

Abstract
Background
We evaluated lipoprotein‐associated phospholipase A2 (Lp‐PLA 2) activity in patients with stable coronary heart disease before and during treatment with darapladib, a selective Lp‐PLA 2 inhibitor, in relation to outcomes and the effects of darapladib in the STABILITY trial.
Methods and Results
Plasma Lp‐PLA 2 activity was determined at baseline (n=14 500); at 1 month (n=13 709); serially (n=100) at 3, 6, and 18 months; and at the end of treatment. Adjusted Cox regression models evaluated associations between Lp‐PLA 2 activity levels and outcomes. At baseline, the median Lp‐PLA 2 level was 172.4 μmol/min per liter (interquartile range 143.1–204.2 μmol/min per liter). Comparing the highest and lowest Lp‐PLA 2 quartile groups, the hazard ratios were 1.50 (95% CI 1.23–1.82) for the primary composite end point (cardiovascular death, myocardial infarction, or stroke), 1.95 (95% CI 1.29–2.93) for hospitalization for heart failure, 1.42 (1.07–1.89) for cardiovascular death, and 1.37 (1.03–1.81) for myocardial infarction after adjustment for baseline characteristics, standard laboratory variables, and other prognostic biomarkers. Treatment with darapladib led to a ≈65% persistent reduction in median Lp‐PLA 2 activity. There were no associations between on‐treatment Lp‐PLA 2 activity or changes of Lp‐PLA 2 activity and outcomes, and there were no significant interactions between baseline and on‐treatment Lp‐PLA 2 activity or changes in Lp‐PLA 2 activity levels and the effects of darapladib on outcomes.
Conclusions
Although high Lp‐PLA 2 activity was associated with increased risk of cardiovascular events, pharmacological lowering of Lp‐PLA 2 activity by ≈65% did not significantly reduce cardiovascular events in patients with stable coronary heart disease, regardless of the baseline level or the magnitude of change of Lp‐PLA 2 activity.
Clinical Trial Registration
URL: https://www.clinicaltrials.gov. Unique identifier: NCT00799903.
Keywords: atherosclerosis, coronary disease, inflammation, lipoprotein, myocardial infarction
Subject Categories: Biomarkers, Coronary Artery Disease, Pharmacology, Treatment, Risk Factors
Introduction
Inflammatory activity is an integral component of the development of atherosclerosis and its complications.1 Biomarkers indicating myocardial dysfunction (eg, troponin, natriuretic peptides), renal dysfunction (eg, cystatin C), and inflammatory activity (eg, C‐reactive protein) are risk indicators of future cardiovascular events in healthy persons and in patients with established coronary heart disease (CHD).2, 3, 4, 5 Higher levels of lipoprotein‐associated phospholipase A2 (Lp‐PLA2) activity have also been associated with an increased risk of coronary events in healthy elderly persons and in patients with stable CHD.6, 7, 8, 9 There is some evidence that Lp‐PLA2 might be part of the atherosclerotic process and may contribute to plaque destabilization through inflammatory activity in the atherosclerotic lesions.10, 11, 12 Consequently, it is important to further evaluate the independent prognostic value of Lp‐PLA2 activity as a risk marker for individual nonfatal and fatal cardiovascular events in large long‐term prospective studies of patients with CHD in addition to considering information provided by other prognostic biomarkers.
Darapladib is a selective Lp‐PLA2 inhibitor13 that reduces Lp‐PLA2 activity in plasma14 and in the atherosclerotic plaque.15, 16 Experiments in hypercholesterolemic diabetic swine17 and in an angiographic and intravascular ultrasound study in patients18 showed that darapladib prevented progression of the necrotic core, assumed to be the vulnerable component of atherosclerotic lesions. In the present STabilization of Atherosclerotic plaque By Initiation of darapLadIb TherapY (STABILITY) trial (ClinicalTrials.gov identifier NCT00799903), darapladib 160 mg daily did not significantly reduce the primary composite end point of cardiovascular death, myocardial infarction (MI), or stroke in patients with stable CHD (hazard ratio [HR] 0.94, 95% CI 0.85–1.03, P=0.20) but nominally reduced the rate of the secondary end point, major coronary events (coronary death, MI, or urgent coronary revascularization; HR 0.90, 95% CI 0.82–1.00, P=0.045).19 The present predefined ancillary study evaluated the independent prognostic value of baseline Lp‐PLA2 activity concerning cardiovascular events, the reduction of Lp‐PLA2 activity by darapladib, and the associations between baseline and changes in Lp‐PLA2 activity and clinical outcomes.
Methods
Trial Design, Patients, Treatments, and Follow‐up
The STABILITY trial was a prospective double‐blind randomized trial evaluating the efficacy and safety of darapladib 160 mg, a specific inhibitor of Lp‐PLA2, compared with placebo concerning cardiovascular outcomes in patients with stable CHD.19 In brief, the study randomized 15 828 patients from 39 countries with stable CHD, defined as prior MI, prior coronary revascularization, or multivessel CHD confirmed by coronary angiography. Patients also had to meet at least 1 of the following cardiovascular risk criteria: age ≥60 years; diabetes mellitus requiring pharmacotherapy; high‐density lipoprotein cholesterol <1.03 mmol/L; current or previous smoker, defined as ≥5 cigarettes per day on average; significant renal dysfunction (estimated glomerular filtration rate ≥30 and <60 mL/min per 1.73 m2 or urine albumin:creatinine ratio of ≥30 mg albumin per gram of creatinine); or polyvascular disease (CHD and cerebrovascular disease or CHD and peripheral arterial disease). The patients were followed with regular outpatient visits for a median of 3.7 years. The primary outcome was the composite of cardiovascular death, nonfatal MI, or nonfatal stroke. Secondary outcomes were the composites of major coronary events (ie, fatal and nonfatal MI, death from CHD, or urgent coronary revascularization for myocardial ischemia) and the composite of total coronary events, which also included all coronary revascularizations. Additional secondary end points were the individual components of the primary composite end point, hospitalization for heart failure, and all‐cause mortality. Components of cardiovascular death included death from unknown cause, fatal MI, fatal stroke, complications of a cardiac procedure, arrhythmia, congestive heart failure or shock, other vascular cause of death, and sudden death. All suspected end points were documented and reported by STABILITY study investigators and were adjudicated by an independent clinical events committee. All participants provided informed consent, and the study was approved by the relevant institutional review committee in each participating country.
Samples and Biochemical Methods
Blood samples were obtained from the majority of patients at baseline and 24±2 hours after the last intake of study drug at months 1, 3, 6, and 18, and at the end of treatment. Plasma aliquots were stored in central repositories at −70°C until biochemical analysis was performed. Measurements of Lp‐PLA2 activity and other biomarkers were performed in 14 500 patients at baseline for whom samples were available and suitable for assay. For 13 709 of these patients, Lp‐PLA2 activity was also measured after 1 month. In addition, the change in Lp‐PLA2 activity over a longer time period was evaluated in a random sample of 100 patients with plasma samples available from all follow‐up visits. The sample size of 100 patients was based on the assumption of a mean difference in percentage change from baseline between treatment groups in Lp‐PLA2 activity of between 25% and 65%, with a common standard deviation of 25%, a 2‐sided type I error rate (α level) of 5% and power of 90%. In total, 100 participants were selected with the goal of having ≈50 participants from each treatment group to describe the long‐term inhibition of Lp‐PLA2 activity by the randomized treatment.
The Lp‐PLA2 activity was measured in an automated enzyme assay system (PLAC Test for Lp‐PLA2 Activity; Diadexus) with a colorimetric substrate that is converted on hydrolysis by the phospholipase enzyme. The analytical sensitivity (limit of quantitation) of the assay is 10 μmol/min per liter. Within‐run and within‐laboratory variability were determined by testing 4 human plasma samples and 2 controls with Lp‐PLA2 activities ranging from 113 to 315 μmol/min per liter. Samples were assayed in duplicate twice a day over 20 days and with 3 kit lots using 1 instrument. Total precision coefficients of variation for each reagent lot and sample were <3%. Several dilution series were prepared from plasma samples with known high and low Lp‐PLA2 activity levels and were tested with 3 kit lots. Linearity with a deviation of ≤10% was demonstrated from 10 to 382 μmol/min per liter, which is considered the measuring range of the assay. The Lp‐PLA2 activity measurements were performed by the manufacturer (Diadexus) on samples blinded for study treatment. The levels of high‐sensitivity cardiac troponin T, N‐terminal proB‐type natriuretic peptide, growth differentiation factor 15 (precommercial assay),20 and cystatin C were determined by electrochemiluminiscence immunoassays using a Cobas Analytics e601 system (Roche Diagnostics), performed at the Uppsala Clinical Research Center Laboratory at Uppsala University in Sweden. High‐sensitivity C‐reactive protein was analyzed using the CardioPhase high‐sensitivity C‐reactive protein (Dade Behring) 2‐site particle‐enhanced immunonephelometry sandwich assay. The routine biochemical analyses and high‐sensitivity C‐reactive protein assay were performed at a central laboratory with standardized methods (Quest Diagnostics Clinical Laboratories, Inc).
Statistics
Lp‐PLA2 activity at baseline
Demographics and other baseline characteristics were summarized by quartile group of the baseline Lp‐PLA2 activity level. The univariable relationships between Lp‐PLA2 activity and background variables were evaluated using the chi‐square and Kruskal–Wallis tests for categorical and continuous variables, respectively. Multivariable analyses were used to evaluate the independent associations between baseline Lp‐PLA2 activity and other variables, using linear regression models in which continuous variables were included as linear or log‐transformed variables, as appropriate. Baseline Lp‐PLA2 activity as a predictor of outcomes was evaluated using Kaplan–Meier estimates of the cumulative risk to the first occurrence of an event for the Lp‐PLA2 quartile groups. The independent associations between baseline Lp‐PLA2 activity levels and outcomes were evaluated using adjusted Cox regression models in which the HR and 95% CI were calculated using the lowest quartile group of Lp‐PLA2 activity (quartile 1) as the reference and performing a trend analysis across quartile groups. The predefined adjustment variables for the statistical analyses were randomized treatment, geographic region, age, sex, body mass index, current smoking, hypertension, diabetes mellitus, prior MI, prior coronary revascularization, multivessel CHD, polyvascular disease, significant renal dysfunction, levels of standard biomarkers (hemoglobin, white blood cell count, estimated glomerular filtration rate [Chronic Kidney Disease Epidemiology Collaboration], low‐density lipoprotein cholesterol, high‐density lipoprotein cholesterol, and triglycerides), and levels of prognostic biomarkers (high‐sensitivity cardiac troponin T, N‐terminal proB‐type natriuretic peptide, growth differentiation factor 15, high‐sensitivity C‐reactive protein, interleukin 6), which were entered as quartiles after log transformation. A Cox proportional hazards model with treatment group (darapladib or placebo), baseline Lp‐PLA2 activity categorized by quartile group, and treatment group Lp‐PLA2 activity interaction as independent variables was used to test whether the treatment effect differed in relation to Lp‐PLA2 activity level. The relevance of the Lp‐PLA2 activity interacting with the effect of treatment was evaluated based on the significance of interaction statistics. Data were handled by complete‐case analyses with no imputation for missing values. No correction for multiplicity was performed. The analyses of associations between the Lp‐PLA2 activity and baseline characteristics, other biomarkers and outcomes, and changes of Lp‐PLA2 activity over time were prespecified in the statistical analysis plans.
Lp‐PLA2 activity over time
The HRs and P values for the association of changes in Lp‐PLA2 activity after 1 month and clinical outcomes were estimated using a Cox proportional hazards regression model with baseline Lp‐PLA2 activity and change from baseline to month 1 in Lp‐PLA2 activity as the covariate, stratified by treatment. In the 100‐participant cohort, Lp‐PLA2 activity over time was characterized using the generalized estimating equations model with participants fitted as a repeated effect, and it included the following terms: treatment, visit, baseline Lp‐PLA2 activity, treatment by visit, and baseline by visit interactions. Point estimates, 95% CIs, and P values for the mean differences between patients treated with darapladib and placebo at months 1, 3, 6, and 18, and at the end of treatment were calculated using this generalized estimating equations model.
Results
For the 14 500 patients with Lp‐PLA2 activity measurements, baseline characteristics were representative of the overall STABILITY population and balanced between the randomized treatment groups (Table 1). There were no differences in rates of outcome events between patients with and without Lp‐PLA2 activity measurements. The distribution of the Lp‐PLA2 activity levels did not deviate from normality (median 172 μmol/min per liter [interquartile range 143–204 μmol/min per liter]). There were statistically significant differences for the majority of the baseline characteristics and levels of other biomarkers when stratifying patients by baseline Lp‐PLA2 quartile groups (Table 1). The following factors showed independent and relevant associations with higher Lp‐PLA2 activity in multivariable analyses: male sex, North American origin, current smoking, higher low‐density lipoprotein cholesterol level, and lower high‐density lipoprotein cholesterol level. Diabetes mellitus was associated with lower Lp‐PLA2 activity (Table 2).
Table 1.
Demographics, Baseline Characteristics, and Biomarker Levels in Relation to Quartile Groups of Baseline Lp‐PLA2 Activity
| Variables | Baseline Lp‐PLA2 Activity | P Value | P Value for Trend | |||
|---|---|---|---|---|---|---|
| Q1 (<143.1) n=3617 | Q2 (143.1–172.4) n=3631 | Q3 (172.4–204.2) n=3615 | Q4 (≥204.2) n=3637 | |||
| Lp‐PLA2, μmol/min/L | 125 (108–135) | 158 (151–166) | 187 (180–195) | 230 (216–253) | <0.0001 | <0.0001 |
| Assigned treatment | ||||||
| Placebo | 1817 (50.2) | 1766 (48.6) | 1779 (49.2) | 1859 (51.1) | 0.1544 | 0.4342 |
| Darapladib | 1800 (49.8) | 1865 (51.4) | 1836 (50.8) | 1778 (48.9) | ||
| Age, y | 65 (60–71)* | 65 (60–71) | 65 (59–71) | 64 (57–71) | <0.0001 | <0.0001 |
| Sex, male | 2490 (68.8) | 2916 (80.3) | 3127 (86.5) | 3291 (90.5) | <0.0001 | <0.0001 |
| Geographic region | <0.0001 | <0.0001 | ||||
| Asia–Pacific | 821 (22.7) | 602 (16.6) | 534 (14.8) | 609 (16.7) | ||
| Eastern Europe | 712 (19.7) | 826 (22.7) | 922 (25.5) | 958 (26.3) | ||
| North America | 873 (24.1) | 910 (25.1) | 961 (26.6) | 1061 (29.2) | ||
| South America | 207 (5.7) | 204 (5.6) | 231 (6.4) | 226 (6.2) | ||
| Western Europe | 1004 (27.8) | 1089 (30.0) | 967 (26.7) | 783 (21.5) | ||
| Weight, kg | 79 (68–91) | 83 (73–94) | 84 (74–95) | 85 (74–96) | <0.0001 | <0.0001 |
| Current smoker | 466 (12.9) | 555 (15.3) | 677 (18.7) | 926 (25.5) | <0.0001 | <0.0001 |
| Hypertension | 2627 (72.6) | 2608 (71.8) | 2569 (71.1) | 2560 (70.4) | 0.1275 | 0.0311 |
| Diabetes mellitus | 1678 (46.4) | 1459 (40.2) | 1272 (35.2) | 1185 (32.6) | <0.0001 | <0.0001 |
| Statin at randomization | 3575 (98.8) | 3577 (98.5) | 3508 (97.0) | 3439 (94.6) | <0.0001 | <0.0001 |
| High‐intensity statin | 268 (7.4) | 270 (7.4) | 242 (6.7) | 227 (6.2) | 0.1271 | 0.0201 |
| Prior myocardial infarction | 2079 (57.5) | 2146 (59.1) | 2167 (59.9) | 2197 (60.4) | 0.0583 | 0.0018 |
| Prior PCI or CABG | 2731 (75.5) | 2735 (75.3) | 2677 (74.1) | 2661 (73.2) | 0.0715 | 0.0035 |
| Polyvascular disease | 491 (13.6) | 535 (14.7) | 529 (14.6) | 649 (17.8) | <0.0001 | <0.0001 |
| LDL cholesterol, mmol/L | 1.7 (1.3–2.1) | 1.9 (1.6–2.3) | 2.2 (1.8–2.7) | 2.6 (2.1–3.3) | <0.0001 | <0.0001 |
| HDL cholesterol, mmol/L | 1.3 (1.1–1.5) | 1.2 (1.0–1.4) | 1.2 (1.0–1.4) | 1.1 (0.9–1.3) | <0.0001 | <0.0001 |
| Triglycerides, mmol/L | 1.3 (1.0–1.8) | 1.5 (1.1–2.0) | 1.6 (1.1–2.2) | 1.7 (1.3–2.4) | <0.0001 | <0.0001 |
| Hemoglobin, g/L | 138 (129–146) | 142 (133–151) | 145 (137–153) | 148 (139–156) | <0.0001 | <0.0001 |
| eGFR, mL/mina | 75 (62–87) | 74 (61–86) | 74 (62–86) | 74 (61–87) | 0.5690 | 0.9025 |
| C‐reactive protein, mg/L | 2.8 (1.3–3.1) | 2.8 (1.4–3.1) | 2.8 (1.5–3.2) | 3.0 (1.5–3.4) | <0.0001 | <0.0001 |
| Interleukin 6, ng/L | 1.2 (0.6–2.9) | 1.3 (0.6–2.9) | 1.3 (0.7–3.1) | 1.6 (0.8–3.6) | <0.0001 | <0.0001 |
| High‐sensitivity troponin T, ng/L | 9.0 (5.9–13.6) | 9.4 (6.4–14.1) | 9.4 (6.3–14.2) | 9.4 (6.2–15.0) | <0.0001 | <0.0001 |
| NT‐proBNP, ng/L | 174 (85–372) | 181 (87–386) | 186 (81–361) | 174 (78–402) | 0.0762 | 0.5699 |
| Cystatin C, mg/L | 1.0 (0.9–1.2) | 1.0 (0.9–1.2) | 1.0 (0.9–1.2) | 1.0 (0.9–1.2) | <0.0001 | <0.0001 |
| GDF‐15, ng/L | 1280 (923–1824) | 1250 (902–1798) | 1216 (905–1746) | 1266 (927–1821) | 0.0003 | 0.0913 |
Categorical variables are shown as number (percentage); continuous variables are presented as median (interquartile range). CABG indicates coronary artery bypass grafting; eGFR, estimated glomerular filtration rate; GDF‐15, growth differentiation factor 15; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; Lp‐PLA2, lipoprotein‐associated phospholipase A2; NT‐proBNP, N‐terminal proB‐type natriuretic peptide; PCI, percutaneous coronary intervention; Q, quartile.
eGFR was estimated using the Chronic Kidney Disease Epidemiology Collaboration formula.
Table 2.
Multivariable Analysis of Demographics, Baseline Characteristics and Biomarkers Significantly Associated With Baseline Lp‐PLA2 Activity
| Background Characteristic | Adjusted Difference in Meana | 95% CIa | P Value |
|---|---|---|---|
| Western Europe vs North America | −19.81 | (−21.58; −18.03) | <0.0001 |
| Eastern Europe vs North America | −17.09 | (−18.97; −15.21) | <0.0001 |
| Asia–Pacific vs North America | −16.42 | (−18.48; −14.36) | <0.0001 |
| South America vs North America | −14.91 | (−17.81; −12.02) | <0.0001 |
| Female vs male | −15.69 | (−17.60; −13.77) | <0.0001 |
| Diabetes mellitus | −6.640 | (−8.092; −5.188) | <0.0001 |
| HDL cholesterol, 0.1 mmol/L increase | −4.333 | (−4.554; −4.113) | <0.0001 |
| Diagnosis of hypertension | −2.066 | (−3.503; −0.6295) | 0.0048 |
| Body mass index, 1 kg/m2 increase | −0.218 | (−0.361; −0.074) | 0.0030 |
| Triglycerides, 0.1 mmol/L increase | −0.071 | (−0.139; −0.003) | 0.0389 |
| GDF‐15, 10% increase | 0.401 | (0.252; 0.551) | <0.0001 |
| Hemoglobin, 10 g/L increase | 0.589 | (0.536; 0.641) | <0.0001 |
| Age, 10‐year increase | 0.908 | (0.005; 1.812) | 0.0489 |
| Cystatin C, 10% increase | 1.159 | (0.771; 1.547) | <0.0001 |
| Polyvascular disease | 1.919 | (0.169; 3.671) | 0.0317 |
| Current smoker vs never smoked | 2.094 | (0.049; 4.139) | 0.0447 |
| LDL cholesterol, 0.1 mmol/L increase | 3.118 | (3.040; 3.196) | <0.0001 |
GDF‐15 indicates growth differentiation factor 15; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; Lp‐PLA2, lipoprotein‐associated phospholipase A2.
Mean and 95% CI for adjusted difference in mean Lp‐PLA2 activity level estimated in relation to the respective change in each background characteristic.
In the present cohort, 661 (4.6%) cardiovascular deaths, 695 (4.8%) MIs, and 280 (1.9%) strokes occurred. Of the 661 cardiovascular deaths, 142 deaths of unknown cause were not verified as noncardiovascular and thus were included among the cardiovascular deaths. In total, there were 1444 (10.0%) first primary outcome events and 1404 (9.7%) major coronary events. The outcome events were accrued at a stable rate during follow‐up (Figure 1). There was a significant difference in event rates between the highest quartile group of baseline Lp‐PLA2 activity in comparison to the lower 3 quartile groups, among which no significant differences in event rates were observed (Figures 1 and 2). The higher event rates in the highest Lp‐PLA2 quartile group were observed for all types of events, namely, the primary composite of cardiovascular death, MI, or stroke; the secondary composite of major coronary events; and the individual events cardiovascular death, MI, stroke, hospitalization for heart failure, and cardiovascular and total mortality. The elevated event rates of all of these events in the highest Lp‐PLA2 quartile group were maintained after adjusting for baseline characteristics and conventional laboratory tests. In addition, after adjustment for the prognostic biomarkers N‐terminal proB‐type natriuretic peptide, high‐sensitivity cardiac troponin T, growth differentiation factor 15, cystatin C, high‐sensitivity C‐reactive protein, and interleukin 6, the increased risks of the composite events, hospitalization for heart failure, cardiovascular death, and total death remained, whereas associations with the individual ischemic events MI or stroke were somewhat attenuated. After complete adjustment, the HRs were 1.50 (95% CI 1.23–1.82) for the primary composite end point, 1.42 (95% CI 1.16–1.74) for major coronary events, 1.95 (95% CI 1.29–2.93) for hospitalization for heart failure, 1.42 (95% CI 1.07–1.89) for cardiovascular death, 1.47 (95% CI 1.17–1.84) for total death, 1.37 (95% CI 1.03–1.81) for MI, and 1.56 (95% CI 1.00–2.44) for stroke when comparing the highest and lowest Lp‐PLA2 quartile groups (Figure 2A). These findings were similar in men and women, without any interaction with sex (Figure 2B and 2C). The addition of Lp‐PLA2 activity levels to the model including all clinical characteristics, risk factors, and other biomarkers provided no significant change of the c‐index and only a modest improvement of the net reclassification index for most events based on better discrimination of patients without increased risk (Table 3).
Figure 1.
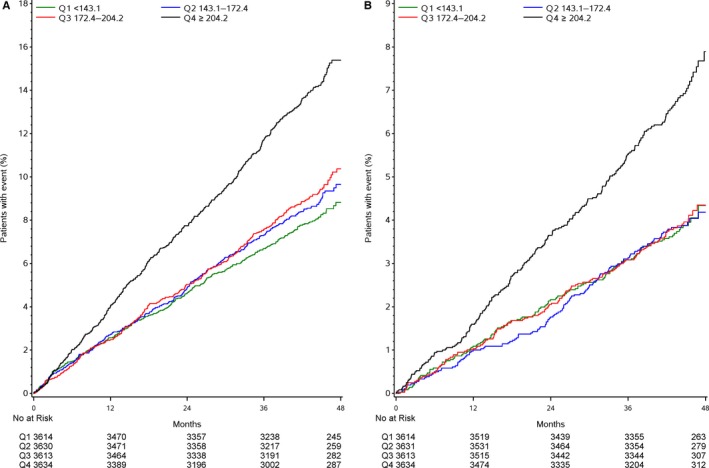
Quartile groups of baseline Lp‐PLA2 activity (in μmol/min per liter) in relation to outcomes by Kaplan–Meier analysis. A, Major adverse cardiac events (cardiovascular death, myocardial infarction, or stroke). B, Cardiovascular death. Q indicates quartile; Lp‐PLA2; lipoprotein‐associated phospholipase A2.
Figure 2.
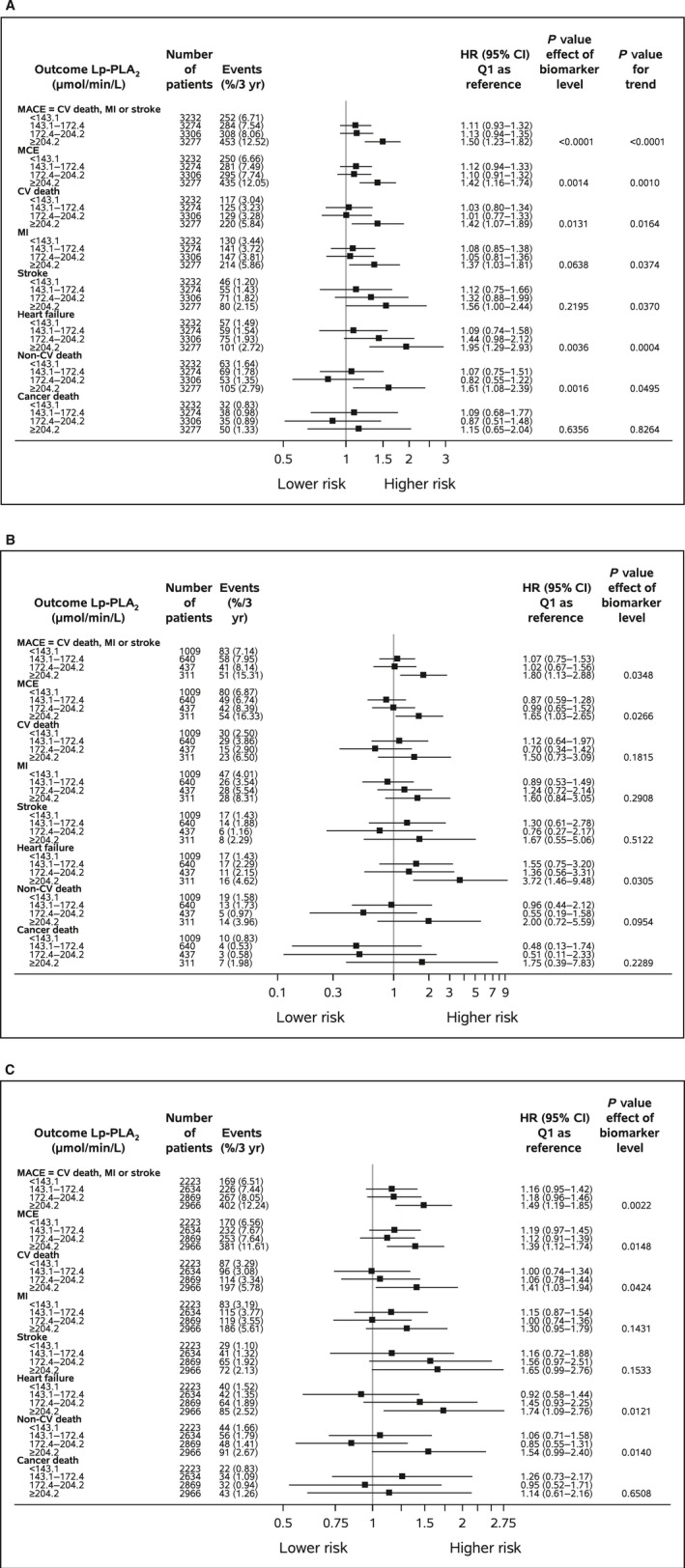
Baseline Lp‐PLA 2 activity (μmol/min per liter) quartile groups in relation to outcomes in (A) all patients, (B) in women, and (C) in men, adjusted for demographics, baseline characteristics, routine biochemical variables, and prognostic biomarkers. The x‐axis presents a logarithmic scale. MACE indicates CV death, MI, or stroke; MCE indicates coronary heart disease death, MI, or urgent coronary revascularization. Event rates are Kaplan–Meier rates. Adjustment variables: randomized treatment, geographic region, age, sex, body mass index, current smoking, hypertension, diabetes mellitus, prior MI, prior coronary revascularization, multivessel coronary heart disease, polyvascular disease, significant renal dysfunction, routine biochemical variables (hemoglobin, white blood cell count, estimated glomerular filtration rate [Chronic Kidney Disease Epidemiology Collaboration], low‐density lipoprotein cholesterol, high‐density lipoprotein cholesterol, and triglycerides), and prognostic biomarkers (N‐terminal proB‐type natriuretic peptide, high‐sensitivity cardiac troponin T, cystatin C, high‐sensitivity C‐reactive protein, and interleukin 6). CV indicates cardiovascular; HR, hazard ratio; Lp‐PLA2, lipoprotein‐associated phospholipase A2; MACE, major adverse cardiac events; MCE, major coronary events; MI, myocardial infarction; Q, quartile.
Table 3.
C‐Indices and Reclassification Statistics When Adding Information on the Lp‐PLA2 Activity Level to the Full Model of Baseline Characteristics and Other Biomarkers
| Outcome | C‐Index (95% CI) −Lp‐PLA2 | C‐Index (95% CI) +Lp‐PLA2 | Event NRI | Nonevent NRI | NRI |
|---|---|---|---|---|---|
| MACE | 0.675 (0.659–0.691) | 0.678 (0.662–0.694) | −0.074 | 0.248 | 0.175 |
| MCE | 0.658 (0.641–0.674) | 0.659 (0.643–0.676) | −0.074 | 0.238 | 0.164 |
| Cardiovascular death | 0.756 (0.733–0.780) | 0.757 (0.734–0.781) | −0.046 | 0.237 | 0.191 |
| MI | 0.665 (0.642–0.687) | 0.667 (0.645–0.689) | −0.099 | 0.239 | 0.140 |
| Stroke | 0.680 (0.646–0.714) | 0.686 (0.652–0.720) | 0.084 | 0.050 | 0.134 |
| Heart failure | 0.832 (0.804–0.860) | 0.833 (0.805–0.861) | 0.167 | 0.081 | 0.249 |
| Non‐cardiovascular death | 0.740 (0.708–0.772) | 0.743 (0.711–0.775) | 0.051 | 0.125 | 0.176 |
| Cancer death | 0.757 (0.716–0.798) | 0.758 (0.717–0.799) | 0.194 | −0.029 | 0.165 |
| Total death | 0.732 (0.714–0.751) | 0.735 (0.716–0.753) | −0.005 | 0.207 | 0.202 |
The full model contained the following data: randomized treatment, geographic region, age, sex, body mass index, current smoking, hypertension, diabetes mellitus, prior myocardial infarction, prior coronary revascularization, multivessel coronary heart disease, polyvascular disease, significant renal dysfunction, routine biochemical variables (hemoglobin, white blood cell count, estimated glomerular filtration rate [Chronic Kidney Disease Epidemiology Collaboration], low‐density lipoprotein cholesterol, high‐density lipoprotein cholesterol, and triglycerides), and prognostic biomarkers (N‐terminal proB‐type natriuretic peptide, high‐sensitivity cardiac troponin T, cystatin C, high‐sensitivity C‐reactive protein, and interleukin 6). Lp‐PLA2 indicates lipoprotein‐associated phospholipase A2; MACE, major adverse cardiac events; MCE, major coronary events; MI, myocardial infarction; NRI, net reclassification index.
At 1 month, the Lp‐PLA2 activity was reduced from a median of 172 to 57 μmol/min per liter (interquartile range 42–75 μmol/min per liter, mean reduction 112 μmol/min per liter, mean percentage reduction 64%) in the darapladib group compared with a median decrease from 173 to 164 μmol/min per liter (interquartile range 136–196 μmol/min per liter, mean reduction 9 μmol/min per liter, mean percentage reduction 4%) in the placebo group. Darapladib treatment produced a persistent ≈65% relative reduction in Lp‐PLA2 activity from the first on‐treatment measurement at 1 month until study termination, whereas the placebo group had no significant change in Lp‐PLA2 activity (Figure 3). The changes in Lp‐PLA2 activity were consistent in all subgroups based on clinical characteristics or biomarkers (Table 4).
Figure 3.
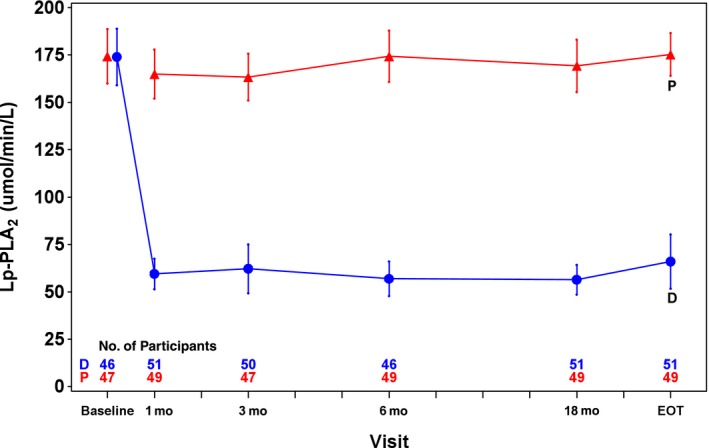
Lp‐PLA 2 activity (μmol/min per liter) over time during treatment with darapladib and placebo in a subset of 100 patients with available plasma samples for Lp‐PLA 2 activity measurements until end of follow‐up. Symbols illustrate means and CIs for the respective darapladib (blue) and placebo (red) groups at baseline; at follow‐up visits at 1, 3, 6, and 18 months; and at the EOT visit. D, darapladib; EOT, end of treatment; Lp‐PLA2, lipoprotein‐associated phospholipase A2; P, placebo.
Table 4.
Change in Lipoprotein‐Associated Phospholipase A2 Activity After 1 Month by Subgroup in Patients Assigned to Darapladib
| Subgroup | Darapladib | ||
|---|---|---|---|
| n | Mean Change | Mean Percentage Inhibition | |
| Prior MI | |||
| Yes | 4063 | −111.8 | 63.0 |
| No | 2773 | −112.9 | 64.3 |
| Prior coronary revascularization | |||
| Yes | 5306 | −112.7 | 64.0 |
| No | 1815 | −111.1 | 62.0 |
| Multivessel CHD | |||
| Yes | 974 | −116.7 | 65.0 |
| No | 5862 | −111.5 | 63.3 |
| Time from CHD event to randomization | |||
| Recent | 1628 | −110.0 | 63.1 |
| Remote | 5229 | −113.0 | 63.7 |
| Age ≥60 yr | |||
| Yes | 5055 | −112.5 | 64.5 |
| No | 1781 | −111.7 | 60.7 |
| Diabetes mellitus requiring pharmaceutical management | |||
| Yes | 2289 | −110.4 | 65.5 |
| No | 4547 | −113.2 | 62.5 |
| HDL‐C <40 mg/dL | |||
| Yes | 2285 | −118.9 | 63.1 |
| No | 4549 | −109.0 | 63.7 |
| Current/previous smoker | |||
| Yes | 1344 | −113.2 | 60.1 |
| No | 5465 | −112.0 | 64.4 |
| Significant renal dysfunction | |||
| Yes | 2068 | −117.5 | 66.3 |
| No | 4768 | −110.0 | 62.3 |
| Polyvascular disease | |||
| Yes | 1045 | −115.3 | 63.9 |
| No | 5791 | −111.7 | 63.4 |
| No. of additional predictors of cardiovascular risk | |||
| 1 | 2376 | −108.4 | 62.0 |
| 2 | 2371 | −112.2 | 63.3 |
| ≥3 | 2066 | −117.0 | 65.5 |
| Race group collapsed | |||
| White | 5533 | −112.2 | 62.6 |
| Nonwhite | 1303 | −112.5 | 67.2 |
| Region | |||
| North America | 1756 | −115.6 | 64.0 |
| Eastern Europe | 1632 | −109.6 | 60.4 |
| Western Europe | 1833 | −110.0 | 63.7 |
| South America | 403 | −114.0 | 62.2 |
| Asia–Pacific | 1212 | −114.0 | 67.2 |
| Group in United States | |||
| US | 1331 | −116.0 | 63.8 |
| Non‐US | 5505 | −111.4 | 63.4 |
| Sex | |||
| Male | 5623 | −114.8 | 63.1 |
| Female | 1213 | −100.8 | 65.4 |
| Blood pressure | |||
| High | 3409 | −113.8 | 64.3 |
| Target | 3425 | −110.8 | 62.8 |
| eGFR, mL/min/1.73 m2 | |||
| <60 | 937 | −121.0 | 67.3 |
| ≥60 | 5888 | −111.0 | 62.9 |
| LDL‐C, mmol/L | |||
| <1.80 | 2401 | −97.1 | 63.3 |
| ≥1.80 to <2.50 | 2723 | −111.8 | 63.6 |
| ≥2.58 | 1805 | −132.5 | 63.6 |
| Baseline Lp‐PLA2 activity | |||
| Tertile 1 | 2273 | −78.3 | 61.4 |
| Tertile 2 | 2342 | −110.4 | 64.0 |
| Tertile 3 | 2221 | −149.0 | 65.2 |
| High‐sensitivity C‐reactive protein, mg/L | |||
| <1.0 | 2516 | −108.9 | 63.9 |
| 1.0 to 3.0 | 2359 | −114.4 | 63.9 |
| >3.0 | 1667 | −115.5 | 62.9 |
| Family history of CHD | |||
| Yes | 1750 | −113.4 | 63.0 |
| No | 5069 | −111.8 | 63.7 |
| Body mass index, kg/m2 | |||
| <25 | 1380 | −113.5 | 66.3 |
| 25 to <30 | 2910 | −113.6 | 63.6 |
| ≥30 | 2536 | −110.1 | 61.9 |
| Waist/hip ratio | |||
| Level 1 | 851 | −110.8 | 65.3 |
| Level 2 | 1681 | −113.9 | 64.9 |
| Level 3 | 4234 | −112.1 | 62.6 |
| Aspirin use | |||
| Yes | 6293 | −111.7 | 63.4 |
| No | 568 | −118.9 | 64.5 |
| Statin use | |||
| Yes | 6657 | −111.7 | 63.5 |
| No | 179 | −133.9 | 64.4 |
| Beta blocker use | |||
| Yes | 5393 | −111.6 | 63.2 |
| No | 1443 | −114.7 | 64.8 |
| P2Y12 use | |||
| Yes | 2267 | −110.4 | 63.9 |
| No | 4569 | −113.2 | 63.3 |
| ACEI/ARB use | |||
| Yes | 5291 | −112.7 | 63.9 |
| No | 1545 | −110.9 | 62.3 |
ACEI indicates angiotensin‐converting enzyme inhibitor; ARB, angiotensin receptor blocker; CHD, coronary heart disease; HDL‐C, high‐density lipoprotein cholesterol; MI, myocardial infarction.
In the highest Lp‐PLA2 quartile group, there was no significant reduction in the primary composite end point of cardiovascular death, MI, or stroke (HR 0.86, 95% CI 0.72–1.03), whereas the secondary composite end point of major coronary events showed a statistically significant reduction (HR 0.82, 95% CI 0.68–0.98); however, there were no significant interactions between quartile groups of baseline Lp‐PLA2 activity and the effects of darapladib (Figures 4 and 5). Finally, there were no significant associations between either the level of Lp‐PLA2 activity at 1 month or the reduction in Lp‐PLA2 activity and any of the outcome events in the trial (Tables 5, 6, 7 through 8).
Figure 4.
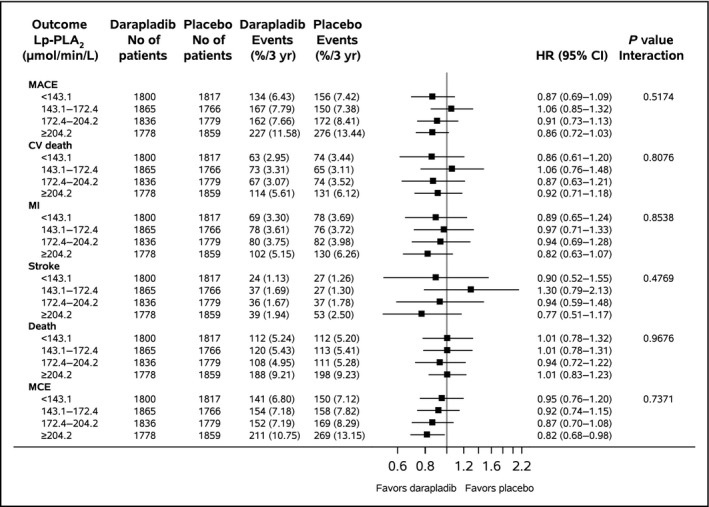
Effect of darapladib compared with placebo in relation to baseline Lp‐PLA 2 activity (μmol/min per liter) quartile groups concerning all outcomes. MACE specifies CV death, MI, or stroke; MCE specifies coronary heart disease death, MI, or urgent coronary revascularization. Event rates are Kaplan–Meier rates. CV indicates cardiovascular; HR, hazard ratio; Lp‐PLA2, lipoprotein‐associated phospholipase A2; MACE, major adverse cardiac events; MCE, major coronary events; MI, myocardial infarction.
Figure 5.
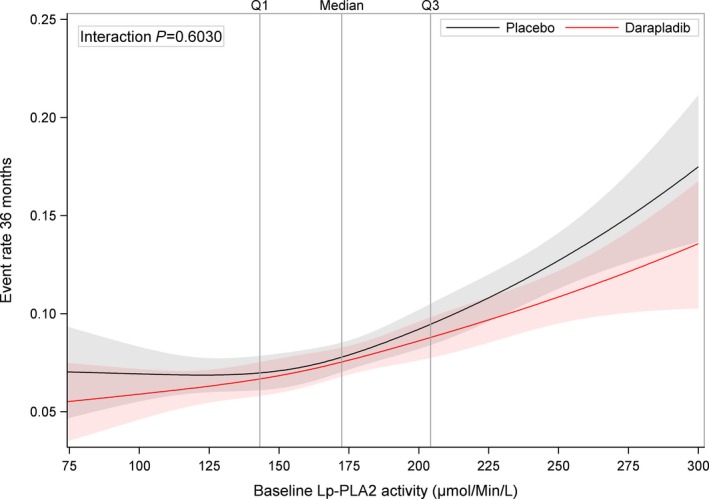
Continuous Lp‐PLA 2 activity (μmol/min per liter) at baseline in relation to major adverse cardiac events (cardiovascular death, myocardial infarction, or stroke) during treatment with darapladib (red line) and placebo (black line) using restricted cubic splines using a Cox proportional hazards model including treatment group, LpPLA 2, and treatment by LpPLA 2 interaction as covariates. Lp‐PLA2, lipoprotein‐associated phospholipase A2; Q, quartile.
Table 5.
Changes in Lp‐PLA2 Activity at 1 Montha Compared With Baseline in Relation to Outcomes in the Respective Placebo and Darapladib Groups
| Outcome Events | Placebo Change | Darapladib Change | Change From Baseline | ||||||
|---|---|---|---|---|---|---|---|---|---|
| n | Mean | % | n | Mean | % | HR for 10‐U Increase (95% CI) | P Value | ||
| MACEb | Yes | 706 | −10.9 | 5.1 | 646 | −121.7 | 65.5 | 0.99 (0.97–1.01) | 0.258 |
| No | 6167 | −8.5 | 4.1 | 6190 | −111.3 | 63.3 | |||
| MCEc | Yes | 699 | −9.7 | 4.4 | 614 | −120.4 | 65.1 | 1.00 (0.98–1.02) | 0.919 |
| No | 6174 | −8.7 | 4.1 | 6222 | −111.5 | 63.3 | |||
| Total coronary events | Yes | 1106 | −9.1 | 4.1 | 976 | −117.2 | 64.3 | 1.01 (0.99–1.02) | 0.451 |
| No | 5767 | −8.7 | 4.2 | 5860 | −111.5 | 63.4 | |||
| Cardiovascular death | Yes | 323 | −12.0 | 5.5 | 291 | −122.4 | 65.4 | 0.99 (0.96–1.01) | 0.335 |
| No | 6550 | −8.6 | 4.1 | 6545 | −111.8 | 63.4 | |||
| Myocardial infarction | Yes | 341 | −9.1 | 4.3 | 311 | −121.6 | 66.0 | 0.99 (0.97–1.02) | 0.571 |
| No | 6532 | −8.8 | 4.2 | 6525 | −111.8 | 63.4 | |||
| Stroke | Yes | 136 | −13.2 | 6.3 | 129 | −117.7 | 64.6 | 0.99 (0.96–1.03) | 0.724 |
| No | 6737 | −8.7 | 4.1 | 6707 | −112.2 | 63.5 | |||
| Hospitalization for heart failure | Yes | 160 | −10.4 | 4.5 | 136 | −127.5 | 66.8 | 0.97 (0.94–1.01) | 0.122 |
| No | 6713 | −8.7 | 4.2 | 6700 | −112.0 | 63.4 | |||
| All‐cause mortality | Yes | 498 | −11.3 | 5.0 | 478 | −120.9 | 65.4 | 0.99 (0.97–1.01) | 0.223 |
| No | 6375 | −8.6 | 4.1 | 6358 | −111.6 | 63.4 | |||
Event rates are Kaplan‐Meier rates. Lp‐PLA2 indicates lipoprotein‐associated phospholipase A2; MACE, major adverse cardiac events; MCE, major coronary events.
The analyses are adjusted for baseline Lp‐PLA2 and stratified by randomized treatment. The changes from baseline results are identical to the Month 1 results as both are adjusted for the baseline value.
MACE: cardiovascular death, myocardial infarction, or stroke.
MCE: coronary heart disease death, myocardial infarction, or urgent coronary revascularization.
Table 6.
Range of Change of Lp‐PLA2 Activity Level From Baseline to 1 Month in All Patients With Lp‐PLA2 Measurements at 1 Month (Including Both Darapladib and Placebo Groups) Per Decile Group
| Decile | Change From Baseline Lp‐PLA2 (μmol/min/L) |
|---|---|
| 1 | ≤−146.1 |
| 2 | >−146.1 to ≤−121.4 |
| 3 | >−121.4 to ≤−101.9 |
| 4 | >−101.9 to ≤−80.9 |
| 5 | >−80.9 to ≤−46.1 |
| 6 | >−46.1 to ≤−21.8 |
| 7 | >−21.8 to ≤−10.9 |
| 8 | >−10.9 to ≤−2.1 |
| 9 | >−2.1 to ≤8.6 |
| 10 | >8.6 |
Lp‐PLA2 indicates lipoprotein‐associated phospholipase A2.
Table 7.
Range of Percentage Change of Lp‐PLA2 Activity Level at 1 Month Compared With Baseline in All Patients With Lp‐PLA2 Measurements at 1 Month (Including Both Darapladib and Placebo Groups) Per Decile Group
| Decile | Percentage Inhibition of Lp‐PLA2 (μmol/min/L) |
|---|---|
| 1 | ≤−5.3 |
| 2 | >−5.3 to ≤1.3 |
| 3 | >1.3 to ≤6.5 |
| 4 | >6.5 to ≤12.3 |
| 5 | >12.3 to ≤25.7 |
| 6 | >25.7 to ≤55.3 |
| 7 | >55.3 to ≤63.4 |
| 8 | >63.4 to ≤69.5 |
| 9 | >69.5 to ≤76.2 |
| 10 | >76.2 |
Lp‐PLA2 indicates lipoprotein‐associated phospholipase A2.
Table 8.
Range of Lp‐PLA2 Activity Level at 1 Month in All Patients With Lp‐PLA2 Measurements at 1 Month (Including Both Darapladib and Placebo Groups) Per Decile Group
| Decile | Levels at 1 Month Lp‐PLA2 (μmol/min/L) |
|---|---|
| 1 | ≤37.9 |
| 2 | >37.9 to ≤50.9 |
| 3 | >50.9 to ≤63.3 |
| 4 | >63.3 to ≤78.5 |
| 5 | >78.5 to ≤106.9 |
| 6 | >106.9 to ≤134.7 |
| 7 | >134.7 to ≤156.4 |
| 8 | >156.4 to ≤178.1 |
| 9 | >178.1 to ≤205.6 |
| 10 | >205.6 |
Lp‐PLA2 indicates lipoprotein‐associated phospholipase A2.
Discussion
This prespecified analysis of the STABILITY trial in patients with stable CHD followed for 3.7 years on a background of optimal medical therapy verified that Lp‐PLA2 activity is a marker of overall cardiovascular risk and total mortality in patients with stable CHD, in accordance with previous studies.6, 7, 8, 9 Dyslipidemia, male sex, current smoking, and living in the North American region were associated with higher Lp‐PLA2 activity, in agreement with other observations.21 After adjusting for both baseline characteristics and other prognostic biomarkers, Lp‐PLA2 activity remained a significant marker of composite cardiovascular events, hospitalization for heart failure, and cardiovascular and total mortality, whereas the associations with new ischemic events such as MI and stroke were attenuated. The finding of an independent association between Lp‐PLA2 activity and composite cardiovascular events is in accordance with most 6, 7, 8, 9, 22, 23, 24, 25, 26, 27 although not all28 previous studies of stable patients at high risk for or with established CHD. The rather weak independent association with a raised risk of cardiovascular events when adjusting for other biomarkers might be a corollary to the lack of association between Lp‐PLA2 activity and outcomes in patients with acute coronary syndromes having pronounced elevations of other prognostic biomarkers.29
Darapladib treatment persistently reduced Lp‐PLA2 activity by ≈65% in all patient groups, consistent with other findings.16, 18, 29 Reduction in Lp‐PLA2 activity was not associated with any significant effect on the primary composite outcome of cardiovascular death, MI, or stroke in the STABILITY trial overall or with any subgroup based on the Lp‐PLA2 activity level at baseline. This lack of effect in the overall trial and in all Lp‐PLA2 activity subgroups is in agreement with the findings from the SOLID trial of patients with recent acute coronary syndromes.29 In addition, in the present study, there were no significant associations between any outcome and the magnitude of reduction in Lp‐PLA2 activity level by darapladib treatment. These accumulated experiences show that although high Lp‐PLA2 activity was associated with an increased risk of cardiovascular events, pharmacological lowering of Lp‐PLA2 activity was not a useful treatment target for prevention of cardiovascular events in patients with stable CHD.
Prior to the current study, several lines of evidence supported Lp‐PLA2 activity as a risk indicator of adverse outcomes both in patients with stable CHD and in the general population in addition to being potentially involved in the development of atherosclerosis and vulnerable plaque at the tissue level.6, 7, 8, 9, 22, 23, 24, 25, 26, 27 Furthermore, in 2 case–control studies, natural deficiency of Lp‐PLA2 activity due to carriage of the V279F‐null allele in the Lp‐PLA2 gene was associated with a lower risk of developing CHD.30 In a recent study in European CHD patients, a single‐nucleotide polymorphism of the Lp‐PLA2 gene was found to be associated with both increased Lp‐PLA2 activity in plasma and a raised risk of MI.31 Lp‐PLA2 has also been shown to be highly concentrated in unstable and ruptured atherosclerotic plaques and to be strongly expressed in macrophages in lesions prone to rupture.16 Direct inhibition of Lp‐PLA2 activity prevented progression to advanced coronary atherosclerotic lesions in diabetic and hypercholesterolemic swine.17 Finally, in a phase II trial, darapladib appeared to stabilize the necrotic core size and plaque burden in coronary atherosclerotic plaques compared with placebo18; therefore, it was a logical hypothesis that inhibition of Lp‐PLA2 activity with darapladib might lead to clinical benefit, especially in patients with high Lp‐PLA2 activity at baseline and/or a pronounced reduction by the treatment.27 Nevertheless, this current study, based on 3 to 5 years darapladib treatment and Lp‐PLA2 activity levels that were available for ≈14 500 patients before and during treatment and with a consistent ≈65% reduction of the Lp‐PLA2 activity in all patient groups, showed no associated effects of darapladib on clinical outcomes. This finding clearly refutes any clinically important effects of darapladib for prevention of cardiovascular events in patients with CHD.
The current findings corroborate other studies showing that patients with stable CHD and high Lp‐PLA2 activity have a raised risk of cardiovascular events and total mortality. The increase in risk with Lp‐PLA2 activity was not linear but was most pronounced in the ≈25% of patients with the highest levels. The increased risk associated with high Lp‐PLA2 activity was independent of demographics, clinical characteristics, and conventional biochemical risk indicators such as dyslipidemia, dysglycemia, anemia, and renal dysfunction. When adjusting for cardiac, inflammatory, and renal function biomarkers, the independent associations between Lp‐PLA2 activity and cardiovascular and total mortality and hospitalization for heart failure remained, whereas the associations with ischemic events were attenuated. Similarly, there was no association between Lp‐PLA2 activity and cardiovascular outcomes in patients with acute coronary syndromes, in which the majority have elevated cardiac and inflammatory markers.29 Without an associated indication for a specific treatment, it is unlikely that Lp‐PLA2 activity will be used for identification of patients at high risk, considering the competition with other already generally available and more specific prognostic biomarkers. In the present study, as in a previous study,32 there was an inverse relationship between Lp‐PLA2 activity and diabetes mellitus. In that study, diabetes mellitus was associated with a redistribution of Lp‐PLA2 activity from low‐ to high‐density lipoprotein particles.32 These complex interactions among Lp‐PLA2 activity, glucose, and lipoprotein metabolism might need to be considered in future studies of the importance of Lp‐PLA2 activity for cardiovascular events in different patient populations.
Conclusions
Based on the experiences from 3 to 5 years of darapladib treatment and available Lp‐PLA2 activity levels before and during treatment in 14 500 patients with stable CHD, high pretreatment Lp‐PLA2 activity was associated with an increased risk of cardiovascular events; however, pharmacological lowering of Lp‐PLA2 activity did not significantly reduce cardiovascular events, regardless of the baseline level or degree of reduction of Lp‐PLA2 activity.
Appendix
Contributors
The authors represent the STABILITY Investigators: Harvey D. White (Green Lane Cardiovascular Service, Auckland City Hospital, and Auckland University, Auckland, NZ), Lars Wallentin (Department of Medical Sciences and Uppsala Clinical Research Center, Uppsala University, Uppsala, SE), Andrzej Budaj (Grochowski Hospital, Warsaw, PL), Christopher P. Cannon (TIMI Study Group, Brigham and Women's Hospital, Boston, MA, US), Robert A. Harrington (Stanford University, Stanford, CA, US), Philippe Gabriel Steg (INSERM‐Unité, AP‐HP; Hôpital Bichat; and Université Paris‐Diderot, Paris, FR; Royal Brompton Hospital, London, UK), Richard Y. Davies (GlaxoSmithKline, King of Prussia, PA, US), Elizabeth Tarka (former employee of GlaxoSmithKline, King of Prussia, PA, US), Diego Ardissino (Azienda Ospedaliero‐Universitaria di Parma, Parma, IT), Paul W. Armstrong (University of Alberta, Edmonton, CA, US), Alvaro Avezum (Dante Pazzanese Institute of Cardiology, São Paulo, BR), Philip E. Aylward (South Australian Health and Medical Research Institute, Flinders University and Medical Centre, Adelaide, AU), Alfonso Bryce (Cardiogolf/Clinica El Golf, Lima, PE), Hong Chen (Peking University People's Hospital, Beijing, CN), Ming‐Fong Chen (National Taiwan University Hospital, Taipei, TW), Ramon Corbalan (Pontificia Universidad Catolica de Chile, Santiago, CL), Anthony J. Dalby (Milpark Hospital, Johannesburg, ZA), Nicolas Danchin (AP‐HP and Université Paris Descartes, Paris, FR), Robbert J. De Winter (University of Amsterdam, Amsterdam, NL), Stefan Denchev (University Hospital Alexandrovska, Sofia, BG), Rafael Diaz (ECLA Estudios Cardiológicos, Latinoamérica, Rosario, AR), Moses Elisaf (University of Ioannina, Ioannina, GR), Marcus D. Flather (University of East Anglia and Norfolk and Norwich University Hospital, UK), Assen R. Goudev (Queen Giovanna University Hospital, Sofia, BG), Christopher B. Granger (Duke University, Medical Center, Durham, NC, US), Liliana Grinfeld (University of Buenos Aires, School of Medicine, Buenos Aires, AR), Claes Held (Department of Medical Sciences, Cardiology, and Uppsala Clinical Research Center, Uppsala University, Uppsala, SE), Judith S. Hochman (NYU Langone Medical Center, New York, NY, US), Steen Husted (Hospital Unit West, Herning/Holstebro, DK), Hyo‐Soo Kim (Seoul National University Hospital, Seoul, KR), Wolfgang Koenig (University of Ulm Medical Center, Ulm, DE), Ales Linhart (Charles University in Prague, Prague, CZ), Eva Lonn (McMaster University, Hamilton, Ontario, CA), José López‐Sendón (Hospital Universitario La Paz, Madrid, ES), Athanasios J. Manolis (Asklepeion Hospital, Athens, GR), Emile R. Mohler, III (University of Pennsylvania, Philadelphia, PA, US), José C. Nicolau (University of São Paulo Medical School, São Paulo, BR), Prem Pais (St. John's Medical Collage, Bangalore, IN), Alexander Parkhomenko (Institute of Cardiology, Kiev, UA), Terje R. Pedersen (University of Oslo and Oslo University Hospital, Oslo, NO), Daniel Pella (PJ Safarik University, Kosice, SK), Marco A. Ramos‐Corrales (San Jose Satelite Hospital, Naucalpan, MX), Mikhail Ruda (Russian Cardiologic Research and Production Complex of Rosmedtechnology, Moscow, RU), Mátyás Sereg (St. George Hospital, Székesfehérvár, HU), Saulat Siddique (Shaikh Zayed Postgraduate Medical Institute, Lahore, PK), Peter Sinnaeve (University Hospitals Leuven, Leuven, BE), Piyamitr Sritara (Mahidol University, Bangkok, TH), Ralph A. H. Stewart (Green Lane Cardiovascular Service, Auckland City Hospital, and Auckland University, Auckland, NZ), Henk P. Swart (Antonius Hospital Sneek, NL), Rody G. Sy (University of the Philippines, Manila, PH), Tamio Teramoto (Teikyo Academic Research Center, Itabashi‐ku, Tokyo, JP), Hung‐Fat Tse (University of Hong Kong, Hong Kong SAR, CN), W. Douglas Weaver (Henry Ford Heart and Vascular Institute, Wayne State University, Detroit, MI, US), Robert Weiss (Maine Research Associates, Auburn, ME, US), Margus Viigimaa, (Tallinn University of Technology, Tallinn, EE), Dragos Vinereanu (University of Medicine and Pharmacy, University and Emergency Hospital, Bucharest, RO), Junren Zhu (Fudan University, Shanghai, CN).
Sources of Funding
The STABILITY trial was funded by GlaxoSmithKline, Philadelphia, PA, USA. The growth differentiation factor 15 assays were provided free of charge by Roche Diagnostics, Penzberg, Germany.
Disclosures
Wallentin reports institutional research grants, honoraria, consultancy fees, travel support and lecture fees from GlaxoSmithKline, during the conduct of the study; institutional research grants, consultancy fees, travel support and lecture fees from AstraZeneca, Bristol‐Myers Squibb/Pfizer; institutional research grants, consultancy fees, and lecture fees from Boehringer Ingelheim; institutional research grants from Merck & Co., Roche; consultancy fees from Abbott, holds 2 patents involving growth differentiation factor 15, outside the submitted work. Held reports institutional research grant from GlaxoSmithKline, during the conduct of the study; institutional research grants from Merck, Roche, Bristol‐Myers Squibb; grants, advisory board member and lecture fees from AstraZeneca, outside the submitted work. Armstrong reports grants and consultant fees from GlaxoSmithKline, during the conduct of the study; grants from Regado Biosciences, Sanofi Aventis, Amylin Pharmaceuticals; consultant fees from Eli Lilly, F. Hoffmann La Roche, Axio Research/Orexigen, GlaxoSmithKline, Bayer, AstraZeneca; grants and consultant fees from Boehringer Ingelheim, Merck, outside the submitted work. Cannon reports research grants and consultant fees from GlaxoSmithKline, during the conduct of the study; research grants and travel support from AstraZeneca; research grants, travel support and consultant fees from Boerhinger Ingelheim; research grants and consultant fees from Takeda, Merck; research grants and personal fees from Accumetrics; research grants from Arisaph, Janssen; consultant fees from Bristol‐Myers Squibb, Alnylam, Pfizer, Essentialis, Kowa, Lipimedix, Regeneron, Sanofi; personal fees from CSL Behring, outside the submitted work. Davies is an employee of and has stock ownership in GlaxoSmithKline. Granger reports grants and consultancy fees from GlaxoSmithKline, during the conduct of the study; grants and consultancy fees from AstraZeneca, Boehringer Ingelheim, Bristol‐Myers Squibb, Daiichi Sankyo, Pfizer, Sanofi‐Aventis, Takeda, The Medicines Company, Janssen, Bayer; grants from Medtronics Foundation, Armetheon; consultancy fees from Hoffmann‐La Roche, Salix Pharmaceuticals, Gilead, Medtronic Inc., outside the submitted work. Hagström reports an institutional research grant from GlaxoSmithKline during the conduct of the study; institutional research grants from AstraZeneca, Amgen, Sanofi; expert committee member for Sanofi, Ariad, MSD; lecture fees from Amgen, outside the submitted work. Harrington reports research grants from GlaxoSmithKline, during the conduct of the study; research grants from AstraZeneca, CSL Behring, Merck, Portola, Regado, Sanofi, TMC; consultant/advisory relationship to Amgen, Gilead, Merck, MyoKardia, TMC, WebMD; stockholder in Element Sciences, Scanadu, Signal Path; non‐commercial relationship with American Heart Association, outside the submitted work. Hochman reports travel reimbursement from GlaxoSmithKline, during the conduct of the study. Koenig reports consultancy fees from GlaxoSmithKline, during the conduct of the study; research grants from Roche Diagnostics, Abbott, Singulex, Beckmann; lecture fees and consultancy fees from Novartis, Amgen, AstraZeneca, Servier; lecture fees from Actavis; consultancy fees from BioInvent, diaDexus, Cerenis, The Medicines Company, Genzyme, Pfizer, Merck Sharpe & Dohme, outside the submitted work. Krug‐Gourley is an employee of and has stock ownership in GlaxoSmithKline. Mohler reports grants and personal fees from GlaxoSmithKline, during the conduct of the study. Siegbahn reports an institutional research grant from GlaxoSmithKline, during the conduct of the study; institutional research grants from AstraZeneca, Boehringer Ingelheim, Bristol‐Myers Squibb/Pfizer, outside the submitted work. Tarka is a former employee of and has stock ownership in GlaxoSmithKline. Steg reports honoraria from GlaxoSmithKline, during the conduct of the study; research grants, honoraria and non‐financial support from Sanofi, Servier; honoraria and non‐financial support from AstraZeneca; honoraria from Amarin, Bayer, Boehringer Ingelheim, Bristol‐Myers Squibb, Daiichi‐Sankyo, Eli Lilly, Merck‐Sharpe‐Dohme, Novartis, Pfizer, Medtronic, Janssen, The Medicines Company, CSL‐Behring, outside the submitted work; Dr Steg is a stockholder in Aterovax, which among other activities, holds a patent for the use of plasma levels of sPLA2 to determine prognosis in patients at risk of cardiovascular events. He received royalties from that company. sPLA2 is distinct but somewhat related to Lp‐PLA2, which is targeted by darapladib, the experimental drug used in this trial. Stewart reports grants and non‐financial support from GlaxoSmithKline, during the conduct of the study. Weiss reports grants from Amgen, Sanofi, Pfizer, Daiichi, outside the submitted work. Östlund reports institutional research grants from GlaxoSmithKline, during the conduct of the study. White reports research grants and personal fees from GlaxoSmithKline, during the conduct of the study; research grants and consultancy fees from Daiichi‐Sankyo Pharma Development; research grants and advisory board member for AstraZeneca; research grants from Sanofi‐Aventis, Eli Lilly, National Institute of Health, Merck Sharp & Dohme, outside the submitted work.
Acknowledgments
Ebba Bergman, PhD, and Sanne Carlsson, BA, BSc, at Uppsala Clinical Research Center provided editorial assistance.
(J Am Heart Assoc. 2016;5:e003407 doi: 10.1161/JAHA.116.003407)
Results of this study were presented in part at the European Society of Cardiology Congress, August 30 to September 3, 2014, in Barcelona, Spain.
References
- 1. Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. [DOI] [PubMed] [Google Scholar]
- 2. Ridker PM, Wilson PW, Grundy SM. Should C‐reactive protein be added to metabolic syndrome and to assessment of global cardiovascular risk? Circulation. 2004;109:2818–2825. [DOI] [PubMed] [Google Scholar]
- 3. Omland T, Pfeffer MA, Solomon SD, de Lemos JA, Rosjo H, Saltyte Benth J, Maggioni A, Domanski MJ, Rouleau JL, Sabatine MS, Braunwald E; PEACE Investigators . Prognostic value of cardiac troponin I measured with a highly sensitive assay in patients with stable coronary artery disease. J Am Coll Cardiol. 2013;61:1240–1249. [DOI] [PubMed] [Google Scholar]
- 4. Kragelund C, Gronning B, Kober L, Hildebrandt P, Steffensen R. N‐terminal pro‐B‐type natriuretic peptide and long‐term mortality in stable coronary heart disease. N Engl J Med. 2005;352:666–675. [DOI] [PubMed] [Google Scholar]
- 5. Jernberg T, Lindahl B, James S, Larsson A, Hansson LO, Wallentin L. Cystatin C: a novel predictor of outcome in suspected or confirmed non‐ST‐elevation acute coronary syndrome. Circulation. 2004;110:2342–2348. [DOI] [PubMed] [Google Scholar]
- 6. The Lp‐PLA2 Studies Collaboration . Lipoprotein‐associated phospholipase A(2) and risk of coronary disease, stroke, and mortality: collaborative analysis of 32 prospective studies. Lancet. 2010;375:1536–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kleber ME, Siekmeier R, Delgado G, Grammer TB, Winkelmann BR, Scharnagl H, Boehm BO, März W. C‐reactive protein and lipoprotein‐associated phospholipase A2 in smokers and nonsmokers of the Ludwigshafen Risk and Cardiovascular Health study. Adv Exp Med Biol. 2015;832:15–23. [DOI] [PubMed] [Google Scholar]
- 8. Di Angelantonio E, Gao P, Pennells L, Kaptoge S, Caslake M, Thompson A, Butterworth AS, Sarwar N, Wormser D, Saleheen D, Ballantyne CM, Psaty BM, Sundström J, Ridker PM, Nagel D, Gillum RF, Ford I, Ducimetiere P, Kiechl S, Koenig W, Dullaart RP, Assmann G, D'Agostino RB Sr, Dagenais GR, Cooper JA, Kromhout D, Onat A, Tipping RW, Gómez‐de‐la‐Cámara A, Rosengren A, Sutherland SE, Gallacher J, Fowkes FG, Casiglia E, Hofman A, Salomaa V, Barrett‐Connor E, Clarke R, Brunner E, Jukema JW, Simons LA, Sandhu M, Wareham NJ, Khaw KT, Kauhanen J, Salonen JT, Howard WJ, Nordestgaard BG, Wood AM, Thompson SG, Boekholdt SM, Sattar N, Packard C, Gudnason V, Danesh J. Lipid‐related markers and cardiovascular disease prediction. JAMA. 2012;307:2499–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kleber ME, Wolfert RL, De Moissl GD, Grammer TB, Dietz S, Winkelmann BR, Boehm BO, März W. Lipoprotein associated phospholipase A2 concentration predicts total and cardiovascular mortality independently of established risk factors (The Ludwigshafen Risk and Cardiovascular Health Study). Clin Lab. 2011;57:659–667. [PubMed] [Google Scholar]
- 10. Mallat Z, Lambeau G, Tedgui A. Lipoprotein‐associated and secreted phospholipases A(2) in cardiovascular disease: roles as biological effectors and biomarkers. Circulation. 2010;122:2183–2200. [DOI] [PubMed] [Google Scholar]
- 11. Hakkinen T, Luoma JS, Hiltunen MO, Macphee CH, Milliner KJ, Patel L, Rice SQ, Tew DG, Karkola K, Ylä‐Herttuala S. Lipoprotein‐associated phospholipase A(2), platelet‐activating factor acetylhydrolase, is expressed by macrophages in human and rabbit atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 1999;19:2909–2917. [DOI] [PubMed] [Google Scholar]
- 12. Liu J, Wang W, Qi Y, Yong Q, Zhou G, Wang M, Sun J, Liu J, Jia Z, Zhao D. Association between the lipoprotein‐associated phospholipase A2 activity and the progression of subclinical atherosclerosis. J Atheroscler Thromb. 2014;21:532–542. [PubMed] [Google Scholar]
- 13. MacPhee CH, Moores KE, Boyd HF, Dhanak D, Ife RJ, Leach CA, Leake DS, Milliner KJ, Patterson RA, Suckling KE, Tew DG, Hickey DM. Lipoprotein‐associated phospholipase A2, platelet‐activating factor acetylhydrolase, generates two bioactive products during the oxidation of low‐density lipoprotein: use of a novel inhibitor. Biochem J. 1999;338(Pt 2):479–487. [PMC free article] [PubMed] [Google Scholar]
- 14. Mohler ER III, Ballantyne CM, Davidson MH, Hanefeld M, Ruilope LM, Johnson JL, Zalewski A; Darapladib Investigators . The effect of darapladib on plasma lipoprotein‐associated phospholipase A2 activity and cardiovascular biomarkers in patients with stable coronary heart disease or coronary heart disease risk equivalent: the results of a multicenter, randomized, double‐blind, placebo‐controlled study. J Am Coll Cardiol. 2008;51:1632–1641. [DOI] [PubMed] [Google Scholar]
- 15. Johnson JL, Shi Y, Snipes R, Janmohamed S, Rolfe TE, Davis B, Postle A, Macphee CH. Effect of darapladib treatment on endarterectomy carotid plaque lipoprotein‐associated phospholipase A2 activity: a randomized, controlled trial. PLoS One. 2014;9:e89034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kolodgie FD, Virmani R, Burke AP, Farb A, Weber DK, Kutys R, Finn AV, Gold HK. Pathologic assessment of the vulnerable human coronary plaque. Heart. 2004;90:1385–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wilensky RL, Shi Y, Mohler ER III, Hamamdzic D, Burgert ME, Li J, Postle A, Fenning RS, Bollinger JG, Hoffman BE, Pelchovitz DJ, Yang J, Mirabile RC, Webb CL, Zhang L, Zhang P, Gelb MH, Walker MC, Zalewski A, Macphee CH. Inhibition of lipoprotein‐associated phospholipase A2 reduces complex coronary atherosclerotic plaque development. Nat Med. 2008;14:1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Serruys PW, Garcia‐Garcia HM, Buszman P, Erne P, Verheye S, Aschermann M, Duckers H, Bleie O, Dudek D, Bøtker HE, von Birgelen C, D'Amico D, Hutchinson T, Zambanini A, Mastik F, van Es GA, van der Steen AF, Vince DG, Ganz P, Hamm CW, Wijns W, Zalewski A; Integrated Biomarker and Imaging Study‐2 Investigators . Effects of the direct lipoprotein‐associated phospholipase A(2) inhibitor darapladib on human coronary atherosclerotic plaque. Circulation. 2008;118:1172–1182. [DOI] [PubMed] [Google Scholar]
- 19. White HD, Held C, Stewart R, Tarka E, Brown R, Davies RY, Budaj A, Harrington RA, Steg PG, Ardissino D, Armstrong PW, Avezum A, Aylward PE, Bryce A, Chen H, Chen MF, Corbalan R, Dalby AJ, Danchin N, De Winter RJ, Denchev S, Diaz R, Elisaf M, Flather MD, Goudev AR, Granger CB, Grinfeld L, Hochman JS, Husted S, Kim HS, Koenig W, Linhart A, Lonn E, López‐Sendón J, Manolis AJ, Mohler ER III, Nicolau JC, Pais P, Parkhomenko A, Pedersen TR, Pella D, Ramos‐Corrales MA, Ruda M, Sereg M, Siddique S, Sinnaeve P, Smith P, Sritara P, Swart HP, Sy RG, Teramoto T, Tse HF, Watson D, Weaver WD, Weiss R, Viigimaa M, Vinereanu D, Zhu J, Cannon CP, Wallentin L. Darapladib for preventing ischemic events in stable coronary heart disease. N Engl J Med. 2014;370:1702–1711. [DOI] [PubMed] [Google Scholar]
- 20. Wallentin L, Zethelius B, Berglund L, Eggers KM, Lind L, Lindahl B, Wollert KC, Siegbahn A. GDF‐15 for prognostication of cardiovascular and cancer morbidity and mortality in men. PLoS One. 2013;8:e78797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gregson J, Stirnadel‐Farrant HA, Doobaree IU, Koro C. Variation of lipoprotein associated phospholipase A2 across demographic characteristics and cardiovascular risk factors: a systematic review of the literature. Atherosclerosis. 2012;225:11–21. [DOI] [PubMed] [Google Scholar]
- 22. Koenig W, Twardella D, Brenner H, Rothenbacher D. Lipoprotein‐associated phospholipase A2 predicts future cardiovascular events in patients with coronary heart disease independently of traditional risk factors, markers of inflammation, renal function, and hemodynamic stress. Arterioscler Thromb Vasc Biol. 2006;26:1586–1593. [DOI] [PubMed] [Google Scholar]
- 23. Corsetti JP, Rainwater DL, Moss AJ, Zareba W, Sparks CE. High lipoprotein‐associated phospholipase A2 is a risk factor for recurrent coronary events in postinfarction patients. Clin Chem. 2006;52:1331–1338. [DOI] [PubMed] [Google Scholar]
- 24. Sabatine MS, Morrow DA, O'Donoghue M, Jablonksi KA, Rice MM, Solomon S, Rosenberg Y, Domanski MJ, Hsia J; PEACE Investigators . Prognostic utility of lipoprotein‐associated phospholipase A2 for cardiovascular outcomes in patients with stable coronary artery disease. Arterioscler Thromb Vasc Biol. 2007;27:2463–2469. [DOI] [PubMed] [Google Scholar]
- 25. Ridker PM, MacFadyen JG, Wolfert RL, Koenig W. Relationship of lipoprotein‐associated phospholipase A(2) mass and activity with incident vascular events among primary prevention patients allocated to placebo or to statin therapy: an analysis from the JUPITER trial. Clin Chem. 2012;58:877–886. [DOI] [PubMed] [Google Scholar]
- 26. O'Donoghue M, Morrow DA, Sabatine MS, Murphy SA, McCabe CH, Cannon CP, Braunwald E. Lipoprotein‐associated phospholipase A2 and its association with cardiovascular outcomes in patients with acute coronary syndromes in the PROVE IT‐TIMI 22 (PRavastatin Or atorVastatin Evaluation and Infection Therapy‐Thrombolysis In Myocardial Infarction) trial. Circulation. 2006;113:1745–1752. [DOI] [PubMed] [Google Scholar]
- 27. White HD, Simes J, Stewart RA, Blankenberg S, Barnes EH, Marschner IC, Thompson P, West M, Zeller T, Colquhoun DM, Nestel P, Keech AC, Sullivan DR, Hunt D, Tonkin A; LIPID Study Investigators . Changes in lipoprotein‐associated phospholipase A2 activity predict coronary events and partly account for the treatment effect of pravastatin: results from the Long‐Term Intervention with Pravastatin in Ischemic Disease study. J Am Heart Assoc. 2013;2:e000360 doi: 10.1161/JAHA.113.000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heart Protection Study Collaborative Group . Lipoprotein‐associated phospholipase A(2) activity and mass in relation to vascular disease and nonvascular mortality. J Intern Med. 2010;268:348–358. [DOI] [PubMed] [Google Scholar]
- 29. O'Donoghue ML, Braunwald E, White HD, Lukas MA, Tarka E, Steg PG, Hochman JS, Bode C, Maggioni AP, Im K, Shannon JB, Davies RY, Murphy SA, Crugnale SE, Wiviott SD, Bonaca MP, Watson DF, Weaver WD, Serruys PW, Cannon CP; SOLID‐TIMI 52 Investigators , Steen DL. Effect of darapladib on major coronary events after an acute coronary syndrome: the SOLID‐TIMI 52 randomized clinical trial. JAMA. 2014;312:1006–1015. [DOI] [PubMed] [Google Scholar]
- 30. Jang Y, Waterworth D, Lee JE, Song K, Kim S, Kim HS, Park KW, Cho HJ, Oh IY, Park JE, Lee BS, Ku HJ, Shin DJ, Lee JH, Jee SH, Han BG, Jang HY, Cho EY, Vallance P, Whittaker J, Cardon L, Mooser V. Carriage of the V279F null allele within the gene encoding Lp‐PLA(2) is protective from coronary artery disease in South Korean males. PLoS One. 2011;6:e18208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maiolino G, Pedon L, Cesari M, Frigo AC, Wolfert RL, Barisa M, Pagliani L, Rossitto G, Seccia TM, Zanchetta M, Rossi GP. Lipoprotein‐associated phospholipase A2 activity predicts cardiovascular events in high risk coronary artery disease patients. PLoS One. 2012;7:e48171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mayer O Jr, Seidlerova J, Filipovsky J, Timoracka K, Bruthans J, Vanek J, Cerná L, Wohlfahrt P, Renata C, Trefil L. Unexpected inverse relationship between impaired glucose metabolism and lipoprotein‐associated phospholipase A2 activity in patients with stable vascular disease. Eur J Intern Med. 2014;25:556–560. [DOI] [PubMed] [Google Scholar]