

Abstract
Background
The Brugada syndrome is an inherited cardiac arrhythmia associated with high risk of sudden death. Although 20% of patients with Brugada syndrome carry mutations in SCN5A, the molecular mechanisms underlying this condition are still largely unknown.
Methods and Results
We combined whole‐exome sequencing and linkage analysis to identify the genetic variant likely causing Brugada syndrome in a pedigree for which SCN5A mutations had been excluded. This approach identified 6 genetic variants cosegregating with the Brugada electrocardiographic pattern within the pedigree. In silico gene prioritization pointed to 1 variant residing in KCNAB2, which encodes the voltage‐gated K+ channel β2‐subunit (Kvβ2‐R12Q). Kvβ2 is widely expressed in the human heart and has been shown to interact with the fast transient outward K+ channel subunit Kv4.3, increasing its current density. By targeted sequencing of the KCNAB2 gene in 167 unrelated patients with Brugada syndrome, we found 2 additional rare missense variants (L13F and V114I). We then investigated the physiological effects of the 3 KCNAB2 variants by using cellular electrophysiology and biochemistry. Patch‐clamp experiments performed in COS‐7 cells expressing both Kv4.3 and Kvβ2 revealed a significant increase in the current density in presence of the R12Q and L13F Kvβ2 mutants. Although biotinylation assays showed no differences in the expression of Kv4.3, the total and submembrane expression of Kvβ2‐R12Q were significantly increased in comparison with wild‐type Kvβ2.
Conclusions
Altogether, our results indicate that Kvβ2 dysfunction can contribute to the Brugada electrocardiographic pattern.
Keywords: Brugada syndrome, cardiac arrhythmia, clinical electrophysiology, genetics, KCNAB2/Kvβ2, potassium ion channels, whole exome sequencing
Subject Categories: Arrhythmias, Electrophysiology, Genetics
Introduction
Sudden cardiac death is a major health burden in industrialized countries, with ≈250 000 events recorded annually in the United States alone.1 Although coronary artery disease remains the first underlying cause of sudden cardiac death, 5% to 10% of events occur in the absence of detectable cardiac structural abnormalities.1 A large proportion of these autopsy‐negative cases are a likely consequence of inherited cardiac arrhythmia disorders.1, 2 Among such disorders, Brugada syndrome (BrS), which affects ≈1 in 2500 individuals, is characterized by an ST‐segment elevation and a negative T wave in the right precordial leads on the electrocardiogram (ECG).3 Because only transient ST‐segment elevation is seen in most patients, sodium channel blocker challenge is widely used to unmask this anomaly in patients with an ECG suggestive of BrS and their relatives.3
Approximately 20% of patients with BrS carry loss‐of‐function mutations in the SCN5A gene,4, 5 which encodes the pore‐forming subunit of the cardiac voltage‐gated Na+ channel (Nav1.5). Although this condition is usually described as a monogenic disease with autosomal dominant transmission, family‐based linkage analysis has most frequently failed to identify disease‐causing genes. Mutations in ≈20 other genes have already been identified in patients with BrS, but ≈70% of cases remain genetically unexplained.4
While common genetic polymorphisms have been recently associated with the risk of BrS,6 familial case studies remain extremely useful to highlight rare variants with strong effect and discover new genes involved in disease susceptibility. In this study, we combined whole‐exome sequencing, comparative genomic hybridization array (array‐CGH), genome‐wide simple nucleotide polymorphism genotyping, cellular electrophysiology, biochemistry, and computer modeling to identify a gain‐of‐function mutation in the KCNAB2 gene, which encodes the voltage‐gated K+ channel β2‐subunit (Kvβ2),7 as involved in this cardiac arrhythmia disorder.
Methods
Clinical Recruitment
Patients with BrS and unaffected relatives were recruited by following the French ethical guidelines for genetic research and under approval from the local ethical committee. Written informed consent was obtained from every patient and family member. ECGs were systematically recorded at baseline and under drug challenge tests, according to consensus criteria.8 A Brugada type I ECG pattern was defined on the basis of a coved type ST elevation at baseline or after a drug challenge test, in ≥1 right precordial leads.8 Holter recording, echocardiographic, and electrophysiological investigations were performed in all patients diagnosed with BrS. Two physicians evaluated each ECG independently.
Linkage Analysis
Simple nucleotide polymorphism genotyping was performed on population‐optimized Affymetrix Axiom Genome‐Wide CEU 1 array plates following the standard manufacturer's protocol. Fluorescence intensities were quantified by using the Affymetrix GeneTitan Multi‐Channel Instrument, and primary analysis was conducted with Affymetrix Power Tools following the manufacturer's recommendations. After genotype calling, all individuals had a genotype call rate >97%. Simple nucleotide polymorphisms with a minor allele frequency (MAF) <10%, a call rate <95%, or with P<1×10−4 when testing for Hardy–Weinberg equilibrium were excluded. We used the MERLIN algorithm9 to detect chromosomal fragments cosegregating with the BrS phenotype, by testing a model of autosomal dominant pattern of inheritance with incomplete penetrance (80%). The maximal theoretical LOD (logarithm of odds) score was 0.82 for this family. The threshold for selecting shared genomic regions was set to 0.7 (P=0.05 uncorrected for multiple testing).
Array‐CGH
Array‐CGH was performed on Agilent whole‐genome 1M microarrays following the manufacturer's recommendations. Variant calling was performed as previously described, after exclusion of probes located within known copy‐number variable intervals.10, 11
Exome Sequencing
Enrichment in coding sequences was performed by using the Agilent SureSelect in‐solution capture protocol, which targeted 39.3 Mb of exonic sequences (GENCODE custom design as previously described12). The enriched library was sequenced on 3 lanes of an Illumina Genome Analyzer IIx, producing 54‐bp paired‐end reads and 13.3 Gb of sequence for the proband. Reads were aligned to the human genome assembly GRCh37 (BWA‐MEM, version 0.7.5a), which led to a mean depth of 114×. Coverage of ≥10× was obtained for 90% of the targeted positions, considering only reads with a mapping quality score >30. Genetic variations (single nucleotide variants and indels) were detected by using Samtools mpileup v0.1.19 and GATK Unified Genotyper v2.8 and were considered for further analyses if found by both algorithms with a minimum quality score of 25 and a minimum mapping quality of 30. Functional consequences were annotated by using Ensembl VEP (Variant Effect Predictor). Variants were considered as having a potential functional consequence if they were annotated with ≥1 of the following SO terms for ≥1 RefSeq transcript: “transcript_ablation” (SO:0001893), “splice_donor_variant” (SO:0001575), “splice_acceptor_variant” (SO:0001574), “stop_gained” (SO:0001587), “frameshift_variant” (SO:0001589), “stop_lost” (SO:0001578), “start_lost” (SO:0002012), “protein_altering_variant” (SO:0001818) “inframe_insertion” (SO:0001821), “inframe_deletion” (SO:0001822), “missense_variant” (SO:0001583), or “transcript_amplification” (SO:0001889). Variants were considered as rare if the MAF was <0.1% in the Exome Aggregation Consortium (ExAC) non‐Finnish European population (http://exac.broadinstitute.org/, version 0.3, 33 370 individuals). Knime4Bio, a set of custom nodes for the interpretation of next‐generation sequencing data with KNIME,13 was used for all merging and filtering steps. Validation experiments and familial segregation analyses were performed by using capillary sequencing on an Applied Biosystems 3730 DNA Analyzer, with standard procedures. Two in silico algorithms—Endeavour14 and Toppgene15—were then applied to rank the putative disease‐causing genes. Both tools were accessed through the Internet by using the default prioritization parameters and based on 21 genes associated with BrS16 as the training gene set.
Targeted Sequencing
We have developed a custom kit to capture and sequence the coding regions of KCNAB2 as well as 45 arrhythmia‐susceptibility genes (Agilent Technologies HaloPlex capture, Illumina sequencing). Among those 45 genes, 21 genes were previously linked to BrS (SCN5A, SCN1B, SCN2B, SCN3B, SCN10A, CACNA1C, CACNB2, CACNA2D1, KCNH2, KCNE3, KCNE1L/KCNE5, KCND3, KCNJ8, ABCC9, TRPM4, HCN4, GPD1L, RANGRF/MOG1, SLMAP, PKP2, and FGF12). In total, 167 BrS cases (unrelated index cases) and 167 control individuals aged >65 years and showing no history of cardiac arrhythmia were sequenced.16 On average, >99.9% of the KCNAB2 coding sequence was covered by at least 10 reads in cases and controls. Public databases of genetic variants, generated by sequencing control or diseased individuals, were interrogated for KCNAB2 variants of interest (1000 Genomes Project, NHLBI GO Exome Sequencing Project and ExAC; accessed September 2015).
Site‐Directed Mutagenesis
Three transcripts are described in the RefSeq database for KCNAB2. Through RT‐PCR experiments on a panel of human tissue types, we identified NM_003636 as the main cardiac isoform (data not shown). We cloned this isoform in a pCMV6 expression plasmid to perform functional experiments. The human WT (wild‐type)‐KCNAB2, R12Q‐KCNAB2, L13F‐KCNAB2, and V114I‐KCNAB2 cDNAs were purchased from OriGene. The constructs were sequenced to ensure that there were no other mutations.
Cellular Electrophysiology and Modeling
The African green monkey kidney fibroblast‐like cell line (COS‐7) was obtained from American Type Culture Collection and cultured as previously described.17 Cells were transfected with 2 μg of DNA complexed with JetPEI (Polyplus‐tranfection) according to the manufacturer's instructions. For Kv4.3 experiments, DNA amounts were 100 ng of pCMV6‐KCND3 (NM_004980.3), 500 ng of WT or mutant pCMV6‐KCNAB2 (NM_003636.2), 250 ng of WT and mutant in the heterozygous condition (Kv4.3:Kvβ2 ratio 1:5), and 1400 ng pEGFP (Clontech). For Nav1.5 experiments, DNA amounts were 200 ng of pCI‐SCN5A (NM_000335.4), 200 ng of pRC‐SCN1B (NM_001037) encoding the cardiac Na+ channel auxiliary subunit Navβ1, 600 ng of WT or mutant pCMV6‐KCNAB2, and 1000 ng pEGFP. Eight hours posttransfection, the cells were isolated and seeded onto plastic Petri dishes at low density. Whole‐cell currents were recorded at room temperature by using the patch‐clamp technique, 24 (Nav1.5) to 48 hours (Nav1.5 or Kv4.3) posttransfection. The cells were continuously superfused with Tyrode solution containing (in mmol/L) NaCl 145, KCl 4, MgCl2 1, CaCl2 1, HEPES 5, and glucose 5, pH adjusted to 7.4 with NaOH. For K+ current recordings, wax‐coated pipettes (tip resistance: 1.8–3 MΩ) were filled with K+ intracellular medium containing (in mmol/L) KCl 150, MgCl2 1, EGTA 5, and HEPES 10, pH adjusted to 7.2 with KOH. During Na+ current recording, the studied cell was locally superfused with extracellular medium containing (in mmol/L) NaCl 145, CsCl 4, CaCl2 1, MgCl2 1, HEPES 5, and glucose 5, pH adjusted to 7.4 with NaOH, and the wax‐coated pipette (tip resistance: 0.8–1.3 MΩ) was filled with Na+ intracellular medium containing (in mmol/L) CsCl 80, gluconic acid 45, NaCl 10, MgCl2 1, CaCl2 2.5, EGTA 5, and HEPES 10, pH adjusted to 7.2 with CsOH. All products were purchased from Sigma. Stimulation, data recording through an A/D converter (Tecmar TM100 Labmaster, Scientific Solutions, or Digidata 1440A, Molecular Devices), and analysis were performed with Acquis1 software (Bio‐Logic) or Axon pClamp 10 (Molecular Devices). All current amplitudes were normalized by cell capacitance. Capacitance and series resistances were compensated to obtain minimal contribution of capacitive transients by using a VE‐2 amplifier (Alembic Instrument) or an Axopatch 200A amplifier (Axon Instruments, Molecular Devices).
Action potentials and pseudo‐ECG were calculated as described in Gima and Rudy,18 by simulating the heterogeneous transmural wedge of the right ventricular outflow tract.
Coimmunoprecipitation of Heterologously Expressed Proteins
Forty‐eight hours after transfection (666 ng of pCMV6‐KCND3 and 1333 ng of pCMV6‐KCNAB2, Kv4.3:Kvβ2 ratio 1:2), COS‐7 cells were washed twice with PBS and lysed in ice‐cold lysis buffer containing 1× PBS, 1% Triton X‐100, and complete mini EDTA‐free protease inhibitor mixture tablet (Roche). Cell lysates were used in immunoprecipitation experiments with 5 μg of anti‐Kv4.3 rabbit polyclonal antibody (RbαKv4.3; Alomone Labs). Before immunoprecipitation, antibodies were bound to 12.5 μL of protein A–magnetic beads (Invitrogen). Cell lysates and antibody‐coupled beads were mixed for 2 hours at 4°C. Magnetic beads were then collected and washed 3 times with ice‐cold lysis buffer, and isolated protein complexes were eluted with 1× SDS sample buffer at 60°C for 10 minutes. Immunoprecipitated proteins were analyzed by Western blotting as described previously.19 The RbαKv4.3 antibody used for Western blotting was purchased from Alomone Labs, and the mouse monoclonal anti‐Kvβ2 and anti–transferrin receptor (TransR) antibodies were purchased from OriGene and Invitrogen, respectively. Goat anti‐rabbit or anti‐mouse horseradish peroxidase–conjugated secondary antibodies were purchased from Santa Cruz Biotechnology.
Cell Surface Biotinylation Assays
Surface biotinylation of transfected COS‐7 cells (Kv4.3:Kvβ2 ratio 1:2) was completed as described previously.19 Briefly, cells were incubated with 0.5 mg/mL EZ‐Link Sulfo‐NHS‐SS‐Biotin (Pierce) in PBS, pH 7.4, for 30 minutes on ice. The biotinylation reaction was quenched with Tris–saline solution (10 mmol/L Tris, pH 7.4, 120 mmol/L NaCl), and detergent‐soluble cell lysates were prepared. Biotinylated cell surface proteins were affinity‐purified by using NeutrAvidin‐conjugated agarose beads (Pierce) and analyzed by Western blotting as described earlier. Bands corresponding to Kv4.3 and Kvβ2 were normalized to bands corresponding to TransR from the same sample. Kv4.3 and Kvβ2 protein expression (total or biotinylated fraction) in cells transfected with pCMV6‐KCNAB2‐R12Q alone or with pCMV6‐KCNAB2‐WT (heterozygous condition) is expressed relative to Kv4.3 or Kvβ2 protein expression (total or biotinylated fraction) in cells transfected with pCMV6‐KCNAB2‐WT alone.
Statistics of Functional Studies
Data are presented as mean±SEM. The statistical analyses were achieved by using SigmaPlot (Systat Software Inc.). The statistical significance of the observed effects was assessed by using a Mann–Whitney rank sum test or a 2‐way ANOVA for repeated measures followed by a Holm–Sidak test for multiple comparisons. A value of P<0.05 was considered statistically significant.
Results
Clinical Report of the Familial Case
The proband (individual II:3; Figure 1A) was diagnosed with symptomatic BrS after having experienced unexplained syncope. He had a permanent type I BrS ECG pattern without conduction disturbance (Figure 2A). Electrophysiological studies confirmed normal conduction with atrium–to–His bundle and His bundle–to–ventricle conduction times of 96 and 57 ms, respectively. Ventricular fibrillation was induced during programmed ventricular stimulation (apical stimulation with 2 extrasystoles). The ventricular refractory period was 210 ms. Holter monitoring revealed nonsustained polymorphic ventricular tachycardia (Figure 2B). No recurrence of syncope or arrhythmia was observed after implantable cardioverter‐defibrillator implantation.
Figure 1.
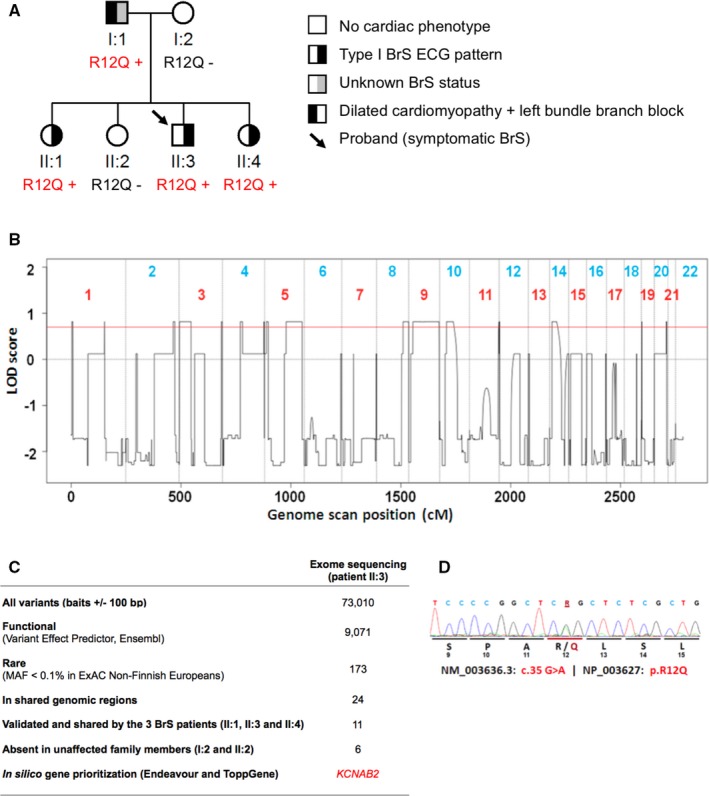
Genetic investigations in a familial case of Brugada syndrome (BrS). A, Pedigree of the Kvβ2‐R12Q family. R12Q + signs represent carriers of the heterozygous R12Q mutation. R12Q − signs represent mutation‐negative family members. B, LOD score profile obtained by genome‐wide linkage analysis. The red line indicates the LOD (logarithm of odds) score threshold of 0.7 used to filter DNA variants of interest (maximum theoretical LOD score 0.82). C, Number of DNA variants selected through successive filtering steps. Variants were considered as having a potential functional consequence if they were annotated as “transcript_ablation,” “splice_donor_variant,” “splice_acceptor_variant,” “stop_gained,” “frameshift_variant,” “stop_lost,” “start_lost,” “protein_altering_variant,” “inframe_insertion,” “inframe_deletion,” “missense_variant,” and/or “transcript_amplification” by Ensembl Variant Effect Predictor. D, Capillary sequencing confirmed that all affected siblings carry a missense variant in the KCNAB2 gene (NM_003636: c.35G>A; NP_003627: p.Arg12Gln; R12Q), inherited from their father who presents with cardiac conduction abnormalities. The variant was not detected in their unaffected mother and sister (as shown in A). MAF indicates minor allele frequency.
Figure 2.
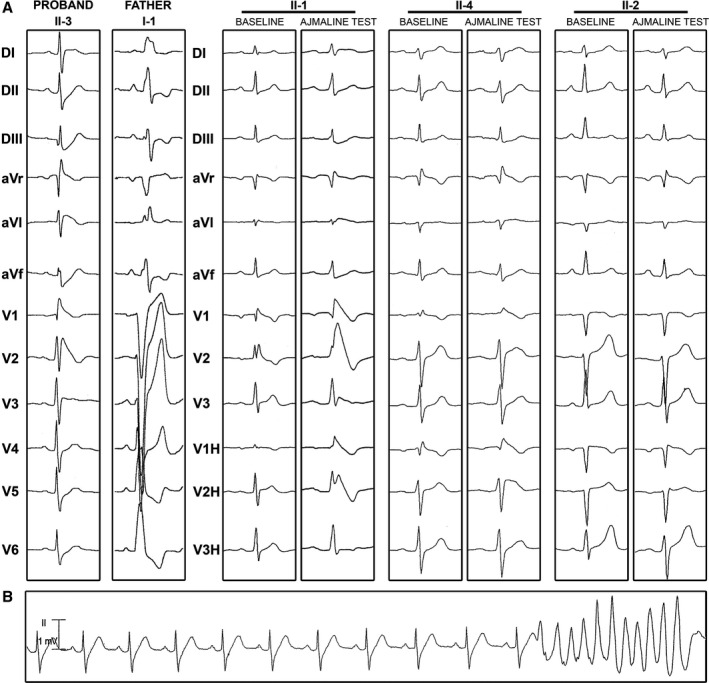
Electrocardiographic patterns of family members. A, Twelve‐lead electrocardiograms of the proband and his relatives at baseline or/and during ajmaline test. B, Event of ventricular tachycardia recorded in the proband.
By family screening, 2 asymptomatic first‐degree relatives (individuals II:1 and II:4) were diagnosed with BrS after the ajmaline test (Figure 2A). Individuals I:2 and II:2 did not present with the ECG pattern typical of BrS, even after the ajmaline test. Because individual I:1 was diagnosed with a complete left bundle‐branch block associated with a dilated cardiomyopathy (Figure 2A), no drug challenge was performed and his phenotype regarding BrS could not be determined. Clinical data for this French family are described in Table S1.
Genetic Investigations
We first checked whether any variation in the SCN5A gene could explain this familial case of BrS. No SCN5A mutation was detected in the proband through capillary sequencing. Moreover, no rare copy‐number variant could be identified by array‐based CGH. We combined whole‐exome sequencing and linkage analysis to identify any novel genetic variant likely to explain this familial case. Whole‐exome sequencing led to the detection of 73 010 simple nucleotide variants (substitutions and indels) in the proband compared with the human reference genome assembly (GRCh37). Only 173 of these simple nucleotide variants were annotated as functional and rare (MAF <0.1% in ExAC non‐Finnish Europeans). In parallel, by linkage analysis on the complete pedigree, we determined which chromosomal intervals contain haplotypes cosegregating with cardiac electrical anomalies. Only 17% of the genome displayed haplotype‐sharing patterns in line with autosomal dominant inheritance (Figure 1B). Within this portion of the genome, we validated 11 rare nonsynonymous variants carried by the 3 BrS‐ affected family members. Among these 11 variants, 6 cosegregated perfectly with Brugada ECG phenotypes (Figure 1C, Table S2). These 6 cosegregating variants were each located in KCNAB2, LAMB2, GRAMD3, HARS2, TXN, and PHF19. In silico gene prioritization using Endeavour14 and ToppGene15 identified the KCNAB2 gene, which encodes the voltage‐gated K+ channel β2‐subunit (Kvβ2), as the most likely susceptibility gene for BrS (see Methods, Figure 1C and Table S3). Kvβ2 has been shown to interact with the Kv4.3 channel, which has itself been involved in the pathogenesis of BrS.20 It is noteworthy that the KCNAB2 variant (NM_003636: c.35G>A; NP_003627: p.R12Q; Figure 1D) was inherited from the father (I.1; Figure 1A), whose BrS phenotype could not be determined, because no drug challenge test could be performed in the presence of dilated cardiomyopathy. This variant was not detected in 308 healthy blood donors and is absent from the European populations reported in public databases. It is reported in 1 of 4291 individuals from East Asia in the ExAC database, with no available phenotype information (ExAC East Asian population MAF 0.01%, total ExAC MAF 8.4×10−6).
Targeted sequencing among 167 unrelated French patients with BrS (including the Kvβ2‐R12Q carrier) identified 2 additional rare missense variants in KCNAB2 carried by 2 unrelated BrS patients. One variant, found in 1 female patient (NM_003636: c.37C>T; NP_003627: p.L13F), is absent from public databases, including ExAC. Her sister did not carry the variant but was diagnosed with BrS after the ajmaline test. The other variant (NM_003636: c.340G>A; NP_003627: p.V114I; rs140319610), found in 2 patients, is reported as very rare in public databases (MAF 4.5×10−5 in the ExAC European population based on data from 66 600 alleles). Both V114I carriers had episodes of vasovagal syncope and were diagnosed after the ajmaline test. In applying the same targeted sequencing strategy, no rare coding variant was detected in KCNAB2 among 167 control individuals showing no history of cardiac arrhythmia.
No additional rare variants were identified in 21 previously reported BrS‐susceptibility genes (see list of genes in the Methods). Regarding other arrhythmia‐susceptibility genes, a rare missense variant in the ANK2 gene, of unknown significance, was detected in the carrier of the Kvβ2‐L13F variant and her affected sister.
Electrophysiological Analyses
Whole‐cell voltage‐clamp analyses were performed in COS‐7 cells coexpressing Kv4.3 and Kvβ2. As shown in Figure 3, the current generated by Kv4.3 in the presence of Kvβ2‐R12Q was 3 times larger than that in the presence of WT Kvβ2. The R12Q variant had no effect on steady‐state activation (G‐V 1/2 WT 1.8±1.3 mV, n=22; G‐V 1/2 R12Q 2.6±1.1 mV, n=17). In contrast, the potential at which half of the channels are inactivated was slightly shifted toward more negative potentials, from −53.4±1.1 mV with WT Kvβ2 (n=9) to −57.5±1.2 mV with Kvβ2‐R12Q (n=5; P<0.05, Student t test). Kinetics of activation, inactivation, and recovery from inactivation were not modified by the variant (not shown). The L13F variant also increased significantly the density of Kv4.3‐generated current, whereas the V114I variant had no effect (Figure 3).
Figure 3.
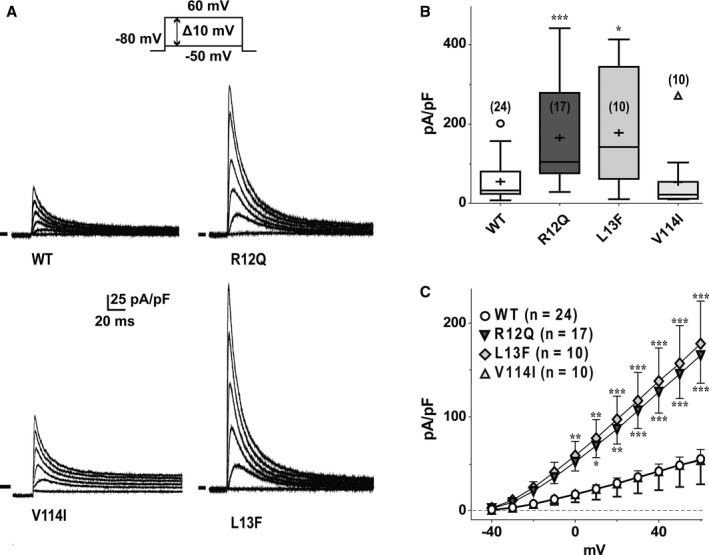
Effects of Kvβ2 mutations on Kv4.3‐generated currents. A, Superimposed representative currents, activated by a 1‐second pulse every 5 seconds (only the first 180 ms are shown) to various potentials (from −50 mV to +60 mV, 10‐mV increments; holding potential: −80 mV; only pulses at −20, 0, +20, +40, and +60 mV are shown) obtained from 4 different COS‐7 cells expressing Kv4.3 and either wild‐type (WT; top left), R12Q (top right), V114I (bottom left), or L13F (bottom right) Kvβ2. B, Tukey plots of peak K+ current densities recorded at +60 mV in cells transfected as in (A). Sample size is indicated in brackets, only outlier values are represented as empty symbols (*P<0.05 and ***P<0.001 versus WT; Mann–Whitney rank sum test). C, Mean current density–voltage relationship of Kv4.3‐generated currents in cells transfected as in (A). *P<0.05, **P<0.01, and ***P<0.001 versus WT (2‐way ANOVA followed by a Holm‐Sidak post‐hoc test for multiple comparison).
Because the major susceptibility gene for BrS identified to date is SCN5A, 16 we also investigated the function of the Nav1.5 channels in the presence of Kvβ2‐R12Q. As shown in Figure S1 and Table S4, the Na+ current was not modified by the presence of the R12Q mutation in Kvβ2.
Biochemical Analyses
To gain insights into the total and cell surface expression of the Kv4.3‐Kvβ2 protein complexes, biochemical experiments were performed in transfected COS‐7 cells. Our results show that Kvβ2 coimmunoprecipitates with Kv4.3 channels and that the R12Q mutation does not prevent the interaction (Figure 4A). In addition, although no significant changes were observed in the total and cell surface protein expression of Kv4.3 (Figure 4B and 4C), the Kvβ2 expression and its presence underneath the plasma membrane were significantly increased in the presence of the R12Q mutation (Figure 4B and 4D). Similar findings were obtained when the Kv4.3 and Kvβ2 constructs were transfected at a 1:5 ratio (data not shown). The same pattern was observed in the absence of Kv4.3 coexpression (Figure S2). On the contrary, no changes in total or cell surface expression of Kv4.3 and Kvβ2 were observed in the presence of the L13F or V114I variant (Figure 4E and 4F).
Figure 4.
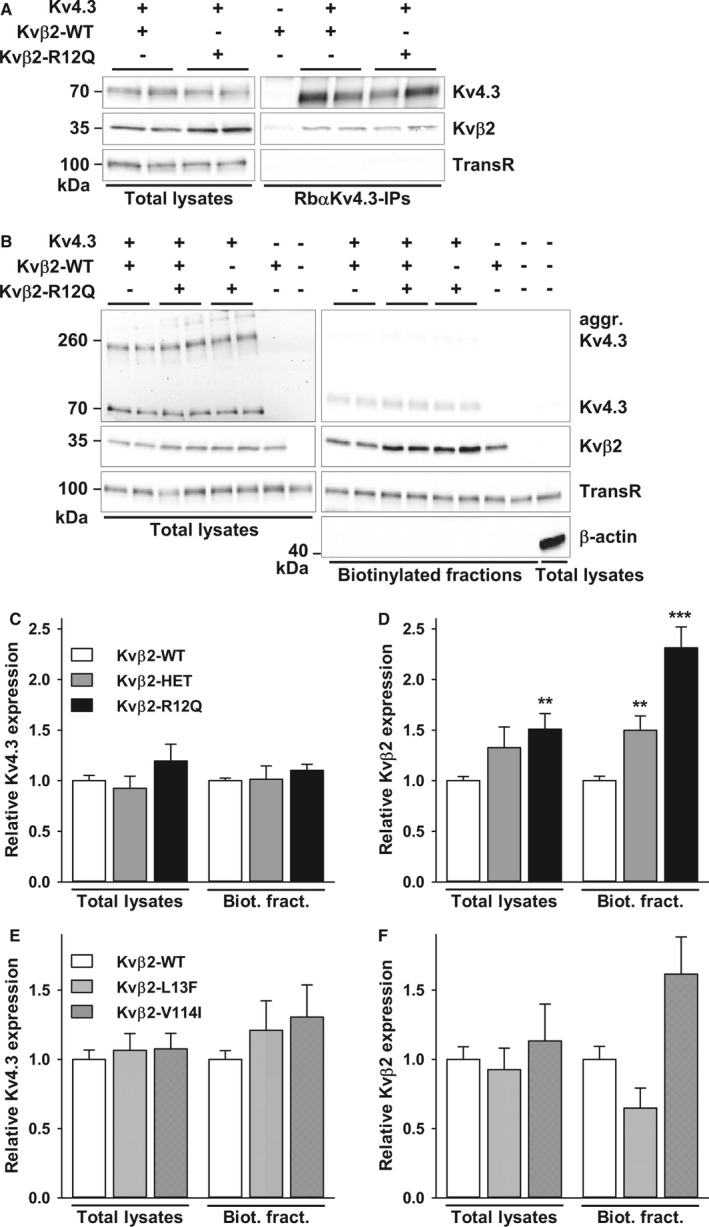
Effects of Kvβ2 variants on Kvβ2 and Kv4.3 expression. A, Western blot analyses of total lysates (left) and RbαKv4.3 immunoprecipitates (IP, right) probed with anti‐Kv4.3, anti‐Kvβ2 and anti–transferrin receptor (TransR) antibodies revealed that the R12Q mutation does not alter the coimmunoprecipitation of Kvβ2 with Kv4.3. B, Representative Western blots of Kv4.3 and Kvβ2 of the total (left) and biotinylated (right) fractions from transfected COS‐7 cells. Western blot analyses of β‐actin confirmed that biotinylated fractions are not contaminated by cytoplasmic proteins. C and D, Kv4.3 and Kvβ2 of both fractions from COS‐7 cells transfected with Kv4.3+Kvβ2‐WT (n=16–17 samples), Kv4.3+Kvβ2‐WT+Kvβ2‐R12Q (heterozygous condition, n=4–5), or Kv4.3+Kvβ2‐R12Q (n=16–17). Expression of Kv4.3 and Kvβ2 in each sample was normalized to the TransR protein in the same blot and then expressed relative to Kv4.3 or Kvβ2 in cells expressing Kv4.3+Kvβ2‐WT. Whereas no changes in Kv4.3 expression were observed, relative Kvβ2 total and submembrane expression was significantly (** P<0.01, *** P<0.001; Mann–Whitney rank sum test) higher in cells transfected with Kv4.3+Kvβ2‐R12Q than in cells expressing Kv4.3+Kvβ2‐WT. Kvβ2 submembrane expression was also significantly (** P<0.01) increased when expressed in the heterozygous condition. On the contrary, no changes in Kv4.3 (E) and Kvβ2 (F) total and cell surface expression were observed in presence of the L13F or V114I variants.
Action Potential and ECG Modeling
By using the Gima–Rudy model, we simulated the right ventricular outflow tract electrical activity, taking into account the gain‐of‐function of the Kvβ2‐R12Q variant on the Kv4.3 currents in the heterozygous condition, as obtained from the electrophysiological data (Figure 5A). The control ECG presented no ST‐segment elevation and an upright T wave. I to,f was then increased by 1.5‐ to 2.5‐fold, corresponding to the lower and average experimental current changes in the heterozygous condition. As a result, the pseudo‐ECG shape was progressively modified. Increasing I to,f from 1.5‐ to 1.9‐fold led to a progressive ST‐segment elevation and inversion of the T wave, both typical for the BrS ECG pattern (Figures S3 and 5C). When Ito,f was increased up to 2.5‐fold, a massive ST‐segment elevation persisted, but the inverted T wave disappeared. To understand the mechanisms underlying the ECG changes, we focused on the 1.9‐ and 2.5‐fold conditions. As illustrated in Figure 5B, when I to,f was increased by 1.9‐fold, the subepicardial action potential showed an exaggerated phase 1 repolarization. This induced a delayed plateau leading to a large heterogeneity in the transmural depolarization gradient responsible for the ST‐segment elevation and inverted T wave (Figure 5B and 5D). The subepicardial delayed plateau was initiated by a late I Na resurgence and sustained by I Ca,L increase (Figure S4). The 2.5‐fold increase in current density induced a loss of the action potential dome in the subepicardium (Figure 5 and Figure S4). The mid‐myocardium plateau exhibited a deeper notch with no major change in duration. These results highlight that because of ventricular transmural heterogeneity of electrical activity, a uniform I to,f increase can cause a nonuniform loss of plateau. This allows late reactivations of more repolarized regions driven by proximal and late appearing plateaus.
Figure 5.
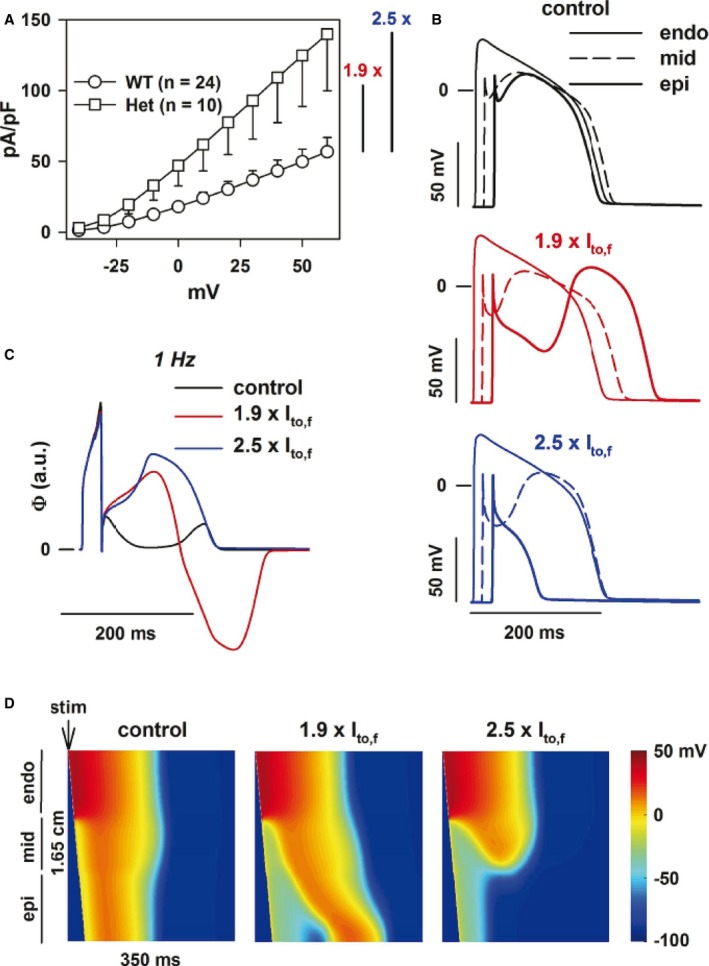
Modeling the effect of the R12Q variant at the heterozygous state. A, The Kv4.3 current density is increased by an averaged factor of 2.5 at +60 mV and by a minimal factor of 1.5 when considering only the lower limit for the heterozygous R12Q condition. B, Action potentials of the subendocardial (endo), midmyocardial (mid), and subepicardial (epi), corresponding, respectively, to cells 15, 80, and 150 in a 165‐cell fiber (thin, dashed, and thick lines, respectively) in control condition (black) and in presence of a 1.9‐fold (red) or a 2.5‐fold (blue) increase in I to,f. Shown is the 10th beat at 1 Hz. C, Corresponding right ventricular outflow tract wedge pseudo‐ECG, in arbitrary units (a.u.). D, Heat map of voltage in space and time (vertical and horizontal axes, respectively) in control condition and in the presence of a 1.9‐ and 2.5‐fold increase in I to,f. Stimulation (stim) was given on cell 1. In all conditions, there was zero I to in subendocardium, and 85% as much I to in midmyocardium as in subepicardium.
Discussion
More than 20 genes have been involved in susceptibility to BrS.4, 5 Gene finding has been largely based so far on candidate gene approaches, and only a few mutations have been shown to segregate in familial cases. Here, we applied a hypothesis‐free approach based on whole‐exome sequencing, to identify a new susceptibility gene for the BrS.
Following this strategy, we detected 6 potential causal variants in a familial case of BrS, all cosegregating with the BrS phenotype in the family. Among these 6 variants, a missense variant in the KCNAB2 gene—causing the Kvβ2‐R12Q substitution—appears as the most likely causal variant. However, one cannot formally exclude that ≥1 of the 5 other variants could also play a role in the occurrence of the familial BrS phenotype or that another variant of interest might have been missed because of low coverage depth for small proportions of coding sequences. Nonetheless, subsequent functional investigations showed that the gain‐of‐function effect of the Kvβ2‐R12Q variant is causally related to the BrS phenotype.
Most of the genetic variants identified so far as causing BrS result in reduction of the cardiac Na+ current (I Na). These mutations are located either in SCN5A,21 encoding the Nav1.5 channel, or genes encoding regulatory subunits of Nav1.5.5 Mutations in SCN10A—which encodes Nav1.8—have also been reported as associated with BrS.22 Other mutations related to BrS are located in genes encoding the α1‐, β2‐, and α2δ1‐subunits of the L‐type Ca2+ channel and reduce the L‐type Ca2+ current (I Ca,L).23, 24 In the presence of the latter variants, the ECG phenotype associates the typical BrS ST‐segment pattern with a short QT interval. Finally, a third group of BrS mutations increases the K+ currents and, in particular, the fast component of the transient outward current, I to,f. They are located in the KCND3 gene, encoding the Kv4.3 channel, and in the KCNE3 gene, encoding a Kv4.3‐interacting β‐subunit.20, 25
Our study confirms the involvement of I to,f in the pathogenesis of BrS. KCNAB2 encodes Kvβ2, a voltage‐gated K+ channel regulatory subunit related to the aldo‐keto reductase superfamily,7 that increases the density of Kv4.3‐generated currents, without affecting measurably the time and/or voltage dependence of the current properties.26 Because KCNAB2 is expressed in human ventricles,27 we hypothesized that the R12Q variant could modulate the properties and/or the expression of Kv4.3 channels. Our results demonstrate that the R12Q substitution in Kvβ2 increases the density of Kv4.3‐generated currents in COS‐7 cells, without affecting Kv4.3 biophysical properties. Biochemical experiments indicate the presence of greater amounts of both total and cell surface Kvβ2‐R12Q without any changes in Kv4.3 expression. These findings suggest that the p.R12Q mutation increases the stability of the Kvβ2 protein, which results, by mass action, in greater density underneath the cell surface. Additionally, although the mutation in Kvβ2 does not seem to alter the interaction with Kv4.3, one could speculate that the increased expression of Kvβ2 increases the probability of interaction between the 2 channel subunits and alters the functioning, either the open probability or the conductance, of the channel. Kvβ2 has been shown to be a functional oxidoreductase, which affects the biophysical properties of Kv1 channels by converting NADPH cofactor to NADP+.28 The relevance of this catalytic activity in regulating Kv channel functioning in cardiomyocytes remains unclear.29 Additional studies will be required to investigate its role in regulating the cardiac Kv4.3‐generated Ito,f channels, as well as the impact of the mutations in this regulation.
Because the major BrS‐susceptibility gene identified so far is SCN5A, we also evaluated the possible effects of Kvβ2 R12Q variant on the Nav1.5 current. We did not detect any changes in Na+ current. Additionally, in silico modeling results strongly suggest that the I to,f changes are sufficient to affect the right ventricular outflow tract action potential shape and ECG.
Screening for KCNAB2 coding variation in additional unrelated patients allowed identification of a second novel missense substitution, Kvβ2‐L13F, carried by 1 index case among 167 patients with BrS. This variant produces a similar increase as Kvβ2‐R12Q in the Kv4.3‐generated current density. The affected sister of the proband, however, does not carry the substitution. Furthermore, in contrast to the results obtained with Kvβ2‐R12Q, we observed no changes of the Kvβ2‐L13F protein fraction underneath the cell membrane. These results suggest that the Kvβ2‐L13F variant may not be associated with the BrS phenotype. Indeed, one cannot exclude that BrS may be triggered by the rare ANK2 variant of unknown significance and carried by both affected sisters or by any other rare and undetected genetic variant. Finally, distinct combinations of genetic determinants, including the Kvβ2‐L13F variant in 1 case, may result in similar ECG anomalies in both affected siblings. This hypothesis is supported by the complex BrS‐inheritance pattern observed in familial cases with SCN5A mutations30 and by the existence of common genetic polymorphism predisposing to this rare disease.6
Overall, while rare coding variants in KCNAB2 were detected in 4 of 167 index cases with BrS, no rare coding variants were found in this gene among a control population of 167 individuals. These results suggest that rare coding variants in KCNAB2 might be more prevalent among patients with BrS. Nevertheless, whether KCNAB2 variants should be considered as the primary cause for BrS remains an open question—and this holds true, also, for previously reported variants in other ion channel subunits. There is, indeed, accumulating evidence that BrS exhibits local structural anomalies31 that could lead to delayed segmented conduction,32, 33, 34 which by itself could underlie the BrS phenotype.35 In this context, a genetically inherited decrease in I Na or I Ca,L or an increase in I to might only unmask or exacerbate the BrS phenotype.
Conclusions
Our results herein indicate that Kvβ2‐mediated deregulation of Kv4.3‐encoded I to,f channels is involved in the pathogenesis of BrS. Together with previous reports, our study further emphasizes the critical role of the I Na/I to balance in BrS. This new finding also expands the list of genes associated with susceptibility to BrS and may contribute to improve molecular diagnosis of this cardiac arrhythmia disorder.
Sources of Funding
This work was supported by grants from the Leducq Foundation (CVD‐05; Alliance against Sudden Cardiac Death), the Fondation pour la Recherche Médicale (DEQ20140329545, to Dr Schott), the Inserm (ATIP‐Avenir program, to Dr Redon), and the French Regional Council of Pays‐de‐la‐Loire. Dr Es‐Salah‐Lamoureux was supported by the Fondation Lefoulon‐Delalande, the Fondation pour la Recherche Médicale, and the Fondation Genavie. Drs Le Scouarnec and Scott were supported by the Wellcome Trust (grant WT098051). Dr Marionneau acknowledges financial support from the Marie Curie 7th Framework Program of the European Commission (NavEx‐256397) and the Fondation Genavie.
Disclosures
None.
Supporting information
Figure S1. Absence of effect of R12Q Kvβ2 mutation on heterologously expressed Nav1.5‐generated current. A, Superimposed representative currents activated by a 50‐ms pulse every 1 second to various potentials (from −80 mV to +60 mV, 5‐mV increments; holding potential: −100 mV; only 1 of 2 pulses are shown for clarity) obtained from COS‐7 cells expressing Nav1.5, Navβ1, and either wild‐type (WT; left) or R12Q (right) Kvβ2. B, Tukey plots of peak current (I Na) densities recorded at −20 mV. Sample size is indicated in brackets, only outlier values are represented as symbols. C, Mean INa density–voltage relationship of Nav1.5‐generated currents in cells expressing Nav1.5, Navβ1, and either WT or R12Q Kvβ2. D, Relative peak conductance (G/Gmax) vs test‐pulse membrane potential (activation) and steady‐state channel availability at −20 mV (I/Imax) vs prepulse potential (inactivation) plots for Nav1.5 channels. Inactivation voltage protocol: 500‐ms polarization to various prepulse potentials from −110 to −50 mV (10‐mV increment) and test pulse at −20 mV for 20 ms (holding potential −100 mV; frequency 0.25 Hz). Curves are Boltzmann fits to the data.
Figure S2. The submembrane expression of the heterologously expressed Kvβ2‐R12Q protein is increased in the absence of Kv4.3. Forty‐eight hours following transfection of COS‐7 cells with cDNA constructs encoding Kvβ2‐WT and/or Kvβ2‐R12Q, cell lysates were prepared and used in cell surface biotinylation assays. Representative western blots of total (left) and biotinylated (right) Kvβ2 fractions from transfected COS‐7 cells show that the R12Q mutation increases Kvβ2 submembrane protein expression in the absence of Kv4.3 and whether expressed alone or coexpressed with Kvβ2‐WT. Western blot analyses of the transferrin receptor (TransR) and β‐actin confirmed equal protein loading and absence of contamination of the biotinylated fractions by cytoplasmic proteins, respectively.
Figure S3. Modeling the effect of the R12Q variant at the heterozygous state. Right ventricular outflow tract wedge pseudo‐ECGs, when the Kv4.3 current density is increased by a factor varying from the minimal value of 1.5, when considering only the lower limit for the heterozygous R12Q condition, to the averaged value of 2.5 (see Figure 5A, a.u.: arbitrary units).
Figure S4. Ion currents and Ca2+ transients underlying the different action potentials (gray) in control condition (black) and in presence of a 1.9‐fold (red) or a 2.5‐fold (blue) increase in Ito,f. See multiple scales for I Na graphs. Arrows indicate the peak currents in control condition.
Table S1. Clinical Characteristics of the Kvβ2‐R12Q Family
Table S2. Rare Variants Cosegregating With the Brugada ECG Phenotype in a Familial Case of Brugada Syndrome
Table S3. In Silico Ranking of Candidate Genes
Table S4. Absence of Effects of R12Q Mutation of Kvβ2 on Sodium Channel Biophysical Parameters
Acknowledgments
The authors are grateful to Aurore Girardeau, Angélique Erraud, Patricia Charpentier (Therassay core facility), and Juan‐Eugenio Sandoval‐Tortosa for technical assistance and to Béatrice Delasalle and Ruben Coronel for fruitful discussions. They acknowledge the members of the genomics and bioinformatics core facilities of Nantes (Biogenouest) for their expert services.
(J Am Heart Assoc. 2016;5:e003122 doi: 10.1161/JAHA.115.003122)
References
- 1. Chugh SS, Reinier K, Teodorescu C, Evanado A, Kehr E, Al Samara M, Mariani R, Gunson K, Jui J. Epidemiology of sudden cardiac death: clinical and research implications. Prog Cardiovasc Dis. 2008;5:213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tester DJ, Ackerman MJ. Postmortem long QT syndrome genetic testing for sudden unexplained death in the young. J Am Coll Cardiol. 2007;49:240–246. [DOI] [PubMed] [Google Scholar]
- 3. Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, Gussak I, LeMarec H, Nademanee K, Perez Riera AR, Shimizu W, Schulze‐Bahr E, Tan H, Wilde A. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659–670. [DOI] [PubMed] [Google Scholar]
- 4. Nielsen MW, Holst AG, Olesen S‐P, Olesen MS. The genetic component of Brugada syndrome. Front Physiol. 2013;4:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Antzelevitch C, Yan G‐X. J‐wave syndromes: Brugada and early repolarization syndromes. Heart Rhythm. 2015;12:1852–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud J‐B, Simonet F, Verkerk AO, Schwartz PJ, Crotti L, Dagradi F, Guicheney P, Fressart V, Leenhardt A, Antzelevitch C, Bartkowiak S, Borggrefe M, Schimpf R, Schulze‐Bahr E, Zumhagen S, Behr ER, Bastiaenen R, Tfelt‐Hansen J, Olesen MS, Kääb S, Beckmann BM, Weeke P, Watanabe H, Endo N, Minamino T, Horie M, Ohno S, Hasegawa K, Makita N, Nogami A, Shimizu W, Aiba T, Froguel P, Balkau B, Lantieri O, Torchio M, Wiese C, Weber D, Wolswinkel R, Coronel R, Boukens BJ, Bézieau S, Charpentier E, Chatel S, Despres A, Gros F, Kyndt F, Lecointe S, Lindenbaum P, Portero V, Violleau J, Gessler M, Tan HL, Roden DM, Christoffels VM, Le Marec H, Wilde AA, Probst V, Schott J‐J, Dina C, Redon R. Common variants at SCN5A‐SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. 2013;45:1044–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gulbis JM, Mann S, MacKinnon R. Structure of a voltage‐dependent K+ channel beta subunit. Cell. 1999;97:943–952. [DOI] [PubMed] [Google Scholar]
- 8. Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang C‐E, Huikuri H, Kannankeril P, Krahn A, Leenhardt A, Moss A, Schwartz PJ, Shimizu W, Tomaselli G, Tracy C. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10:1932–1963. [DOI] [PubMed] [Google Scholar]
- 9. Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. [DOI] [PubMed] [Google Scholar]
- 10. Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, Fitzgerald T, Hu M, Ihm CH, Kristiansson K, Macarthur DG, Macdonald JR, Onyiah I, Pang AWC, Robson S, Stirrups K, Valsesia A, Walter K, Wei J; Wellcome Trust Case Control Consortium , Tyler‐Smith C, Carter NP, Lee C, Scherer SW, Hurles ME. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Crooijmans RPMA, Fife MS, Fitzgerald TW, Strickland S, Cheng HH, Kaiser P, Redon R, Groenen MAM. Large scale variation in DNA copy number in chicken breeds. BMC Genomics. 2013;14:398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Coffey AJ, Kokocinski F, Calafato MS, Scott CE, Palta P, Drury E, Joyce CJ, Leproust EM, Harrow J, Hunt S, Lehesjoki A‐E, Turner DJ, Hubbard TJ, Palotie A. The GENCODE exome: sequencing the complete human exome. Eur J Hum Genet. 2011;19:827–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lindenbaum P, Le Scouarnec S, Portero V, Redon R. Knime4Bio: a set of custom nodes for the interpretation of next‐generation sequencing data with KNIME. Bioinformatics. 2011;27:3200–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tranchevent L‐C, Barriot R, Yu S, Van Vooren S, Van Loo P, Coessens B, De Moor B, Aerts S, Moreau Y. ENDEAVOUR update: a web resource for gene prioritization in multiple species. Nucleic Acids Res. 2008;36(Web Server issue):W377–W384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen J, Xu H, Aronow BJ, Jegga AG. Improved human disease candidate gene prioritization using mouse phenotype. BMC Bioinformatics. 2007;8:392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Le Scouarnec S, Karakachoff M, Gourraud J‐B, Lindenbaum P, Bonnaud S, Portero V, Duboscq‐Bidot L, Daumy X, Simonet F, Teusan R, Baron E, Violleau J, Persyn E, Bellanger L, Barc J, Chatel S, Martins R, Mabo P, Sacher F, Haïssaguerre M, Kyndt F, Schmitt S, Bézieau S, Le Marec H, Dina C, Schott J‐J, Probst V, Redon R. Testing the burden of rare variation in arrhythmia‐susceptibility genes provides new insights into molecular diagnosis for Brugada syndrome. Hum Mol Genet. 2015;24:2757–2763. [DOI] [PubMed] [Google Scholar]
- 17. Loussouarn G, Park K‐H, Bellocq C, Baró I, Charpentier F, Escande D. Phosphatidylinositol‐4,5‐bisphosphate, PIP2, controls KCNQ1/KCNE1 voltage‐gated potassium channels: a functional homology between voltage‐gated and inward rectifier K+ channels. EMBO J. 2003;22:5412–5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gima K, Rudy Y. Ionic current basis of electrocardiographic waveforms: a model study. Circ Res. 2002;90:889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marionneau C, Carrasquillo Y, Norris AJ, Townsend RR, Isom LL, Link AJ, Nerbonne JM. The sodium channel accessory subunit Navβ1 regulates neuronal excitability through modulation of repolarizing voltage‐gated K+ channels. J Neurosci. 2012;32:5716–5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Giudicessi JR, Ye D, Tester DJ, Crotti L, Mugione A, Nesterenko VV, Albertson RM, Antzelevitch C, Schwartz PJ, Ackerman MJ. Transient outward current (I(to)) gain‐of‐function mutations in the KCND3‐encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm. 2011;8:1024–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, Brugada P, Fressart V, Guerchicoff A, Harris‐Kerr C, Kamakura S, Kyndt F, Koopmann TT, Miyamoto Y, Pfeiffer R, Pollevick GD, Probst V, Zumhagen S, Vatta M, Towbin JA, Shimizu W, Schulze‐Bahr E, Antzelevitch C, Salisbury BA, Guicheney P, Wilde AAM, Brugada R, Schott J‐J, Ackerman MJ. An international compendium of mutations in the SCN5A‐encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hu D, Barajas‐Martínez H, Pfeiffer R, Dezi F, Pfeiffer J, Buch T, Betzenhauser MJ, Belardinelli L, Kahlig KM, Rajamani S, DeAntonio HJ, Myerburg RJ, Ito H, Deshmukh P, Marieb M, Nam G‐B, Bhatia A, Hasdemir C, Haïssaguerre M, Veltmann C, Schimpf R, Borggrefe M, Viskin S, Antzelevitch C. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol. 2014;64:66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, Gelber P, Bonaros EP, Burashnikov E, Wu Y, Sargent JD, Schickel S, Oberheiden R, Bhatia A, Hsu L‐F, Haïssaguerre M, Schimpf R, Borggrefe M, Wolpert C. Loss‐of‐function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST‐segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Burashnikov E, Pfeiffer R, Barajas‐Martinez H, Delpón E, Hu D, Desai M, Borggrefe M, Häissaguerre M, Kanter R, Pollevick GD, Guerchicoff A, Laiño R, Marieb M, Nademanee K, Nam G‐B, Robles R, Schimpf R, Stapleton DD, Viskin S, Winters S, Wolpert C, Zimmern S, Veltmann C, Antzelevitch C. Mutations in the cardiac L‐type calcium channel associated with inherited J‐wave syndromes and sudden cardiac death. Heart Rhythm. 2010;7:1872–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Delpón E, Cordeiro JM, Núñez L, Thomsen PEB, Guerchicoff A, Pollevick GD, Wu Y, Kanters JK, Larsen CT, Hofman‐Bang J, Burashnikov E, Christiansen M, Antzelevitch C. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol. 2008;1:209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang EK, Alvira MR, Levitan ES, Takimoto K. Kvbeta subunits increase expression of Kv4.3 channels by interacting with their C termini. J Biol Chem. 2001;276:4839–4844. [DOI] [PubMed] [Google Scholar]
- 27. Gaborit N, Varro A, Le Bouter S, Szuts V, Escande D, Nattel S, Demolombe S. Gender‐related differences in ion‐channel and transporter subunit expression in non‐diseased human hearts. J Mol Cell Cardiol. 2010;49:639–646. [DOI] [PubMed] [Google Scholar]
- 28. Weng J, Cao Y, Moss N, Zhou M. Modulation of voltage‐dependent Shaker family potassium channels by an aldo‐keto reductase. J Biol Chem. 2006;281:15194–15200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kilfoil PJ, Tipparaju SM, Barski OA, Bhatnagar A. Regulation of ion channels by pyridine nucleotides. Circ Res. 2013;112:721–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Probst V, Wilde AAM, Barc J, Sacher F, Babuty D, Mabo P, Mansourati J, Le Scouarnec S, Kyndt F, Le Caignec C, Guicheney P, Gouas L, Albuisson J, Meregalli PG, Le Marec H, Dina C, Schott J‐J, Probst V, Redon R. SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ Cardiovasc Genet. 2009;2:552–557. [DOI] [PubMed] [Google Scholar]
- 31. Catalano O, Antonaci S, Moro G, Mussida M, Frascaroli M, Baldi M, Cobelli F, Baiardi P, Nastoli J, Bloise R, Monteforte N, Napolitano C, Priori SG. Magnetic resonance investigations in Brugada syndrome reveal unexpectedly high rate of structural abnormalities. Eur Heart J. 2009;30:2241–2248. [DOI] [PubMed] [Google Scholar]
- 32. Ikeda T, Sakurada H, Sakabe K, Sakata T, Takami M, Tezuka N, Nakae T, Noro M, Enjoji Y, Tejima T, Sugi K, Yamaguchi T. Assessment of noninvasive markers in identifying patients at risk in the Brugada syndrome: insight into risk stratification. J Am Coll Cardiol. 2001;37:1628–1634. [DOI] [PubMed] [Google Scholar]
- 33. Postema PG, van Dessel PFHM, de Bakker JMT, Dekker LRC, Linnenbank AC, Hoogendijk MG, Coronel R, Tijssen JGP, Wilde AAM, Tan HL. Slow and discontinuous conduction conspire in Brugada syndrome: a right ventricular mapping and stimulation study. Circ Arrhythm Electrophysiol. 2008;1:379–386. [DOI] [PubMed] [Google Scholar]
- 34. Postema PG, van Dessel PFHM, Kors JA, Linnenbank AC, van Herpen G, Ritsema van Eck HJ, van Geloven N, de Bakker JMT, Wilde AAM, Tan HL. Local depolarization abnormalities are the dominant pathophysiologic mechanism for type 1 electrocardiogram in Brugada syndrome a study of electrocardiograms, vectorcardiograms, and body surface potential maps during ajmaline provocation. J Am Coll Cardiol. 2010;55:789–797. [DOI] [PubMed] [Google Scholar]
- 35. Hoogendijk MG, Potse M, Vinet A, de Bakker JMT, Coronel R. ST segment elevation by current‐to‐load mismatch: an experimental and computational study. Heart Rhythm. 2011;8:111–118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Absence of effect of R12Q Kvβ2 mutation on heterologously expressed Nav1.5‐generated current. A, Superimposed representative currents activated by a 50‐ms pulse every 1 second to various potentials (from −80 mV to +60 mV, 5‐mV increments; holding potential: −100 mV; only 1 of 2 pulses are shown for clarity) obtained from COS‐7 cells expressing Nav1.5, Navβ1, and either wild‐type (WT; left) or R12Q (right) Kvβ2. B, Tukey plots of peak current (I Na) densities recorded at −20 mV. Sample size is indicated in brackets, only outlier values are represented as symbols. C, Mean INa density–voltage relationship of Nav1.5‐generated currents in cells expressing Nav1.5, Navβ1, and either WT or R12Q Kvβ2. D, Relative peak conductance (G/Gmax) vs test‐pulse membrane potential (activation) and steady‐state channel availability at −20 mV (I/Imax) vs prepulse potential (inactivation) plots for Nav1.5 channels. Inactivation voltage protocol: 500‐ms polarization to various prepulse potentials from −110 to −50 mV (10‐mV increment) and test pulse at −20 mV for 20 ms (holding potential −100 mV; frequency 0.25 Hz). Curves are Boltzmann fits to the data.
Figure S2. The submembrane expression of the heterologously expressed Kvβ2‐R12Q protein is increased in the absence of Kv4.3. Forty‐eight hours following transfection of COS‐7 cells with cDNA constructs encoding Kvβ2‐WT and/or Kvβ2‐R12Q, cell lysates were prepared and used in cell surface biotinylation assays. Representative western blots of total (left) and biotinylated (right) Kvβ2 fractions from transfected COS‐7 cells show that the R12Q mutation increases Kvβ2 submembrane protein expression in the absence of Kv4.3 and whether expressed alone or coexpressed with Kvβ2‐WT. Western blot analyses of the transferrin receptor (TransR) and β‐actin confirmed equal protein loading and absence of contamination of the biotinylated fractions by cytoplasmic proteins, respectively.
Figure S3. Modeling the effect of the R12Q variant at the heterozygous state. Right ventricular outflow tract wedge pseudo‐ECGs, when the Kv4.3 current density is increased by a factor varying from the minimal value of 1.5, when considering only the lower limit for the heterozygous R12Q condition, to the averaged value of 2.5 (see Figure 5A, a.u.: arbitrary units).
Figure S4. Ion currents and Ca2+ transients underlying the different action potentials (gray) in control condition (black) and in presence of a 1.9‐fold (red) or a 2.5‐fold (blue) increase in Ito,f. See multiple scales for I Na graphs. Arrows indicate the peak currents in control condition.
Table S1. Clinical Characteristics of the Kvβ2‐R12Q Family
Table S2. Rare Variants Cosegregating With the Brugada ECG Phenotype in a Familial Case of Brugada Syndrome
Table S3. In Silico Ranking of Candidate Genes
Table S4. Absence of Effects of R12Q Mutation of Kvβ2 on Sodium Channel Biophysical Parameters