

Abstract
Background
Reports on soluble interleukin‐6 (IL‐6) receptor (sIL‐6R) and glycoprotein 130 (sgp130) in ST‐elevation myocardial infarction (STEMI) are few and include a small number of patients. The aim of this study was to investigate the possible association between levels of these biomarkers in the acute phase of STEMI and future cardiovascular events.
Methods and Results
Circulating IL‐6, sgp130, sIL‐6R, and C‐reactive protein (CRP) were measured in 989 STEMI patients during 2007–2011, and cardiovascular events were recorded during follow‐up. The primary endpoint was composite of all‐cause mortality, myocardial infarction, stroke, unscheduled revascularization, or rehospitalization for heart failure. Cox regression models were used to estimate hazard ratios (HRs) for cardiovascular events in relation to biomarker levels. Median levels of sIL‐6R, sgp130, IL‐6, and CRP measured 24 hours (median) after symptom onset were 39.2 ng/mL, 240 ng/mL, 18.8 pg/mL, and 13.7 mg/L, respectively. During a median follow‐up time of 4.6 years, 200 patients (20.2%) experienced a primary endpoint, and 82 patients (8.3%) died. Patients with sIL‐6R levels in the upper quartile (>47.7 ng/mL) had significantly higher risk of future adverse events (primary endpoint) and mortality compared to patients with lower levels (adjusted HR, 1.54 [1.08, 2.21]; P=0.02 and 1.81 [1.04, 3.18]; P=0.04, respectively). Neither IL‐6 nor sgp130 levels were related to future events, but patients with CRP levels in the upper quartile (>31.5 mg/L) had higher risk of death.
Conclusion
High levels of sIL‐6R were associated with future cardiovascular events and mortality in STEMI patients, suggesting an important role of the IL‐6 signaling system.
Keywords: inflammation, interleukins, myocardial infarction, prognosis
Subject Categories: Biomarkers, Inflammation, Ischemia
Introduction
Inflammation in the vessel wall is associated with cardiovascular disease (CVD) and activation of the interleukin‐6 (IL‐6) signaling pathway has been shown to be important in the atherosclerotic process, plaque building and plaque destabilization.1, 2 Importantly, human genetic studies have suggested a causal association between IL‐6 receptor (IL‐6R) signaling and CVD, and IL‐6R blockade seems to be a possible therapeutic approach in these patients.3, 4 Interestingly, tocilizumab, a monoclonal antibody acting as an IL‐6R antagonist, is currently available for clinical use in patients with rheumatoid arthritis.5 Inflammation involving the IL‐6 pathway has also been shown to play a major role in the myocardial remodeling process occurring after an acute ST‐elevation myocardial infarction (STEMI), a process known to be important for heart failure development and long‐term mortality.6, 7
The associations between IL‐6 and C‐reactive protein (CRP), which is induced by IL‐6, and acute myocardial infarction (AMI)8, 9 and heart failure,10, 11, 12 respectively, are well described. Additionally, both markers have been associated with higher mortality in CVD patients as well as with later clinical events.9, 13, 14 In contrast, studies on the relatively novel members of the IL‐6 signaling cascade, soluble IL‐6R and soluble glycoprotein 130 (sgp130) in STEMI are few and include a small number of patients. Elevated sIL‐6R levels were reported in patients with AMI, compared to stable coronary artery disease (CAD) patients and healthy controls,15, 16 whereas no differences in sgp130 levels were found between AMI, CAD, and control patients.15
We have recently reported results on the associations between myocardial necrosis, left ventricular impairment, and levels of IL‐6, sgp130, sIL‐6R, and CRP in STEMI patients undergoing acute percutaneous intervention (PCI).17 The aim of the present study was to investigate possible associations between serum levels of members of the IL‐6 trans‐signaling system in the acute phase of STEMI and future clinical events, with special emphasis on sgp130 and sIL‐6R.
Methods
Study Population
This study was an observational cohort study of STEMI patients admitted to Oslo University Hospital Ullevål (Oslo, Norway), all treated with primary PCI. From June 2007 to August 2011, a total of 1026 patients with diagnosed STEMI were included during weekdays after written informed consent was obtained. Patients below age 18 years and patients unable (clinically unstable or sedated patients) or unwilling to give written informed consent were not included. Hospital records at admission and questionnaires were used for clinical information. The study was approved by the Regional Committee for Medical and Health Research Ethics.
STEMI was defined as electrocardiographic ST‐segment elevation ≥2 mm in 2 or more contiguous chest leads or ≥1 mm in 2 or more limb leads or new onset of left bundle‐branch block, together with chest pain or other typical symptoms and elevated troponin levels >99th percentile.
Laboratory Methods
Blood samples were collected at a median of 24 hours after symptom onset and 18 hours after the PCI procedure, between 8:00 and 10:00 am the following morning, except those with infarction during the weekend, who were included the following workday morning. In order to standardize blood sampling, all samples were taken after an overnight fast. Serum was prepared by centrifugation for 10 minutes at 2000g and samples were stored at −80°C until analyzed. Circulating levels of sgp130, sIL‐6R, and IL‐6 were determined by commercial ELISA (R&D Systems, Abingdon, UK) and CRP with kits from DRG Instruments (Marburg/Lahn, Germany). In our laboratory, the interassay coefficients of variation were 5.2%, 3.6%, 10.5%, and <5%, respectively. Routine blood samples were analyzed by use of conventional methods.
Left Ventricular Ejection Fraction
Left ventricular ejection fraction (LVEF) was measured by echocardiography ad Modum Simpson or by visual judgment during the index hospital stay or at the local hospital within 3 months after the index myocardial infarction. In case of rehospitalization between the index infarction and the examination, measurements of LVEF were excluded.
Definition of Clinical Endpoints
The primary endpoint was a composite of death, myocardial infarction, stroke, unscheduled revascularization ≥3 months after the index infarction, or rehospitalization for heart failure, whichever occurred first. The secondary endpoint was all‐cause mortality. Scheduled PCI treatment or coronary artery bypass grafting as a consequence of the index infarction or new coronary angiography without PCI were not registered as new clinical events.
Follow‐up
The patients were followed from inclusion until December 31, 2013. Clinical endpoints were recorded by telephone contact from June 2013 until December 2013, and the patients not reached by telephone were contacted by mail. For each endpoint, the hospital records were collected and then further evaluated and adjudicated by an Endpoint Committee before analyzing the results. Mortality endpoints were obtained from the Cause of Death Registry, administered by the Norwegian Institute of Public Health.
Statistical Analysis
Comparisons of continuous variables were performed by the 2‐sample Student t test or, for markedly skewed variables, the Mann–Whitney U test. Chi‐squared tests were used to compare categorical variables.
To investigate the effect of IL‐6, sgp130, sIL‐6R, and CRP on time to endpoint, the variables were divided into quartiles (Qs), because linear effect could not be assumed. First, Qs 2, 3, and 4 were compared to Q1 in univariate Cox regression models. Next, Q4 was compared to Q1 to Q3 (grouped). Because of the explorative nature of the study, correction of multiple comparisons was not performed. Relevant clinical or biochemical variables (Table 1) were assessed as potential confounders. Variables with a P<0.2 in univariate analyses were included in the multivariate Cox regression model.
Table 1.
Baseline Characteristics of the Total Study Population Stratified According to the Primary Endpoint (Composite of First Clinical Event)
| All Patients (n=989) | Primary Endpoint + (n=200) | Primary Endpoint − (n=789) | P Value | |
|---|---|---|---|---|
| Age, ya | 61.0 (24/94) | 64.5 (29/94) | 60 (24/90) | <0.0001 |
| Male | 794 (80.1) | 157 (78.1) | 637 (80.6) | 0.48 |
| Body mass index, weight/height2 | 27.0 (±4.28) | 27.1 (±4.81) | 27.0 (±4.14) | 0.81 |
| Systolic blood pressure, mm Hg | 139 (±27.5) | 139 (±29.4) | 139 (±27.0) | 0.74 |
| Smokers | 463 (46.7) | 90 (44.8) | 373 (47.2) | 0.57 |
| Previous disorders | ||||
| Myocardial infarction | 121 (12.2) | 36 (17.9) | 85 (10.8) | 0.008 |
| Hypertension | 343 (34.6) | 82 (40.8) | 261 (33.0) | 0.05 |
| Diabetes mellitus | 130 (13.1) | 36 (17.9) | 94 (11.9) | 0.03 |
| Heart failure | 20 (2.02) | 9 (4.48) | 11 (1.39) | 0.01 |
| Cerebrovascular disease | 45 (4.54) | 17 (8.46) | 28 (3.54) | 0.005 |
| Malignancy | 46 (4.64) | 15 (7.46) | 31 (3.92) | 0.05 |
| Use of statin | 225 (22.7) | 57 (28.4) | 168 (21.3) | 0.04 |
| Biochemical analyses at admission | ||||
| Admission cholesterol, mmol/L | 4.85 (±1.12) | 4.69 (±1.22) | 4.89 (±1.10) | 0.03 |
| Admission glucose, mmol/L | 8.12 (±2.80) | 8.60 (±2.95) | 8.00 (±2.75) | 0.008 |
| NT‐proBNP, ng/Lb | 33.0 (11.0, 124) | 58.0 (18.5, 224) | 29.0 (10.0 112) | <0.0001 |
| Creatinine, μmol/L | 76.4 (±27.8) | 80.7 (±26.2) | 75.3 (±28.1) | 0.01 |
| Peak troponin T, ng/Lb | 3840 (1730, 7140) | 3900 (1430, 7565) | 3840 (1810, 7085) | 0.33 |
| Levels of cytokines | ||||
| Interleukin‐6, pg/mLb | 18.8 (14.1, 30.0) | 18.7 (14.5, 30.1) | 18.8 (14.0, 29.9) | 0.64 |
| sgp130, ng/mLb | 240 (219, 260) | 246 (223, 268) | 239 (218, 259) | 0.002 |
| sIL‐6R, ng/mLb | 39.2 (31.0, 47.7) | 41.1 (32.0, 51.2) | 38.9 (30.7, 46.8) | 0.01 |
| hsCRP, mg/Lb | 13.7 (7.05, 31.5) | 15.6 (7.59, 40.6) | 13.2 (6.91, 29.3) | 0.09 |
| Echocardiography | ||||
| LVEF (%) | 49.2 (±9.26) | 46.7 (±11.4) | 49.8 (±8.55) | 0.002 |
Continuous data are presented as mean (SD) and categorical data as numbers (%), if not otherwise stated. Comparisons of continuous variables were performed by the 2‐sample Student t test or, for markedly skewed variables, the Mann–Whitney U‐test. Chi‐squared tests were used to compare categorical variables. BNP indicates brain natriuretic peptide; hsCRP, high‐sensitivity C‐reactive protein; LVEF, left ventricular ejection fraction; sgp130, soluble glycoprotein 130; sIL‐6R, soluble interleukin‐6 receptor.
Mean (range).
Median (25th, 75th percentiles).
Kaplan–Meier survival curves were used to estimate the survival status for patients in Q4 and Q1 to Q3, and log rank tests were used to compare the survival curves.
All analyses were performed by IBM SPSS software (version 21.0 for Windows; SPSS, Inc., Chicago, IL). A 2‐sided P<0.05 was considered to be statistically significant.
Results
A total of 1026 patients were included in the study. Data were obtained from 989 of 1026 patients (96.4%) with a median follow‐up time of 4.6 years. Of the remaining 37 patients, 32 were not reached by telephone or repeated mails and 5 were excluded from the database because of a final diagnosis other than STEMI. Baseline characteristics and serum levels of the measured cytokines of the 989 patients included in the final study population are shown in Table 1. Median age was 61 years, 80% were male, and peak troponin T (median, 25th, 75th percentiles) was 3840 (1730, 7140) ng/L.
Median time to first new clinical event was 16 months (1.3 years) after the index infarction, and 250 endpoints were registered. A total of 200 of 989 patients experienced a primary endpoint. Among these were 66 deaths (33%), 61 reinfarctions (31%), 6 strokes (3%), 52 urgent unscheduled PCI procedures (26%), and 15 (7.5%) readmissions for heart failure. In addition, 16 patients died after first having experienced a nonfatal event. Thus, the total number of deaths was 82. Patients who experienced the primary endpoint were significantly older, had higher incidence of previous myocardial infarction, cerebrovascular disease, diabetes mellitus, and heart failure compared to patients without. They also presented with slightly higher levels of admission glucose, N‐terminal probrain natriuretic peptide (NT‐proBNP), and creatinine and had significantly lower LVEF. Peak troponin T was, however, not significantly different in patients with or without events. Patients who experienced events had significantly lower levels of cholesterol, but more of these patients were using cholesterol‐lowering medications at baseline. Additionally, sex was associated with total mortality, given that female patients had a reduced risk of dying during the follow‐up period.
Levels of sIL‐6R and sgp130 were significantly elevated in patients experiencing the composite primary endpoint compared to patients without (P=0.01, 0.002, respectively; Table 1). Levels of CRP also tended to be higher in the endpoint group, whereas IL‐6 levels were similar in the 2 groups. The highest quartile (Q4) of sIL6‐R, sgp130, and CRP, compared to both Q1 and Q1 to Q3 (grouped), were significantly associated with the primary endpoint in univariate analyses (Table 2). Univariate associations between clinical covariates and the composite endpoint of first clinical event are given in Table 3. After adjustment for relevant covariates, Q4 of sIL‐6R, compared to both the lowest and the grouped quartiles (Q1–Q3), was still significantly associated with the primary endpoint with a hazard ratio (HR) of 1.67 (95% CI, 1.04, 2.67; P=0.03) and 1.54 (95% CI, 1.08, 2.21; P=0.02), respectively (Table 2).
Table 2.
Associations Between Members of the Interleukin‐6 Signaling Cascade Measured in STEMI Patients and the Composite Primary Endpoint of First New Clinical Event
| Unadjusted (n=989) | P Value | Adjusteda (n=989) | P Value | |
|---|---|---|---|---|
| HR (95% CI) | HR (95% CI) | |||
| sIL‐6R | ||||
| Q4 vs Q1 | 1.69 (1.14, 2.50) | 0.009 | 1.67 (1.04, 2.67) | 0.03 |
| Q4 vs Q1 to Q3 | 1.45 (1.08, 1.95) | 0.01 | 1.54 (1.08, 2.21) | 0.02 |
| sgp130 | ||||
| Q4 vs Q1 | 1.88 (1.26, 2.80) | 0.002 | 1.37 (0.85, 2.21) | 0.20 |
| Q4 vs Q1 to Q3 | 1.55 (1.16, 2.09) | 0.004 | 1.39 (0.96, 2.01) | 0.08 |
| Interleukin‐6 | ||||
| Q4 vs Q1 | 1.16 (0.78, 1.73) | 0.45 | 0.82 (0.51, 1.32) | 0.41 |
| Q4 vs Q1 to Q3 | 1.11 (0.80, 1.52) | 0.54 | 0.87 (0.58, 1.30) | 0.50 |
| hsCRP | ||||
| Q4 vs Q1 | 1.52 (1.04, 2.23) | 0.03 | 1.03 (0.63, 1.70) | 0.91 |
| Q4 vs Q1 to Q3 | 1.48 (1.10, 2.00) | 0.01 | 1.11 (0.75, 1.64) | 0.60 |
Hazard ratios (HRs) and 95% CIs obtained from Cox regression models. hsCRP indicates high‐sensitivity C‐reactive protein; Q1, lowest quartile; Q1 to Q3, the 3 lowest quartiles; Q4, highest quartile; sgp130, soluble glycoprotein 130; sIL‐6R, soluble interleukin‐6 receptor; STEMI, ST‐elevation myocardial infarction.
Adjusted for age, sex, previous disease (hypertension, diabetes mellitus, cardiovascular disease, malignancy, chronic obstructive pulmonary disease), left ventricular ejection fraction, peak troponin T, admission glucose, N‐terminal probrain natriuretic peptide, admission cholesterol, and admission creatinine.
Table 3.
Univariate Associations Between Covariates and the Composite Primary Endpoint of First New Clinical Event
| Composite Endpoint HR (95% CI) | P Value | |
|---|---|---|
| Sex, female | 0.864 (0.618, 1.207) | 0.4 |
| Age | 1.029 (1.017, 1.041) | <0.0001 |
| Body mass index | 0.999 (0.967, 1.032) | 0.97 |
| Smoker | 0.911 (0.690, 1.204) | 0.5 |
| Peak troponin T | 1.017 (0.992, 1.042) | 0.2 |
| Admission cholesterol | 0.874 (0.770, 0.991) | 0.04 |
| Admission glucose | 1.060 (1.017, 1.103) | 0.005 |
| Fasting glucose | 1.092 (1.017, 1.173) | 0.02 |
| HbA1c | 1.090 (0.965, 1.231) | 0.2 |
| NT‐proBNP | 1.001 (1.000, 1.001) | <0.0001 |
| Admission creatinine | 1.004 (1.001, 1.007) | 0.02 |
| Previous myocardial infarction | 1.632 (1.138, 2.340) | 0.008 |
| Previous cerebrovascular disease | 2.256 (1.373, 3.709) | 0.001 |
| Previous diabetes mellitus | 1.487 (1.037, 2.132) | 0.03 |
| Previous hypertension | 1.395 (1.053, 1.848) | 0.02 |
| Previous COPD | 1.715 (0.957, 3.073) | 0.07 |
| LVEF | 0.967 (0.950, 0.984) | <0.0001 |
| Previous hyperlipidemia | 0.859 (0.561, 1.316) | 0.5 |
| Previous heart failure | 2.831 (1.451, 5.526) | 0.002 |
| Previous renal failure | 0.922 (0.295, 2.884) | 0.9 |
| Malignancy | 1.738 (1.027, 2.943) | 0.04 |
| Time, PCI to blood sampling | 0.995 (0.983, 1.008) | 0.4 |
Hazard ratios (HRs) and 95% CIs obtained from Cox regression models. COPD indicates chronic obstructive pulmonary disease; HbA1c, glycated hemoglobin; LVEF, left ventricular ejection fraction; NT‐proBNP, N‐terminal probrain natriuretic peptide; PCI, percutaneous coronary intervention.
When analyzing for the associations with all‐cause mortality, the Q4 of sIL‐6R versus the grouped Q1 to Q3 was significantly associated with mortality, also after adjusting for covariates (HR, 1.81; 95% CI, 1.04, 3.18; P=0.04; Table 4). A similar pattern was observed for CRP, whereas sgp130 was not associated with all‐cause mortality in multivariate analyses (Table 4). Univariate associations between clinical covariates and all‐cause mortality are given in Table 5. Survival curves demonstrate the difference between Q4 and Q1 to Q3 of sIL‐6R both in time to first event (P=0.01; Figure 1) and mortality (P=0.02; Figure 2).
Table 4.
Associations Between Members of the Interleukin‐6 Signaling Cascade Measured in STEMI Patients and All‐Cause Mortality
| Unadjusted (n=989) | P Value | Adjusteda (n=989) | P Value | |
|---|---|---|---|---|
| HR (95% CI) | HR (95% CI) | |||
| sIL‐6R | ||||
| Q4 vs Q1 | 1.63 (0.91, 2.91) | 0.10 | 1.49 (0.73, 3.02) | 0.28 |
| Q4 vs Q1 to Q3 | 1.70 (1.08, 2.67) | 0.02 | 1.81 (1.04, 3.18) | 0.04 |
| sgp130 | ||||
| Q4 vs Q1 | 4.23 (2.04, 8.81) | <0.0001 | 2.45 (1.01, 5.95) | 0.05 |
| Q4 vs Q1 to Q3 | 2.37 (1.53, 3.68) | <0.0001 | 1.56 (0.88, 2.74) | 0.13 |
| Interleukin‐6 | ||||
| Q4 vs Q1 | 1.52 (0.82, 2.80) | 0.18 | 0.90 (0.43, 1.90) | 0.78 |
| Q4 vs Q1 to Q3 | 1.37 (0.85, 2.21) | 0.19 | 0.90 (0.49, 1.66) | 0.74 |
| hsCRP | ||||
| Q4 vs Q1 | 2.88 (1.55, 5.33) | 0.001 | 3.02 (1.25, 7.33) | 0.01 |
| Q4 vs Q1 to Q3 | 2.59 (1.67, 4.01) | <0.0001 | 2.09 (1.16, 3.75) | 0.01 |
Hazard ratios (HRs) and 95% CIs obtained from Cox regression models. hsCRP indicates high‐sensitivity C‐reactive protein; Q1, lowest quartile; Q1 to Q3, the 3 lowest quartiles; Q4, highest quartile; sgp130, soluble glycoprotein 130; sIL‐6R, soluble interleukin‐6 receptor; STEMI, ST‐elevation myocardial infarction.
Adjusted for age, sex, previous disease (hypertension, diabetes mellitus, cardiovascular disease, malignancy, and chronic obstructive pulmonary disease), left ventricular ejection fraction, peak troponin T, admission glucose, N‐terminal probrain natriuretic peptide, admission cholesterol, and admission creatinine.
Table 5.
Univariate Associations Between Covariates and All‐Cause Mortality
| All‐Cause Mortality | P Value | |
|---|---|---|
| HR (95% CI) | ||
| Sex, female | 0.443 (0.282, 0.696) | <0.0001 |
| Age | 1.092 (1.069, 1.114) | <0.0001 |
| Body mass index | 0.975 (0.924, 1.029) | 0.4 |
| Smoker | 0.875 (0.567, 1.351) | 0.5 |
| Peak troponin T | 1.031 (0.995, 1.069) | 0.09 |
| Admission cholesterol | 0.710 (0.580, 0.868) | 0.001 |
| Admission glucose | 1.109 (1.052, 1.168) | <0.0001 |
| Fasting glucose | 1.183 (1.078, 1.299) | <0.0001 |
| HbA1c | 1.278 (1.100, 1.484) | 0.001 |
| NT‐proBNP | 1.001 (1.001, 1.001) | <0.0001 |
| Admission creatinine | 1.007 (1.003, 1.010) | <0.0001 |
| Previous myocardial infarction | 2.223 (1.332, 3.710) | 0.002 |
| Previous cerebrovascular disease | 3.952 (2.143, 7.289) | <0.0001 |
| Previous diabetes mellitus | 2.260 (1.378, 3.707) | 0.001 |
| Previous hypertension | 2.198 (1.428, 3.381) | <0.0001 |
| Previous COPD | 4.199 (2.226, 7.920) | <0.0001 |
| LVEF | 0.947 (0.923, 0.972) | <0.0001 |
| Previous hyperlipidemia | 0.916 (0.473, 1.774) | 0.8 |
| Previous heart failure | 5.578 (2.571, 12.105) | <0.0001 |
| Previous renal failure | 1.605 (0.395, 6.531) | 0.5 |
| Malignancy | 3.203 (1.652, 6.211) | 0.001 |
| Time PCI to blood sampling | 0.995 (0.975, 1.016) | 0.6 |
Hazard ratios (HRs) and 95% CIs obtained from Cox regression models. COPD indicates chronic obstructive pulmonary disease; HbA1c, glycated hemoglobin; LVEF, left ventricular ejection fraction; NT‐proBNP, N‐terminal probrain natriuretic peptide; PCI, percutaneous coronary intervention.
Figure 1.
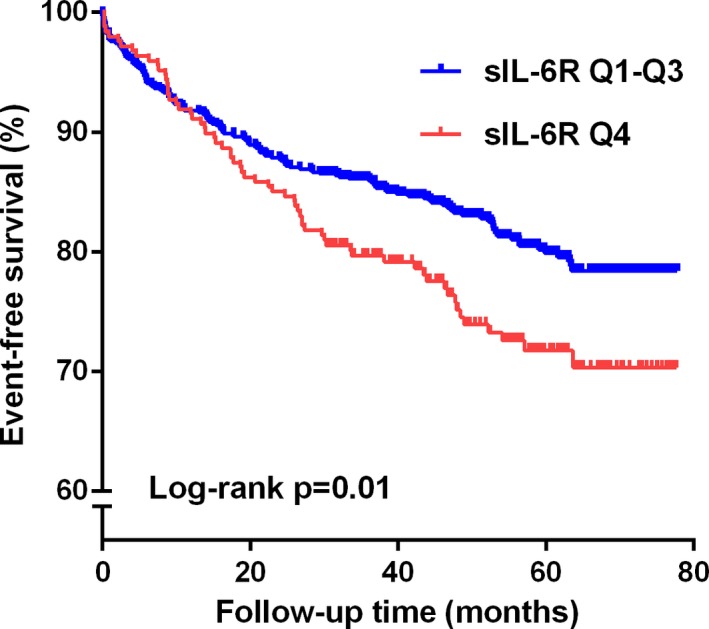
Time‐to‐event curves for the highest quartile (Q4) of soluble interleukin‐6 receptor (sIL‐6R) vs the 3 lowest quartiles (Q1–Q3) measured in 989 STEMI patients according to the primary endpoint. The log rank test was used to compare the survival curves. STEMI indicates ST‐elevation myocardial infarction.
Figure 2.
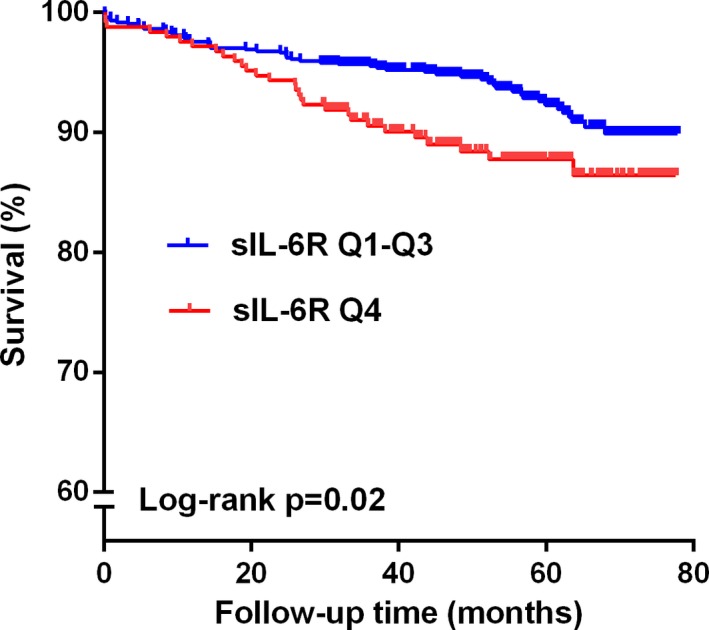
Time‐to‐event curves for the highest quartile (Q4) of soluble interleukin‐6 receptor (sIL‐6R) vs the 3 lowest quartiles (Q1–Q3) measured in 989 STEMI patients according to the secondary endpoint. The log rank test was used to compare the survival curves. STEMI indicates ST‐elevation myocardial infarction.
Discussion
The main result of the present study was that high levels of sIL‐6R measured in the acute phase of STEMI were significantly associated with future cardiovascular events in a large cohort of STEMI patients followed for a median of 4.6 years. Levels of both sIL‐6R and sgp130 were significantly higher in the group with new events; however, sgp130 was not related to new events after adjustments for covariates. The highest quartiles of sIL‐6R and CRP were also significantly related to all‐cause mortality. The strengths of the study are the relatively large cohort of STEMI patients, long‐term follow‐up, and that measurements of several components of the IL‐6 signaling were performed in the acute phase.
IL‐6 intracellular signaling acts through 2 mechanisms: (1) IL‐6 interacts with the common signal transducing receptor, gp130, when bound to the IL‐6‐specific surface receptor (classical signaling) or (2) by binding to sIL‐6R in a complex that enables interaction with cell surface gp130 and further activation of intracellular signaling pathways (trans‐signaling).18 The components involved in the trans‐signaling pathway seem to be more important for the proinflammatory effect of IL‐6 than the classical one.19 Additionally, sgp130 acts by forming multicomponent receptor complexes for the IL‐6 family20 and is probably the natural inhibitor of the trans‐signaling system.21
Our results, although probably reflecting mainly the acute phase, indicate that sIL‐6R plays an important role in the excessive inflammatory response accompanying the remodeling process and repair of ischemic myocardium in STEMI patients.7 The association between sIL‐6R/sgp130 and future cardiovascular events in STEMI patients may reflect involvement of mechanisms that are not directly related to myocardial necrosis. Such mechanisms may be related to inflammation of the vessel wall, destabilization of atherosclerotic plaques, increased platelet reactivity, as well as myocardial repair and remodeling of the left ventricle. The present results on patients with relatively small‐to‐moderate infarct size judged by troponin levels indicate that other important mechanisms are involved in the risk of new cardiovascular events during the first years after an myocardial infarction. Our results can be discussed in line with the recent report of Moreno Velasquez et al.,22 showing that high levels of sIL‐6R, although measured in a stable phase after an STEMI, were related to the occurrence of future myocardial infarction. It has also been reported that high levels of sIL‐6R were associated with reduced myocardial reperfusion in STEMI patients undergoing acute PCI,23 indicating a role of sIL‐6R and inflammation in ischemia‐reperfusion injury.
So far, attempts to treat patients with CVD with anti‐inflammatory drugs have failed; howewer, recent preliminary results from a phase II trial on patients with acute coronary syndrome showed that IL‐6R inhibition with tocilizumab reduced the inflammatory response as well as troponin levels.24 Tocilizumab is supposed to inhibit both soluble and membrane bound IL‐6R25 and increased understanding of the role of sIL‐6R in patients with myocardial infarction is therefore warranted. Furthermore, randomized, clinical trials exploring the effect of IL‐6R antagonists on clinical endpoints would be of interest.
To our knowledge, an association between high levels of sIL‐6R and long‐term mortality in patients with STEMI has not been reported previously.
Levels of sgp130 were not associated with the composite primary endpoint or total mortality after adjustments for covariates in our population. sgp130 regulation depends on a diversity of other factors and is probably not only reflecting an adverse immune response in patients with myocardial infarction. sgp130 has been proposed to have pleiotropic properties and may have anti‐inflammatory effects by serving as an inhibitor of the sIL‐6R/IL‐6 complex.21 In a recent study, high levels of sgp130 were suggested to be beneficial and protective in myocardial infarction,22 which differs somewhat from our results. The diverging results may be attributed to the different time points of blood sampling. In our study, blood samples were taken in the acute phase early after STEMI, whereas in the study by Moreno Velasquez et al.22 sgp130 was analyzed in samples collected 3 months after the index infarction, probably reflecting a more‐chronic inflammation. Interestingly, however, increased levels of sgp130 have been reported to be associated with clinical events in patients with chronic heart failure.26
CRP was significantly related to all‐cause mortality, which has been described by others.27, 28 Levels of IL‐6, however, were not associated with the composite of clinical events or all‐cause mortality in the present population, in contrast to other reports.9, 29 We have earlier described significant associations in the present cohort of STEMI patients between IL‐6 and CRP and myocardial necrosis defined as peak troponin T,17 whereas sIL‐6R and sgp130 did not show any associations with peak troponin T. Why high levels of IL‐6 or troponin T were not related to clinical events or all‐cause mortality in this cohort of PCI‐treated patients is unclear, but a similar lack of findings between peak troponin levels and subsequent clinical events have also been reported from other STEMI cohorts.9, 29
Limitations
The lack of repeated blood sampling in order to study fluctuations over time in levels of the measured biomarkers is a major limitation. We do not know the temporal profile of sIL‐6R and sgp130 after an acute myocardial infarction and may have missed the peak value. It has, however, been shown, that levels of sIL‐6R were relatively stable during the first 24 hours in patients with AMI.16 Additionally, the time from symptom onset to blood sampling was not associated with the primary endpoint or all‐cause mortality in the present study (Tables 3 and 5), indicating that the time of sampling may be representative for the IL‐6 response in STEMI patients with regard to future clinical events.
Although performing multivariate analyses adjusting for potential confounders, important unmeasured variables may have been missed.
Conclusion
High levels of sIL‐6R were significantly associated with a composite endpoint of first clinical event as well as total mortality during long‐term follow‐up of STEMI patients. The associations persisted after adjustment for clinically relevant covariates. High levels of CRP were associated with mortality only. Our results suggest that components of the IL‐6 trans‐signaling pathway may play an important role both in the inflammatory response pattern accompanying an acute STEMI as well as in the risk of developing new cardiovascular events; however, further studies are needed to validate our results.
Sources of Funding
This work was supported by Stein Erik Hagen Foundation for Clinical Heart Research, Oslo, Norway.
Disclosures
None.
Acknowledgments
We thank the study nurses and the staff at the Intensive Cardiac Care Unit and Center for Clinical Heart Research for excellent assistance and medical technologist Sissel Åkra for laboratory analyses. The study was a part of the Biobanking in myocardial infarction (BAMI) project at Oslo University Hospital Ullevål, which is led by a steering committee including Reidar Bjørnerheim, as well as the following authors: Seljeflot, Arnesen (Chair), Eritsland, Halvorsen, and Andersen.
(J Am Heart Assoc. 2016;5:e003014 doi: 10.1161/JAHA.115.003014)
The results of this study were presented, in part, at the European Society of Cardiology Scientific Congress, August 29 to September 2, 2015, in London, United Kingdom.
References
- 1. Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. [DOI] [PubMed] [Google Scholar]
- 2. Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. [DOI] [PubMed] [Google Scholar]
- 3. The Interleukin‐6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium . The interleukin‐6 receptor as a target for prevention of coronary heart disease: a Mendelian randomisation analysis. Lancet. 2012;379:1214–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. IL6R Genetics Consortium Emerging Risk Factors Collaboration . Interleukin‐6 receptor pathways in coronary heart disease: a collaborative meta‐analysis of 82 studies. Lancet. 2012;379:1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dhillon S. Intravenous tocilizumab: a review of its use in adults with rheumatoid arthritis. BioDrugs. 2014;28:75–106. [DOI] [PubMed] [Google Scholar]
- 6. Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. [DOI] [PubMed] [Google Scholar]
- 7. Huang M, Yang D, Xiang M, Wang J. Role of interleukin‐6 in regulation of immune responses to remodeling after myocardial infarction. Heart Fail Rev. 2015;20:25–38. [DOI] [PubMed] [Google Scholar]
- 8. Orn S, Manhenke C, Ueland T, Damas JK, Mollnes TE, Edvardsen T, Aukrust P, Dickstein K. C‐reactive protein, infarct size, microvascular obstruction, and left‐ventricular remodelling following acute myocardial infarction. Eur Heart J. 2009;30:1180–1186. [DOI] [PubMed] [Google Scholar]
- 9. Tan J, Hua Q, Li J, Fan Z. Prognostic value of interleukin‐6 during a 3‐year follow‐up in patients with acute ST‐segment elevation myocardial infarction. Heart Vessels. 2009;24:329–334. [DOI] [PubMed] [Google Scholar]
- 10. Aukrust P, Ueland T, Lien E, Bendtzen K, Muller F, Andreassen AK, Nordoy I, Aass H, Espevik T, Simonsen S, Froland SS, Gullestad L. Cytokine network in congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 1999;83:376–382. [DOI] [PubMed] [Google Scholar]
- 11. Karpinski L, Plaksej R, Kosmala W, Witkowska M. Serum levels of interleukin‐6, interleukin‐10 and C‐reactive protein in relation to left ventricular function in patients with myocardial infarction treated with primary angioplasty. Kardiol Pol. 2008;66:1279–1285. [PubMed] [Google Scholar]
- 12. Hirota H, Izumi M, Hamaguchi T, Sugiyama S, Murakami E, Kunisada K, Fujio Y, Oshima Y, Nakaoka Y, Yamauchi‐Takihara K. Circulating interleukin‐6 family cytokines and their receptors in patients with congestive heart failure. Heart Vessels. 2004;19:237–241. [DOI] [PubMed] [Google Scholar]
- 13. Kishimoto T, Akira S, Narazaki M, Taga T. Interleukin‐6 family of cytokines and gp130. Blood. 1995;86:1243–1254. [PubMed] [Google Scholar]
- 14. Juillière Y, Cambou JP, Bataille V, Mulak G, Galinier M, Gibelin P, Benamer H, Bouvaist H, Meneveau N, Tabone X, Simon T, Danchin N. Heart failure in acute myocardial infarction: a comparison between patients with or without heart failure criteria from the FAST‐MI registry. Rev Esp Cardiol. 2012;65:326–333. [DOI] [PubMed] [Google Scholar]
- 15. Anderson DR, Poterucha JT, Mikuls TR, Duryee MJ, Garvin RP, Klassen LW, Shurmur SW, Thiele GM. IL‐6 and its receptors in coronary artery disease and acute myocardial infarction. Cytokine. 2013;62:395–400. [DOI] [PubMed] [Google Scholar]
- 16. Kanda T, Inoue M, Kotajima N, Fujimaki S, Hoshino Y, Kurabayashi M, Kobayashi I, Tamura J. Circulating interleukin‐6 and interleukin‐6 receptors in patients with acute and recent myocardial infarction. Cardiology. 2000;93:191–196. [DOI] [PubMed] [Google Scholar]
- 17. Ritschel VN, Seljeflot I, Arnesen H, Halvorsen S, Weiss T, Eritsland J, Andersen GO. IL‐6 signalling in patients with acute ST‐elevation myocardial infarction. Results Immunol. 2014;4:8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schuett H, Luchtefeld M, Grothusen C, Grote K, Schieffer B. How much is too much? Interleukin‐6 and its signalling in atherosclerosis. Thromb Haemost. 2009;102:215–222. [DOI] [PubMed] [Google Scholar]
- 19. Scheller J, Chalaris A, Schmidt‐Arras D, Rose‐John S. The pro‐ and anti‐inflammatory properties of the cytokine interleukin‐6. Biochim Biophys Acta. 2011;1813:878–888. [DOI] [PubMed] [Google Scholar]
- 20. Luchtefeld M, Preuss C, Ruhle F, Bogalle EP, Sietmann A, Figura S, Muller W, Grote K, Schieffer B, Stoll M. Gp130‐dependent release of acute phase proteins is linked to the activation of innate immune signaling pathways. PLoS One. 2011;6:e19427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jostock T, Mullberg J, Ozbek S, Atreya R, Blinn G, Voltz N, Fischer M, Neurath MF, Rose‐John S. Soluble gp130 is the natural inhibitor of soluble interleukin‐6 receptor transsignaling responses. Eur J Biochem. 2001;268:160–167. [DOI] [PubMed] [Google Scholar]
- 22. Moreno Velasquez I, Golabkesh Z, Kallberg H, Leander K, de Faire U, Gigante B. Circulating levels of interleukin 6 soluble receptor and its natural antagonist, sgp130, and the risk of myocardial infarction. Atherosclerosis. 2015;240:477–481. [DOI] [PubMed] [Google Scholar]
- 23. Groot HE, Hartman MH, Gu YL, de Smet BJ, van den Heuvel AF, Lipsic E, van der Harst P. Soluble interleukin 6 receptor levels are associated with reduced myocardial reperfusion after percutaneous coronary intervention for acute myocardial infarction. Cytokine. 2015;73:207–212. [DOI] [PubMed] [Google Scholar]
- 24. Kleveland O, Kunszt G, Bratlie M, Ueland T, Amundsen B, Aakhus S, Damaas JK, Aukrust P, Wiseth R, Gullestad L. The interleukin‐6 receptor antagonist tocilizumab reduces inflammation and myocardial damage in non‐ST elevation myocardial infarction ‐a randomized, double‐blind, placebo controlled study. Eur Heart J. 2015;36(Abstract suppl):336. [Google Scholar]
- 25. Ohsugi Y, Kishimoto T. The recombinant humanized anti‐IL‐6 receptor antibody tocilizumab, an innovative drug for the treatment of rheumatoid arthritis. Expert Opin Biol Ther. 2008;8:669–681. [DOI] [PubMed] [Google Scholar]
- 26. Askevold ET, Nymo S, Ueland T, Gravning J, Wergeland R, Kjekshus J, Yndestad A, Cleland JG, McMurray JJ, Aukrust P, Gullestad L. Soluble glycoprotein 130 predicts fatal outcomes in chronic heart failure: analysis from the controlled rosuvastatin multinational trial in heart failure (CORONA). Circ Heart Fail. 2013;6:91–98. [DOI] [PubMed] [Google Scholar]
- 27. Leu HB, Lin CP, Lin WT, Wu TC, Chen JW. Risk stratification and prognostic implication of plasma biomarkers in nondiabetic patients with stable coronary artery disease: the role of high‐sensitivity C‐reactive protein. Chest. 2004;126:1032–1039. [DOI] [PubMed] [Google Scholar]
- 28. Karadeniz M, Duran M, Akyel A, Yarlioglues M, Ocek AH, Celik IE, Kilic A, Yalcin AA, Ergun G, Murat SN. High sensitive CRP level is associated with intermediate and high syntax score in patients with acute coronary syndrome. Int Heart J. 2015;56:377–380. [DOI] [PubMed] [Google Scholar]
- 29. Jeong YH, Lee SW, Lee CW, Hong MK, Kim JJ, Park SW, Park SJ, Park DW, Kim YH. Biomarkers on admission for the prediction of cardiovascular events after primary stenting in patients with ST‐elevation myocardial infarction. Clin Cardiol. 2008;31:572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]