directed against the macrophage activation marker, neopterin, human leukocyte antigen–antigen D related (HLA‐DR; cell activation marker), fascin (DCs), tryptase (mast cells), myeloperoxidase (MPO), matrix metallopeptidase 8 (MMP8; neutrophils), and eosinophil granule monoclonal antibody (EG2; eosinophils). Details and antibodies used for immunohistochemical staining are listed in Table 1. Antigen retrieval was heat‐induced using either citrate (pH 6) or Tris‐EDTA (pH, 9.2) buffers or proteolytic digestion with trypsin, when necessary. Antibody binding was visualized by a polymer‐based HRP substrate (EnVision; DakoCytomation A/S, Glostrup, Denmark) with diaminobenzidine (DAB) as the chromogen and Gill's hematoxylin as counterstain. Tissue sections from human tonsils and abdominal aortic aneurysms were used for positive controls whereas negative controls were performed by omitting the primary antibody.
Table 1.
Antibodies Used in the Present Study
| Antibody, Clone | Host Isotype; Subclass | Specificity | Pretreatment | pH | Dilution | Reference/Source |
|---|---|---|---|---|---|---|
| Apolipoprotein B | Goat (polyclonal), IgG | Apolipoprotein B | Tris/EDTA | 9.2 | 1:4000 | Abcam |
| CD4 | Rabbit (polyclonal) | T‐helper cells | Citrate | 6.0 | 1:200 | Abcam |
| CD68, KP1 | Mouse (monoclonal), IgG1 | Pan Macrophage | Tris/EDTA | 9.2 | 1:7000 | DakoCytomation |
| CD163, 10d6 | Mouse (monoclonal) IgG1 | “M2” Macrophages (used in triple stains) | Tris/EDTA | 9.2 | 1:200 | Thermo Scientific/neomarkers |
| CCR7/CD197 | Rabbit (polyclonal), IgG | “M1” Macrophages (used in triple stains) | Tris/EDTA | 9 | 1:300 | Bioss Inc. |
| CD206, 685645 | Mouse (monoclonal), IgG2 | “M2” Macrophages (used in triple stains) | Tris/EDTA | 9 | 1:1000 | R&D Systems |
| Dectin‐1, NBP2‐13845 | Rabbit (polyclonal) | “M2” Macrophages (used in triple stains) | Tris/EDTA | 9.2 | 1:50 | Novus Biologicals |
| EG2 | Mouse | Activated eosinophils | Trypsin | — | 1:600 | Pharmacia Diagnostics |
| Fascin, 55k‐2 | Mouse (monoclonal), IgG1 | Dendritic cells | Citrate | 6.0 | 1:800 | DakoCytomation |
| HLA‐DR, TAL.1B5 | Mouse (monoclonal), IgG1 | Antigen‐presenting cells (APC) | Citrate | 6.0 | 1:200 | DakoCytomation |
| IL‐6, NYRhIL6 | Mouse (monoclonal), IgG2 | “M1” Macrophages (used in triple stains) | Citrate | 6.0 | 1:400 | Santa Cruz Biotechnology |
| iNOS | Rabbit (polyclonal), IgG | “M1” Macrophages (used in triple stains) | Tris/EDTA | 6.0 | 1:400 | Abcam |
| Myeloperoxidase | Rabbit (polyclonal) | Neutrophils | — | — | 1:5000 | DakoCytomation |
| MMP8, MAB9081 | Mouse (monoclonal) IgG2a | Neutrophils | Trypsin | — | 1:1000 | R&D Systems |
| Neopterin | Rabbit (polyclonal) | Activated macrophages | Citrate | 6.0 | 1:500 | Biogenesis |
| T‐bet | Rabbit (polyclonal) | NK cells/T‐helper 1 cells | Tris/EDTA | 9.2 | 1:200 | Santa Cruz Biotechnology |
| Tryptase, AA1 | Mouse (monoclonal), IgG1 | Mast cells | Citrate | 6.0 | 1:3000 | DakoCytomation |
AA1 indicates mast cell tryptase antibody; EG2, eosinophil granule monoclonal antibody; HLA‐DR, human leukocyte antigen–antigen D related; IgG, immunoglobulin G; IL‐6, interleukin‐6; iNOS, inducible nitric oxide synthase; MMP8, matrix metalloproteinase 8; NK, natural killer.
Double‐labeling immunohistochemistry for visualizing NK cells and low‐density lipoprotein with elastin
NK cells (T‐bet+/CD4−) were identified by double labelling against T‐bet (NK cells and T‐helper1 cells) and CD4 (T‐helper cells) with DAB and Vina Green chromogens, respectively, as described previously.22 Dual staining for low‐density lipoprotein (LDL) and elastin was performed with an antibody directed against apolipoprotein B100 (ApoB100) with DAB and counterstained by fuchsin.
Triple‐labeling immunohistochemistry for M1 and M2 macrophage subpopulations
Details of all antibodies are listed in Table 2. Primary markers included CD68 (pan‐MΦ macrophage marker with iNOS (inducible nitric oxide synthase; M1) and CD163 (macrophage scavenger receptor; M2). Macrophage phenotype was further validated with interleukin‐6 (IL‐6; M1) and dectin‐1 (M2; major β‐glucan receptor on macrophages). Antibody reactions were detected using an EnVision+System—HRP‐labeled polymer antimouse/antirabbit functioned as the secondary antibody (DakoCytomation) and positive reactions were visualized with DAB and Vina Green chromogen or 4plus Biotinylated Universal Goat link kit (Biocare Medical, Concord, CA) in combination with 4plus streptavidin AP label for Ferangi Blue and Warp Red visualizations. For triple stainings, a second heat‐induced citrate antigen retrieval was used after incubation with denature solution (Biocare Medical) to inactivate the preceding labels.
Table 2.
Demographic Data of the 110 Studied Aortic Samples
| Male | Female | |
|---|---|---|
| N | 60 | 50 |
| Mean age, y (SD) | 46.9 (19.8) | 47.0 (15.7) |
| Mean length, cm (SD) | 178.9 (11.7) | 165.6 (11.9) |
| Mean weight, kg (SD) | 80.0 (16.2) | 63.9 (14.5) |
| Mean BMI, kg/m2 (SD) | 24.9 (3.0) | 22.5 (4.3) |
| No. of patients with known history of nicotine abuse (%) | 21 (35) | 17 (34) |
| No. of patients with known history of hypertensiona (%) | 13 (21.7) | 10 (20) |
| No. of patients with known diabetes (%) | 0 (0.0) | 1 (2) |
| Cause of death, n (%) | ||
| Severe head trauma | 11 (18.3) | 7 (14.0) |
| Cerebral vascular accident | 8 (13.3) | 10 (20.0) |
| Subarachnoid bleeding | 15 (25) | 14 (28.0) |
| Cardiac arrest | 7 (11.6) | 0 (0.0) |
| Trauma | 1 (1.6) | 1 (2.0) |
| Other | 6 (10) | 3 (6.0) |
| Unknown | 12 (20) | 15 (30) |
| Medication, n (%) | ||
| Antihypertensives | 11 (18.3) | 6 (12.0) |
| Statins | 1 (1.6) | 1 (2.0) |
| Anticoagulants | 1 (1.6) | 2 (4.0) |
| Other | 5 (8.3) | 8 (16.0) |
| None | 31 (51.7) | 22 (44.0) |
| Unknown | 11 (18.3) | 9 (18.0) |
BMI indicates body mass index.
Known antihypertensive medication or systolic blood pressure >140 mm Hg and diastolic >90 mm Hg in the period preceding death.
Quantification of Immunostained Cells
Aortic lesion area was defined as the region between the lumen and first elastic lamina over a circumferential width of 1 mm. For fibroathermatous plaques, the lateral distance was expanded to include flanking regions of the necrotic core incorporating shoulder regions. Regions of interest (ROIs) were selected from the central region of the plaque and lateral shoulder regions with corresponding areas of underlying media and the adventitia (Figure 1). Three representative areas within separate ROIs involving the intima, media, and adventitia were imaged at ×200 magnification (Axiovision 4.6.3)—a total of 9 images per atherosclerotic lesion, which were each analyzed.
Figure 1.
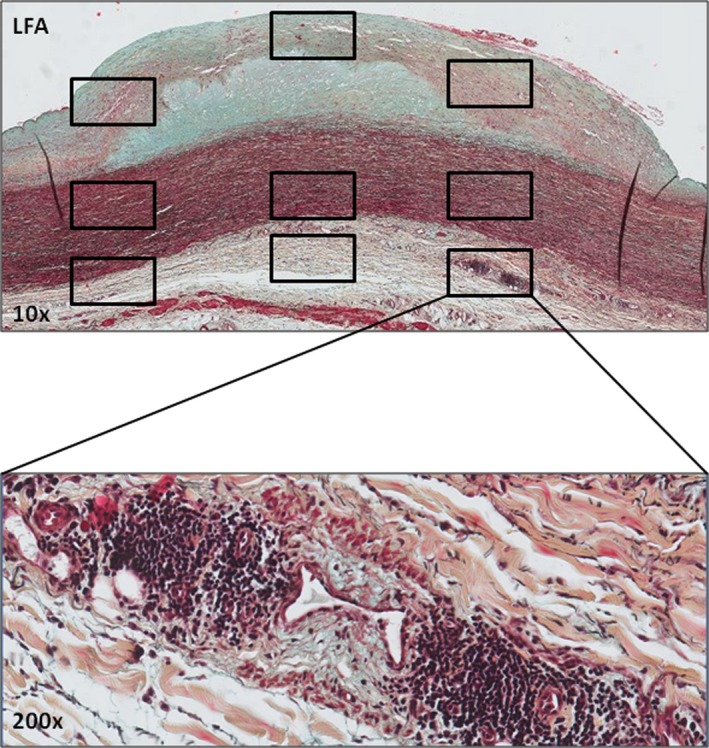
Defining the 9 regions of interest (ROIs) within atherosclerotic lesions. Early aortic fibroatheroma stained by Movat pentachrome with ROIs (boxed areas) selected within the fibrous cap, flanking shoulders, and underlying media and the adventitia. Immunostaining for select inflammatory markers within ROIs was quantitatively assessed with an image processing program (ImageJ; plug‐in Cellcounter). An additional high‐resolution image of an adventitial inflammatory infiltrate at ×200 magnification is shown to illustrate the cellular detail. LFA indicates late fibroatheroma.
Positive staining for mast cells (tryptase), NK cells (T‐bet+/CD4−), DCs (fascin), and neutrophils (myeloperoxidase and MMP8) was quantified using ImageJ Colour Deconvolution. Tissue sections stained for macrophages (CD68), neopterin, and HLA‐DR were quantified and presented as a percentage of plaque area.
Triple immunostains for macrophage subtypes were analyzed using the Nuance multispectral imaging system, FX,23 based on unmixed color spectra from DAB, Warp Red, and Vina Green or Ferangi Blue. Total numbers of M1 macrophages (CD68+/iNOS+, CD68+/IL6+, and CD68+/CD197+) and M2 macrophages (CD68+/CD163+, CD68+/Dectin‐1+, and CD68+/CD206+) are expressed as a percentage of colocalized CD68 as well as the percentage of the triple‐positive immunostaining (IL‐6/dectin‐1/CD68 or iNOS/CD163/CD68) in addition to the percentage of CD68 without any colocalization.
Statistical Analysis
Data are presented as means±SEM, stratified by the aortic layers (intima, media, and adventitia). Means of cellular distribution were compared using one‐way ANOVA. Second, lesion types were grouped in 5 categories, and mean values were pairwise compared with the Wilcoxon‐Mann‐Whitney test for the two adjacent lesion types. The Kruskal‐Wallis test was used to control for a type 1 error. To assess whether there was an association between the mean value of positive cells and the 5 lesion types within the aortic layers (intima, media, and adventitia), Spearman's correlation test was used. Unless specifically otherwise reported, the coefficient for this test was significant (P<0.05). Because only four pair‐wise comparisons were made per layer, we did not consider a post‐hoc method to control for multiple (pairwise) comparisons; a value of P<0.05 was considered statistically significant (version 20.0; SPSS, Inc., Chicago, IL).
Results
Study Population
The male/female ratio among donors was 54.5% male, and mean age was ≈47 years. There was a strong relationship between donor age and stage of atherosclerosis19 morphology, whereby normal aortic wall samples had a mean age of 17 years, whereas those with end‐stage fibrocalcific plaques were 60 years of age. Cardiovascular risk factors of smoking were known in 34%, hypertension in 15%, whereas 2 donors received statin therapy (Table 2).
Cellular Aspects of the Innate Immune Response
Normal and nonprogressive lesions
The intima and media of the normal aortic wall are devoid of LDL, macrophages, DCs, NK cells, mast cells, and neutrophils.
Early, nonprogressive atherosclerotic lesions inclusive of AIT and IX showed traces of LDL (ApoB100) within the intima (Figure 2). By definition, IX lesions showed extensive infiltration of (CD68+) macrophage foam cells (Figure 3), which was also heterogeneous, as shown by additional markers for M1 and M2 macrophages. Spectral imaging analysis of macrophage subtypes showed a predominance of “M1” (CD68+/iNOS+/CD163−) over “M2” (CD68+/iNOS−/CD163+) macrophages (25% and 13%, respectively; Figure 4) with ≈17% of the macrophages coexpressing both (M1) iNOS and CD163 (M2) markers, with the remaining only positive for CD68. Comparable results were obtained with a second and third set of validation markers, IL‐6 and CD197 (M1) and dectin‐1 and CD206 (M2; Figure 4B through 4D).
Figure 2.
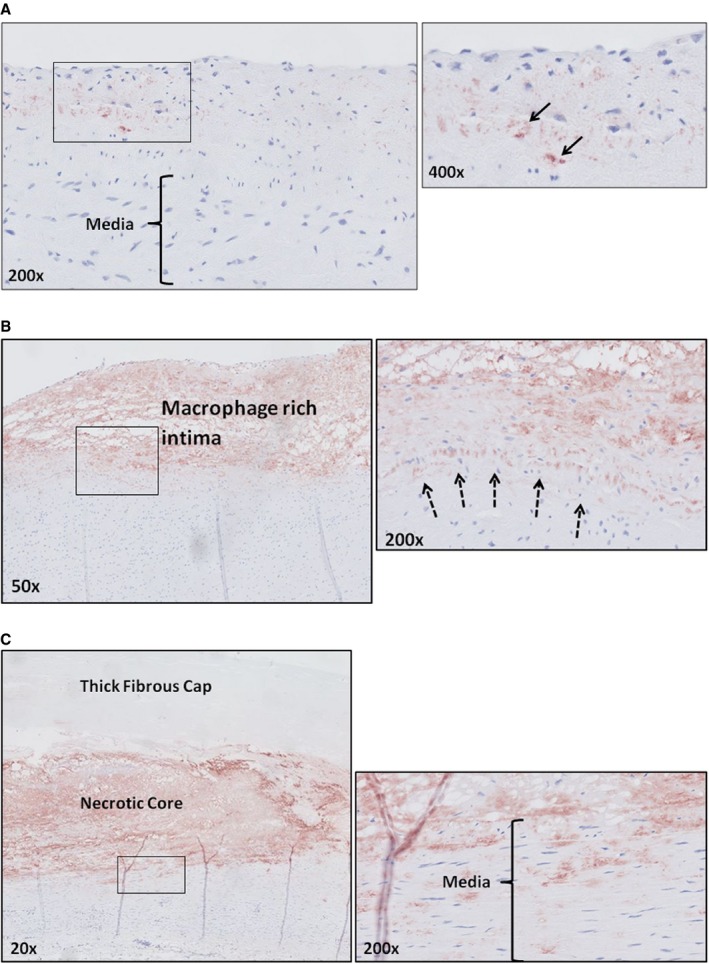
Apolipoprotein B100 (ApoB100) (low‐density lipoprotein) deposition. A, Representative image (×200) of an aorta with adaptive intimal thickening stained for ApoB100 with a high‐resolution image at a ×400 magnification. ApoB100 accumulation is confined to the intima (black arrow). B, Representative image (×50) of an intimal xanthoma stained for ApoB100 with a high‐resolution image at a ×200 magnification. Increase in intimal ApoB100 deposition within the intima. Accumulation of ApoB100 is restricted to the intima (dotted arrows represent the internal elastic lamina). C, Representative image (×20) of a late fibroatheroma stained for ApoB100 with a high‐resolution image at a ×200 magnification at the intimal‐medial border. This stage of atherosclerosis is characterized by accumulation of ApoB100 within the media, coinciding with loss of integrity of the internal elastic lamina. All ApoB100 samples were visualized with diaminobenzidine (DAB) and counterstained with hematoxylin.
Figure 3.
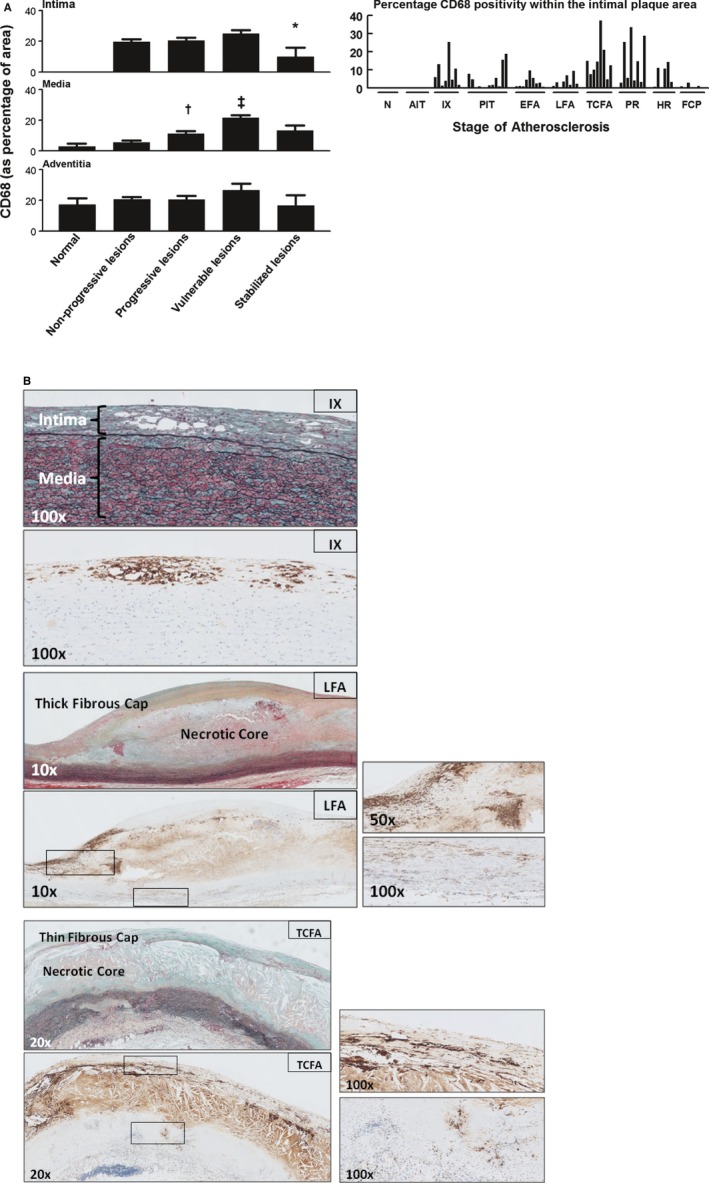
Macrophage (CD68+) distribution in normal, nonprogressive, progressive, vulnerable, and stable atherosclerotic plaques. A, Mean percentage of CD68‐positive area within various regions of interest (intima, media, and adventitia) plotted by lesion morphology (±SEM). An additional plot of the intima illustrates the percentage of CD68 positivity for individual plaque stratified by the stage of atherosclerosis. CD68 is seen from the stage of intimal xanthoma (IX). CD68 positivity increases during disease progression and is maximal for vulnerable plaques, followed by a significant decrease when the lesions stabilize (*P<0.005; compared to vulnerable lesions). CD68 positivity in the media increases with progressive atherosclerotic lesions and vulnerable lesions († P<0.004; ‡ P<0.001; compared to nonprogressive lesions and progressive lesions, respectively) in contrary to the adventitia, which remained relatively constant except for a slight decrease for stabilizing lesions. Spearman's rho correlation coefficient is not significant in the intima, media, and adventitia. B, Representative images of IX, late fibroatheroma (LFA), and a thin‐cap fibroatheroma (TCFA) stained by Movat pentachrome with corresponding immunostain for macrophages (CD68; KP‐1). Macrophages and macrophage foam cells essentially form the IX. The necrotic core in LFA stains highly positive for CD68, representing accumulation of macrophage remnants and therefore was excluded during morphometric analysis. Whereas the thick fibrous cap of the LFA is mainly negative for CD68, the shoulder regions show infiltration by macrophages and macrophage foam cells (see the ×50 magnification) in addition to a notable presence within the media (see the ×100 magnification). More‐advanced TCFAs demonstrate increased macrophage accumulation in the cap. Total number of cases in (A): 85—normal 9, nonprogressive lesions 18 (viz AIT [10] and IX [8]), progressive lesions 29 (viz PIT [12], EFA [9] and LFA [8]), vulnerable lesions 16 (viz TCFA [8] and PR [8]), and stabilized lesions 13 (viz HR [7] and FCP [6]). AIT indicates adaptive intimal thickening; EFA, early fibroatheroma; FCP, fibrotic calcified plaque; HR, healed rupture; N, normal; PIT, pathological intimal thickening; PR, plaque rupture. For a detailed description concerning the classification, see the Material and Methods section.
Figure 4.
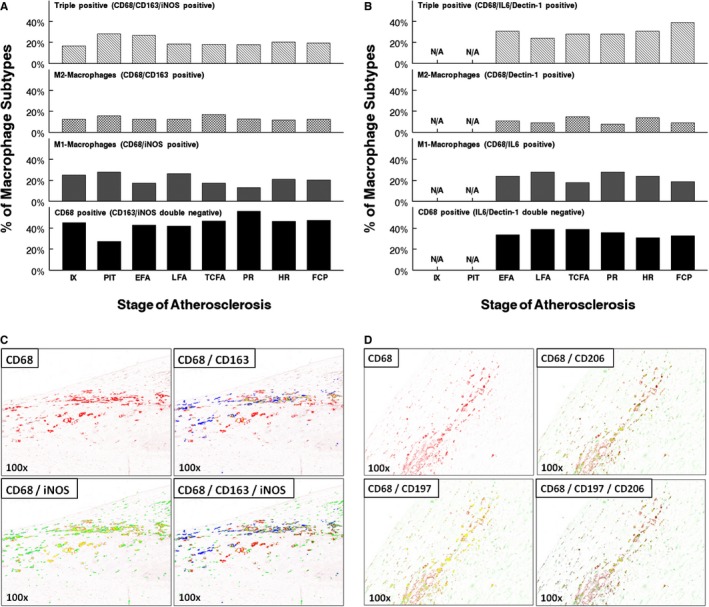
Macrophage subclass distribution (intima) during aortic atherosclerosis. A, Lesional macrophages were identified by the pan macrophage marker, CD68, and respective subclasses identified based on the expression of inducible nitric oxide synthase (iNOS; M1) and CD163 (M2). Various macrophage subtypes are plotted as a relative percentage of the total amount of CD68 cells for each atherosclerotic stage. As lesions progress toward a vulnerable phase (ie, TCFA and PR), there is a slight decreases in the extent of M1 macrophages whereas the M2 lineage increases, resulting in a 1:1 ratio. Within healed and stabilized FCP, the M1:M2 ratio is ≈2:1. Around 20% to 30% of the macrophages stain positive for both iNOS and CD163 and over 40% of the macrophages are double negative for the selective markers. B, Confirmation of M1 (iNOS) and M2 (CD163) expressing macrophages within various progressive, vulnerable lesions, and stable aortic lesions substituting IL6 and Dectin‐1 as respective “M1” and “M2” markers. Nearly the same percentages and distribution of M1 and M2 macrophages were observed when compared with lesions triple stained for iNOS and CD163, where ≈20% to 30% of the macrophages express both IL6 and dectin‐1 and nearly 40% of the macrophages are double negative for both markers. C, Representative images of a shoulder region at a ×100 magnification triple stained with CD68 (pan macrophage marker, Warp Red chromogen; spectral image color red), iNOS (inducible nitric oxide synthase; M1 macrophages, diaminobenzidine (DAB); spectral image color green) and CD163 (macrophage scavenger receptor; M2 macrophages, Ferangi Blue; spectral image color blue). Yellow indicates the amount of colocalization between the various macrophage markers. D, Representative images of a shoulder region at a ×100 magnification triple stained with CD68 (pan macrophage marker, Vina Green chromogen; spectral image color red), CD197 (M1 macrophages, Vulcan Red; spectral image color green) and CD206 (M2 macrophages, Ferangi Blue; spectral image color blue). Yellow indicates the amount of colocalization between the various macrophage markers. EFA indicates early fibroatheroma; FCP, fibrotic calcified plaque; HR, healed rupture; IX, intimal xanthoma; LFA, late fibroatheroma; N/A, not applicable; PIT, pathological intimal thickening; PR, plaque rupture; TCFA, thin‐cap fibroatheroma.
IXs were further assessed for the general cellular activation marker, HLA‐DR, and the macrophage‐specific marker, neopterin (Figures 5 and 6). The profuse macrophage infiltration seen in IX is paralleled by increased expression of HLA‐DR and neopterin in this early stage of the atherosclerotic process.
Figure 5.
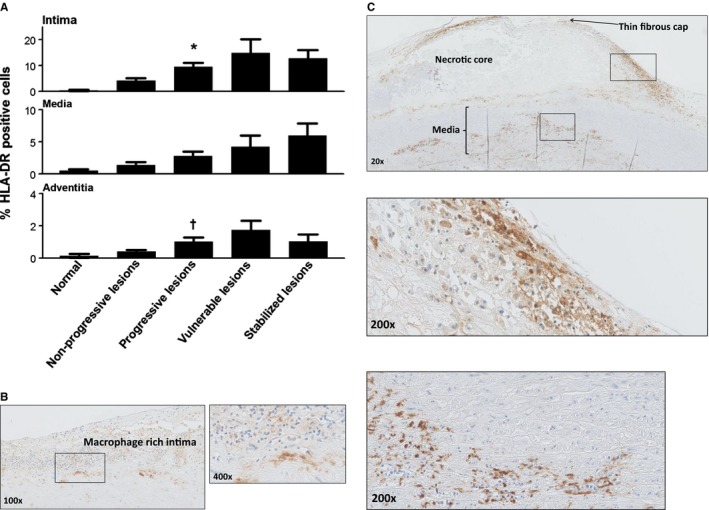
Human leukocyte antigen–antigen D related (HLA‐DR) expression during aortic atherosclerosis. A, Mean percentage of HLA‐DR expression (intima, media, and adventitia) based on lesion morphology. HLA‐DR expression increases within all the vascular layers (significant within the intima and the adventitia with progressive atherosclerosis; *P<0.002; † P<0.01; compared to nonprogressive lesions) and tend to decrease in the stabilized phase. B, Representative image of an intimal xanthoma stained for HLA‐DR at ×100 magnification with a high‐resolution image at a ×400 magnification. HLA‐DR staining is clearly seen in macrophage rich areas located in the intima. C, Representative image of a thin‐cap fibroatheroma stained for HLA‐DR at ×20 magnification with more detail (×200 magnification) of the macrophage foam cell‐rich shoulder region and media. Expression of HLA‐DR is more extensive in advanced and vulnerable stages of atherosclerosis. Total number of cases in (A): 89 (normal 8, nonprogressive lesions 24, progressive lesions 34, vulnerable lesions 14, and stabilized lesions 9). Large solid bars in (A): represent the mean percentage of HLA‐DR within the aortic wall section per atherosclerotic phase±SEM. All sections were developed with diaminobenzidine (DAB) and counterstained with Mayer's hematoxylin.
Figure 6.
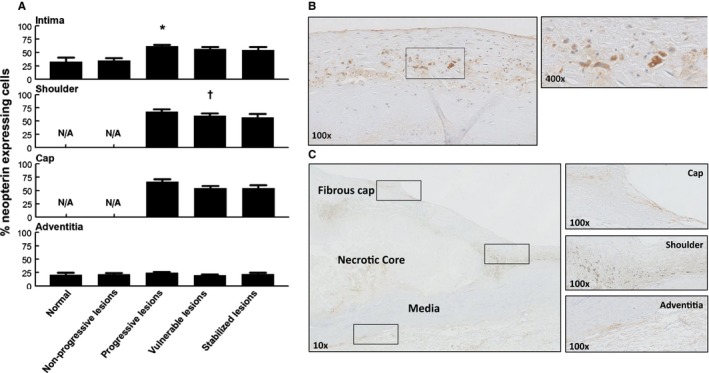
Neopterin expression during aortic atherosclerosis. A, Mean percentage of neopterin (intima, media, and adventitia) based on lesion morphology. Neopterin expression is minimal in early atherosclerosis, but increases during progressive disease (*P<0.0007; compared to nonprogressive lesions); afterward, remains relatively unchanged for stable healed plaque ruptures and fibrocalcific plaques. There is a small, but significant, decrease in neopterin expression within the shoulder regions of vulnerable plaques († P<0.043; compared to progressive lesions). Adventitial neopterin staining is relatively minimal and stable throughout all lesion morphologies. Spearman's rho correlation coefficient is not significant in the intima, shoulder, cap, and adventitia. B, Representative image of an intimal xanthoma stained for neopterin with a high‐resolution image at a ×400 magnification. Neopterin expression is seen in areas containing macrophage foam cells. C, Representative image of a healed rupture stained for neopterin with high‐resolution details of the cap, shoulder, and adventitia at a ×100 magnification. Neopterin expression is present within the various areas of the atherosclerotic lesion. Total number of cases in (A): 102 (normal 7, nonprogressive lesions 23, progressive lesions 35, vulnerable lesions 22, and stabilized lesions 15). Large solid bars in (A) represent the mean percentage of neopterin expression within the aortic wall section per atherosclerotic phase±SEM. All sections were developed with diaminobenzidine (DAB) and counterstained with Mayer's hematoxylin. N/A indicates not applicable.
Mast cells (Figure 7) are also reported to express CD68 and may thus (partly) account for the CD68 double‐negative population. Tryptase/CD68 double staining indeed showed strong CD68 reactivity on mast cells, although given the sparse number of mast cells in the intimal layer, the overall effects are likely minimal, unlike the interpretation of the macrophage data for the media and adventitia where mast cells populations are more prominent (Figure 7E). NK cells (Figure 8), DCs (Figure 9), and neutrophils (Figure 10) are not identified in the intimal layer of normal aorta and nonprogressive lesions. Moreover, the NK cells and DCs that are seen in the media are located at the medial‐adventitial border. No eosinophils (EG2+) were identified in the normal and nonprogressive lesions.
Figure 7.
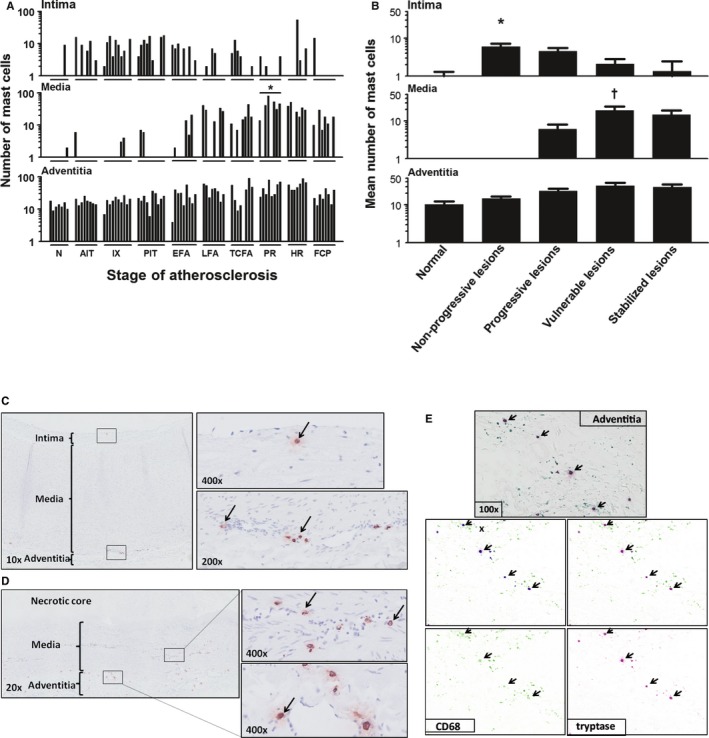
Mast cell (tryptase+) distribution during aortic atherosclerosis. A, Mast cells are constitutively present in the intima and adventitia in the aortic wall. The media practically remains devoid of mast cells until the progressive lesions. Significantly more mast cells are seen in the media of ruptured plaques (*P<0.01; compared to TCFA). B, The highest number of mast cells within the intima is present in the nonprogressive phase (P<0.01) while showing a gradual decrease with lesion progression achieving a minimum for stable fibrocalcific plaques. The media remains almost devoid of mast cells in the normal aorta's and in the nonprogressive lesions, but significantly increases in vulnerable plaques († P<0.0001; compared to progressive lesions) in contrast to adventitial mast cells, which remain relatively stable throughout the atherosclerotic process. C, Representative image of a normal aorta stained for tryptase with high‐resolution images of the intima (×400 magnification) and adventitia (×200 magnification), as indicated by black arrows. Note that adventitial mast cells are scattered and mainly located near vasa vasorum. D, Representative image of a late fibroatheroma with a large necrotic core stained for Tryptase at ×20 magnification with high‐resolution images of media and adventitia at a ×400 magnification. Mast cells are relatively more and remain in close proximity to the vasa vasorum. E, Representative image of the adventitia of an intimal xanthoma double stained for CD68 (Vina Green) and tryptase (Warp Red). CD68‐positive mast cells are identified (arrows), and using the Nuance multispectral imaging system FX, the colocalization can be separately analyzed. Total number of cases in (A and B): 83—normal 7, nonprogressive lesions 18 (viz AIT [8] and IX [10]), progressive lesions 27 (viz PIT [10], EFA [9], and LFA [8]), vulnerable lesions 16 (viz TCFA [8] and PR [8]), and stabilized lesions 15 (viz HR [7] and FCP [8]). In (A), the vertical axis of the intima, media, and adventitia is presented as a log scale. Each solid bar in (A) represents the number of positively stained mast cells within the intima, media, and adventitia of 1 aortic plaque. Large solid bars in (B) represent the mean total number of mast cells within the entire aortic wall per atherosclerotic phase±SEM. For abbreviations and a detailed description concerning the classification, see the Material and Methods section. All sections were developed with diaminobenzidine (DAB) and counterstained with Mayer's hematoxylin. AIT indicates adaptive intimal thickening; EFA, early fibroatheroma; FCP, fibrotic calcified plaque; HR, healed rupture; IX, intimal xanthoma; LFA, late fibroatheroma; N, normal; PIT, pathological intimal thickening; PR, plaque rupture; TCFA, thin cap fibroatheroma.
Figure 8.
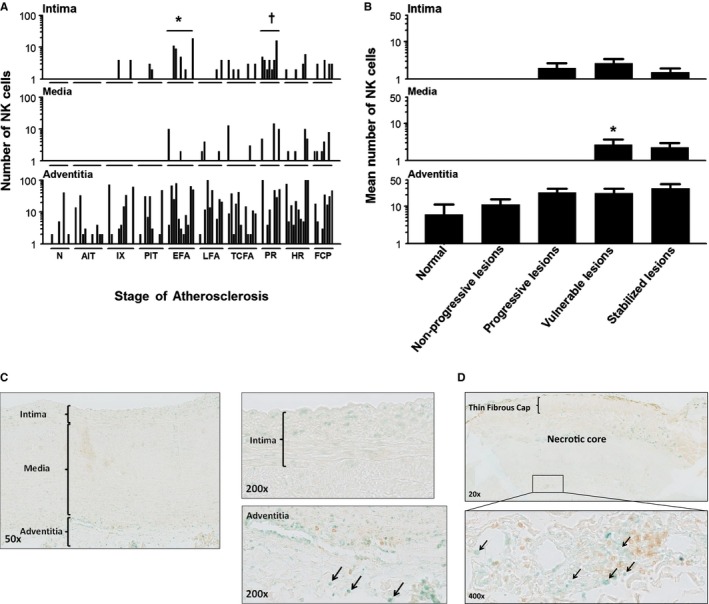
Natural killer (NK) cell (T‐bet+/CD4−) distribution during aortic atherosclerosis. A, Significantly more NK cells are seen in the intima in EFA (*P<0.01; compared to PIT) and in ruptured plaques († P<0.011; compared to TCFA). The media practically remains devoid of NK cells in progressive atherosclerosis. NK cells are constitutively and similarly present in the adventitia throughout the disease process. B, NK cells are minimally present in the aortic intima and media. NK cells are largely confined to the adventitia and the medial‐adventitial border zone, and the number of NK cells increases during the atherosclerotic process. A small, but significant, increase in NK cells in the media is seen in the vulnerable phase (viz TCFA and PR). (*P<0.001; compared to progressive lesions). Spearman's rho correlation coefficient is not significant in the intima, media, and adventitia. C, Illustrative images of a nonprogressive lesion (AIT) showing T‐Bet+/CD4– cells (methylgreen; black arrows). Note T‐helper cells (CD4+ single positive) cells in the adventitia (brown; diaminobenzidine chromogen) and the T‐helper 1 cells (CD4 and T‐bet double‐positive cells). D, Representative low‐power image of a TCFA dual immunostained for CD4/T‐bet with a high‐resolution image of the adventitia at ×20 magnification. Note the predominant adventitial location of NK cells (Vinagreen; black arrows). The lesion is a consecutive slide from the Movat and CD68 shown in Figure 3B. Total number of cases in (A and B): 100—normal 8, nonprogressive lesions 23 (viz AIT [12] and IX [11]), progressive lesions 31 (viz PIT [10], EFA [11] and LFA [10]), vulnerable lesions 20 (viz TCFA [12] and PR [8]), and stabilized lesions 18 (viz HR [10] and FCP [8]). Each solid bar in (A) represents the number of positively stained NK cells within the intima, media, and adventitia of a single lesion whereas the large solid bars in (B) represent the mean total number of NK cells within the aortic wall section per atherosclerotic phase±SEM. For abbreviations and a detailed description concerning the classification, see the Material and Methods section. All sections were developed with Vina green and diaminobenzidine (DAB) and counterstained with Mayer's hematoxylin. AIT indicates adaptive intimal thickening; EFA, early fibroatheroma; FCP, fibrotic calcified plaque; HR, healed rupture; IX, intimal xanthoma; LFA, late fibroatheroma; N, normal; PIT, pathological intimal thickening; PR, plaque rupture; TCFA, thin cap fibroatheroma.
Figure 9.
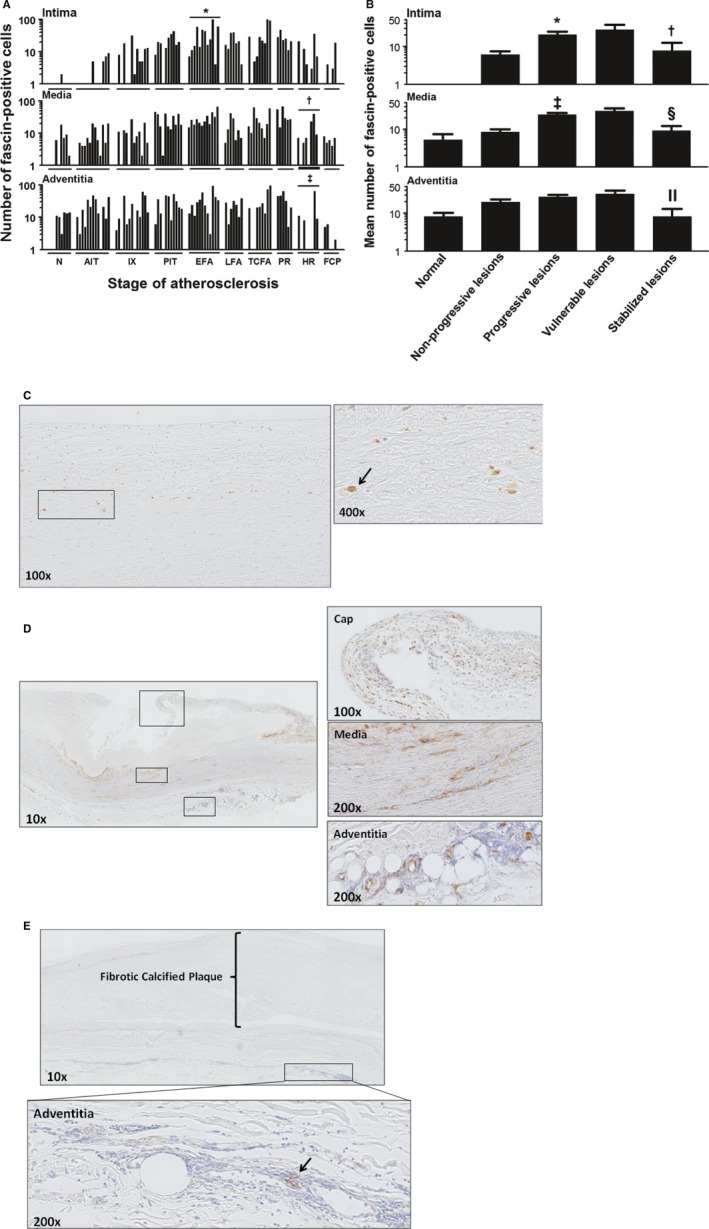
Dendritic cell (DC; fascin+) distribution during aortic atherosclerosis. A, DCs are mainly localized to the adventitia near the medial border and gradually increase in number with lesion progression. DCs are minimally present in the normal intima and intima of nonprogressive lesions and significantly more DCs are seen in EFA (*P<0.046; compared to PIT). On the contrary, significantly less DCs are seen in stabilizing lesions († P<0.001 and ‡ P<0.029; compared to PR). B, The amount of DCs in the intima and media increases significantly in the progressive phase (*P<0.0004; ‡ P<0.0001; compared to nonprogressive lesions). In postruptured stabilized atherosclerotic lesions, the number of fascin‐positive cells decrease in all the vascular layers († P<0.015, § P<0.0004, and ║ P<0.002; compared to vulnerable lesions). C, Representative image (×100) of a nonprogressive lesion (IX) stained for fascin. Only a few DCs are identified in the deeper intima. D, Representative image (×20) of TCFA stained for fascin. This example of a TCFA is from a consecutive slide as the example seen in Figure 6C. Note the abundance of fascin‐positive cells in the intima, media, and adventitia. E, Illustrative image (×10) of a FCP stained for fascin with a high‐resolution image of the adventitia (×200) showing a relative decrease in fascin‐positive cells in all vascular layers. Total number of cases in (A and B): 92—normal 8, nonprogressive lesions 26 (viz AIT [13] and IX [13]), progressive lesions 30 (viz PIT [11], EFA [12] and LFA [7]), vulnerable lesions 15 (viz TCFA [9] and PR [6]), and stabilized lesions 13 (viz HR [8] and FCP [5]). The vertical axis of the intima, media, and adventitia in (A and B) is presented as a log‐scale. Each solid bar in (A) represents the number of fascin‐positive cells within the intima, media, and adventitia of 1 aortic plaque. The large solid bars in (B) represent the mean total number of fascin‐positive cells within the aortic wall section per atherosclerotic phase±SEM. For abbreviations and a detailed description concerning the classification, see the Material and Methods section. All sections were developed with diaminobenzidine (DAB) and counterstained with Mayer's hematoxylin. AIT indicates adaptive intimal thickening; EFA, early fibroatheroma; FCP, fibrotic calcified plaque; HR, healed rupture; IX, intimal xanthoma; LFA, late fibroatheroma; N, normal; PIT, pathological intimal thickening; PR, plaque rupture; TCFA, thin cap fibroatheroma.
Figure 10.
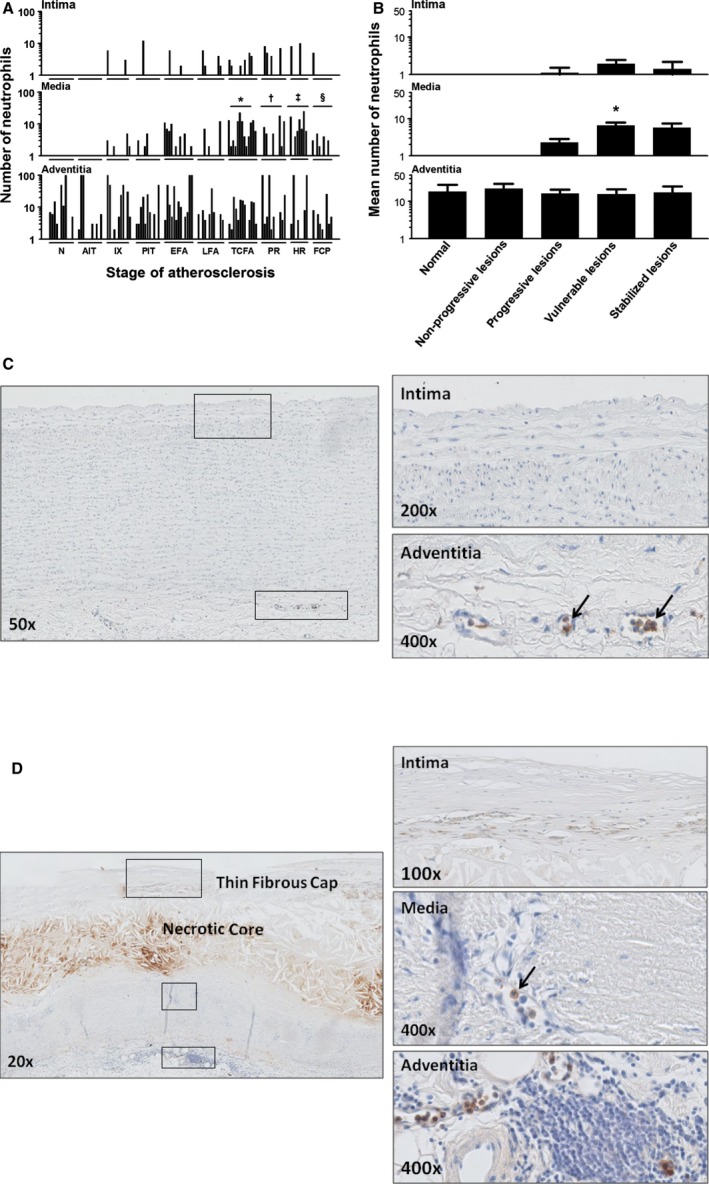
Neutrophil (myeloperoxidase+) distribution during aortic atherosclerosis. A, Minimal presence of intimal neutrophils during the atherosclerotic process. There are significantly more medial located neutrophils in TCFA and PR (*P<0.0002; † P<0.033; compared to LFA and TCFA, respectively) and healing ruptures (‡ P<0.007; compared to PR) and significantly less in FCP (§ P<0.0002; compared to HR). B, Overall, the mean amount of neutrophils within the media significantly increase in vulnerable lesions (*P<0.0005; compared to progressive lesions). Spearman's rho correlation coefficient is not significant in the intima and adventitia. C, Illustrative image (×50) of a normal aorta stained for myeloperoxidase (MPO) with high‐resolution images of the intima (×200) and the adventitia (×400). Identified neutrophils are all intravascular located in the vaso vasorum (black arrows). D, Representative image of a TCFA stained for MPO with a high‐resolution image of the intima, media, and adventitia. This example of a TCFA is again a consecutive section of the Movat and CD68 provided in Figures 3B and 8D. Neutrophils in the media and adventitia are located within the infiltrating vaso vasorum (black arrows). Total number of cases in (A and B): 108—normal 11, nonprogressive lesions 22 (viz AIT [11] and IX [11]), progressive lesions 35 (viz PIT [11], EFA [13] and LFA [11]), vulnerable lesions 24 (viz TCFA [13] and PR [11]), and stabilized lesions 16 (viz HR [7] and FCP [9]). The vertical axis of the intima, media, and adventitia in (A and B) is presented as a log scale. Each solid bar in (A) represents the number of neutrophils within the intima, media, and adventitia of one aortic plaque. Large solid bars in (B) represent the mean total number of neutrophils within the aortic wall section per atherosclerotic phase±SEM. For abbreviations and a detailed description concerning the classification, see the Material and Methods section. All sections were developed with diaminobenzidine (DAB) and counterstained with Mayer's hematoxylin. AIT indicates adaptive intimal thickening; EFA, early fibroatheroma; FCP, fibrotic calcified plaque; HR, healed rupture; IX, intimal xanthoma; LFA, late fibroatheroma; N, normal; PIT, pathological intimal thickening; PR, plaque rupture; TCFA, thin cap fibroatheroma.
Progressive atherosclerotic lesions
Progressive lesions of PIT, EFA, and LFA are characterized by loss of integrity of the intimal medial border zone, as hallmarked by increasing ApoB100 infiltration in the inner media layer (Figure 2C), along with a major increase in the macrophage content both in the intima and adjacent media (Figures 3 and 4). In parallel, we observed increased levels of macrophage activation, as indicated by HLA‐DR and neopterin expression. The percentage of M1 macrophages increased in late fibroatheroma (≈27%) and slightly decreased, as the lesions progressed toward a vulnerable TCFA or PR. On the other hand, M2 macrophages slightly increased as early and late fibroatheromas (≈13%) progressed toward a TCFA (≈17%) whereas the number of monocytes and macrophages double positive for the M1 and M2 marker remained stable (Figure 4A and 4B). CD68+ cells negative for M1 and M2 markers slightly tended to increase in number as lesions progressed toward PR, an observation that may (partly) reflect a major increase in the number of mast cells within the adventitia (Figure 7).
Progressive numbers of NK cells were found within the media and adventitia of these more‐advanced lesions (Figure 8). Fascin‐positive cells significantly increased in number in the intima and media (*P<0.0004 and ‡ P<0.0001) and remained stable within the adventitia.
Only 1 or 2 neutrophils were present in the intima (≈8% of total) and the neutrophils identified in the media were located intravascular in the infiltrating vasa vasorum (Figure 10B and 10C).
Vulnerable atherosclerotic lesions
TCFAs and PRs were characterized by a further increase in macrophage content in the intima and media, but the M1/M2 ratio and level of macrophage activation (HLA‐DR and neopterin) remained unchanged when compared to stable lesions (Figure 4). Increased numbers of mast cells and NK cells and fascin‐positive cells were found in all the vascular layers. Increasing numbers of intravascular neutrophils were observed in the media and absent within the neointima and areas outside vessels. Overall, eosinophils were absent in the progressive and vulnerable stages.
Healed and stabilized lesions
HRs and FCPs associated with vessel stabilization showed a marked reduction in the number of macrophages and decreased in cell activation, as indicated by reduced HLA‐DR positivity and intimal neopterin expression. M1 macrophages increased in number whereas the M2 lineage remained stable. NK cells, DCs, and neutrophils significantly decreased in number as lesions stabilized. Eosinophils remained absent.
Discussion
To a large extent, murine studies have emphasized the pivotal role of the innate immune system in the development and progression of atherosclerosis,11 although there are fundamental metabolic, inflammatory, and immunological differences that exist between various mouse strains and, more important, between mice and humans. As such, definitive evidence on how observations of lipoprotein oxidation, inflammation, and immunity translate to human atherosclerotic disease is still lacking.24 Consequently, it has been pointed out that studies in the human atherosclerotic process are essential to ensure the data obtained from experimental murine atherosclerosis models are relevant.17, 25 This explorative study is based on histological observations from a unique biobank of human aorta, which allows evaluation of the full spectrum of atherosclerotic disease.
Our findings confirm a predominance of macrophages. Multiplex staining for M1/M2 macrophages failed to show a significant predominance in M1 macrophages in the foam cell‐rich areas, which is inconsistent with a dichotomous classification system for macrophage phenotype differentiation,26 as has been described for murine models where M2 macrophages dominate the later stages of atherosclerotic disease. Moreover, quantitative findings for mast cells, NK cells, eosinophils, and neutrophils do not imply these cell types as critical contributors to plaque destabilization.
LDL and Innate Immunity in Lesion Progression
Accumulation of LDL fragments is the primary trigger for the atherosclerotic process and is directly linked to activation and perpetuation of the innate immune response.27 In the present study, ApoB100 was used to visualize proatherogenic lipoprotein particles mainly consistent with LDL accumulation, although antibody staining is not specific for oxidation products. In an earlier study, we extensively reported on oxidation specific epitopes, ApoB100 and apo(a), in a wide range of human coronary and carotid atherosclerotic lesions and demonstrated that plaques in early atherosclerotic lesions are enriched in ApoB100.28
ApoB100 expression was limited to the intimal layer in aortic wall samples classified as normal, adaptive intimal thickening, and IX. On the contrary, transition to pathological intimal thickening, the earliest form of progressive atherosclerosis, is characterized by accumulation of ApoB100 within the medial wall, which, in fact, is much earlier than previously reported.29 Demonstration that the transition to irreversible atherosclerosis is associated with a loss of internal elastic lamina integrity hallmarks the beginning of medial and adventitial involvement in atherosclerosis, a phenomenon that warrants further investigation.30, 31
Macrophage Subtypes
Macrophages play a critical role in innate and acquired immunity and are, in varying degrees, involved in all phases of atherosclerotic lesion development, particularly in regard to lipid core formation, lesion remodeling, and degradation of the fibrous cap, as observed in advanced symptomatic disease.32 Macrophage accumulation in the intimal layer of the vessel wall is observed in early, nonprogressive atherosclerotic lesions and is thought to be essentially driven by accumulation of oxidized LDL.33, 34 In the present study, the overall number of CD68‐positive macrophages increased with advancing disease, in concordance with previous studies. Among the various lesion types, macrophage infiltration was most prominent in TCFAs, particularly in regions of the fibrous cap, and least for HRs and FCPs.19 It is generally assumed that macrophages, through release of matrix metalloproteinases, directly contribute to plaque destabilization.7, 32 The overall amount of lesional macrophages as recognized in the present study, however, is less pronounced compared with murine atherosclerosis7 and affords a different perspective on the complexity of human plaques,19 particularly in reference to animal models.
Contrary to murine atherosclerosis, we found no evidence of a macrophage subclass shift during initiation, progression, rupture, and ultimate stabilization of human atherosclerotic lesions. Overall, ≈20% to 30% of macrophages in early to advanced plaques are double positive for both M1 and M2 markers, challenging the paradigm of a clear M1 to M2 dichotomy. This finding was further confirmed using alternative M1 and M2 markers (IL‐6/dectin‐1 and CD197/CD206), respectively, in combination with CD68 to exclude the possibility that the iNOS/CD163/CD68 combination was inadequate in identifying all M1/M2 macrophages. Moreover, ≈40% of the intimal macrophages were double negative for iNOS/CD163 or alternatively, IL‐6/dectin‐1. CD68 was equally expressed in tryptase‐positive mast cells, which may partially contribute to macrophages double negative for M1 or M2 markers.
Taken together, a simple dichotomous classification scheme as proposed for macrophage differentiation in mice falls short in the biological context and complexity of human disease. Clearly, more than 2 subtypes of macrophages with presumably functionally different roles might be involved in human atherosclerosis.8 As mentioned, the current immunostaining techniques likely fall short in identifying known and unknown macrophage subsets in human disease for lack of specificity. Single‐cell analyses could provide invaluable insights into studying macrophage heterogeneity, but is technically challenging and beyond the scope of this article. Along these same lines, a functional equivalence to murine macrophages remains unclear where caution is warranted with directly extrapolation of findings from mouse to humans or vice versa.
DCs
DCs are professional antigen‐presenting cells (APCs) and constitute a unique interface between innate and adaptive immune systems.35 The identification of DCs in human and mouse vascular wall has stimulated interest in the role of these cells in the pathogenesis of several acute and chronic vascular disorders, including atherosclerosis.36 How DCs influence the initiation and progression of atherosclerosis remains unclear. On basis of their pivotal role in antigen presentation, activating T cells, and secreting cytokines, and perhaps also their ability to become foam cells, it has been suggested that DCs are critically involved in the pathogenesis of atherosclerosis.37, 38
Unfortunately, attempts to visualize DC subtype markers (CD80, CD86, DC lamp, and DC sign) failed on formalin‐fixed, paraffin‐embedded material, as demonstrated in a previous study.39 As such, DCs could only be recognized by the general DC marker, fascin, which has been shown to correlate with DC maturation into APCs.40
There was a strong positive relationship between progressive disease and fascin‐positive DCs in all vascular layers. The accumulation of DCs within atherosclerosis‐prone areas of normal aorta further supports the involvement of immune mechanisms from very early stages of the disease.41 DCs residing in the media and adventitia were mainly located near the infiltrating vaso vasorum and adventitial infiltrates of T cells. DCs are known to patrol this region of the plaque, sampling antigens and presenting them to T lymphocytes. Our previous study regarding the adaptive immune response in human atherosclerosis identified such organized tertiary lymphoid structures in the adventitia.22
The current observations indicate a prominent role of DCs in human atherosclerosis. However, different roles for DCs in atherogenesis or plaque progression have been identified in mouse models whereas a clear attribution to DCs and the exact molecular mechanisms engaged remain unresolved. Identification of human homologs of mouse DC subsets and further work in this area will aid to promote translational approaches of single‐cell studies.
Mast Cells
Abundance of mast cells in the shoulder region of human coronary atheromas spurred interest in a possible role of these cells in the atherosclerotic process.42 Although a role for mast cells is supported by a rodent studies, a role in human atherosclerosis remains unclear with the current knowledge.43, 44
Mast cells were most prominent in the media and adventitial layers and were independent of the phase of atherosclerosis. Similarly, intimal mast cell infiltration constitutes an early event in the atherosclerotic process, which subsequently plateaus. Specifically, our data do not imply an association between mast cell content and plaque destabilization and, on the contrary, suggest an apparent decrease in progressive and vulnerable atherosclerotic lesions. Moreover, we did not find extracellular mast cell granules associated with fibrous cap thinning, in contrary to the proposed active participation in substantial degradation of the extracellular matrix and local weakening of inflamed atherosclerotic lesions, a situation well documented in diseases affecting the connective tissue.45, 46, 47 Altogether, these findings fail to point to a direct relationship between intimal mast cell content and cap destabilization.
Considering medial and adventitial mast cells were typically located within, or in close proximity to, the infiltrating vaso vasorum, it is conceivable that these cells are involved in plaque neovascularization in advancing and vulnerable atherosclerotic lesions. Yet, an alternative explanation would be that mast cells enter through, and home in the proximity of, the infiltrating vaso vasorum.26, 36 Accumulation of mast cells in shoulder regions of human coronary atheromas may be similarly secondary to neovascularization. Overall, these observational data neither support nor refute a role for mast cell in progression and complications of atherosclerosis. Such a role can only be assessed in clinical studies. Although aggressive immunosuppression for abdominal aortic aneurysms appeared to have no effect on resident mast cells, including macrophages and smooth muscle cells, a similar study on atherosclerosis related endpoints is urgently missing.48
NK Cells
NK cells are cytotoxic lymphocytes that are part of the innate immune system. Apart from their cytotoxic action, they are an important early source of interferon‐γ and tumor necrosis factor in response to acute injury and infections. NK cells can prime macrophages to secrete proinflammatory cytokines and play a key role in viral defense, tumor surveillance, and tolerance during pregnancy.49 In the context of atherosclerosis, it is demonstrated in vitro that NK cells infiltrate the vessel wall and promote atherosclerotic lesion development in murine models of the disease.50 Along these lines, deficiency of functional NK cells has been found to reduce lesion size.51 At this point, a role of NK cells in clinical atherosclerosis still remains unclear.
We tested several classical NK cell markers (CD56, NKp30, and the PEN5 epitope) in order to visualize NK cells, yet all these markers failed on formalin‐fixed, paraffin‐embedded material and therefore selected an alternative strategy of applied double staining for the T‐helper 1 (Th1)/NK cell‐specific transcription factor, T‐bet, in combination with CD4 staining. CD4−/T‐bet+ cells classified as “NK cells” were abundant in abdominal aortic aneurysm tissue, but minimal in atherosclerotic samples. The few NK cells present in the intima were most common to advanced and vulnerable stages of atherosclerosis. Whereas CD56 immunostaining clearly identified aortic ganglia (and NK cells in tumor tissue), samples of aortic atherosclerosis were negative. In essence, NK cells failed to present as a major local factor contributing to human atherosclerotic disease despite previous indications suggesting marginally decreased systemic NK cell activity in advanced atherosclerosis in the elderly.52
Neutrophils
Neutrophils are emerging as a potential new player in the atherosclerotic process primarily based on murine studies, suggesting their involvement in the initiation of the atherosclerotic process, as well as plaque angiogenesis in later stages of the disease.53, 54 Moreover, neutrophil infiltration at the level of a disrupted plaque could potentially contribute to plaque progression attributed to release high amounts of reactive oxygen species and the pro‐oxidant enzyme, MPO.55 Yet, a role for neutrophils in human atherogenesis, atheroprogression, and atherosclerotic plaque destabilization is still unclear based on available data.56
Selective immunostaining suggested that neutrophils were mainly present in the vasa vasorum, or in the posthemorrhagic hematoma attributed to surgical manipulation where the specific neutrophil markers, MPO or MMP8, failed to identify tissue‐infiltrating neutrophils with no association of atherosclerotic progression or plaque vulnerability. The failure to detect neutrophil infiltrates within atherosclerotic tissue, however, may be related to the short half‐life of these cells, although our earlier protein‐based studies showed negligible MMP8 and chemokine (C‐X‐C motif) ligand 8 expression, thus indicating an absence of neutrophil remnants within the atherosclerotic aorta wall.57, 58 To summarize, these observational data do not support a primary role of neutrophils in human atherosclerosis, but rather indicates that neutrophil presence is related to surgical manipulation of the aorta during removal.59
Eosinophils
Eosinophils were fully absent during the entire process of human atherosclerosis.
Limitations
Numerous inflammatory cell phenotypes identified within well‐defined morphological stages of atherosclerosis emphasize the complexity of innate immune response and atherosclerotic disease. This study was performed on aortic sections of deceased individuals, where continuous data represent incidental findings from a large series of patients and therefore may not necessarily reflect longitudinal data. Another limitation of the study is the fact all the findings are based on immunohistochemistry using paraffin‐embedded tissue sections, which precludes use of other relevant and confirmatory markers, particularly those for dendritic cells. Although immunohistochemistry has the advantage of showing the spatial relationships, multiple staining is challenging and allows only limited marker sets. Consequently, findings in this study should be considered in this context. Moreover, differences in plaque inflammation may also exist among various vascular beds, although our previous work clearly shows that aortic atherosclerosis follow a similar pattern of disease progression as for coronary tissue.19, 20, 21 As such, we cannot exclude that the findings for the aorta may not fully translate to the coronary and other vascular beds. Furthermore, macrophage subtyping relies on the M1 and M2 submarker. A more functional classification is missing, such as the Mox macrophages.60 We observed a large number of double M1/M2‐positive macrophages. In this observational study, we are unable to distinguish between a real “M1 to M2” phenotype and/or phagocytosis of M1 macrophages by M2 macrophages and vice versa.
In conclusion, the presence of various macrophage subsets and fascin‐positive DCs are strongly associated with atherosclerotic disease of human aorta, particularly for progressive and vulnerable fibroatheromatous plaques. A less‐prominent role, however, is suggested for mast cells and NK cells, whereas a direct relation with neutrophils and eosinophils was not established. More interestingly, macrophage heterogeneity is far more complex than the current paradigm predicated on murine data and supports the involvement of additional (poorly defined) macrophage subtypes or perhaps a greater dynamic range of macrophage plasticity. Clearly, there is a divergence in observational data between murine models of atherosclerosis from our human atherosclerosis studies, which point to a limited translational aspect of animal findings.
Sources of Funding
This study was, in part, funded by the European Commission (Cartardis, FP7 HEALTH.2013.2.4.2‐1).
Disclosures
None.
(J Am Heart Assoc. 2016;5:e002860 doi: 10.1161/JAHA.115.002860)
References
- 1. Fernández‐Velasco M, González‐Ramos S, Boscá L. Involvement of monocytes/macrophages as key factors in the development and progression of cardiovascular diseases. Biochem J. 2014;458:187–193. [DOI] [PubMed] [Google Scholar]
- 2. Douaiher J, Succar J, Lancerotto L, Gurish MF, Orgill DP, Hamilton MJ, Krilis SA, Stevens RL. Development of mast cells and importance of their tryptase and chymase serine proteases in inflammation and wound healing. Adv Immunol. 2014;122:211–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ma Y, Yabluchanskiy A, Lindsey ML. Neutrophil roles in left ventricular remodeling following myocardial infarction. Fibrogenesis Tissue Repair. 2013;6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hansson GK, Libby P, Schönbeck U, Yan ZQ. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res. 2002;91:281–291. [DOI] [PubMed] [Google Scholar]
- 5. Peled M, Fisher EA. Dynamic aspects of macrophage polarization during atherosclerosis progression and regression. Front Immunol. 2014;5:579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang XP, Zhang W, Liu XQ, Wang WK, Yan F, Dong WQ, Zhang Y, Zhang MX. Arginase I enhances atherosclerotic plaque stabilization by inhibiting inflammation and promoting smooth muscle cell proliferation. Eur Heart J. 2014;35:911–919. [DOI] [PubMed] [Google Scholar]
- 7. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chinetti‐Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 2015;12:10–17. [DOI] [PubMed] [Google Scholar]
- 9. Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, Elliott MR, Gruber F, Han J, Chen W, Kensler T, Ravichandran KS, Isakson BE, Wamhoff BR, Leitinger N. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res. 2010;107:737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johnson JL, Newby AC. Macrophage heterogeneity in atherosclerotic plaques. Curr Opin Lipidol. 2009;20:370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. [DOI] [PubMed] [Google Scholar]
- 12. Martinez FO, Helming L, Milde R, Varin A, Melgert BN, Draijer C, Thomas B, Fabbri M, Crawshaw A, Ho LP, Ten Hacken NH, Cobos Jiménez V, Kootstra NA, Hamann J, Greaves DR, Locati M, Mantovani A, Gordon S. Genetic programs expressed in resting and IL‐4 alternatively activated mouse and human macrophages: similarities and differences. Blood. 2013;121:e57–e69. [DOI] [PubMed] [Google Scholar]
- 13. Bryant CE, Monie TP. Mice, men and the relatives: cross‐species studies underpin innate immunity. Open Biol. 2012;2:120015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Colucci F, Di Santo JP, Leibson PJ. Natural killer cell activation in mice and men: different triggers for similar weapons? Nat Immunol. 2002;3:807–813. [DOI] [PubMed] [Google Scholar]
- 15. Ylä‐Herttuala S, Bentzon JF, Daemen M, Falk E, Garcia‐Garcia HM, Herrmann J, Hoefer I, Jukema JW, Krams R, Kwak BR, Marx N, Naruszewicz M, Newby A, Pasterkamp G, Serruys PW, Waltenberger J, Weber C, Tokgözoglu L. Stabilisation of atherosclerotic plaques. Position paper of the European Society of Cardiology (ESC) Working Group on Atherosclerosis and Vascular Biology. Thromb Haemost. 2011;106:1–19. [DOI] [PubMed] [Google Scholar]
- 16. Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald‐Smith GP, Gao H, Hennessy L, Finnerty CC, López CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller‐Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2013;110:3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–2738. [DOI] [PubMed] [Google Scholar]
- 18. Jiang X, Shen C, Yu H, Karunakaran KP, Brunham RC. Differences in innate immune responses correlate with differences in murine susceptibility to Chlamydia muridarum pulmonary infection. Immunology. 2010;129:556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van Dijk RA, Virmani R, von der Thüsen JH, Schaapherder AF, Lindeman JH. The natural history of aortic atherosclerosis: a systematic histopathological evaluation of the peri‐renal region. Atherosclerosis. 2010;210:100–106. [DOI] [PubMed] [Google Scholar]
- 20. Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;10:1262–1275. [DOI] [PubMed] [Google Scholar]
- 21. Yahagi K, Kolodgie FD, Otsuka F, Finn AV, Davis HR, Joner M, Virmani R. Pathophysiology of native coronary, vein graft, and in‐stent atherosclerosis. Nat Rev Cardiol. 2016;13:79–98. [DOI] [PubMed] [Google Scholar]
- 22. van Dijk RA, Duinisveld AJ, Schaapherder AF, Mulder‐Stapel A, Hamming JF, Kuiper J, de Boer OJ, der van Wal AC, Kolodgie FD, Virmani R, Lindeman JH. A change in inflammatory footprint precedes plaque instability: a systematic evaluation of cellular aspects of the adaptive immune response in human atherosclerosis.J Am Heart Assoc. 2015;4:e001403 doi: 10.1161/JAHA.114.001403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van der Loos CM. Multiple immunoenzyme staining: methods and visualizations for the observation with spectral imaging. J Histochem Cytochem. 2008;56:313–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen YC, Peter K. Determining the characteristics of human atherosclerosis: a difficult but indispensable task providing the direction and proof of concept for pioneering atherosclerosis research in animal models. Atherosclerosis. 2015;241:595–596. [DOI] [PubMed] [Google Scholar]
- 25. Watanabe H, Numata K, Ito T, Takagi K, Matsukawa A. Innate immune response in Th1‐ and Th2‐dominant mouse strains. Shock. 2004;22:460–466. [DOI] [PubMed] [Google Scholar]
- 26. David MM, Justin PE. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yan ZQ, Hansson GK. Innate immunity, macrophage activation, and atherosclerosis. Immunol Rev. 2007;219:187–203. [DOI] [PubMed] [Google Scholar]
- 28. van Dijk RA, Kolodgie F, Ravandi A, Leibundgut G, Hu PP, Prasad A, Mahmud E, Dennis E, Curtiss LK, Witztum JL, Wasserman BA, Otsuka F, Virmani R, Tsimikas S. Differential expression of oxidation‐specific epitopes and apolipoprotein(a) in progressing and ruptured human coronary and carotid atherosclerotic lesions. J Lipid Res. 2012;53:2773–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moreno PR, Purushothaman KR, Fuster V, O'Connor WN. Intimomedial interface damage and adventitial inflammation is increased beneath disrupted atherosclerosis in the aorta: implications for plaque vulnerability. Circulation. 2002;105:2504–2511. [DOI] [PubMed] [Google Scholar]
- 30. Libby P, Hansson GK. Inflammation and immunity in diseases of the arterial tree: players and layers. Circ Res. 2015;116:307–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Otsuka F, Kramer MCA, Woudstra P, Yahagi K, Ladich E, Finn AV, de Winter RJ, Kolodgie FD, Wight TN, Davis HR, Joner M, Virmani R. Natural progression of atherosclerosis from pathologic intima thickening to late fibroatheroma in human coronary arteries: a pathology study. Atherosclerosis. 2015;241:772–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47:C13–C18. [DOI] [PubMed] [Google Scholar]
- 33. Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999;340:115–126. [DOI] [PubMed] [Google Scholar]
- 34. Glass CK, Witztum JL. Atherosclerosis the road ahead. Cell. 2001;104:503–516. [DOI] [PubMed] [Google Scholar]
- 35. Bobryshev YV. Dendritic cells in atherosclerosis: current status of the problem and clinical relevance. Eur Heart J. 2005;26:1700–1704. [DOI] [PubMed] [Google Scholar]
- 36. Choi JH, Do Y, Cheong C, Koh H, Boscardin SB, Oh YS, Bozzacco L, Trumpfheller C, Park CG, Steinman RM. Identification of antigen‐presenting dendritic cells in mouse aorta and cardiac valves. J Exp Med. 2009;206:497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hansson GK, Libby P. The immune response in atherosclerosis: a double‐edged sword. Nat Rev Immunol. 2006;6:508–519. [DOI] [PubMed] [Google Scholar]
- 38. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. [DOI] [PubMed] [Google Scholar]
- 39. Van Vré EA, Bosmans JM, Van Brussel I, Maris M, De Meyer GR, Van Schil PE, Vrints CJ, Bult H. Immunohistochemical characterisation of dendritic cells in human atherosclerotic lesions: possible pitfalls. Pathology. 2011;43:239–247. [DOI] [PubMed] [Google Scholar]
- 40. Al‐Alwan MM, Rowden G, Lee TD, West KA. Fascin is involved in the antigen presentation activity of mature dendritic cells. J Immunol. 2001;166:338–345. [DOI] [PubMed] [Google Scholar]
- 41. Zernecke A. Dendritic cells in atherosclerosis: evidence in mice and humans. Arterioscler Thromb Vasc Biol. 2015;35:763–770. [DOI] [PubMed] [Google Scholar]
- 42. Holmes DR Jr, Savage M, LaBlanche JM, Grip L, Serruys PW, Fitzgerald P, Fischman D, Goldberg S, Brinker JA, Zeiher AM, Shapiro LM, Willerson J, Davis BR, Ferguson JJ, Popma J, King SB III, Lincoff AM, Tcheng JE, Chan R, Granett JR, Poland M. Results of Prevention of REStenosis with Tranilast and its Outcomes (PRESTO) trial. Circulation. 2002;106:1243–1250. [DOI] [PubMed] [Google Scholar]
- 43. Spinas E, Kritas SK, Saggini A, Mobili A, Caraffa A, Antinolfi P, Pantalone A, Tei M, Speziali A, Saggini R, Conti P. Role of mast cells in atherosclerosis: a classical inflammatory disease. Int J Immunopathol Pharmacol. 2014;27:517–521. [DOI] [PubMed] [Google Scholar]
- 44. Kaartinen M, Penttilä A, Kovanen PT. Accumulation of activated mast cells in the shoulder region of human coronary atheroma, the predilection site of atheromatous rupture. Circulation. 1994;90:1669–1678. [DOI] [PubMed] [Google Scholar]
- 45. Bromley M, Fisher WD, Woolley DE. Mast cells at site of cartilage erosion in the rheumatoid joint. Ann Rheum Dis. 1984;43:76–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Loke P, Gallagher I, Nair MG, Zang X, Brombacher F, Mohrs M, Allison JP, Allen JE. Alternative activation is an innate response to injury that requires CD4+ T cells to be sustained during chronic infection. J Immunol. 2007;179:3926–3936. [DOI] [PubMed] [Google Scholar]
- 47. Coussens LM, Raymond WW, Bergers G, Laig‐Webster M, Behrendtsen O, Werb Z, Caughey GH, Hanahan D. Inflammatory mast cells up‐regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999;13:1382–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lindeman JH, Rabelink TJ, van Bockel JH. Immunosuppression and the abdominal aortic aneurysm: Doctor Jekyll or Mister Hyde?. Circulation. 2011;124:e463–e465. [DOI] [PubMed] [Google Scholar]
- 49. O'Shea JJ, Murray PJ. Cytokine signaling modules in inflammatory responses. Immunity. 2008;28:477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stöger JL, Gijbels MJ, van der Velden S, Manca M, van der Loos CM, Biessen EA, Daemen MJ, Lutgens E, de Winther MP. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis. 2012;225:461–468. [DOI] [PubMed] [Google Scholar]
- 51. Whitman SC, Rateri DL, Szilvassy SJ, Yokoyama W, Daugherty A. Depletion of natural killer cell function decreases atherosclerosis in low‐density lipoprotein receptor null mice. Arterioscler Thromb Vasc Biol. 2004;24:1049–1054. [DOI] [PubMed] [Google Scholar]
- 52. Bruunsgaard H, Pedersen AN, Schroll M, Skinhøj P, Pedersen BK. Decreased natural killer cell activity is associated with atherosclerosis in elderly humans. Exp Gerontol. 2001;37:127–136. [DOI] [PubMed] [Google Scholar]
- 53. Weber C, Zernecke A, Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol. 2008;8:802–815. [DOI] [PubMed] [Google Scholar]
- 54. Soehnlein O. Multiple roles for neutrophils in atherosclerosis. Circ Res. 2012;110:875–888. [DOI] [PubMed] [Google Scholar]
- 55. Drechsler M, Megens RT, van Zandvoort M, Weber C, Soehnlein O. Hyperlipidemia‐triggered neutrophilia promotes early atherosclerosis. Circulation. 2010;122:1837–1845. [DOI] [PubMed] [Google Scholar]
- 56. Döring Y, Drechsler M, Soehnlein O, Weber C. Neutrophils in atherosclerosis: from mice to man. Arterioscler Thromb Vasc Biol. 2015;35:288–295. [DOI] [PubMed] [Google Scholar]
- 57. Lindeman JH, Abdul‐Hussien H, Schaapherder AF, Van Bockel JH, Von der Thüsen JH, Roelen DL, Kleemann R. Enhanced expression and activation of pro‐inflammatory transcription factors distinguish aneurysmal from atherosclerotic aorta: IL‐6‐ and IL‐8‐dominated inflammatory responses prevail in the human aneurysm. Clin Sci. 2008;114:687–697. [DOI] [PubMed] [Google Scholar]
- 58. Abdul‐Hussien H, Soekhoe RG, Weber E, der von Thüsen JH, Kleemann R, Mulder A, van Bockel JH, Hanemaaijer R, Lindeman JH. Collagen degradation in the abdominal aneurysm: a conspiracy of matrix metalloproteinase and cysteine collagenases. Am J Pathol. 2007;170: 809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. 2013;38:1092–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Leitinger N, Schulman IG. Phenotypic polarization of macrophages in atherosclerosis. Arterioscler Thromb Vasc Biol. 2013;33:1120–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]