

Abstract
Background
The postnatal development of myofibrillar mechanics, a major determinant of heart function, is unknown in pediatric patients with tetralogy of Fallot and related structural heart defects. We therefore determined the mechanical properties of myofibrils isolated from right ventricular tissue samples from such patients in relation to the developmental changes of the isoforms expression pattern of key sarcomere proteins involved in the contractile process.
Methods and Results
Tissue samples from the infundibulum obtained during surgery from 25 patients (age range 15 days to 11 years, median 7 months) were split into half for mechanical investigations and expression analysis of titin, myosin heavy and light chain 1, troponin‐T, and troponin‐I. Of these proteins, fetal isoforms of only myosin light chain 1 (ALC‐1) and troponin‐I (ssTnI) were highly expressed in neonates, amounting to, respectively, 40% and 80%, while the other proteins had switched to the adult isoforms before or around birth. ALC‐1 and ssTnI expression subsequently declined monoexponentially with a halftime of 4.3 and 5.8 months, respectively. Coincident with the expression of ssTnI, Ca2+ sensitivity of contraction was high in neonates and subsequently declined in parallel with the decline in ssTnI expression. Passive tension positively correlated with Ca2+ sensitivity but not with titin expression. Contraction kinetics, maximal Ca2+‐activated force, and the fast phase of the biphasic relaxation positively correlated with the expression of ALC‐1.
Conclusions
The developmental changes in myofibrillar biomechanics can be ascribed to fetal‐to‐adult isoform transition of key sarcomeric proteins, which evolves regardless of the specific congenital cardiac malformations in our pediatric patients.
Keywords: cardiac myofibrils, contractile function, contractile proteins, force kinetics, heart development, human myocardium, sarcomere physiology, tetralogy of Fallot
Subject Categories: Congenital Heart Disease, Contractile function, Basic Science Research, Proteomics
Introduction
Birth leads to major adaptions of the normal right ventricle (RV) of the heart because of the reduction in the mechanical load.1 The physiological adaptations are, however, impeded in pediatric patients with congenital heart defects that impose a persistent hemodynamic overload on the RV.2, 3 Animal models have revealed that the developmental adaptations at the organ level are paralleled by developmental changes at the subcellular scale, which includes changes in the sarcomere protein isoform pattern starting before birth and extending well into the postnatal period.4 A large body of evidence supports the notion that processes intrinsic to the sarcomere rather than the kinetics of the systolic and diastolic Ca2+ fluxes determine the duration of systole, the rate of isovolumic relaxation, and the diastolic extensibility.5 Hence, developmental isoform shifts of sarcomeric proteins will impart the mechanical function of the developing heart. Key sarcomeric proteins that affect mechanics and undergo such a developmental shift are titin, which determines passive stiffness6, 7; the heavy (MyHC) and essential light chains (LC‐1) of myosin,8, 9 which determine cross‐bridge turnover kinetics; and the subunits of the regulatory troponin complex, TnI4, 7, 10 and TnT,11, 12 which confer calcium regulation. Each of these proteins switches at a species‐specific time during development, and the switch is not synchronized between the various sarcomeric proteins not even for subunits of protein complexes such as myosin and Tn (cf. Siedner et al4 and references therein). Thus, isoform expression pattern of the sarcomere changes continuously in a species‐specific manner with respective functional consequences until the mature sarcomere is reached (eg, Siedner and Reiser and colleagues4, 13).
Several studies have addressed the postnatal fetal‐to‐adult transition of these proteins in hearts from mainly patients with tetralogy of Fallot (TOF), with most of them investigating only 1 or 2 proteins with only a few time points and, to our knowledge, none included functional investigations.10, 14, 15, 16, 17 The functional impact of certain fetal sarcomere protein isoforms has been studied in human left ventricular fetal myofibrils (gestational age <134 days)18 and hypertrophied adult hearts including those from patients with TOF,8, 19, 20, 21 as these hearts reexpress some, but not all, fetal sarcomere protein isoforms (cf. Sasse et al16). However, the consequences of protein isoform switching on steady state and dynamic biomechanical function of the developing postnatal human myocardium are unknown.
We therefore mapped the postnatal development of RV myofibrillar mechanical performance in conjunction with protein isoform analysis of the earlier mentioned key sarcomeric proteins in patients with conotruncal malformations, transposition of the great arteries (TGA), and hypoplastic left heart syndrome (HLHS). For ethical reasons, it is nearly impossible to obtain time‐matched healthy tissue from neonates and infants and, thus, the normal development will remain elusive. Nevertheless, such a study will provide valuable information for the clinician regarding the postnatal development, that is, maturation, of myofibrillar sarcomeric biomechanics, the major determinant of pump function, in these diseased hearts. These different congenital disease entities have in common that they are usually well tolerated before birth but lead to pressure and/or volume overload of the RV after birth. Our developmental study builds on 2 recent technological advances: (1) the progress in pediatric cardiac surgery allowing early postnatal repair and (2) the development of methods to fully characterize the biomechanical performance of myofibrils isolated from minute tissue samples with sufficient material for parallel biochemical analysis of protein expression. Compared with skinned fibers, myofibrils are ideal for biomechanical characterization of the sarcomere, because the myofibrils are not diffusion limited and their biomechanics is not confounded by extracellular matrix. We found that Ca2+ sensitivity was high in the neonate and declined thereafter monoexponentially in parallel with the decline in expression of fetal ssTnI and ALC‐1 and upregulation of their mature isoforms. Moreover, the kinetics of contraction and relaxation correlated with the expression levels of ALC‐1. Interestingly, passive stiffness correlated with Ca2+ sensitivity but not with titin isoform expression. We propose that the sarcomeric protein phenotype and its respective mechanical properties are highly adaptive and its postnatal fetal‐to‐adult transition proceeds regardless of the specific structural congenital defect of the hearts.
Materials and Methods
Ethics
All studies on human and animal samples were reviewed and approved by the local institutional ethics committees and were conducted in accordance with the Declaration of Helsinki and National Institutes of Health guidelines, respectively. All subjects′ parents gave informed consent, as it was explained to them that a piece of RV myocardium would be removed in order to relieve the obstruction or to create an opening to place a conduit.
Human Tissue Samples
Samples of the RV outflow tract were obtained from 25 patients (aged 4 days to 3 years and an 11‐year‐old patient) undergoing repair surgery for congenital heart disease, including TOF (n=7), double‐outlet RV (DORV, n=3), pulmonary stenosis (PS, n=6), pulmonary atresia (PA, n=4), TGA (n=3), and HLHS (n=3). In all patients, muscular tissue was removed from the RV outflow tract. This was almost always a full‐thickness specimen; in patients with TOF, where a transannular incision was made, more tissue from the endocardial aspect was obtained than from the epicardial side. In a subset of TOF patients, corrective surgery required additional removal of muscular tissue from the moderator band in the RV. Immediately after excision, tissue samples were divided into 2 portions; one was shock frozen in liquid nitrogen and stored at −80°C for protein analysis, and the other was placed in ice‐cold skinning solution for 4 hours for mechanical experiments.
Mechanical Experiments
Subcellular myofibrillar bundles were prepared immediately before the mechanical experiment by homogenizing the skinned tissues at 4°C for 5 to 15 seconds with a blender at maximum speed.22, 23 Relaxing (pCa 7.5) and maximally activating (pCa 4.5) solutions contained (in mmol/L) 10 imidazole, 1 K2Cl2‐Na2Mg‐ATP, 3 MgCl2, 47.7 Na2CrP, 2 dithiothreitol, and either 3 K4Cl2‐EGTA (relaxing solution) or 3 K4Cl2Ca‐EGTA (activating solution), adjusted to pH 7.0 at 10°C. Solutions of intermediate [Ca2+] calculated as in24 were obtained by mixing the relaxing and activating solutions in the appropriate ratio. The techniques for the force measurement were described previously.23 The myofibrillar bundles suspended in 500 μL of relaxing solution were pipetted into a thermostated chamber (10°C) that was mounted on the stage of an inverted microscope. After sedimentation of the myofibrils to the chamber bottom for 1 hour, a myofibrillar bundle was mounted between the tips of a stiff tungsten needle and an atomic force cantilever that had been precoated with a mixture of nitrocellulose and a silicon adhesive (50% v:v). Bundles used in experiments had diameters of 1.6 to 6.5 μm, lengths of 30 to 70 μm, and slack sarcomere lengths of 1.93±0.01 μm (mean±SEM). The slack sarcomere length, the overall length, and the diameter of the bundles were measured by using a CCD camera under phase contrast at 90‐fold magnification. The bundles were then stretched to a sarcomere length (SL) of 2.3 μm before force recording. Rapid activation and relaxation were induced with the rapid movement of a theta style micropipette (TGC150–15; Clark Electromedical Instruments) that produced 2 laminar streams of solution, with one stream containing the activating and the other the relaxing solution. The movement of the micropipette was driven by a piezoactuator (P289.40; Physik Instrumente) and produced a rapid solution change at the myofibrillar bundle that was completed within 10 ms. To determine the passive and active force at 2.3 μm SL, release–restretch protocols were applied to the myofibrillar bundle during relaxation at pCa 7.5 and during Ca2+ activation by different [Ca2+] values from pCa 6.16 to 4.53. Force was determined by recording the deflection of a laser beam that was microfocused onto the back side of the cantilever. The cantilevers had a stiffness of 2 to 4 nN/nm and a resonance frequency of 70 to 90 kHz.
Expression Analysis of Sarcomeric Proteins
Samples were homogenized in Laemmli buffer and protein concentration was determined according to Bradford.25 Isoform compositions of TnI and TnT were analyzed using, respectively, 12.5% SDS‐PAGE (37.5:1 acrylamide:bisacrylamide) and 9% SDS‐PAGE (27.3:1 acrylamide:bisacrylamide) followed by Western blotting. The separated proteins were transferred to nitrocellulose membranes (0.2 mm pore size, Schleicher and Schuell), which were blocked with 5% (w/v) fat‐free dried milk in TBS and incubated overnight at 4°C with either a monoclonal mouse anti‐TnI antibody (H86550M; Dunn Technologies) that recognizes both ssTnI and cTnI, or with a monoclonal mouse anti‐TnT antibody (MA5‐12960; Thermo Scientific). The membranes were washed and incubated for 1 hour with donkey anti‐mouse IgG, peroxidase‐conjugated secondary antibodies (Jackson ImmunoResearch). Immunoreactive bands were detected with the use of enhanced chemiluminescence (Amersham). The relative amounts of TnI and TnT isoforms were determined by densitometric scanning of the chemiluminograms with use of an EPSON scanner and Phoretix software.
Isoforms of titin, myosin heavy chain isoforms (MyHC), and myosin light chains were separated according to published protocols.20, 26, 27 In brief, MyHC isoforms were separated by using 6% SDS‐PAGE (50:1 acrylamide:bisacrylamide) with 3.3% (50:1 acrylamide:bisacrylamide) stacking gels, and the gels were run at 7 mA for 14 hours. Expression of light chains (LC‐1) was analyzed by using 2‐dimensional PAGE. Isoelectric focusing was performed with a pH gradient of 4.5 to 5.4 as described in the literature.20 The gels were run overnight at 600 V constant. The second dimension was a 15% SDS‐PAGE (37.5:1 acrylamide:bisacrylamide). To determine titin isoforms, a 1.5 mm thick 1% agarose gel was run on a Hoefer SE600 system at 8°C either overnight at 3.5 mA or at 15 mA for 4 hours. For identification of MyHC and LC‐1 and titin isoforms, gels were stained with Coomassie Brilliant Blue or Silver according to the manufacturer's instructions. The relative amounts of titin and LC‐1 isoforms were determined with densitometric scans of the stained gels by using Phoretix software (Biostep).
Data Analysis and Statistics
Because of the tiny size of the specimens, in most cases only 1 to 3 determinations for each protein per patient could be performed, and not all proteins could be analyzed in each patient. Means for each patient were plotted versus age in months and fitted by a monoexponential function. This function [y=(A−B)×exp(−x×ln(2)/t ½)+B] yields the expression level at birth (A), the final expression level (B), and the halftime of the postnatal isoform change in months (t ½).
Mechanical experiments were performed with 2 to 12 individual myofibrils per patient, and the mean±SEM for the different mechanical parameters was calculated for each patient. Ca2+ sensitivity given by the pCa (−log[Ca2+]) required for half‐maximal activation (pCa50) was determined by fitting force–pCa relations from individual myofibrils with the Hill equation.22 To determine kinetic parameters of contraction and relaxation, respectively, force transients were fitted either by a monoexponential function yielding the rate constant of Ca2+‐induced force development k ACT or by a function consisting of a linear and an exponential term yielding the parameters k LIN, t LIN, and k REL of the relaxation kinetics.23 For each patient, the mean±SEM was calculated for the different mechanical parameters. Statistical analysis was performed by using ANOVA followed by the Bonferroni posttest or Student unpaired t test as appropriate. Correlations were computed with the Pearson correlation coefficient based on the mean parameter values for patients. P<0.05 was considered significant. Unless otherwise noted, all data are reported as mean±SEM of n myofibrils.
Results
Clinical Data
Patients’ clinical characteristics are available online (Table S1). All patients with TGA or HLHS and 1 patient with PA underwent surgery within the first month after birth. All other patients had surgery at a later age, and timing was independent of diagnosis (Figure S1). Some patients underwent palliative surgery before repair surgery was performed (Table S1).
Development of Sarcomeric Protein Composition
To relate the mechanical performance to postnatal development of sarcomeric protein phenotype, we determined the isoform expression of MyHC, TnI, TnT, LC‐1, and titin in patients aged 0.1 to 38.2 months. Unfortunately, because of the limited sample size, titin isoform analysis was possible in only 7 of 25 samples, including an 8‐day‐old patient. In this patient, expression of the compliant fetal N2BA titin isoform predominated with ≈60% compared with ≈40% of the stiffer N2B isoform. In contrast, in the 6 samples from the older patients, the average titin composition was 42±3% N2BA and 58±3% N2B (n=6) (Figure 1A, Table S2). Regarding TnT isoforms, the adult isoform of TnT, TnT3, was the predominant isoform in our samples (Figure 1B, Table S2), whereas the fetal isoform, TnT4, was detected in only 7 of 19 samples at very low expression levels (Figure 1B, Table S2). We detected only a single MyHC protein band corresponding to the adult β‐MyHC isoform, even in the youngest patients (4 and 8 days old) (Figure 1C). Our results, therefore, indicate that the mature MyHC and TnT isoforms were already present at birth. Our results further demonstrated that the expression levels of fetal TnI (ssTnI) and LC‐1 isoforms (ALC‐1) were still high after birth and declined monoexponentially within the first 10 months after birth (Figure 2A through 2D). In neonates (age <2 weeks), the fetal ssTnI isoform was 76±4% of total TnI, (n=7). Fetal ssTnI expression thereafter declined and was replaced by the adult cTnI isoform with a halftime of 4.3 months (Figure 2B, Figure S2A). It was still detectable in 24‐ to 38.2‐month‐old patients (4±1%, n=4; Figure 2B) but was completely replaced by the adult cTnI isoform in the 11‐year‐old TOF patient (Figure 2B). The expression level of ALC‐1 in neonates was 42±2% of LC‐1total (n=7) and subsequently declined in a monoexponential manner with a halftime of 5.8 months approaching 9±3% (n=4) in children older than 24 months and was replaced by VLC‐1 (Figure 2D, Figure S2B). The ratio of LC‐1 to LC‐2 was 1.01±0.03 (mean±SEM), indicating preserved stoichiometry.
Figure 1.
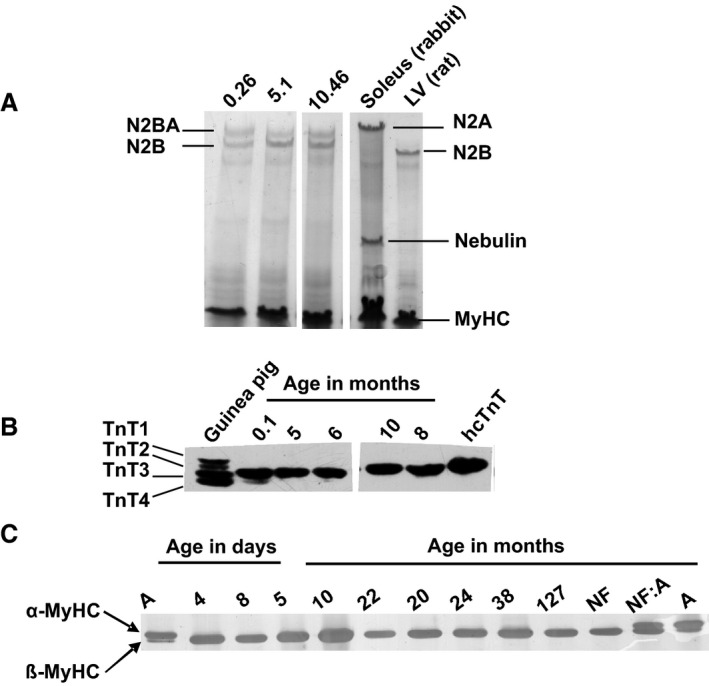
Development of titin, myosin heavy chain (MyHC) and troponin T (TnT) isoform expression. A, Representative Coomassie‐stained 1% SDS‐agarose gel of titin isoforms in RV samples obtained at the indicated ages; rabbit soleus and rat left ventricle (LV) were used as markers. B, Western blot probing for TnT, guinea pig myocardium expressing 4 isoforms, and human recombinant (hcTnT) are shown for comparison; in all human samples the predominant isoform is TnT3. C, Representative SDS‐PAGE of separation of MyHC isoforms, markers for α‐ and β‐MyHC isoforms: atrium (A), nonfailing adult heart (NF), and comigration (NF:A). Pediatric patients express only the β‐MyHC.
Figure 2.
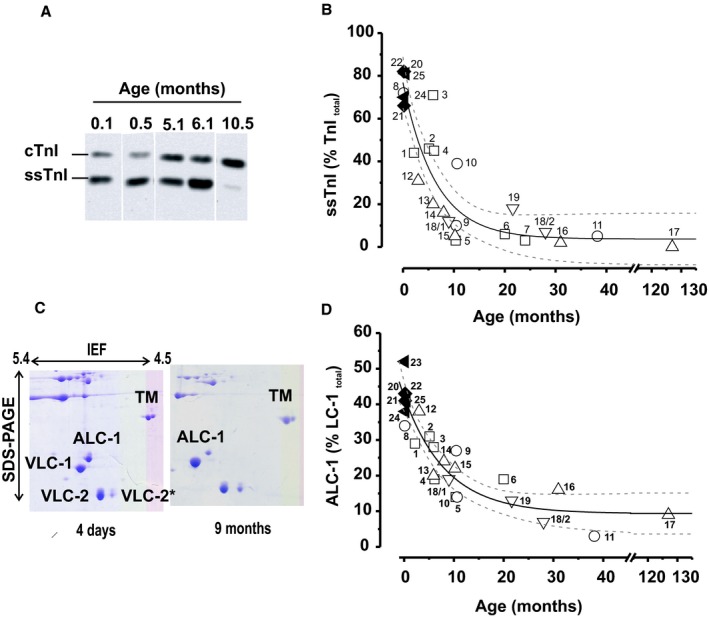
Postnatal expression of ssTnI and ALC‐1 in RV. A, Representative Western blots of TnI expression at different ages, showing the reciprocal signal intensities of ssTnI and cTnI. B, Time course of postnatal decline in ssTnI expression in percentage of total TnI=ssTnI+cTnI. C, Representative 2‐dimensional PAGE from 2 patients aged 4 days and 9 months; ALC‐1 and VLC‐1 indicate atrial and ventricular LC‐1 isoforms; VLC‐2 and VLC‐2*, ventricular regulatory LC isoforms; TM, tropomyosin. D, Relative ALC‐1 expression vs patient age (LC‐1total=ALC‐1+VLC‐1). Symbols in (B and D): hypoplastic left heart syndrome, closed triangles; transposition of the great arteries, closed diamonds; tetralogy of Fallot, open squares; pulmonary atresia, open circles; pulmonary stenosis, open upward triangles; double‐outlet right ventricle, open downward triangles; each symbol represents 1 patient, and numbers are patient numbers from Table S1. Solid and dotted lines show monoexponential fit with 95% confidence limit (ALC‐1: r 2=0.56, P<0.05; ssTnI; r 2=0.46, P<0.05).
The variability in the developmental decline of ssTnI expression was higher than that of ALC‐1 for as yet unknown reasons (Figure S2). Except for HLHS and TGA, protein expression levels were independent of the structural defect (Figure S2C and S2D). Still, we believe that age rather than diagnosis determined the developmental isoform transition of TnI and LC‐1 because the values of these proteins in HLHS and TGA patients, which were operated on at a much younger age than the others, fell on the regression curves (Figure 2B and 2D). Of note, one patient (No. 18) was operated on twice at 9 and 28 months. Fetal protein isoform expression in this patient declined from the first to the second operation as predicted by the regression curve. Protein expression in the infundibulum was similar to that in the moderator band of the RV in patients in whom tissues from both regions could be obtained (Figure S3A and S3B). There was no correlation between ssTnI or ALC‐1 expression and the pressure gradient between the RV and the pulmonary artery, or arterial oxygen saturation (Figure S4A through S4D). Thus, developmental isoform switching occurs in the infant hearts regardless of the structural malformation and the pleiotropic effects of the disease.
Steady State Force Parameters and Ca2+ Sensitivity
Of the 26 samples collected from the 25 patients (1 patient was operated on twice), 14 had subcellular myofibrillar bundles suitable for mechanical investigation. The remaining samples yielded no suitable myofibrils because of overcontraction or a high degree of tissue fibrosis precluding isolation of myofibrils. Figure 3 shows a myofibrillar bundle mounted in the myograph and typical force recordings. From the force transients, we obtained the passive force, the active steady state force, and the kinetic parameters of Ca2+‐induced (k ACT) and mechanically induced contraction (k TR) and relaxation (k LIN, t LIN, and k REL) summarized in Table S3.
Figure 3.
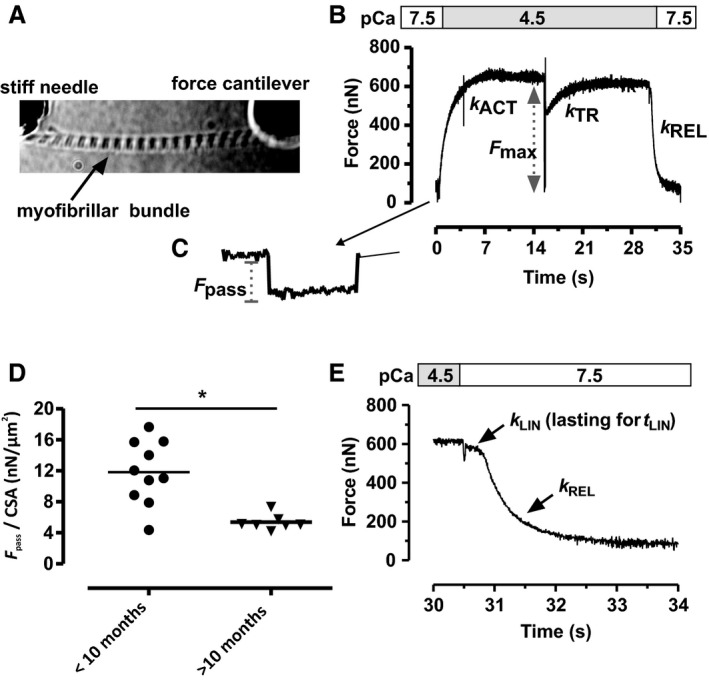
Force transients and passive force of right ventricular myofibrillar bundles. A, Image of a mounted myofibrillar bundle. B, Representative contraction–relaxation transient induced by switching (within 10 ms) from pCa 7.5 to 4.5 and back. Raising [Ca2+] and a release–restretch maneuver applied at the force plateau result in monoexponential force development with respective rate constants k ACT and k TR. C, Passive force per cross‐sectional area (F pass/CSA) at 2.3 μm SL was determined before activation by slackening the myofibril by 20% of slack length. D, F pass/CSA was significantly higher (*P<0.05) in younger (aged <10 months) than in older (>10 months) infants. E, Switching pCa back 7.5 leads to a biphasic relaxation that was fitted by a biphasic function (Methods) yielding k LIN, t LIN, and k REL.
Interestingly, passive tension (F pass/CSA) measured at 2.3 μm SL was significantly higher in younger patients (age range 4 days to 10 months) with 13±1 nN/μm2 (n=6) compared than in older patients (>10 months old) with 5±0.3 nN/μm2 (Figure 3D). The higher F pass/CSA values were not associated with a shorter slack SL (Figure S5A) or with changes in titin isoform expression (Table S2).
Maximum Ca2+ activated tension (F max/CSA) was similar at all ages (Table S3). To determine force–pCa relations, myofibrils were subjected to several activation–relaxation cycles with different activating [Ca2+] values (pCa 5.8–4.5, Figure 4A). Representative force–pCa relations in a neonate and a 10‐month‐old child (Figure 4B) showed that the curve was shifted to the right in the neonate compared with the infant, while the steepness of the force–pCa relations (n H) did not change with age (Table S3). From these curves, we calculated pCa50 as a measure of Ca2+ sensitivity, which declined from pCa ≈5.95 shortly after birth to ≈5.4 at age >3 years (Figure 4C). Of note, Ca2+ sensitivity declined in parallel with both ALC‐1 and ssTnI expression, as reflected by the positive correlations between pCa50 and expression levels of these proteins (Figure 4D and 4E).
Figure 4.
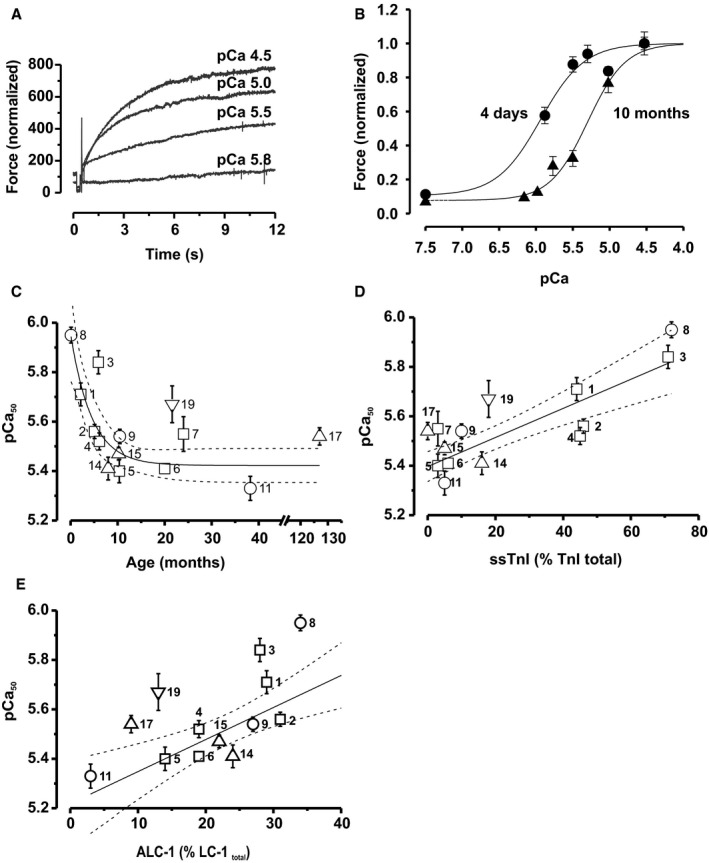
Postnatal change in Ca2+ sensitivity of force development. A, Original force tracings of contractions elicited by the indicated pCa values. B, Force–pCa relations collected from 5 myofibril bundles of a newborn (patient No. 8) and 6 myofibril bundles of a 10‐month‐old child (patient No. 5), illustrating the representative rightward shift with this increase of age. The curves represent Hill functions fitted to the data, which were normalized to F max at pCa 4.5. C, pCa50 declines monoexponentially with age (r 2=0.59, P<0.05). D and E, pCa50 positively correlated with ssTnI (r 2=0.81, P<0.0001) and ALC‐1 (r 2=0.62, P<0.05) expression. Solid and dotted lines represent the monoexponential (C) and linear fits (D and E) and the respective 95% confidence limits. The pCa50 values were determined from 2 to 12 myofibrils per patient and are given as mean±SEM. Symbols in (C through E): tetralogy of Fallot, open squares; pulmonary atresia, open circles; pulmonary stenosis, open upward triangles; double‐outlet right ventricle, open downward triangles. Each symbol represents 1 patient, and numbers are patient numbers from Table S1.
Kinetics of Contraction and Relaxation
Both ALC‐1 and ssTnI have been proposed to modulate the dynamics of the mechanical output.20, 28, 29 We, therefore, investigated whether force kinetic parameters correlated with the expression of these proteins. Ca2+‐induced force development following rapid switching from relaxing (pCa 7.5) to fully activating solution (pCa 4.5) and mechanically induced force redevelopment following a period of unloaded shortening occur monoexponentially with rate constants k ACT and k TR, respectively (Figure 3B). Relaxation of myofibrils induced by rapidly switching back to pCa 7.5 is biphasic, starting with a slow linear force decay with a rate constant k LIN lasting for a time t LIN followed by a rapid monoexponential force decay with a rate constant k REL (Figure 3E). Previous investigations revealed that sarcomeres remain isometric during the slow phase, whereas the fast phase starts when the weakest sarcomere lengthens (see review30). Neither of the kinetic parameters correlated with ssTnI expression at expression levels >10% (Figure 5A through 5E). ALC‐1 expression had no effect on the rate constant k LIN and the time t LIN of the initial, slow, linear phase of relaxation, but the rate constants of the force rises k ACT and k TR, as well as the rate constant k REL of the rapid exponential relaxation phase, significantly declined with decreasing expression levels of ALC‐1 (Figure 6).
Figure 5.
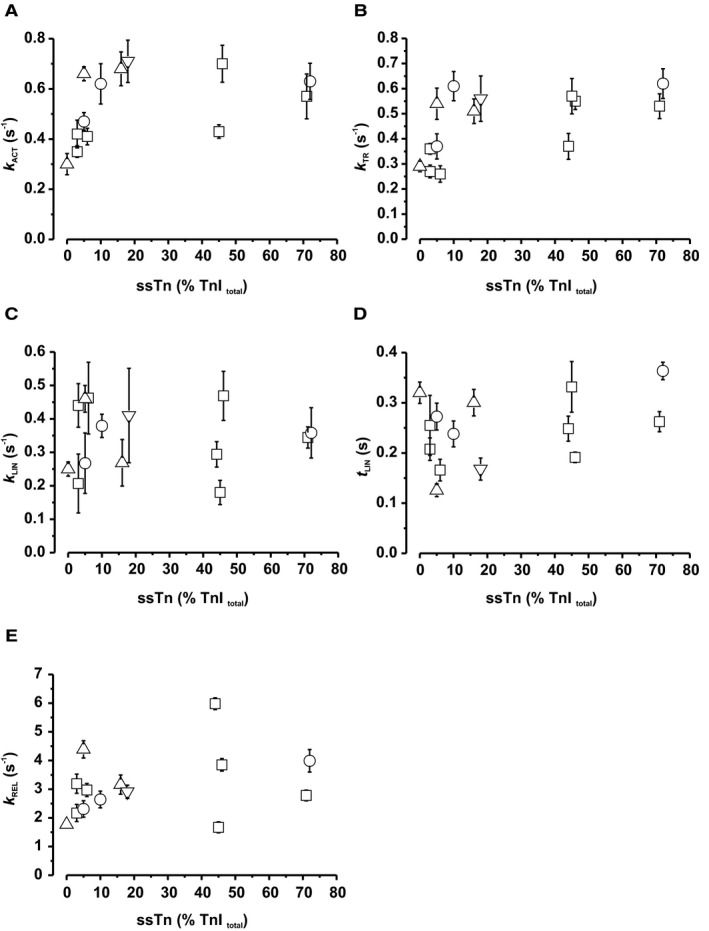
Effect of ssTnI expression on kinetic parameters of contraction and relaxation. A, Rate constant of Ca2+‐induced force development; B, Rate constant of mechanically‐induced force redevelopment; C, Rate constant of initial slow linear relaxation phase; D, Duration of initial slow linear relaxation phase; E, Rate constant of subsequent rapid exponential relaxation phase. For determination of rate constants of contraction (k ACT and k TR) and relaxation (k LIN, t LIN, k REL), cf. Figure 3. Symbols: tetralogy of Fallot, open squares; pulmonary atresia, open circles; pulmonary stenosis, open upward triangles; double‐outlet right ventricle, open downward triangles. Each symbol represents the mean±SEM of 2 to 12 myofibrils of a patient.
Figure 6.
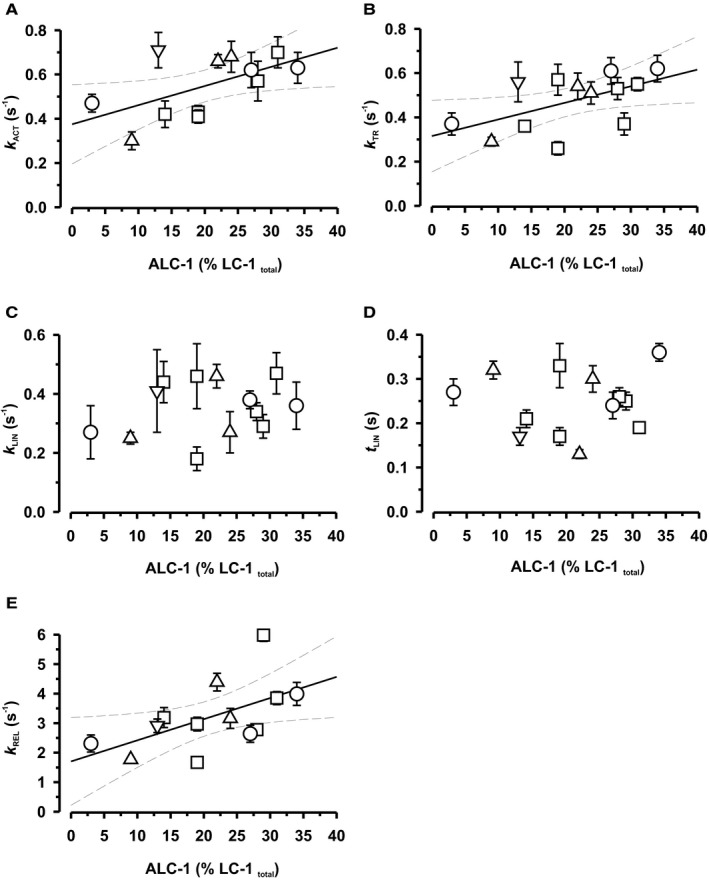
Effect of ALC‐1 expression on kinetic parameters of contraction and relaxation. A, Rate constant of Ca2+‐induced force development; B, Rate constant of mechanically‐induced force development; C, Rate constant of initial slow linear relaxation phase; D, Duration of initial slow linear relaxation phase; E, Rate constant of subsequent rapid exponential relaxation phase. For determination of rate constants of contraction (k ACT and k TR) and relaxation (k LIN, t LIN, k REL), cf. Figure 3. Symbols: tetralogy of Fallot, open squares; pulmonary atresia, open circles; pulmonary stenosis, open upward triangles; double‐outlet right ventricle, open downward triangles. Each symbol represents the mean±SEM of 2 to 12 myofibrils of a patient. Solid and dotted lines represent linear correlations and 95% confidence limits (Pearson correlation coefficients: A, r 2=0.58, P<0.05; B, r 2=0.55, P<0.05; E, r 2=0.57, P<0.05).
Discussion
The present developmental study is, to our knowledge, the first comprehensive assessment of the mechanical performance of human RV myofibrillar bundles in conjunction with the isoform expression pattern of key sarcomeric proteins determined in the same patient. Of the sarcomeric proteins we selected for analysis in this study, only the fetal isoforms of TnI and LC‐1 were still expressed in significant amounts in the newborn, before gradually being replaced by their adult counterparts in parallel with the respective changes in Ca2+ sensitivity of contraction and kinetic parameters. Our data indicate that the expression levels of the isoforms of TnI and LC‐1 and the corresponding biomechanical parameters were determined primarily by postnatal age rather than by the individual patient's specific structural defect and complex hemodynamic situation. Based on these results, we propose that the investigated congenital heart diseases perhaps modulate the time course but do not abrogate the evolution of the developmental program. Consistent with the progressively developing secondary, hemodynamic overload‐induced hypertrophy, end points in ALC‐1 expression are reached that are comparable to those seen in hemodynamically stressed adult hearts.20, 31
Titin Isoform Expression and Passive Force
Because of the very limited sample availability, neonatal titin isoform expression has not been studied in detail yet. Thus, our study is among the first to describe a high expression level of fetal N2BA titin in an 8‐day‐old patient. Unfortunately, it was not possible to obtain mechanical data in this patient. Nevertheless, this is an important observation as it confirms that in human hearts, titin isoform switching occurs in the early perinatal period, just as it does in different animal models.6, 7 In infants >1 month old, titin expression was similar to that in adults.32 Despite this, we noted a shift in passive tension at 2.3 μm SL from high values in 1‐ to 10‐month‐old infants to lower values in older patients. Such shifts in passive tension unrelated to titin expression have been observed before21, 33 and were related, among others, to covalent modification of titin (see review32). Another possibility is that the very high Ca2+ sensitivity in neonates causes residual activation of cross‐bridges at basal [Ca2+] (pCa 7.5).34 Indeed, we observed a significant positive correlation between passive tension (F pass) and pCa50 (Figure S5B), indicating that part of the so‐called passive tension at pCa 7.5 might be caused by Ca2+‐activated processes. We are aware that this conclusion is limited by the low number of titin gels precluding a detailed structure–function relation for human heart development. Thus, a more focused future investigation has to evaluate the precise relation between titin expression and passive stiffness and whether titin modification or residual Ca2+‐activated processes or both contribute to the low diastolic compliance of the fetal/neonatal heart (cf. Krüger et al7 and references therein).
Postnatal Transition of Fetal to Adult Isoforms of TnI and LC‐1
Ca2+‐dependent regulation of contraction and cross‐bridge turnover kinetics depend primarily on the isoforms of the subunits TnI and TnT of the regulatory troponin complex and those of the myosin motor, respectively. Of these proteins, only expression levels of the fetal TnI and LC‐1 isoforms were high in neonates and then declined monoexponentially coincident with upregulation of the adult isoforms. In some patients, the ssTnI/cTnI switch appeared to be delayed compared with the majority of patients for unknown reasons. The fetal‐to‐adult protein isoform transition of ssTnI and ALC‐1 has been studied before, showing that expression of fetal protein isoforms was high in neonates and declined thereafter,10, 14, 15, 16 but the exact time course was known only for ACL‐1,14 and our results are consistent with this report (Figure S2A and S2B). The time course of the transition of ssTnI/cTnI remained elusive because of the few time points investigated in these earlier studies.10, 15, 16 Our investigation revealed that ssTnI expression correlated positively with ALC‐1 expression during development. This is interesting given that the developmental regulation of the isoform expression of many sarcomeric proteins is not synchronized.
As yet, the developmental regulation of ssTnI and ALC‐1 expression is poorly understood. Studies in genetically modified mice indicate that ssTnI is set to switch off even in cTnI knock‐out mice, albeit at a slower rate.35 In our samples, ssTnI expression was negligible in infants older than 10 months (as in15), consistent with the observation that ssTnI was not reexpressed in hemodynamically stressed human adult hearts.16 In contrast, ALC‐1, which was absent in the normal ventricles of 2.5‐year‐old infants,14 was detected in our patients >2 years old at an expression level (9%) similar to that reported for hypertrophic adult ventricles (≈12%).20 It is unclear at present whether expression was never completely switched off or whether it was turned on again by the progressively developing secondary RV hypertrophy.
Postnatal Changes in Ca2+ Sensitivity of Contraction in Relation to Protein Expression
Of all mechanical parameters investigated, Ca2+ sensitivity showed the most prominent and clear‐cut age dependence and declined by nearly 0.6 pCa unit from pCa 5.95 shortly after birth to ≈5.4 in >3‐year‐old patients. The latter value is comparable to that reported for adult human left ventricular (LV) myofibrils.23, 36 Based on a large body of experimental evidence in animal models (4, 7 and references therein), the most likely underlying cause is the decline in ssTnI expression. Whether ALC‐1 expression affects Ca2+‐sensitivity is controversial.20, 37 We cannot dismiss such an effect of ALC‐1 expression on Ca2+ sensitivity, because we found that Ca2+ sensitivity correlated positively not only with ssTnI but also with ALC‐1 expression, especially at low expression levels of ssTnI (<10%) consistent with previous reports in humans31 and mice.29 This was not unexpected given the parallel decline in ALC‐1 and ssTnI expression, but the overall correlation was weaker than that for ssTnI (Figure 4D and 4E). The age dependence of Ca2+ sensitivity exhibited a greater variability than protein expression, suggesting that protein isoforms of TnI and LC‐1 might not be the sole determinant of Ca2+ sensitivity. It is well established that Ca2+ sensitivity is modulated by protein kinase A–mediated phosphorylation of myosin binding protein C38 and cTnI,39 whereas ssTnI is not a substrate. However, we found that the pCa values in the subset of patients (7 of 24, Table S1), who were treated with β‐blockers did not systematically deviate from the regression curves and fell within the 95% CIs. Therefore, we concluded that the major determinant of the developmental decline in Ca2+ sensitivity is the isoform shift. This notion is also supported by experiments in fetal/neonatal mice.4
Downregulation of ssTnI and ALC‐1 Expression and Kinetic Parameters
Forced expression of ssTnI in mice resulted in impaired diastolic relaxation of cardiomyocytes.28 Such an impaired relaxation was not observed in the myofibrils from our patients who coexpress ALC‐1. Rather ALC‐1 expression, but not ssTnI (expression levels >10%), positively correlated with the rates of the fast phase of relaxation (k REL) and contraction (k ACT, k TR); that is, these rates were high in neonates at high expression levels of ALC‐1. Our results are consistent with the notion that force kinetics is primarily governed by intrinsic kinetics of myosin interaction with actin rather than by the dynamics of Ca2+‐controlled switch of the Tn complex (see 40 for review5). We consider a contribution of MyHC expression unlikely as in all our samples, β‐MyHC was the predominant isoform, as is already the case in the human fetal heart.18 The increased contraction kinetics observed at high expression levels of ALC‐1 was associated with a higher active maximum tension (F max, Figure S6A), thus relating ALC‐1 expression with enhanced contractile force at the myofibrillar level for the first time, to our knowledge. This provides a mechanistic explanation for the observation that ALC‐1 expression enhances contractility in the hypertrophied human heart41 and is consistent with a previous study in skinned fibers from predominantly adult patients with TOF.20 The increased rates of k ACT and k TR, and F max can be best explained if ALC‐1 promoted force generation by increasing the rate by which cross‐bridges enter force‐generating states, reflected by the rate constant f app 30 (cf. legend to Figure S6 for a detailed discussion). Noteworthy, F max positively correlated not only with contraction kinetics but also with Ca2+ sensitivity (Figure 6B), suggesting that not only thick filament properties (ALC‐1) but also thin filament properties improve myofibrillar contractility in the neonatal human RV.
The positive correlation between ALC‐1 expression and V MAX found by Morano and coworkers20 is likely related to an effect of the LC‐1 isoform on the maximum rate of cross‐bridge detachment, g MAX, occurring when cross‐bridges become rapidly unstrained during unloaded sarcomere shortening. The positive correlation between ALC‐1 and the rate constant of the rapid relaxation phase (k REL) of isometrically held myofibrils can also be attributed to such an effect of ALC‐1 on g MAX. This is because of the considerable intersarcomere dynamics observed during this phase, where some sarcomeres lengthen and others shorten, perturbing cross‐bridge strain (discussed in2). In contrast, ALC‐1 expression did not affect cross‐bridge detachment at high loads, as k LIN reflecting g app did not change with ALC‐1 expression level.
Our study is the first to demonstrate an effect of ALC‐1 expression on sarcomere dynamics. Moreover, we believe that relaxation of isometrically held myofibrils reflects isovolumic diastolic relaxation of the heart better than unloaded shortening velocity, which does not occur at the organ level. Based on our results, we propose that the fast relaxation is supported by the fetal LC‐1 isoform (Figure 6E) and counteracts potential slowing effects of ssTnI.28 To summarize, ALC‐1 enhances active force generation during high‐loaded contractions via increasing f app, whereas at low loads, it accelerates shortening and relaxation by promoting cross‐bridge detachment via g MAX. These kinetic effects obtained in our study are consistent with published results in a transgenic adult mouse model overexpressing ALC‐1 in the ventricles, which manifested at the organ scale as, respectively, an increased +dP/dtmax and −dP/dt.29
Study Limitations
This was an observational study from the perspective of clinical diagnosis and age at surgery. Although our data indicated that postnatal age rather than the specific clinical diagnosis is the major determinant of the fetal‐to‐adult protein isoform switch, we cannot exclude the possibility that the rates of these transitions may be modified in these patients or in subgroups compared with healthy children. Further, the subject‐specific postnatal development and change of ssTnI and ALC‐1 expression in RV are not known. This would require a study with longitudinal data, which could be extremely difficult to conduct. Therefore, the results of the study could be partly attributed to between‐subject variability observed at different ages and not really the underlying development process. For obvious ethical reasons, it is virtually impossible to obtain fresh age‐matched normal control tissue in this age group. We refrained from using donor hearts, which is widely used as healthy control, for several reasons: (1) fresh age‐matched pediatric donor hearts are not readily available, (2) donor hearts from biobanks undergo a freezing/rethawing process, and (3) given that there is a significant incidence of early posttransplantation dysfunction, with isolated RV dysfunction being the leading cause of early mortality and morbidity,42 donor hearts at best approximate the in vivo situation of normal RV function.43 Because the size of tissue specimens was solely determined by the demand of the surgical correction of the small neonatal hearts, it was not possible to obtain samples for histology or determine the phosphorylation status or other posttranslational modifications from the tiny specimens. The phosphoproteome of rat neonatal hearts differed from that of adult rat hearts.44 Important candidates for developmental changes in phosphorylation revealed by the rat study were sites on myosin binding protein C, myosin light chain‐2, and tropomyosin, thus on proteins that reveal a stable developmental isoform pattern.45 Additionally, cTnI phosphorylation, especially at low levels of ssTnI expression, might affect Ca2+ sensitivity. Although we find in our correlational study that changes in mechanical parameters in these nonfailing hearts could be reasonably well explained by protein isoform changes, posttranslational modifications may account for the somewhat greater variability in developmental progression of Ca2+ sensitivity and force kinetics compared with the isoform expression of TnI and LC‐1. Future studies are thus required to address the early postnatal development of posttranslational modification in these congenital heart diseases. Finally, most of the tissue samples were obtained from the infundibular region, which may not be representative for the whole ventricle. However, in the few patients in whom it was possible to also take samples of the moderator band of the RV, ssTnI and ALC‐1 expression was not different from that in the infundibulum, suggesting that protein expression in the infundibular region is representative for endocardial cardiomyocytes.
Conclusions and Clinical Relevance
The most important finding of our study is that the developmental gene program that regulates developmental maturation of the sarcomere continues to evolve largely regardless of the various structural but hemodynamically related cardiac malformations, which impose abnormal pressure and volume load on the RV, albeit isoform switching may be slowed down.14 The expressions of fetal protein isoforms of titin, TnT, and MyHC appear to be switched off before or around birth. Expressions of the fetal isoforms, ssTnI and ALC‐1, which are still high at birth, is gradually downregulated, while that of adult isoforms is upregulated at the age of 1 year. We propose that the coexisting high expression levels of ssTnI and ALC‐1 after birth are highly adaptive and may in fact be beneficial for the hemodynamically stressed RV in our patients. This is because ssTnI protects from the cardiodepressant effects of intracellular acidosis, to which cyanotic patients may be prone.46, 47, 48 The coexpression of ALC‐1, on the other hand, counteracts the shortcomings of high levels of ssTnI expression, namely slowed diastolic relaxation. Such a diastolic dysfunction would be detrimental because the high heart rate of neonates and infants requires fast relaxation. Our study suggests that the coexisting expression of the fetal LC‐1 isoform is important for sustaining fast diastolic relaxation as long as the fetal TnI isoform is still expressed in the neonatal human heart. Further, ALC‐1 expression increased isometric force production, thus allowing the buildup of ventricular pressure to overcome the obstruction. Our results justify the conclusion that the developmental maturation of the sarcomere continues despite abnormal load and structure of the RV. Future studies have to delineate whether and, if so, by which mechanism the TnI and LC‐1 isoform transitions rates are modified in these patients. This requires more‐detailed knowledge about regulation of these genes.
Sources of Funding
This work was funded by the Medical Faculty of the University Cologne (Köln Fortune) to Drs Iorga, Stehle, and Pfitzer.
Disclosures
None.
Supporting information
Figure S1. Relation between the age of the patients at the surgery and their clinical diagnosis. Patients with HLHS and TGA were operated at a significantly lower age whereas in the other patients the time of surgery did not depend on the diagnosis. Lines represent means. DORV indicates double‐outlet right ventricle; HLHS, hypoplastic left heart syndrome; PA, pulmonary atresia; PS, pulmonary stenosis; TGA, transposition of the great arteries; TOF, tetralogy of Fallot. Exept for HLHS and TGA, no significant differences were observed, indicating that the diagnosis does not affect timing of surgery.
Figure S2. Dependence of ssTnI and ALC‐1 expression vs age (A and B) and vs clinical diagnosis (C and D). Replot of expression levels of ALC‐1 (A) as in Auckland et al (1986) Cardiovasc Res. 20:828‐836, as well as ssTnI (B) for different age groups. The decline in ALC‐1 expression in our cohort is consistent with the published results. Patients with HLHS and TGA expressed significantly higher levels of the fetal ssTnI (C) and ALC‐1 (D) isoforms than the other patients. There were no significant differences in these parameters between the other patients, indicating that in these patients the diagnosis does not affect protein expression levels. The high variability in expression levels relates to the large age range at which repair surgery was performed; lines represent means. DORV indicates double‐outlet right ventricle; HLHS, hypoplastic left heart syndrome; PA, pulmonary atresia; PS, pulmonary stenosis; TGA, transposition of the great arteries; TOF, tetralogy of Fallot.
Figure S3. Expression levels of ssTnI (A) and ALC‐1 (B) in tissues from the infundibulum and the moderator band from the right ventricle taken from the same hearts. Open symbols. infundibulum; closed symbols, moderator band; circles, pulmonary atresia; squares, tetralogy of Fallot. Each symbol represents 1 measurement.
Figure S4. The expression levels of ALC‐1 and ssTnI correlate with neither the oxygen saturation (A and B) nor the pressure gradient along the right ventricular outflow tract (C and D). The pressure gradient and oxygen saturation were assessed before repair surgery.
Figure S5. Interdependence of passive tension, slack sarcomere length (A), and Ca2+ sensitivity (B) for individual myofibrillar bundles. While there was no effect of slack sarcomere length on passive tension (P=0.766), passive tension positively correlated with Ca2+ sensitivity of force generation (r 2=0.13, P=0.003). Solid and dotted lines represent linear correlations and 95% confidence limits. Each data point represents determination from 1 myofibrillar bundle.
Figure S6. Relation between maximal force per cross‐sectional area (F max/CSA) and k ACT (A) or pCa50 (B), respectively. The maximal tension correlated significantly with the Ca2+‐induced contraction kinetics (r 2=0.11, P=0.0025) and with the Ca2+ sensitivity of force (r 2=0.06, P=0.044). The rate constant k ACT reflects the sum of apparent rate constants by which the cross‐bridges enter and leave the force‐generating state; that is, k ACT=f app+g app. Further, F max is proportional to f app/(f app+g app). Because F max is positively and not negatively correlated with k ACT, increased maximal force relates to an increase in f app rather than to a decrease in g app. Of note, k ACT would also increase if g app is increased. However, this would result in a decreased F max. Thus, increased isometric force generation and contraction kinetics at high ALC1‐expression levels can be explained by an increased f app, that is, by the increased probability of cross‐bridges to form force‐generating interactions with actin. Solid and dotted lines represent linear correlations and 95% confidence limits. Each data point represents determination from 1 myofibrillar bundle.
Table S1. Clinical Characteristics of Patients
Table S2. Expression Levels of Sarcomeric Protein Isoforms in RV Samples
Table S3. Summary of Functional Characterization
Acknowledgments
We are grateful to the surgical and pediatric teams involved in collecting the tissue and clinical data and to the parents of the little patients who allowed us to use the tissue samples. The expert technical assistance of D. Metzler and S. Zittrich and the language editing of an earlier version of the manuscript by Dr C. Habicht, Heidelberg, are gratefully acknowledged.
(J Am Heart Assoc.2016;5:e003699 doi: 10.1161/JAHA.116.003699)
References
- 1. Anderson PA, Glick KL, Manring A, Crenshaw C Jr. Developmental changes in cardiac contractility in fetal and postnatal sheep: in vitro and in vivo. Am J Physiol. 1984;247:H371–H379. [DOI] [PubMed] [Google Scholar]
- 2. Bailliard F, Anderson RH. Tetralogy of Fallot. Orphanet J Rare Dis. 2009;4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Towbin JA, Belmont J. Molecular determinants of left and right outflow tract obstruction. Am J Med Genet. 2000;97:297–303. [DOI] [PubMed] [Google Scholar]
- 4. Siedner S, Kruger M, Schroeter M, Metzler D, Roell W, Fleischmann BK, Hescheler J, Pfitzer G, Stehle R. Developmental changes in contractility and sarcomeric proteins from the early embryonic to the adult stage in the mouse heart. J Physiol (London). 2003;548:493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hinken AC, Solaro RJ. A dominant role of cardiac molecular motors in the intrinsic regulation of ventricular ejection and relaxation. Physiology (Bethesda). 2007;22:73–80. [DOI] [PubMed] [Google Scholar]
- 6. Lahmers S, Wu Y, Call DR, Labeit S, Granzier H. Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ Res. 2004;94:505–513. [DOI] [PubMed] [Google Scholar]
- 7. Krüger M, Kohl T, Linke WA. Developmental changes in passive stiffness and myofilament Ca2+ sensitivity due to titin and troponin‐I isoform switching are not critically triggered by birth. Am J Physiol Heart Circ Physiol. 2006;291:H496–H506. [DOI] [PubMed] [Google Scholar]
- 8. Reiser PJ, Portman MA, Ning XH, Schomisch Moravec C. Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. Am J Physiol Heart Circ Physiol. 2001;280:H1814–H1820. [DOI] [PubMed] [Google Scholar]
- 9. Lompre AM, Mercadier JJ, Wisnewsky C, Bouveret P, Pantaloni C, D'Albis A, Schwartz K. Species‐ and age‐dependent changes in the relative amounts of cardiac myosin isoenzymes in mammals. Dev Biol. 1981;84:286–290. [DOI] [PubMed] [Google Scholar]
- 10. Hunkeler NM, Kullman J, Murphy AM. Troponin I isoform expression in human heart. Circ Res. 1991;69:1409–1414. [DOI] [PubMed] [Google Scholar]
- 11. Gomes AV, Venkatraman G, Davis JP, Tikunova SB, Engel P, Solaro RJ, Potter JD. Cardiac troponin T isoforms affect the Ca2+ sensitivity of force development in the presence of slow skeletal troponin I: insights into the role of troponin T isoforms in the fetal heart. J Biol Chem. 2004;279:49579–49587. [DOI] [PubMed] [Google Scholar]
- 12. Sabry MA, Dhoot GK. Identification of and changes in the expression of troponin T isoforms in developing avian and mammalian heart. J Mol Cell Cardiol. 1989;21:85–91. [DOI] [PubMed] [Google Scholar]
- 13. Reiser PJ, Westfall MV, Schiaffino S, Solaro RJ. Tension production and thin‐filament protein isoforms in developing rat myocardium. Am J Physiol. 1994;267:H1589–H1596. [DOI] [PubMed] [Google Scholar]
- 14. Auckland LM, Lambert SJ, Cummins P. Cardiac myosin light and heavy chain isotypes in tetralogy of Fallot. Cardiovasc Res. 1986;20:828–836. [DOI] [PubMed] [Google Scholar]
- 15. Bhavsar PK, Dhoot GK, Cumming DV, Butler‐Browne GS, Yacoub MH, Barton PJ. Developmental expression of troponin I isoforms in fetal human heart. FEBS Lett. 1991;292:5–8. [DOI] [PubMed] [Google Scholar]
- 16. Sasse S, Brand NJ, Kyprianou P, Dhoot GK, Wade R, Arai M, Periasamy M, Yacoub MH, Barton PJ. Troponin I gene expression during human cardiac development and in end‐stage heart failure. Circ Res. 1993;72:932–938. [DOI] [PubMed] [Google Scholar]
- 17. Saba Z, Nassar R, Ungerleider RM, Oakeley AE, Anderson PA. Cardiac troponin T isoform expression correlates with pathophysiological descriptors in patients who underwent corrective surgery for congenital heart disease. Circulation. 1996;94:472–476. [DOI] [PubMed] [Google Scholar]
- 18. Racca AW, Klaiman JM, Pioner JM, Cheng Y, Beck AE, Moussavi‐Harami F, Bamshad MJ, Regnier M. Contractile properties of developing human fetal cardiac muscle. J Physiol (London). 2016;594:437–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H. Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail Rev. 2007;12:331–343. [DOI] [PubMed] [Google Scholar]
- 20. Morano M, Zacharzowski U, Maier M, Lange PE, Alexi‐Meskishvili V, Haase H, Morano I. Regulation of human heart contractility by essential myosin light chain isoforms. J Clin Invest. 1996;98:467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chaturvedi RR, Herron T, Simmons R, Shore D, Kumar P, Sethia B, Chua F, Vassiliadis E, Kentish JC. Passive stiffness of myocardium from congenital heart disease and implications for diastole. Circulation. 2010;121:979–988. [DOI] [PubMed] [Google Scholar]
- 22. Elhamine F, Radke MH, Pfitzer G, Granzier H, Gotthardt M, Stehle R. Deletion of the titin N2B region accelerates myofibrillar force development but does not alter relaxation kinetics. J Cell Sci. 2014;127:3666–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stehle R, Kruger M, Scherer P, Brixius K, Schwinger RH, Pfitzer G. Isometric force kinetics upon rapid activation and relaxation of mouse, guinea pig and human heart muscle studied on the subcellular myofibrillar level. Basic Res Cardiol. 2002;97:I127–I135. [DOI] [PubMed] [Google Scholar]
- 24. Fabiato A, Fabiato F. Calculator programs for computing the composition of the solutions containing multiple metals and ligands used for experiments in skinned muscle cells. J Physiol (Paris). 1979;75:463–505. [PubMed] [Google Scholar]
- 25. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal Biochem. 1976;72:248–254. [DOI] [PubMed] [Google Scholar]
- 26. Warren CM, Krzesinski PR, Campbell KS, Moss RL, Greaser ML. Titin isoform changes in rat myocardium during development. Mech Dev. 2004;121:1301–1312. [DOI] [PubMed] [Google Scholar]
- 27. Talmadge RJ, Roy RR. Electrophoretic separation of rat skeletal muscle myosin heavy‐chain isoforms. J Appl Physiol (1985). 1993;75:2337–2340. [DOI] [PubMed] [Google Scholar]
- 28. Fentzke RC, Buck SH, Patel JR, Lin H, Wolska BM, Stojanovic MO, Martin AF, Solaro RJ, Moss RL, Leiden JM. Impaired cardiomyocyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart. J Physiol (London). 1999;517:143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fewell JG, Hewett TE, Sanbe A, Klevitsky R, Hayes E, Warshaw D, Maughan D, Robbins J. Functional significance of cardiac myosin essential light chain isoform switching in transgenic mice. J Clin Invest. 1998;101:2630–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stehle R, Solzin J, Iorga B, Gomez D, Blaudeck N, Pfitzer G. Mechanical properties of sarcomeres during cardiac myofibrillar relaxation: stretch‐induced cross‐bridge detachment contributes to early diastolic filling. J Muscle Res Cell Motil. 2006;27:423–434. [DOI] [PubMed] [Google Scholar]
- 31. Morano I, Hadicke K, Haase H, Bohm M, Erdmann E, Schaub MC. Changes in essential myosin light chain isoform expression provide a molecular basis for isometric force regulation in the failing human heart. J Mol Cell Cardiol. 1997;29:1177–1187. [DOI] [PubMed] [Google Scholar]
- 32. Linke WA, Hamdani N. Gigantic business: titin properties and function through thick and thin. Circ Res. 2014;114:1052–1068. [DOI] [PubMed] [Google Scholar]
- 33. Rain S, Handoko ML, Trip P, Gan CT, Westerhof N, Stienen GJ, Paulus WJ, Ottenheijm CA, Marcus JT, Dorfmuller P, Guignabert C, Humbert M, Macdonald P, Dos Remedios C, Postmus PE, Saripalli C, Hidalgo CG, Granzier HL, Vonk‐Noordegraaf A, van der Velden J, de Man FS. Right ventricular diastolic impairment in patients with pulmonary arterial hypertension. Circulation. 2013;128:2016–2025. [DOI] [PubMed] [Google Scholar]
- 34. Iorga B, Blaudeck N, Solzin J, Neulen A, Stehle I, Lopez Davila AJ, Pfitzer G, Stehle R. Lys184 deletion in troponin I impairs relaxation kinetics and induces hypercontractility in murine cardiac myofibrils. Cardiovasc Res. 2008;77:676–686. [DOI] [PubMed] [Google Scholar]
- 35. Huang X, Pi Y, Lee KJ, Henkel AS, Gregg RG, Powers PA, Walker JW. Cardiac troponin I gene knockout: a mouse model of myocardial troponin I deficiency. Circ Res. 1999;84:1–8. [DOI] [PubMed] [Google Scholar]
- 36. Piroddi N, Belus A, Scellini B, Tesi C, Giunti G, Cerbai E, Mugelli A, Poggesi C. Tension generation and relaxation in single myofibrils from human atrial and ventricular myocardium. Pflugers Arch. 2007;454:63–73. [DOI] [PubMed] [Google Scholar]
- 37. van der Velden J, Papp Z, Zaremba R, Boontje NM, de Jong JW, Owen VJ, Burton PB, Goldmann P, Jaquet K, Stienen GJ. Increased Ca2+‐sensitivity of the contractile apparatus in end‐stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res. 2003;57:37–47. [DOI] [PubMed] [Google Scholar]
- 38. Moss RL, Fitzsimons DP, Ralphe JC. Cardiac MyBP‐C regulates the rate and force of contraction in mammalian myocardium. Circ Res. 2015;116:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Solaro RJ, Henze M, Kobayashi T. Integration of troponin I phosphorylation with cardiac regulatory networks. Circ Res. 2013;112:355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Solzin J, Iorga B, Sierakowski E, Gomez Alcazar DP, Ruess DF, Kubacki T, Zittrich S, Blaudeck N, Pfitzer G, Stehle R. Kinetic mechanism of the Ca2+‐dependent switch‐on and switch‐off of cardiac troponin in myofibrils. Biophys J. 2007;93:3917–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ritter O, Luther HP, Haase H, Baltas LG, Baumann G, Schulte HD, Morano I. Expression of atrial myosin light chains but not alpha‐myosin heavy chains is correlated in vivo with increased ventricular function in patients with hypertrophic obstructive cardiomyopathy. J Mol Med (Berl). 1999;77:677–685. [DOI] [PubMed] [Google Scholar]
- 42. Stoica SC, Satchithananda DK, White PA, Parameshwar J, Redington AN, Large SR. Noradrenaline use in the human donor and relationship with load‐independent right ventricular contractility. Transplantation. 2004;78:1193–1197. [DOI] [PubMed] [Google Scholar]
- 43. Jweied E, deTombe P, Buttrick PM. The use of human cardiac tissue in biophysical research: the risks of translation. J Mol Cell Cardiol. 2007;42:722–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yuan C, Sheng Q, Tang H, Li Y, Zeng R, Solaro RJ. Quantitative comparison of sarcomeric phosphoproteomes of neonatal and adult rat hearts. Am J Physiol Heart Circ Physiol. 2008;295:H647–H656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gautel M, Furst DO, Cocco A, Schiaffino S. Isoform transitions of the myosin binding protein C family in developing human and mouse muscles: lack of isoform transcomplementation in cardiac muscle. Circ Res. 1998;82:124–129. [DOI] [PubMed] [Google Scholar]
- 46. Martin AF, Ball K, Gao LZ, Kumar P, Solaro RJ. Identification and functional significance of troponin I isoforms in neonatal rat heart myofibrils. Circ Res. 1991;69:1244–1252. [DOI] [PubMed] [Google Scholar]
- 47. Wolska BM, Vijayan K, Arteaga GM, Konhilas JP, Phillips RM, Kim R, Naya T, Leiden JM, Martin AF, de Tombe PP, Solaro RJ. Expression of slow skeletal troponin I in adult transgenic mouse heart muscle reduces the force decline observed during acidic conditions. J Physiol. 2001;536:863–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Metzger JM, Michele DE, Rust EM, Borton AR, Westfall MV. Sarcomere thin filament regulatory isoforms. Evidence of a dominant effect of slow skeletal troponin I on cardiac contraction. J Biol Chem. 2003;278:13118–13123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Relation between the age of the patients at the surgery and their clinical diagnosis. Patients with HLHS and TGA were operated at a significantly lower age whereas in the other patients the time of surgery did not depend on the diagnosis. Lines represent means. DORV indicates double‐outlet right ventricle; HLHS, hypoplastic left heart syndrome; PA, pulmonary atresia; PS, pulmonary stenosis; TGA, transposition of the great arteries; TOF, tetralogy of Fallot. Exept for HLHS and TGA, no significant differences were observed, indicating that the diagnosis does not affect timing of surgery.
Figure S2. Dependence of ssTnI and ALC‐1 expression vs age (A and B) and vs clinical diagnosis (C and D). Replot of expression levels of ALC‐1 (A) as in Auckland et al (1986) Cardiovasc Res. 20:828‐836, as well as ssTnI (B) for different age groups. The decline in ALC‐1 expression in our cohort is consistent with the published results. Patients with HLHS and TGA expressed significantly higher levels of the fetal ssTnI (C) and ALC‐1 (D) isoforms than the other patients. There were no significant differences in these parameters between the other patients, indicating that in these patients the diagnosis does not affect protein expression levels. The high variability in expression levels relates to the large age range at which repair surgery was performed; lines represent means. DORV indicates double‐outlet right ventricle; HLHS, hypoplastic left heart syndrome; PA, pulmonary atresia; PS, pulmonary stenosis; TGA, transposition of the great arteries; TOF, tetralogy of Fallot.
Figure S3. Expression levels of ssTnI (A) and ALC‐1 (B) in tissues from the infundibulum and the moderator band from the right ventricle taken from the same hearts. Open symbols. infundibulum; closed symbols, moderator band; circles, pulmonary atresia; squares, tetralogy of Fallot. Each symbol represents 1 measurement.
Figure S4. The expression levels of ALC‐1 and ssTnI correlate with neither the oxygen saturation (A and B) nor the pressure gradient along the right ventricular outflow tract (C and D). The pressure gradient and oxygen saturation were assessed before repair surgery.
Figure S5. Interdependence of passive tension, slack sarcomere length (A), and Ca2+ sensitivity (B) for individual myofibrillar bundles. While there was no effect of slack sarcomere length on passive tension (P=0.766), passive tension positively correlated with Ca2+ sensitivity of force generation (r 2=0.13, P=0.003). Solid and dotted lines represent linear correlations and 95% confidence limits. Each data point represents determination from 1 myofibrillar bundle.
Figure S6. Relation between maximal force per cross‐sectional area (F max/CSA) and k ACT (A) or pCa50 (B), respectively. The maximal tension correlated significantly with the Ca2+‐induced contraction kinetics (r 2=0.11, P=0.0025) and with the Ca2+ sensitivity of force (r 2=0.06, P=0.044). The rate constant k ACT reflects the sum of apparent rate constants by which the cross‐bridges enter and leave the force‐generating state; that is, k ACT=f app+g app. Further, F max is proportional to f app/(f app+g app). Because F max is positively and not negatively correlated with k ACT, increased maximal force relates to an increase in f app rather than to a decrease in g app. Of note, k ACT would also increase if g app is increased. However, this would result in a decreased F max. Thus, increased isometric force generation and contraction kinetics at high ALC1‐expression levels can be explained by an increased f app, that is, by the increased probability of cross‐bridges to form force‐generating interactions with actin. Solid and dotted lines represent linear correlations and 95% confidence limits. Each data point represents determination from 1 myofibrillar bundle.
Table S1. Clinical Characteristics of Patients
Table S2. Expression Levels of Sarcomeric Protein Isoforms in RV Samples
Table S3. Summary of Functional Characterization