


Abstract
Gross chromosomal rearrangements (including translocations, deletions, insertions and duplications) are a hallmark of cancer genomes and often create oncogenic fusion genes. An obligate step in the generation of such gross rearrangements is the formation of DNA double-strand breaks (DSBs). Since the genomic distribution of rearrangement breakpoints is non-random, intrinsic cellular factors may predispose certain genomic regions to breakage. Notably, certain DNA sequences with the potential to fold into secondary structures [potential non-B DNA structures (PONDS); e.g. triplexes, quadruplexes, hairpin/cruciforms, Z-DNA and single-stranded looped-out structures with implications in DNA replication and transcription] can stimulate the formation of DNA DSBs. Here, we tested the postulate that these DNA sequences might be found at, or in close proximity to, rearrangement breakpoints. By analyzing the distribution of PONDS-forming sequences within ±500 bases of 19 947 translocation and 46 365 sequence-characterized deletion breakpoints in cancer genomes, we find significant association between PONDS-forming repeats and cancer breakpoints. Specifically, (AT)n, (GAA)n and (GAAA)n constitute the most frequent repeats at translocation breakpoints, whereas A-tracts occur preferentially at deletion breakpoints. Translocation breakpoints near PONDS-forming repeats also recur in different individuals and patient tumor samples. Hence, PONDS-forming sequences represent an intrinsic risk factor for genomic rearrangements in cancer genomes.
INTRODUCTION
Genomic instability is a hallmark of most types of cancer (1). Somatic genetic instability, leading to the generation of translocations, gross insertions, deletions and duplications, not only reshapes cancer genomes, but also serves to create de novo fusion genes whose functions may endow the cell with oncogenic potential and/or support tumor progression (2–5). Well described examples include the recurrent t(14;18)(q32;q21) translocation in follicular lymphoma, which fuses the BCL2 gene on chromosome 18 to the transcriptional enhancer of the IgH locus on chromosome 14 (3,6–8); the t(12;16) and t(12;22) translocations generating FUS-CHOP and EWS-CHOP fusion genes in myxoid liposarcoma (9); recurrent MAGI3-AKT3 translocations complemented by MAGI3 hemizygous deletions in breast cancer, which combine the loss of function of a tumor suppressor gene (PTEN) with the activation of an oncogene (AKT3) (10); gene fusions involving the RAF family of serine/threonine protein kinases in pediatric low-grade astrocytomas (11); and a common translocation found in Burkitt lymphoma, t(8;14)(q24;q32), that fuses MYC with an immunoglobulin heavy chain (12).
Key to the generation of chromosomal aberrations are breaks in the continuity of the DNA double helix followed by error-generating repair processing, which may join two noncontiguous segments of a chromosome (deletions), insert novel sequences (insertions), or fuse two different chromosomes (translocations) (1,2,13). Interestingly, two major DNA repair pathways currently known to act upon DNA double-strand breaks (DSBs): (i) non-homologous end joining (NHEJ), which is active throughout the cell cycle and does not require sequence homology; and (ii) homologous recombination (HR), which is active in S phase and G2 and uses homologous sequences from sister chromatids to restore chromosome continuity, are relatively error-free and appear not to be frequently involved in cancer instability (14–17). Indeed, sequence analyses of whole cancer genomes, detailed characterization of the sequence contexts at the points of DSB fusion (referred to as breakpoints), and the finding that HR is often compromised in cancer cells, provide mounting support for the idea that somatic chromosomal aberrations involve DNA repair pathways that play minor or back-up roles in normal cells (15,16,18). Consistent with this notion is the observation that the HR-deficient genetic signature noted in many breast cancers correlates strongly with >3 bp insertions and deletions; this, together with the presence of overlapping microhomologies at the breakpoints, is inconsistent with NHEJ and points instead to a role for replication-based mechanisms of DNA repair (2,18). Two pathways, microhomology-mediated end joining (MMEJ), also referred to as alternative NHEJ (alt-NHEJ), and single-strand annealing (SSA) share with HR the initial steps of end processing and end resection, but diverge at subsequent steps and use either minimal (generally fewer than a dozen bases for MMEJ) or substantial (>30 bases for SSA) homology to complete repair (14,15,18). Hence, replication fidelity issues appear to play a pivotal role in cancer-related genomic instability (11), although tissue-specific mechanisms, such as ectopic V(D)J recombination in hematologic malignancies, are also involved (19).
Replication forks may stall, resulting in fork collapse, following a number of different insults, such as bulky base adducts, pre-existing strand breaks, and DNA crosslinks (2,3,18); indeed, current cancer therapeutics are motivated in part by targeting replication through crosslinking agents, topoisomerase inhibitors and high-dose radiation. However, other mechanisms that lead to replication arrest have recently emerged, including head-on collision with transcription and unresolved DNA secondary structures, commonly referred to as non-B DNA (16,20–23). The possibility that non-B DNA can form in chromosomal DNA, further block replication and cause genomic instability in cancer is particularly intriguing for many reasons. First, several types of potential non-B DNA structure (PONDS)-forming sequence are mutagenic, resulting in DSBs that are then processed into large-scale deletions, rearrangements and translocations (23–28). Second, the sequences in the human genome that can fold into PONDS, such as quadruplexes, triplexes (or H-DNA), hairpin/cruciform, slipped conformations and left-handed Z-DNA, number in the hundreds of thousands (29). Third, an increasing number of hereditary neurological diseases are linked to DNA repeats that expand in length following their folding into PONDS, which then represent aberrant substrates for DNA repair factors (30–32); likewise, PONDS-forming repeats have been associated not only with nonsense and missense mutations but also microinsertions and microdeletions causing human inherited disease (33). Fourth, segments of the genome that are known to be hotspots for genomic rearrangements in cancer genomes, such as common fragile sites, harbor an unusually high density of PONDS-forming sequences (34–39). Indeed, a physical association between the location of rearrangement breakpoints and the occurrence of PONDS-forming repeats has been suggested (9,27,40–44). However, the lack of well-defined criteria for the identification of PONDS-forming repeats, coupled with the absence not only of large sets of genome-wide data with single base-pair resolution for the breakpoint positions but also matching sets of appropriate controls, have until now hampered a robust objective assessment of the role of non-B DNA in genomic instability in cancer.
Herein, we report an unbiased analysis in which we compare the physical distance of two distinct sets of ∼20 000 control genomic positions, ∼20 000 translocation breakpoints and ∼46 000 deletion breakpoints mapped at single base-pair resolution in human cancer genomes with the occurrence (within ±500 bases of the breakpoints) of five types of PONDS-forming repeats (direct repeats, inverted repeats, homo(purine•pyrimidine) tracts with mirror repeat symmetry, alternating purine-pyrimidine runs, and G-quartets), which may form slipped structures, hairpin/cruciforms, triplex (H-DNA), left-handed Z-DNA, and quadruplex DNA (G4-DNA), respectively. Strikingly, we show that for all types of repeat, the aggregate number of bases peaks exactly at the breakpoint positions for translocations and deletions, decreasing with distance from the breakpoints. Statistical analyses reveal a strong correlation between PONDS-forming repeats and rearrangement breakpoints, particularly for translocations. Specific types of sequence combinations, such as AT-rich inverted repeats and homo(purine•pyrimidine) tri- and tetra-nucleotides occur most often at translocation breakpoints, whereas A-tracts are most strongly associated with deletion breakpoints. The association between PONDS-forming repeats and breakpoints observed here is further supported by the observation that rearrangements tend to recur at near-identical genomic positions in different patient and tumor samples. These data provide compelling support for the notion that sequences with the potential to fold into non-B DNA structures merit attention as an intrinsic risk factor for the occurrence of translocations and deletions in cancer genomes.
MATERIALS AND METHODS
Datasets
The dataset of translocation and deletion breakpoint coordinates in cancer genomes was obtained from the Catalogue Of Somatic Mutations In Cancer (COSMIC) at http://cancer.sanger.ac.uk/cosmic/) (file CosmicStructExport_v70_100814.tsv). A first control dataset (Contr1) of simulated genomic breakpoint positions was built using SAMtools (http://samtools.sourceforge.net). A second control dataset (Contr2) comprised all genomic coordinates located 3000 bp upstream from the translocation breakpoint coordinates. The list of L1 retrotransposons was downloaded from the European database of L1-HS retrotransposon insertions in humans (euL1db) at http://eul1db.unice.fr/db/ (file ReferenceL1HS.txt). The dataset of microRNA gene coordinates was downloaded from miRBase, the microRNA database at http://mirbase.smith.man.ac.uk (file hsa.gff3).
Repeat searches
The sequences of genomic intervals (1-kb bins) centered at the translocation, deletion or control (Contr1 and Contr2) breakpoint coordinates were retrieved from the hg19.2bit file using the utility twoBitToFa from http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/. When needed, as with the mir gene list, genomic coordinates were transformed from one assembly to another with liftOver. Any bin containing undefined bases (N) was excluded from subsequent analysis. PONDS-forming repeats were obtained using custom scripts (bash, gawk and C++) using the criteria listed in Table 1. To avoid retrieving overlapping strings of different lengths, motif searches started from the upper bound lengths, breaking the loops after a hit was found and relocating the searches at the end of substrings. Only uninterrupted motifs were sought. All work was performed on Linux clusters at the Texas Advanced Computing Center (https://www.tacc.utexas.edu).
Table 1. Density of PONDS-forming repeats.
| Repeat type | Length (bp) | Spacer (bp) | Contr1 (n/kb) | Contr2 (n/kb) | Trans (n/kb) | Delet (n/kb) |
|---|---|---|---|---|---|---|
| DR | 3 – 100 | 0 | 0.2483 | 0.2678 | 0.3446 | 0.2766 |
| IR | 7 – 30 | 0 – 7 | 1.0639 | 1.0381 | 1.2008 | 1.1944 |
| H-DNA | 6 – 50 | 0 – 7 | 1.2274 | 1.2704 | 1.4444 | 1.2288 |
| G4-DNA | 15 – 90 | 1 – 7 | 0.1234 | 0.1457 | 0.1576 | 0.1316 |
| Z-DNA | 10 – 120 | 0 | 0.1120 | 0.1162 | 0.1239 | 0.1221 |
| Sum | 2.7750 | 2.8382 | 3.2713 | 2.9535 |
DR, direct repeats; IR, inverted repeats; H-DNA, triplex-forming homopurine•homopyrimidine runs with mirror repeat symmetry; G4-DNA, G-quartet-forming sequences of ≥4 runs of GGG each separated by 1–7 bases, but excluding homoG•homoC runs; Z-DNA, alternating purine-pyrimidine motifs (pure or mixed A-C, G-C, G-T runs). Length, min and max lengths of repeats. For DR, length refers to the length of each unit, for IR and H-DNA it signifies the length of each of the two stems, for G4-DNA it indicates the total length of a tract including spacer sequences between the G runs, and for Z-DNA it includes the total number of bases. For DR, the minimum number of repeat units was set to 5. Spacer, number of bases separating two units. Contr1, 1-kb bins flanking 20 222 randomly generated genomic coordinates; Contr2, 1-kb bins flanking 19 935 genomic coordinates, each located 3000 bp upstream (lower genomic coordinate; N-containing bins were excluded) of their respective translocation breakpoints; Trans, 1-kb bins flanking 19 947 translocation breakpoints; Delet, 1-kb bins flanking 46 365 deletion breakpoints; n/kb, density of motifs in number per kb; Sum, sum of all densities.
Statistics
To perform statistical tests, we linked our C++ codes to the BOOST libraries (http://www.boost.org). When assuming unequal variance for the data, the two-sample Student's t-test implemented a Welch-Satterthwaite approximation, which affords real number degrees-of-freedom parameters, and hence high accuracy. For curve fitting, we used SigmaPlot12 (http://www.sigmaplot.com). The program Circos was obtained from http://circos.ca; Perl modules were downloaded from CPAN at http://search.cpan.org.
RESULTS
Translocation and deletion breakpoints occur near PONDS-forming repeats
A primary goal of this work was to robustly ascertain whether DNA strand breaks leading to translocations and deletions in cancer genomes occur preferentially at sites that are capable of adopting alternative DNA structures; such structures are known to be formed by several types of repeating sequence (broadly termed PONDS-forming repeats). These include tandem repeats, inverted repeats, homopurine•homopyrimidine runs with mirror repeat symmetry, four or more GGG repeats separated by a ‘spacer’ of 1–7 bases, and alternating purine-pyrimidine tracts; these elements may give rise to slipped single-stranded loops, cruciforms, triplex DNA, quadruplex and left-handed Z-DNA structures, respectively. We applied defined criteria (Table 1 and Materials and Methods) to search for uninterrupted PONDS-forming repeats of specific length ranges occurring within ±500 bases of 19 947 translocation and 46 365 deletion breakpoints in cancer genomes derived from the COSMIC dataset. We then compared the results with those obtained from two sets of controls: a dataset comprising 20 282 randomly generated genomic positions (Contr1) and a dataset of 19 935 positions (Contr2), each located 3-kb upstream from its corresponding translocation breakpoint, which would capture any regional bias in sequence context in which these rearrangements took place. This bias might for example include a higher GC content at translocations than the genome-wide average (see below). The distribution of translocation (and deletion) breakpoints did not however display a preference for gene regions relative to Contr1 (Supplementary Figure S1A), implying underlying stochastic mechanisms for their occurrence, undetectable levels of selection genome-wide, and a high likelihood that many of these lesions represent passenger mutations.
The number of repeats per kb (repeat density) in the 1-kb bins varied by ∼10-fold, from 0.1/kb for G4-DNA and Z-DNA, to >1/kb for H-DNA and IR (Table 1). However, the density of both individual repeat types and their sum followed a consistent trend, being at their highest near translocation breakpoints (3.27/kb), lower near deletion breakpoints (2.95/kb) and at their lowest (2.77/kb and 2.84/kb) in the controls (Table 1). Although accurate statistical analyses were confounded by the fact that most sequences populated multiple repeat types [e.g. (GGAA)n is both a DR-forming and an H-DNA-forming motif], these results suggested that both translocation and deletion breakpoints tend to occur near PONDS-forming repeats.
Repeats associate more strongly with translocations than with deletions
Next, we assessed the distribution of PONDS-forming motifs with respect to the controls, near translocation and deletion breakpoints by computing the total number of bases belonging to each type of repeat within the range −500 to +500 bp from the breakpoint positions (Figure 1A; 1-kb bin), and comparing these distributions after normalization. Visual inspection of the graphs (Figure 1B–F and Supplementary Figure S1B–F) revealed that the number of repeats was highest for the translocation breakpoint-containing bins for all five types of PONDS-forming repeats, and that in all cases repeat numbers peaked precisely at the breakpoint position. A similar trend, albeit less pronounced, was evident for the deletion breakpoint-containing bins, whereas for the controls the number of repeats oscillated monotonically around average values. For translocations, the peak area was broad for H-DNA, DR and IR (Figure 1B–D), extending approximately from −200 to +200; it was very sharp for Z-DNA (approximately −50 to +50, Figure 1F) and least well defined for G4-DNA (Figure 1E). Thus, with the exception of IR, for which both the abundance and peak area of the repeats were similar for translocations and deletions, PONDS-forming repeats are frequently found exactly at, or in close proximity to (±200 bp), translocation breakpoints in cancer genomes.
Figure 1.

Translocation and deletion breakpoints occur near PONDS-forming motifs. (A) Schematic of a 1 kb-bin showing the breakpoint at position 0 and three sections: left from −500 to −177; middle from −176 to 176; and right from 177 to 500. (B) Number of DNA triplex-forming repeats (H-DNA) for 10 000 bins found near translocation (red), deletion (green) and Contr1 (black) breakpoints. (C) Same as in B, but for cruciform-forming inverted repeats (IR). (D) Same as in B, but for loop DNA-forming tandem repeats (DR). (E) Same as in B, but for quadruplex-forming repeats (G4-DNA). (F) Same as in B, but for left-handed DNA-forming repeats (Z-DNA). Numbers refers to the counts of bases belonging to each repeat type at every position; for H-DNA and IR, any bases separating a pair of repeats were excluded from the count.
To determine whether the associations of PONDS-forming repeats with translocation and deletion breakpoints were statistically significant, we applied Student's t-tests, assuming unequal variance for the data. Since the numbers of PONDS-forming repeats peaked at the breakpoint sites and fell sharply toward the edges of the range (i.e. close to ±-500), the data were compared separately for three distinct sections of the graphs: left, from positions −500 to −167; middle, from positions −166 to +166; and right, from positions +167 to +500 (Figure 1A). P-values were ranked and corrected for multiple testing to determine the threshold of significance (Supplementary Table S1A). The comparisons between left (or right) and middle sections were strongly affected by end-effects, which gave rise to P-values of up to 5.2 × 10−10 (for H-DNA repeats in the control dataset; Supplementary Table S1A). We therefore limited the analyses to the middle sections, which contained the breakpoint sites and therefore may be the most relevant from a biological standpoint.
P-values derived from comparisons between translocations and controls were significant for all five types of PONDS-forming repeats, and spanned more than 175 orders of magnitude, being most pronounced for IR (3.8 × 10−179 and 1.4 × 10−180) the strongest associations of all comparisons), H-DNA (1.4 × 10−142 and 4.9 × 10−146), DR (6.1 × 10−107 and 4.2 × 10−100), G4-DNA (1.6 × 10−107 and 3.9 × 10−39), but weakest for Z-DNA (1.1 × 10−5 and 1.6 × 10−13) (Table 2). For deletions versus Contr1, P-values were significant for four distinct repeat types, viz. IR, where the significance level was most pronounced (1.5 × 10−147), DR (7.8 × 10−116), G4-DNA (8.8 × 10−27) and H-DNA (9.7 × 10−11), but were not significant for Z-DNA (Table 2). P-values were also significant for all five repeat types between translocations and deletions, as expected from the fact that more repeats were found near translocation breakpoints than deletion breakpoints (Figure 1B–F).
Table 2. P-values for middle sections.
| Rank | Trans vs. Contr1 | Trans vs. Contr2 | Delet vs. Contr1 | Trans vs. Delet | ||||
|---|---|---|---|---|---|---|---|---|
| Repeat | P-value | Repeat | P-value | Repeat | P-value | Repeat | P-value | |
| 1 | IR | 3.8E−179 | IR | 1.4E−180 | IR | 1.5E−147 | H-DNA | 1.9E−137 |
| 2 | H-DNA | 1.4E−142 | H-DNA | 4.9E−146 | DR | 7.8E−116 | G4-DNA | 4.0E−074 |
| 3 | G4-DNA | 1.6E−107 | DR | 4.2E−100 | G4-DNA | 8.8E−027 | DR | 6.6E−058 |
| 4 | DR | 6.1E−107 | G4-DNA | 3.9E−037 | H-DNA | 9.7E−011 | IR | 1.1E−018 |
| 5 | Z-DNA | 1.1E−005 | Z-DNA | 1.6E−013 | Z-DNA | 7.3E−002 | Z-DNA | 3.2E−002 |
P-values of Student's t-tests for differences in the number of PONDS-forming repeats in the middle sections of translocation, deletion, Contr1 and Contr2 breakpoints after Bonferroni correction for n multiple testing (n = 20 000).
The H-DNA motifs were characterized by a more frequent occurrence of long tracts within translocation bins than within control and deletion bins (Figure 2A; P-values from t-tests on log-log linear regression slopes: 0.0012 for translocations versus controls; 0.0021 for translocations versus deletions; cf. 0.36 for deletions versus controls), whereas the density distribution of DR within bins was greater in the sequence contexts of translocations (breakpoints and Contr2) than for deletion and Contr1 bins (Figure 2B; P-values from t-tests on log-normal linear regression initial (x-axis from 1 to 6) slopes: 0.0023 for translocations versus Contr1; 0.0007 for translocations versus deletions; cf. 0.13 for deletions versus Contr1). These data establish that all types of PONDS-forming repeat are associated with the occurrence, in their immediate vicinity, of translocation events in cancer genomes. A weaker but still significant association also exists between 4/5 types of PONDS-forming repeat (IR, DR, H-DNA and G4-DNA) and deletion junctions.
Figure 2.
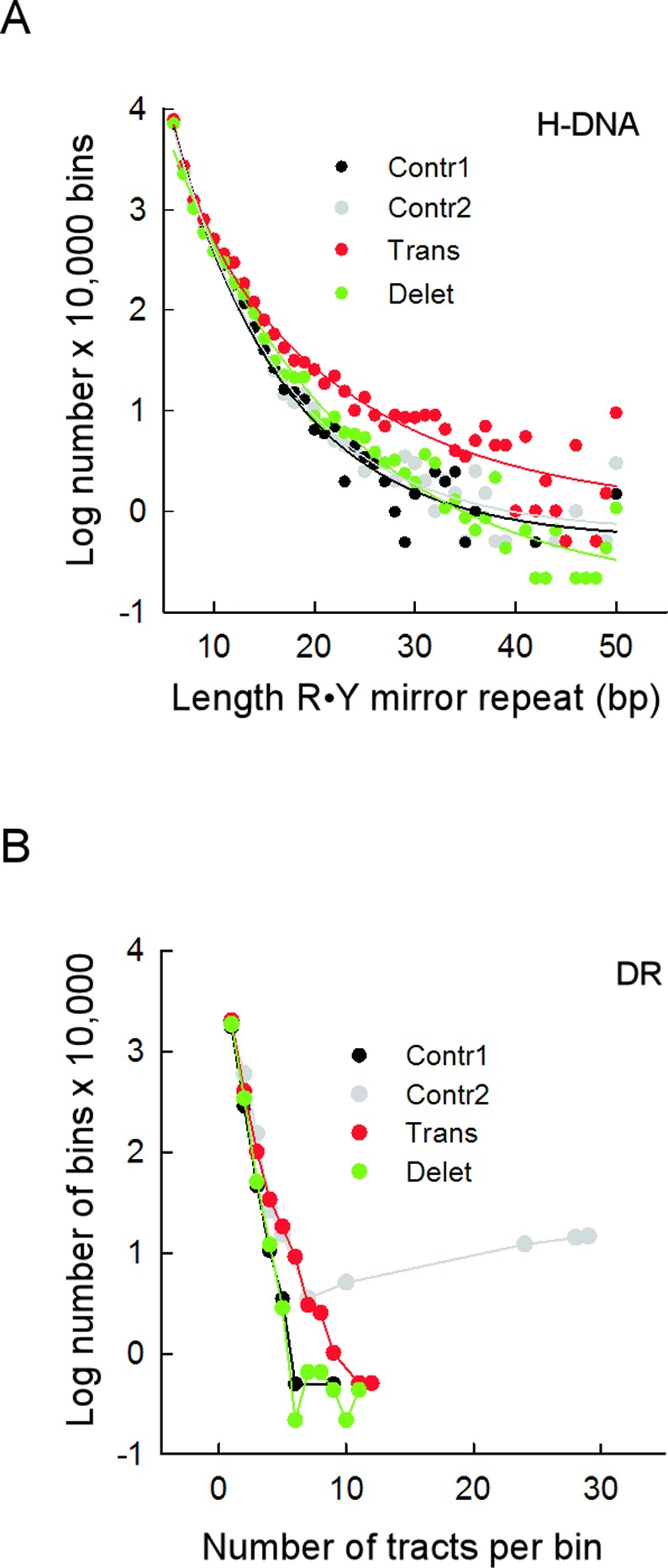
Translocation breakpoints occur near long H-DNA-forming and closely-spaced DR-forming tracts. (A) Length distribution of R•Y mirror repeat tracts in 1-kb bins containing translocation (red), deletion (green), Contr1 (black) and Contr2 (gray) breakpoints. Length refers to the number of bp in each of the two mirror repeats, not including the intervening sequences separating them. (B) Distribution of the number of DR tracts in the 1-kb bins (density) for translocation (red), deletion (green), Contr1 (black) and Contr2 (gray) breakpoints.
Repeat type supersedes genome-wide dependencies on GC content
The fraction of G+C bp (GC content) along genomic DNA deviates from the average near chromosomal rearrangements in cancer genomes, being higher at translocation sites and lower at sites of deletion (45–48), although complex co-dependencies with other genomic features, such as replication timing, transcription, cytosine methylation, and DNA repair processing have been noted (49). We assessed the average GC content at each position along the 1-kb bins (Figure 1A) for translocation, deletion and Contr1 breakpoints, both for the full COSMIC dataset and for the PONDS-forming repeats within the 1-kb bins. For the full dataset, the average GC content was consistently higher for translocations (0.415 ± 0.004; mean ± SD) than for deletions (0.409 ± 0.002) or Contr1 (0.408 ± 0.004), with P-values of 1.0 × 10−138, 2.6 × 10−130 and 2.0 × 10−89 relative to Contr1 for the right, left and middle sections (Figure 1A), respectively (Figure 3A, Table 3 and Supplementary Figure S1B). Hence, we find that translocations in cancer genomes tend to occur within GC-rich regions, thereby supporting and extending previous observations (45–48).
Figure 3.

GC content is repeat-type specific and can vary substantially at translocation and deletion breakpoints. (A) Average GC content at each position along 1-kb bins and running average of the data using 0.100 of sampling proportions for the full COSMIC dataset of translocation (red) and deletion (green) breakpoints and for the Contr1 dataset (black). (B) Average GC content for H-DNA repeats (any sequence separating two mirror repeats was not included) at every position along 1-kb bins and running average of the data using 0.100 of sampling proportions. (C) Same as in B, but for IR (any sequence separating two IR sequences was not included). (D) Same as in B, but for DR. (E) Same as in B, but for G4-DNA. (F) Same as in B, but for Z-DNA.
Table 3. Selected statistics on GC content for middle sections.
| Type | Pair | Means | SD | P-value | |||
|---|---|---|---|---|---|---|---|
| Total | Trans | Contr1 | 0.415 | 0.408 | 0.004 | 0.004 | 2.0E−089 |
| IR | Trans | Contr1 | 0.219 | 0.282 | 0.025 | 0.026 | 8.1E−133 |
| DR | Trans | Contr1 | 0.222 | 0.161 | 0.030 | 0.031 | 4.3E−099 |
| H-DNA | Trans | Delet | 0.272 | 0.232 | 0.022 | 0.024 | 1.0E−077 |
Means, SD and P-values of Student's t-tests after Bonferroni correction for GC content of the most significant differences between translocations (Trans), deletions (Delet) and controls (Contr1) for the middle sections of 1-kb bins for the full COSMIC dataset (Total) and the IR, DR, and H-DNA PONDS-forming repeats.
For H-DNA, IR and DR, the GC content was lower (∼0.12–0.28) than average, irrespective of whether they flanked translocation, deletion or Contr1 breakpoints, whereas for G4-DNA and Z-DNA it was higher (∼0.78 and ∼0.54, respectively). Surprisingly, for the low GC content repeats, significant changes were noted at the breakpoint sites for both translocations and deletions. For example, the GC content for IR fell by almost 0.1 unit at the translocation breakpoint positions relative to the flanking positions, with mean running-average values decreasing from 0.281 ± 0.028 to 0.219 ± 0.025 when proceeding from the left to the middle sections. These differences cannot be explained by end-effects alone, since the P-values between translocations and controls (which are expected to cancel out end-effects) strengthened from non-significant or barely significant (0.0028) to 8.1 × 10−133 when shifting from the left (or right) to the middle sections (Table 3 and Supplementary Table S1B).
For DR, the GC content at translocations increased steadily as it approached the breakpoints from either the left or right sections, with mean running-average values of 0.1667 ± 0.043 for the left section, 0.173 ± 0.034 for the right section, and 0.222 ± 0.030 for the middle section. Again, the difference between translocations and Contr1 (0.161 ± 0.031) increased from non-significant to highly significant (P-value 4.3 × 10−99) when moving from the flanking to the middle sections (Table 3 and Supplementary Table S1B). Finally, for H-DNA, the GC content at translocation and deletion breakpoints displayed a contrasting trend, peaking at the breakpoint positions for translocations (mean running-average for sections: left, 0.245 ± 0.027; right, 0.257 ± 0.032; middle, 0.272 ± 0.022) but reaching the lowest points at the breakpoint positions for deletions (left, 0.246 ± 0.019; right, 0.249 ± 0.020; middle, 0.231 ± 0.024), thereby yielding a marked difference between the two middle sections (P-value 1.0 × 10−77, Table 3). As noted for G4-DNA and Z-DNA, differences between sections were not evident, and there were no differences in GC content between translocations, deletions and Contr1 (Figure 2). We conclude that in cancer genomes, PONDS-forming repeats override the association of translocations with high-GC content genome-wide, and instead set new dependencies that not only apply to both translocations and deletions but are also repeat-specific.
Culprit repeats
The results depicted in Figure 3 suggested that specific DNA sequence combinations might be found near translocation and deletion breakpoints (i.e. in the middle sections), which are expected to elicit genomic rearrangements with the highest frequencies. Thus, we examined the most frequently occurring repeats (top ten) for each repeat type. For IR at translocations, the middle section was characterized by an unusually high number (9/10) of (AT)n dinucleotide repeats, relative to the left (4/10) and right (5/10) sections (Figure 4A), which together comprised 16.1% of all IR, compared to 3.8% (P < 0.001; alpha power at 0.05 = 1.000; z-test) for the left and 6.2% (P < 0.001; alpha power at 0.05 = 1.000; z-test) for the right sections. This result coincides with the sharp fall in GC content at IR translocation breakpoints (Figure 3C), and suggests that (AT)n dinucleotide repeats could be potent inducers of translocation. Consistent with this postulate, a comparison of all IR sequences between translocations and Contr1 revealed that IR stems with no C•G bp [i.e. (AT)n dinucleotides] were vastly overrepresented within the middle section of translocations at the expense of stems with 1–6 C•G bp (Figure 4B and Supplementary Figure S2A). Additional analyses of microRNA genes genome-wide, which are known to comprise imperfect IR motifs, revealed no noticeable association with translocation breakpoints. Hence, we conclude that AT-rich IR play a particularly prominent role in inducing translocations in cancer genomes.
Figure 4.

Specific sequence combinations are strongly associated with translocation and deletion breakpoints. (A) Top ten IR sequences most frequently found near translocation breakpoints. Bars, fractions relative to all IR present in the respective sections, left, middle and right. Color distinguishes between mixed-type sequences (black) and pure (A•T)-containing motifs (red). Sequence corresponds to the upstream (lowest genomic coordinates) repeat, excluding any intervening sequence. Stem, sequence of predicted stem-loop cruciform structures. (B) For each upstream (lowest genomic coordinate) IR sequence containing from zero to six C|G bases, the fraction of the total number of IR found in the left, middle and right sections was computed for the translocation and Contr1 1-kb bins. The fractions obtained for Contr1 were subtracted from those obtained for the translocations and the differences were plotted separately for each section. Negative values indicate overrepresentation of IR sequences in the control bins, whereas positive values indicate overrepresentation in translocation bins. Data for the middle section (dark green) are distinguished from the left and right sections (cyan). (C) Top ten DR sequences most frequently found in the left and middle sections of translocation breakpoints. Bars, fractions relative to all DR present in the respective section. All sequences are (A•T)n mononucleotides, with n ranging from 15 to 30. X-axis, sequence composition of hg19 reference genome sequence, top strand. (D) For DR, the fractions of mono-, di-, tri-, tetra-, penta-, hexa- and >hexa-nucleotides were computed separately for the translocation left and middle sections. Data plotted for the left section were subtracted from those of the middle section. Negative values indicate underrepresentation in the middle section, and vice versa. (E) For DR found in either the left, middle or right sections of the translocation, deletion and Contr1 1-kb bins, the fraction of tetra-nucleotides whose strand sequence composition contained only purines (or pyrimidines, i.e. R•Y tracts) relative to all tetra-nucleotides in the respective section was computed and plotted. The green bar highlights the overrepresentation of R•Y-containing tetranucleotides in the middle section of translocations. (F) For H-DNA, the fraction of repeats containing from zero to six C|G bases in the upstream (lower genomic coordinates) R•Y mirror repeat unit (stem of putative triplex structures) was taken for the middle sections of translocation and deletion 1-kb bins and plotted as a function of C|G occurrences. Note that a value of 0 refers to (A•T)n mononucleotide repeats and that C|G bases could be either contiguous or not. Mean, data for the combined distributions. Pink and green backgrounds highlight the shift in overrepresentation occurring between 1 and 2 C|G.
For DR, A-tracts represented all of the top 10 sequences in 7/9 sections (three for translocations; three for deletions and two for Contr1) and 9/10 sequences in the remaining 2/9 sections. However, the combined fraction (relative to all DR in the corresponding section) was lowest (38.6%) in the middle section of translocations (range 53.0–59.4% for all other sections; P-value of 4.79 × 10−8, 1-sample Student's t-test), again consistent with the sharp increase in GC content observed in this region (Figure 3D). The most abundant A-tracts [(A•T)15, (A•T)20 and (A•T)18] were also the most underrepresented (Figure 4C); A-tract underrepresentation in the translocation middle section was compensated for by an increase in other microsatellites, particularly tetra-nucleotides (Figure 4D and Supplementary Figure S2B). Thus, of all DR, A-tracts appear to be the weakest inducers of translocation in cancer genomes. For the di-, tri- and tetra-nucleotide repeats, the fractions of those whose sequence composition only contained (G|A)•(T|C) bp (i.e. R•Y tracts capable of triplex formation), were also highest in the middle section of translocations (Figure 4E, Supplementary Figure S2C and S2D). Furthermore, among the R•Y-containing DR (for the combined tri- and tetra-nucleotides), the fraction of A-rich sequences, i.e. (GAA)n and (GAAA)n, was also at its highest in the middle section of translocations: 0.74 versus 0.39–0.62 for the other sections (P-value of 1.25 × 10−4, one-sample Student's t-test). Indeed, 197.5/10,000 bins (394 total) (GAA)n and (GAAA)n-containing DR were found in the middle section of translocations as compared to 38.6 ± 2.1 for the left and right sections, 24.1 ± 5.7 for Contr1 and 19.1 ± 3.3 for deletions (P-values of ∼1.90 × 10−10; one-sample Student's t-test). These data provide compelling support for the contention that (GAA)n and (GAAA)n-containing DR are triggers of translocation in cancer genomes, and that the guanine within the otherwise monotonic A-stretches (i.e. (GAA)n and (GAAA)n) plays a key (and indispensable) role in conferring such potency.
For H-DNA, the characteristic decrease in GC content in the middle section of deletions (Figure 3B) was consistent with an enriched fraction of R•Y stems comprising short A-tracts (0.37 versus 0.33 ± 0.02 for the other eight sections; P-value 3.53 × 10−4, one-sample Student's t-test; Supplementary Table S2) and a concomitant decrease in R•Y stems with ≥2 C•G bp (P-values 1.49 × 10−1–1.03 × 10−4; one-sample Student's t-tests; Figure 4F and Supplementary Table S2). The opposite pattern was noted for the middle section of translocations (Figure 4F), which was characterized by the lowest fraction of (A•T)n-containing stems (0.29 versus 0.34 ± 0.01 for the other eight sections; P-value 3.34 × 10−5; one-sample Student's t-test; Supplementary Table S2) and the highest fractions of stems with ≥2 C•G bp (P-values 6.27 × 10−1–1.26 × 10−5; one-sample Student's t-tests; Figure 4F and Supplementary Table S2). These results are consistent with the DR data described above [(A•T)n-containing tracts were retrieved by both DR and H-DNA searches], and indicate that a significant proportion of deletion breakpoints in cancer genomes occurred within a short distance (±250 bp) of A-tracts.
Translocation breakpoints recur at PONDS-forming repeats in different patients
Next, we asked if the co-localization of PONDS-forming repeats with translocation breakpoints was sufficiently potent to recur at or near the same genomic locations in different individuals or tumor samples. In the Contr1 dataset, the number of simulated breakpoints occurring within ±250 bp of any PONDS-forming repeat (its boundaries) increased linearly from 72 to 4821 as the distance between any two breakpoints increased from 500 to 50 kb, thereby confirming the random nature of the distribution (Figure 5A, inset). By contrast, in the translocation dataset, the number of breakpoints occurring within ±250 bp of any PONDS-forming repeat increased sharply from 721 to 3583 in the range from 10 bp to 5 kb, and then followed a rate of increase similar to that of the control dataset (Figure 5A, Inset).
Figure 5.

Clusters of translocation breakpoints occur near both PONDS-forming repeats and L1 retrotransposons. (A) Inset. Total number of breakpoints (y-axis) located within 10 bp to 50 kb (x-axis) from one another. Black circles, subset of breakpoints within ±250 bp of a PONDS-forming repeat present in the Contr1 dataset. Solid red circles, subset of breakpoints within ±250 bp of a PONDS-forming repeat present in the translocation dataset. Open red circles, subset of breakpoints in the COSMIC dataset (total) left after the data from ‘solid red circles’ were subtracted. Main panel, same as inset displaying clustered breakpoints separated by 10–100 bps. (B) Circos plot showing the two main clusters (distance separating any two breakpoints, ≤100 bps) of recurrent translocation (note that rather than being translocations, these may be transductions) events in the COSMIC dataset involving the 3′-end tail of two L1HS transposons, one at 22q12.1 (red links) and the other at Xp22.2 (blue links). Outer circle (green bars on pink background), the 2349 clustered translocation breakpoints in the COSMIC dataset (distance separating any two breakpoints, ≤100 bps); middle circle (orange bars on grey background), the 1586 clustered translocation breakpoints in the COSMIC dataset that are within ±250 bp of a PONDS-forming repeat; inner circle (black and red bars on yellow background), the 311 full-length L1HS transposons mapped on to the hg19 reference human genome assembly; long red bars on thin cyan background, the eight L1HS transposons with a 3′-end tail within ±1-kb of clustered translocation breakpoints. (C) Expansion of the genomic region containing the largest (100 events) translocation cluster breakpoints in the COSMIC dataset (total) on 22q12.1. x-axis, 200 bp tick intervals highlighting (light blue) the direction of TCC28 gene transcription; vertical black bars, individual breakpoints; cyan box, L1HS 3′-end region; green box, zone of highest regional DNaseI hypersensitivity; red bars, numbers and sequences, location and sequence of PONDS-forming repeats. (D) Expansion of the genomic region containing the second largest (23 events) translocation cluster breakpoints in the COSMIC dataset (total) on Xp22.2. Legends are as in panel C. (E) Plot displaying the distribution of the number of breakpoint translocation clusters present in the COSMIC dataset (distance separating any two breakpoints, ≤100 bps; y-axis) containing increasing numbers of events (x-axis). Orange, number of clusters found within ±1-kb of L1HS 3′-end tails and P-value obtained from z-tests. Asterisks, z-test on combined single clusters with >4 events each. Upward and downward arrows signify over or underrepresentation, respectively. (F) Fractions of the main cancer types represented in the full (total) COSMIC dataset (light gray) and in the major translocation breakpoint cluster on 22q12.1 (dark gray). UAT, upper aerodigestive tract.
The initial sharp increase was not specific to the breaks occurring near PONDS-forming sequences, since it was also observed with those breakpoints located outside PONDS regions, obtained by subtracting the breakpoints located within ±250 bp of PONDS-forming repeats from the total number of breakpoints. However, the number of breakpoints recurring within the shortest genomic interval examined (i.e. 10 bp) was greater near PONDS-forming repeats than in more distant regions (721 versus 349), and also increased more rapidly (within short intervals, i.e. ≤50 bp) (Figure 5A, main panel). These data clearly reveal that although cancer translocation breakpoints generally tend to recur in different patients or tissue samples at specific locations in the genome, recurrence is more frequent if a PONDS-forming sequence is present in the vicinity. In other words, PONDS-forming repeats appear to be sufficiently potent in terms of inducing translocations that their impact is evident from the recurrence of chromosomal breaks at near-exact positions in different patient/tumor samples. As revealed by comparison with the Contr1 set, this result is most unlikely to be attributable to chance alone.
LINE-1 (L1) retrotransposition has been reported to be an efficient process leading to genomic rearrangements, although it has occasionally been difficult to distinguish genomic translocation from L1 transduction events (50–52). We assessed the extent to which L1 retrotransposition, rather than (or in conjunction with) PONDS-forming motifs, might have been responsible for the recurrence of translocation breakpoints. A total of 2349 translocation breakpoints were present in the COSMIC dataset whose individual exemplars were within 100 bp of one other member, 1586 of which were within ±250 bp of a PONDS-forming repeat (Figure 5B). For L1HS retrotransposon elements, 311 have been mapped in the reference human genome (hg19); however, only in eight cases was the 3′-end close (±1 kb) to any of the 2349 ‘clustered’ translocation breakpoints (sequences downstream of L1HS 3′-ends have been used to identify transduction events (51); Figure 5B).
Despite this paucity, an L1HS source element located at 22q12.1 (within intron 1 of the TTC28 gene) previously noted for its strong transduction activity in cancer genomes (50–54), was associated with the largest cluster of translocation breakpoints, both in the COSMIC dataset (100 instances) and in the set of breakpoints near PONDS-forming repeats (43 instances; Figure 5B and C). In similar vein, an intergenic L1HS source element located at Xp22.2 was found to be in close proximity to three translocation clusters, the third of which was the second largest cluster (23 instances) in both the COSMIC and PONDS-associated datasets (Figure 5B and D). The remaining 6 L1HS elements were located near translocation clusters that were larger than expected based on their count distribution (Figure 5E). With regard to the tissues in which these genomic alterations occurred, cancers of the pancreas were found to be particularly prominent (Figure 5F). No obvious feature, including the presence of DNaseI hypersensitive elements, transcription factor binding sites, intragenic versus intergenic location or PONDS-forming elements, appeared to play a role in the observed association between L1HS elements and translocation clusters. We conclude that a very small number of L1HS elements may be responsible for at least some of the most common recurrent translocation events present in the COSMIC dataset. By contrast, the vast majority of recurrent translocation breakpoints appear to be related to the presence, in their immediate vicinity, of PONDS-forming motifs, thereby further emphasizing our general conclusion that repetitive sequences are highly likely to be involved in inducing genomic instability in cancer genomes.
DISCUSSION
PONDS form structural alternatives to B-form DNA and often have key regulatory functions in DNA replication and transcription (44,55). However, these DNA structures have the potential to stimulate genetic instability that has not yet been methodically examined by robust statistical analyses. Our bioinformatics approach supports a physical association between translocation and deletion breakpoints in cancer genomes and sequences known to form alternative secondary DNA structures in vitro. To our knowledge, this is the most comprehensive study of its kind performed to date. Moreover, the results of the statistical tests applied are consistent with a strong association between the presence of PONDS-forming repeats and the occurrence of translocations and deletions in human cancer genomes. We confirm that translocations, but not deletions, tend to occur in GC-rich regions of the genome, even though the sequences of three of the five PONDS-forming repeats most frequently found at translocation and deletion breakpoints are highly, if not exclusively, AT-rich. These include (AT)n dinucleotide repeats, (GAA)n trinucleotides and (GAAA)n tetranucleotides at translocation breakpoints, and mononucleotides [i.e. A-tracts] at deletion breakpoints. Furthermore, we show that translocations tend to recur at preferred genomic positions in different patients and patient samples, irrespective of the tumor type, and that such recurrence is enhanced at positions at or near PONDS-forming repeats. In the context of recurring breakpoints, our data concur with earlier reports (50–54) that a very small number of L1HS retrotransposons may be highly active in inducing transductions, which may then be incorrectly scored as translocation events.
The two strongest associations were observed for the co-occurrence of IR at translocation and deletion breakpoints, with (AT)n dinucleotide repeats being most frequently found at the sites of translocations. Co-localization of genomic rearrangements in cancer with high densities of the AT:AT dinucleotide step has been noted previously for common fragile sites (34,35,38). The mechanisms that render common fragile sites hubs for genomic instability in cancer remain elusive; however, peaks of high flexibility and prominent DNA secondary structures, which would be predicted to exacerbate difficulties in completing DNA replication within regions sparsely populated with replication origins (56,57), and cleavage by structure-specific nucleases (23), may play a role.
The extent of the reported relationship between DNA flexibility and genomic instability is currently unclear because the ranking of flexible base-pair steps used in the earlier analyses of common fragile sites (38,58–60) is inconsistent with more recent findings (61–64). Indeed, early thermodynamic calculations of base-pair flexibility in the absence of phosphate backbones indicated that the AT•AT dinucleotide step underwent the largest fluctuations in twist angles (>25°) and was therefore the point of greatest flexibility in duplex DNA (60). However, more recent molecular dynamics determinations based on sugar puckering and rotations around the ζ/ϵ and α/γ torsion angles suggest that the CG•CG, CA•TG and TA•TA dinucleotide steps constitute favorable hinges for global bending and twisting under resting conditions, whereas AT•AT and GC•GC are stiff points for deformation (61). Studies of DNA curvature and flexibility at A-tracts also suggest that the pyrimidine-purine dinucleotide steps represent flexible hinge points and sites of DNA bending (62,63). Analyses of large sets of DNA duplexes by solution NMR and x-ray diffraction data aimed at evaluating backbone conversion between the BI (angles ϵ – ζ <0°) and BII (angles ϵ – ζ >0°) states as a measure of flexibility, also indicate that the AT:AT dinucleotide is least flexible (score of 0), whereas the CG:CG, CA:TG and GG:CC dinucleotides are the most flexible (scores of 43, 42 and 42, respectively) (64).
Thus, we propose that the observed association of (AT)n repeats with translocation breakpoints arises from the propensity of such sequences to fold into intramolecular hairpin and cruciform structures, rather than from their intrinsically high flexibility, although a contribution from low thermal stability and duplex destabilization cannot be excluded (65). An unbiased analysis of the potential of overlapping 300-bp windows along chromosome 10 to fold into looped-out secondary DNA structures revealed a direct correlation between low negative free energy values (i.e. stable secondary structure prediction) and aphidicolin-induced common fragile sites (39). Importantly, the regions of low free energy values were predominantly GC-rich and overlapped with genes known to undergo rearrangements (deletion and amplification) in several cancer types (39). A role for cruciform-forming AT-rich repeats in stimulating chromosomal breaks has also been suggested for several constitutional translocations, including the recurrent t(11;22)(q23;q11.2), t(17;22)(q11.2;q11.2), and t(8;22)(q24.1;q11.2), the non-recurrent t(4;22)(q35.1;q11.2) and t(1;22)(p21.1;q11.2) (66); the t(8;22)(q24.13;q11.2) (67), the t(3;8)(p14.2;q24.2) associated with inherited predisposition to renal cell carcinoma (68), deletions and t(17;22)(q11.2;q11.2) translocations of the NF1 gene causing neurofibromatosis type I (69–71), and a balanced t(8;22)(q24.13;q11.2) translocation that disrupted the TRC8 tumor-suppressor gene and was associated with dysgerminoma (72). Sequence resolution of rearrangement breakpoints in specific inherited human diseases (73), in targeted reporter systems in cell culture (23), mouse (74,75), yeast (23,76,77), and during evolutionary diversification in fungi (78), also supports the conclusion that the genomic instability promoted by IR is due to their tendency to fold into hairpin and cruciform structures.
The next strongest correlation was found in relation to the presence of H-DNA forming-repeats at translocation breakpoints, with (GAA)n and (GAAA)n microsatellites being the most overrepresented. Studies of the structural properties of the (GAA)n trinucleotide repeat have been motivated in part by its relevance to Friedreich ataxia, a recessively inherited neurological disorder caused by a (GAA)n expansion in the first intron of the FXN gene (79). At the lengths relevant to our study, n < 17, (GAA)n repeats have been shown by multiple techniques, including chemical and enzymatic probing (80–83), 2D-gel electrophoresis (80,81), atomic force microscopy (80), UV melting (82,84), CD spectra (84), positive-ion electrospray mass spectrometry (85) and high-resolution NMR (82,86), to adopt both of the possible triplex conformers, i.e. the R:R•Y (: denotes Hoogsteen pairing; • denotes Watson-Crick pairing) and the Y:R•Y conformers. The (GAAA)n repeat is also expected to form triplex DNA, and both types of repeat share additional features, including the ability to form parallel duplex DNA, and highly structured helices via the purine-rich single-strands due to strong stacking interactions (87). Hence, (GAA)n repeats have been found to represent impediments to transcription owing to the formation of recombinogenic R-loops, stable RNA:DNA hybrids caused by the persistent association of the nascent RNA with the template DNA strand (88).
Direct repeats, and in particular A-tracts, displayed the strongest association with deletion breakpoints. A-tracts possess unique structural determinants, including the generation of static bending (89–91), a high degree of stiffness imparted by water coordination along the minor groove (92,93), directional narrowing of the minor groove (94,95), and flexible junctions, which appear to have been responsible for generating preferred sites for short (<200 bp) indels in the human population (95). A-tracts may form slipped structures as a result of misalignment during replication or transcription, as well as triplex DNA. However, we suggest that the association of A-tracts with deletion breakpoints in cancer genomes is likely to reflect the propensity of base-pairs flanking duplex A-tracts to break as a result of their intrinsic high flexibility (95), rather than by the formation of slipped and triplex DNA, although such structure-forming sequences are known to stimulate genetic instability via the formation of DSBs, resulting in deletions, rearrangements and/or translocations (23–28).
That distinct types of repeat motifs were associated with either translocation [(AT)n and (GAA)n/(GAAA)n repeats] or deletion [A-tracts] breakpoints raises the question as to whether these sequences may influence downstream repair events in addition to increasing the frequency of DNA breakage. Cruciforms have been shown to represent substrates for endonucleases, including XRCC1/XPF (23) and GEN1 (28,96), whereas the high number of A-tracts genome-wide provides an opportunity for frequent homology-mediated repair and high rates of oxidative damage at the flexible hinges (95). Hence, it is possible that ‘clean’ ends generated by endonuclease cleavage might be preferred substrates for translocation events (97) at IR, whereas end-processing of ‘un-ligatable’ ends and microhomology might yield predominantly deletions at A-tracts (2).
Our results strengthen previous conclusions (41) that G4-DNA motifs are significantly associated with translocation breakpoints in cancer genomes, and extend their association to deletion breakpoints. Finally, translocation but not deletion breakpoints occurred at a significantly high frequency near Z-DNA-forming repeats, although the strength of the association was weakest among all PONDS-forming repeats. For G4-DNA and Z-DNA, the association is expected to arise in part from their propensity to form quadruplex and left-handed Z-DNA, respectively (44). In addition, a number of Z-DNA-forming (CA)n repeats may trigger genomic instability by promoting ectopic V(D)J recombination. For example, the sequencing of deletion breakpoints in acute lymphoblastic leukemia has identified a recurrent hotspot in the CDKN2A gene on chromosome 9p21, also referred to as BCS-LL2, at a (CA)n repeat ending with 5′-CACAGTA-3′, which is very similar to the consensus heptamer V(D)J recognition signal sequence (5′CACAGTG-3′) (42,98,99). Whether left-handed Z-DNA stimulates recombination at such hybrid sites remains to be determined.
Factors that determine DNA breakage and their observed frequency in cancer genomes probably interact combinatorially, and include DNA sequence (RAG1/2 substrates, CpG islands, CpG methylation, Alu elements, fragile sites, secondary structures), physical torsional stress, chromatin structure and histone modification (transcription, H3K4 methylation) [reviewed in (34)]. However, attempts to determine the relative contribution of each factor have been few. H3K4 methylation alone has been shown to induce a net 0.2–0.3% increase in NPM1/ALK translocation upon ionizing radiation in anaplastic large cell lymphoma cells (100), a considerable effect displayed by a single factor. Here, we find that ∼5% of control sites overlapped with a PONDS-forming sequence, as opposed to ∼10% for translocation breakpoints (Figure 1), suggesting that such repeats may have contributed up to 5% of DNA breakage events leading to translocations in these tumor samples. This contribution is likely to be an underestimate since rearrangements in highly repetitive regions of the genome are currently unmappable.
Overall, our large-scale retrospective study suggests that the association between PONDS-forming repeats and chromosomal rearrangements in cancer genomes arises from structural and physical components that are characteristics of both the entire set of repeats as well as those of individual types of sequence motif, such as A-tracts. DNA secondary structures are known for their ability to create topological barriers to replication and transcription, and to trigger a DNA damage response as a result of strand breaks that derive from arrested replication forks and/or from aberrant repair processing, often resulting from head-on collisions between transcription and replication (21). The link between topological conflicts and genomic instability has also been suggested for GC-rich fragile sites in early replicating regions associated with chromosomal rearrangements in B-cell lymphoma, coinciding with highly transcribed and duplicated genes with convergent or divergent transcription (20). Consistent with a role for non-B DNA in inducing genetic instability during DNA replication and transcription, a yeast screen for single-gene deletion mutants that exacerbate gross chromosomal rearrangements induced by (GAA)n repeats, revealed several candidates comprising the replisome core (Mre11-Rad50-Xrs2, Sae2), repair of stalled replication forks (Rad27, Rtt101-Mms1-Mms22), replication-pausing checkpoint surveillance (Tof1-Csm3-Mrc1) and transcription initiation (TFIIA,B,D,F) (101). In addition to conflicts between replication and transcription, strand breaks and the ensuing genomic instability have also been shown to arise from the cleavage of non-B DNA structures by repair enzymes, including mismatch repair and the nucleotide excision repair (30,102).
R-loops, which as already mentioned may be generated by certain motifs such as H-DNA and DR (88,101,103), are increasingly being recognized as a source of genomic instability in cancer (104,105). An intriguing observation in the context of persistent single-strand DNA during transcription is the observation that the pyrimidine-rich strands of synthetic triplexes function as effective baits for the pull-down of transcription-associated splicing factors (106). If the transcription-coupled splicing machinery were to engage in stable interactions in the context of R-loops, it might stall the transcriptional apparatus and block an incoming replication fork, thereby causing DSBs. Nevertheless, the extent of these effects in the rearrangement datasets examined here appears to be minor, since there is no apparent increase of breakpoints at transcribed regions genome-wide (Supplementary Figure S1A).
Strand breaks are additionally generated by oxidation reactions (107), which are expected to occur at higher rates within certain types of repeat motif as a result of sequence context-dependency effects. These effects include a lowering of the energy required to abstract an electron from the guanine residues at (GAA)n, (GAAA)n and G4-DNA motifs as a result of electron delocalization (108,109), and high flexibilities at A-tract (95) and Z-DNA junctions (110,111). On the other hand, the implied impact of PONDS on genomic instability also has implications for the key repair-independent functions of Fanconi anemia, RAD51, and BRCA1/2 proteins in protecting stalled replication forks from degradation by MRE11 and other nucleases (112,113), as fork stalling is likely to be PONDS-related. While providing firm evidence that PONDS-forming repeats promote genomic rearrangements in cancer genomes, our study also raises several new questions, one of the most intriguing being that most identified motifs are more strongly associated with translocation rather than with deletion breakpoints. Whether this bias originates from a choice in the repair pathways acting on stalled forks, the recognition of DNA secondary structures by repair proteins, the processing of R-loops during transcription, the repair of oxidative lesions, failed fork protection or other hitherto unidentified factors, will be important to elucidate.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Qiagen Inc. through a License Agreement with Cardiff University (to D.N.C.); National Institutes of Health [CA093729 to K.M.V.]; National Institutes of Health and National Cancer Institute [CA092584 to J.A.T.]; National Science Foundation [ACI-1134872 to the Texas Advanced Computing Center]. J.A.T. is supported by a Robert A. Welch Distinguished Chair in Chemistry. Funding for open access charge: National Institutes of Health [CA092584].
Conflict of interest statement. None declared.
REFERENCES
- 1.Forbes S.A., Beare D., Gunasekaran P., Leung K., Bindal N., Boutselakis H., Ding M., Bamford S., Cole C., Ward S., et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43:D805–D811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aparicio T., Baer R., Gautier J. DNA double-strand break repair pathway choice and cancer. DNA Repair. 2014;19:169–175. doi: 10.1016/j.dnarep.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsai A.G., Lu H., Raghavan S.C., Muschen M., Hsieh CL, Lieber M.R. Human chromosomal translocations at CpG sites and a theoretical basis for their lineage and stage specificity. Cell. 2008;135:1130–1142. doi: 10.1016/j.cell.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shortt J., Johnstone R.W. Oncogenes in cell survival and cell death. Cold Spring Harb. Perspect. Biol. 2012;4:a009829. doi: 10.1101/cshperspect.a009829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mertens F., Johansson B., Fioretos T., Mitelman F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer. 2015;15:371–381. doi: 10.1038/nrc3947. [DOI] [PubMed] [Google Scholar]
- 6.Gu K., Chan WC, Hawley R.C. Practical detection of t(14;18)(IgH/BCL2) in follicular lymphoma. Arch. Pathol. Lab. Med. 2008;132:1355–1361. doi: 10.5858/2008-132-1355-PDOBIF. [DOI] [PubMed] [Google Scholar]
- 7.Osborne C.S. Molecular pathways: transcription factories and chromosomal translocations. Clin. Cancer Res. 2014;20:296–300. doi: 10.1158/1078-0432.CCR-12-3667. [DOI] [PubMed] [Google Scholar]
- 8.D'Achille P., Seymour J.F., Campbell L.J. Translocation (14;18)(q32;q21) in acute lymphoblastic leukemia: a study of 12 cases and review of the literature. Cancer Genet. Cytogenet. 2006;171:52–56. doi: 10.1016/j.cancergencyto.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 9.Xiang H., Wang J., Hisaoka M., Zhu X. Characteristic sequence motifs located at the genomic breakpoints of the translocation t(12;16) and t(12;22) in myxoid liposarcoma. Pathology. 2008;40:547–552. doi: 10.1080/00313020802320424. [DOI] [PubMed] [Google Scholar]
- 10.Banerji S., Cibulskis K., Rangel-Escareno C., Brown K.K., Carter S.L., Frederick A.M., Lawrence M.S., Sivachenko A.Y., Sougnez C., Zou L., et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405–409. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lawson A.R., Hindley G.F., Forshew T., Tatevossian R.G., Jamie G.A., Kelly G.P., Neale G.A., Ma J., Jones T.A., Ellison D.W., et al. RAF gene fusion breakpoints in pediatric brain tumors are characterized by significant enrichment of sequence microhomology. Genome Res. 2011;21:505–514. doi: 10.1101/gr.115782.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dalla-Favera R., Bregni M., Erikson J., Patterson D., Gallo R.C., Croce C.M. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc. Natl. Acad. Sci. U.S.A. 1982;79:7824–7827. doi: 10.1073/pnas.79.24.7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y., McCord R.P., Ho Y.J., Lajoie B.R., Hildebrand D.G., Simon A.C., Becker M.S., Alt F.W., Dekker J. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 2012;148:908–921. doi: 10.1016/j.cell.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghezraoui H., Piganeau M., Renouf B., Renaud J.B., Sallmyr A., Ruis B., Oh S., Tomkinson A.E., Hendrickson E.A., Giovannangeli C., et al. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol. Cell. 2014;55:829–842. doi: 10.1016/j.molcel.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sfeir A., Symington L.S. Microhomology-mediated end joining: a back-up survival mechanism or dedicated pathway? Trends Biochem. Sci. 2015;40:701–714. doi: 10.1016/j.tibs.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abbas T., Keaton M.A., Dutta A. Genomic instability in cancer. Cold Spring Harb. Perspect. Biol. 2013;5:a012914. doi: 10.1101/cshperspect.a012914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizuno K., Miyabe I., Schalbetter S.A., Carr A.M., Murray J.M. Recombination-restarted replication makes inverted chromosome fusions at inverted repeats. Nature. 2013;493:246–249. doi: 10.1038/nature11676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ceccaldi R., Rondinelli B., D'Andrea A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2015;26:52–64. doi: 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishana M., Raghavan S.C. A non-B DNA can replace heptamer of V(D)J recombination when present along with a nonamer: implications in chromosomal translocations and cancer. Biochem J. 2012;448:115–125. doi: 10.1042/BJ20121031. [DOI] [PubMed] [Google Scholar]
- 20.Barlow J.H., Faryabi R.B., Callen E., Wong N., Malhowski A., Chen H.T., Gutierrez-Cruz G., Sun H.W., McKinnon P., Wright G., et al. Identification of early replicating fragile sites that contribute to genome instability. Cell. 2013;152:620–632. doi: 10.1016/j.cell.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yadav P., Harcy V., Argueso J.L., Dominska M., Jinks-Robertson S., Kim N. Topoisomerase, I. plays a critical role in suppressing genome instability at a highly transcribed G-quadruplex-forming sequence. PLoS Genet. 2014;10:e1004839. doi: 10.1371/journal.pgen.1004839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamanishi A., Yusa K., Horie K., Tokunaga M., Kusano K., Kokubu C., Takeda J. Enhancement of microhomology-mediated genomic rearrangements by transient loss of mouse Bloom syndrome helicase. Genome Res. 2013;23:1462–1473. doi: 10.1101/gr.152744.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu S., Wang G., Bacolla A., Zhao J., Spitser S., Vasquez K.M. Short inverted repeats are hotspots for genetic instability: relevance to cancer genomes. Cell Rep. 2015;10:1674–1680. doi: 10.1016/j.celrep.2015.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang G., Vasquez K.M. Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2004;101:13448–13453. doi: 10.1073/pnas.0405116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang G., Christensen L.A., Vasquez K.M. Z-DNA-forming sequences generate large-scale deletions in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2006;103:2677–2682. doi: 10.1073/pnas.0511084103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang G., Carbajal S., Vijg J., DiGiovanni J., Vasquez K.M. DNA structure-induced genomic instability in vivo. J. Natl. Cancer Inst. 2008;100:1815–1817. doi: 10.1093/jnci/djn385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nambiar M., Goldsmith G., Moorthy B.T., Lieber M.R., Joshi M.V., Choudhary B., Hosur R.V., Raghavan S.C. Formation of a G-quadruplex at the BCL2 major breakpoint region of the t(14;18) translocation in follicular lymphoma. Nucleic Acids Res. 2011;39:936–948. doi: 10.1093/nar/gkq824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inagaki H., Ohye T., Kogo H., Tsutsumi M., Kato T., Tong M., Emanuel B.S., Kurahashi H. Two sequential cleavage reactions on cruciform DNA structures cause palindrome-mediated chromosomal translocations. Nat. Commun. 2013;4:1592. doi: 10.1038/ncomms2595. [DOI] [PubMed] [Google Scholar]
- 29.Cer R.Z., Donohue D.E., Mudunuri U.S., Temiz N.A., Loss M.A., Starner N.J., Halusa G.N., Volfovsky N., Yi M., Luke B.T., et al. Non-B DB v2.0: a database of predicted non-B DNA-forming motifs and its associated tools. Nucleic Acids Res. 2013;41:D94–D100. doi: 10.1093/nar/gks955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iyer R.R., Pluciennik A., Napierala M., Wells R.D. DNA triplet repeat expansion and mismatch repair. Annu. Rev. Biochem. 2015;84:199–226. doi: 10.1146/annurev-biochem-060614-034010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goula A.V., Merienne K. Abnormal base excision repair at trinucleotide repeats associated with diseases: a tissue-selective mechanism. Genes. 2013;4:375–387. doi: 10.3390/genes4030375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jonson I., Ougland R., Larsen E. DNA repair mechanisms in Huntington's disease. Mol. Neurobiol. 2013;47:1093–1102. doi: 10.1007/s12035-013-8409-7. [DOI] [PubMed] [Google Scholar]
- 33.Kamat M.A., Bacolla A., Cooper D.N., Chuzhanova N. A role for non-B DNA forming sequences in mediating micro-lesions causing human inherited disease. Hum. Mutat. 2016;37:65–73. doi: 10.1002/humu.22917. [DOI] [PubMed] [Google Scholar]
- 34.Roukos V., Burman B., Misteli T. The cellular etiology of chromosome translocations. Curr. Opin. Cell. Biol. 2013;25:357–364. doi: 10.1016/j.ceb.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bignell G.R., Greenman C.D., Davies H., Butler A.P., Edkins S., Andrews J.M., Buck G., Chen L., Beare D., Latimer C., et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463:893–898. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Durkin S.G., Glover T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007;41:169–192. doi: 10.1146/annurev.genet.41.042007.165900. [DOI] [PubMed] [Google Scholar]
- 37.Kumari D., Hayward B., Nakamura A.J., Bonner W.M., Usdin K. Evidence for chromosome fragility at the frataxin locus in Friedreich ataxia. Mutat. Res. 2015;781:14–21. doi: 10.1016/j.mrfmmm.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burrow A.A., Williams L.E., Pierce L.C., Wang Y.H. Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. BMC Genomics. 2009;10:59. doi: 10.1186/1471-2164-10-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dillon L.W., Pierce L.C., Ng M.C., Wang Y.H. Role of DNA secondary structures in fragile site breakage along human chromosome 10. Hum. Mol. Genet. 2013;22:1443–1456. doi: 10.1093/hmg/dds561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raghavan S.C., Swanson P.C., Wu X., Hsieh C.L., Lieber M.R. A non-B-DNA structure at the Bcl-2 major breakpoint region is cleaved by the RAG complex. Nature. 2004;428:88–93. doi: 10.1038/nature02355. [DOI] [PubMed] [Google Scholar]
- 41.Katapadi V.K., Nambiar M., Raghavan S.C. Potential G-quadruplex formation at breakpoint regions of chromosomal translocations in cancer may explain their fragility. Genomics. 2012;100:72–80. doi: 10.1016/j.ygeno.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 42.Novara F., Beri S., Bernardo M.E., Bellazzi R., Malovini A., Ciccone R., Cometa A.M., Locatelli F., Giorda R., Zuffardi O. Different molecular mechanisms causing 9p21 deletions in acute lymphoblastic leukemia of childhood. Hum. Genet. 2009;126:511–520. doi: 10.1007/s00439-009-0689-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Javadekar S.M., Raghavan S.C. Snaps and mends: DNA breaks and chromosomal translocations. FEBS J. 2015;282:2627–2645. doi: 10.1111/febs.13311. [DOI] [PubMed] [Google Scholar]
- 44.Zhao J., Bacolla A., Wang G., Vasquez K.M. Non-B DNA structure-induced genetic instability and evolution. Cell. Mol. Life Sci. 2010;67:43–62. doi: 10.1007/s00018-009-0131-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abeysinghe S.S., Chuzhanova N., Krawczak M., Ball E.V., Cooper D.N. Translocation and gross deletion breakpoints in human inherited disease and cancer I: Nucleotide composition and recombination-associated motifs. Hum. Mutat. 2003;22:229–244. doi: 10.1002/humu.10254. [DOI] [PubMed] [Google Scholar]
- 46.Fisher A.M., Strike P., Scott C., Moorman A.V. Breakpoints of variant 9;22 translocations in chronic myeloid leukemia locate preferentially in the CG-richest regions of the genome. Genes Chrom. Cancer. 2005;43:383–389. doi: 10.1002/gcc.20196. [DOI] [PubMed] [Google Scholar]
- 47.Albano F., Anelli L., Zagaria A., Coccaro N., Casieri P., Rossi A.R., Vicari L., Liso V., Rocchi M., Specchia G. Non random distribution of genomic features in breakpoint regions involved in chronic myeloid leukemia cases with variant t(9;22) or additional chromosomal rearrangements. Mol. Cancer. 2010;9:120. doi: 10.1186/1476-4598-9-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng S., Fu J., Vegesna R., Mao Y., Heathcock L.E., Torres-Garcia W., Ezhilarasan R., Wang S., McKenna A., Chin L., et al. A survey of intragenic breakpoints in glioblastoma identifies a distinct subset associated with poor survival. Genes Dev. 2013;27:1462–1472. doi: 10.1101/gad.213686.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Drier Y., Lawrence M.S., Carter S.L., Stewart C., Gabriel S.B., Lander E.S., Meyerson M., Beroukhim R., Getz G. Somatic rearrangements across cancer reveal classes of samples with distinct patterns of DNA breakage and rearrangement-induced hypermutability. Genome Res. 2013;23:228–235. doi: 10.1101/gr.141382.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Doucet-O'Hare T.T., Rodic N., Sharma R., Darbari I., Abril G., Choi J.A., Young Ahn J., Cheng Y., Anders R.A., Burns K.H., et al. LINE-1 expression and retrotransposition in Barrett's esophagus and esophageal carcinoma. Proc. Natl. Acad. Sci. U.S.A. 2015;112:E4894–E4900. doi: 10.1073/pnas.1502474112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tubio J.M., Li Y., Ju Y.S., Martincorena I., Cooke S.L., Tojo M., Gundem G., Pipinikas C.P., Zamora J., Raine K., et al. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 2014;345:1251343. doi: 10.1126/science.1251343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pitkanen E., Cajuso T., Katainen R., Kaasinen E., Valimaki N., Palin K., Taipale J., Aaltonen L.A., Kilpivaara O. Frequent L1 retrotranspositions originating from TTC28 in colorectal cancer. Oncotarget. 2014;5:853–859. doi: 10.18632/oncotarget.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mader M., Simon R., Kurtz S. FISH Oracle 2: a web server for integrative visualization of genomic data in cancer research. J. Clin. Bioinforma. 2014;4:5. doi: 10.1186/2043-9113-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Network CGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lemmens B., van Schendel R., Tijsterman M. Mutagenic consequences of a single G-quadruplex demonstrate mitotic inheritance of DNA replication fork barriers. Nat. Commun. 2015;6:8909. doi: 10.1038/ncomms9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Le Tallec B., Koundrioukoff S., Wilhelm T., Letessier A., Brison O., Debatisse M. Updating the mechanisms of common fragile site instability: how to reconcile the different views? Cell. Mol. Life Sci. 2014;71:4489–4494. doi: 10.1007/s00018-014-1720-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thys R.G., Lehman C.E., Pierce L.C., Wang Y.H. DNA secondary structure at chromosomal fragile sites in human disease. Curr. Genomics. 2015;16:60–70. doi: 10.2174/1389202916666150114223205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mishmar D., Rahat A., Scherer S.W., Nyakatura G., Hinzmann B., Kohwi Y., Mandel-Gutfroind Y., Lee J.R., Drescher B., Sas D.E., et al. Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proc. Natl. Acad. Sci. U.S.A. 1998;95:8141–8146. doi: 10.1073/pnas.95.14.8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fungtammasan A., Walsh E., Chiaromonte F., Eckert K.A., Makova K.D. A genome-wide analysis of common fragile sites: what features determine chromosomal instability in the human genome? Genome Res. 2012;22:993–1005. doi: 10.1101/gr.134395.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sarai A., Mazur J., Nussinov R., Jernigan R.L. Sequence dependence of DNA conformational flexibility. Biochemistry. 1989;28:7842–7849. doi: 10.1021/bi00445a046. [DOI] [PubMed] [Google Scholar]
- 61.Perez A., Lankas F., Luque F.J., Orozco M. Towards a molecular dynamics consensus view of B-DNA flexibility. Nucleic Acids Res. 2008;36:2379–2394. doi: 10.1093/nar/gkn082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perez A., Noy A., Lankas F., Luque F.J., Orozco M. The relative flexibility of B-DNA and A-RNA duplexes: database analysis. Nucleic Acids Res. 2004;32:6144–6151. doi: 10.1093/nar/gkh954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beveridge D.L., Dixit S.B., Barreiro G., Thayer K.M. Molecular dynamics simulations of DNA curvature and flexibility: helix phasing and premelting. Biopolymers. 2004;73:380–403. doi: 10.1002/bip.20019. [DOI] [PubMed] [Google Scholar]
- 64.Heddi B., Oguey C., Lavelle C., Foloppe N., Hartmann B. Intrinsic flexibility of B-DNA: the experimental TRX scale. Nucleic Acids Res. 2010;38:1034–1047. doi: 10.1093/nar/gkp962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Benham C.J. Duplex destabilization in superhelical DNA is predicted to occur at specific transcriptional regulatory regions. J. Mol. Biol. 1996;255:425–434. doi: 10.1006/jmbi.1996.0035. [DOI] [PubMed] [Google Scholar]
- 66.Kato T., Kurahashi H., Emanuel B.S. Chromosomal translocations and palindromic AT-rich repeats. Curr. Opin. Genet. Dev. 2012;22:221–228. doi: 10.1016/j.gde.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mishra D., Kato T., Inagaki H., Kosho T., Wakui K., Kido Y., Sakazume S., Taniguchi-Ikeda M., Morisada N., Iijima K., et al. Breakpoint analysis of the recurrent constitutional t(8;22)(q24.13;q11.21) translocation. Mol. Cytogenet. 2014;7:55. doi: 10.1186/s13039-014-0055-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kato T., Franconi C.P., Sheridan M.B., Hacker A.M., Inagakai H., Glover T.W., Arlt M.F., Drabkin H.A., Gemmill R.M., Kurahashi H., et al. Analysis of the t(3;8) of hereditary renal cell carcinoma: a palindrome-mediated translocation. Cancer Genet. 2014;207:133–140. doi: 10.1016/j.cancergen.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hsiao M.C., Piotrowski A., Alexander J., Callens T., Fu C., Mikhail F.M., Claes K.B., Messiaen L. Palindrome-mediated and replication-dependent pathogenic structural rearrangements within the NF1 gene. Hum. Mutat. 2014;35:891–898. doi: 10.1002/humu.22569. [DOI] [PubMed] [Google Scholar]
- 70.Kurahashi H., Shaikh T., Takata M., Toda T., Emanuel B.S. The constitutional t(17;22): another translocation mediated by palindromic AT-rich repeats. Am. J. Hum. Genet. 2003;72:733–738. doi: 10.1086/368062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kehrer-Sawatzki H., Haussler J., Krone W., Bode H., Jenne D.E., Mehnert K.U., Tummers U., Assum G. The second case of a t(17;22) in a family with neurofibromatosis type 1: sequence analysis of the breakpoint regions. Hum. Genet. 1997;99:237–247. doi: 10.1007/s004390050346. [DOI] [PubMed] [Google Scholar]
- 72.Gimelli S., Beri S., Drabkin H.A., Gambini C., Gregorio A., Fiorio P., Zuffardi O., Gemmill R.M., Giorda R., Gimelli G. The tumor suppressor gene TRC8/RNF139 is disrupted by a constitutional balanced translocation t(8;22)(q24.13;q11.21) in a young girl with dysgerminoma. Mol. Cancer. 2009;8:52. doi: 10.1186/1476-4598-8-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kurahashi H., Inagaki H., Ohye T., Kogo H., Kato T., Emanuel B.S. Palindrome-mediated chromosomal translocations in humans. DNA Repair. 2006;5:1136–1145. doi: 10.1016/j.dnarep.2006.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Akgun E., Zahn J., Baumes S., Brown G., Liang F., Romanienko P.J., Lewis S., Jasin M. Palindrome resolution and recombination in the mammalian germ line. Mol. Cell Biol. 1997;17:5559–5570. doi: 10.1128/mcb.17.9.5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cunningham L.A., Cote A.G., Cam-Ozdemir C., Lewis S.M. Rapid, stabilizing palindrome rearrangements in somatic cells by the center-break mechanism. Mol. Cell. Biol. 2003;23:8740–8750. doi: 10.1128/MCB.23.23.8740-8750.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lobachev K.S., Gordenin D.A., Resnick M.A. The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell. 2002;108:183–193. doi: 10.1016/s0092-8674(02)00614-1. [DOI] [PubMed] [Google Scholar]
- 77.Lewis S.M., Cote A.G. Palindromes and genomic stress fractures: bracing and repairing the damage. DNA Repair. 2006;5:1146–1160. doi: 10.1016/j.dnarep.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 78.Seidl V., Gamauf C., Druzhinina I.S., Seiboth B., Hartl L., Kubicek C.P. The Hypocrea jecorina (Trichoderma reesei) hypercellulolytic mutant RUT C30 lacks a 85 kb (29 gene-encoding) region of the wild-type genome. BMC Genomics. 2008;9:327. doi: 10.1186/1471-2164-9-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pandolfo M. Molecular pathogenesis of Friedreich ataxia. Arch. Neurol. 1999;56:1201–1208. doi: 10.1001/archneur.56.10.1201. [DOI] [PubMed] [Google Scholar]
- 80.Potaman V.N., Oussatcheva E.A., Lyubchenko Y.L., Shlyakhtenko L.S., Bidichandani S.I., Ashizawa T., Sinden R.R. Length-dependent structure formation in Friedreich ataxia (GAA)n*(TTC)n repeats at neutral pH. Nucleic Acids Res. 2004;32:1224–1231. doi: 10.1093/nar/gkh274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shimizu M., Hanvey J.C., Wells R.D. Intramolecular DNA triplexes in supercoiled plasmids. I. Effect of loop size on formation and stability. J. Biol. Chem. 1989;264:5944–5949. [PubMed] [Google Scholar]
- 82.LeProust E.M., Pearson C.E., Sinden R.R., Gao X. Unexpected formation of parallel duplex in GAA and TTC trinucleotide repeats of Friedreich's ataxia. J. Mol. Biol. 2000;302:1063–1080. doi: 10.1006/jmbi.2000.4073. [DOI] [PubMed] [Google Scholar]
- 83.Bergquist H., Nikravesh A., Fernandez R.D., Larsson V., Nguyen C.H., Good L., Zain R. Structure-specific recognition of Friedreich's ataxia (GAA)n repeats by benzoquinoquinoxaline derivatives. Chembiochem. 2009;10:2629–2637. doi: 10.1002/cbic.200900263. [DOI] [PubMed] [Google Scholar]
- 84.Jain A., Rajeswari M.R., Ahmed F. Formation and thermodynamic stability of intermolecular (R*R*Y) DNA triplex in GAA/TTC repeats associated with Freidreich's ataxia. J. Biomol. Struct. Dyn. 2002;19:691–699. doi: 10.1080/07391102.2002.10506775. [DOI] [PubMed] [Google Scholar]
- 85.Mariappan S.V., Cheng X., van Breemen R.B., Silks L.A., Gupta G. Analysis of GAA/TTC DNA triplexes using nuclear magnetic resonance and electrospray ionization mass spectrometry. Anal. Biochem. 2004;334:216–226. doi: 10.1016/j.ab.2004.07.036. [DOI] [PubMed] [Google Scholar]
- 86.Mariappan S.V., Catasti P., Silks L.A., 3rd, Bradbury E.M., Gupta G. The high-resolution structure of the triplex formed by the GAA/TTC triplet repeat associated with Friedreich's ataxia. J. Mol. Biol. 1999;285:2035–2052. doi: 10.1006/jmbi.1998.2435. [DOI] [PubMed] [Google Scholar]
- 87.Bacolla A., Larson J.E., Collins J.R., Li J., Milosavljevic A., Stenson P.D., Cooper D.N., Wells R.D. Abundance and length of simple repeats in vertebrate genomes are determined by their structural properties. Genome Res. 2008;18:1545–1553. doi: 10.1101/gr.078303.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McIvor E.I., Polak U., Napierala M. New insights into repeat instability: role of RNA*DNA hybrids. RNA Biol. 2010;7:551–558. doi: 10.4161/rna.7.5.12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Haran T.E., Mohanty U. The unique structure of A-tracts and intrinsic DNA bending. Q. Rev. Biophys. 2009;42:41–81. doi: 10.1017/S0033583509004752. [DOI] [PubMed] [Google Scholar]
- 90.Stefl R., Wu H., Ravindranathan S., Sklenar V., Feigon J. DNA A-tract bending in three dimensions: solving the dA4T4 vs. dT4A4 conundrum. Proc. Natl. Acad. Sci. U.S.A. 2004;101:1177–1182. doi: 10.1073/pnas.0308143100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stellwagen E., Peters J.P., Maher L.J., 3rd, Stellwagen N.C. DNA A-tracts are not curved in solutions containing high concentrations of monovalent cations. Biochemistry. 2013;52:4138–4148. doi: 10.1021/bi400118m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Goodsell D.S., Kaczor-Grzeskowiak M., Dickerson R.E. The crystal structure of C-C-A-T-T-A-A-T-G-G. Implications for bending of B-DNA at T-A steps. J. Mol. Biol. 1994;239:79–96. doi: 10.1006/jmbi.1994.1352. [DOI] [PubMed] [Google Scholar]
- 93.Zhu X., Schatz G.C. Molecular dynamics study of the role of the spine of hydration in DNA A-tracts in determining nucleosome occupancy. J. Phys. Chem. B. 2012;116:13672–13681. doi: 10.1021/jp3084887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sprous D., Young M.A., Beveridge D.L. Molecular dynamics studies of axis bending in d(G5-(GA4T4C)2-C5) and d(G5-(GT4A4C)2-C5): effects of sequence polarity on DNA curvature. J. Mol. Biol. 1999;285:1623–1632. doi: 10.1006/jmbi.1998.2241. [DOI] [PubMed] [Google Scholar]
- 95.Bacolla A., Zhu X., Chen H., Howells K., Cooper D.N., Vasquez K.M. Local DNA dynamics shape mutational patterns of mononucleotide repeats in human genomes. Nucleic Acids Res. 2015;43:5065–5080. doi: 10.1093/nar/gkv364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wyatt H.D., Sarbajna S., Matos J., West S.C. Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol. Cell. 2013;52:234–247. doi: 10.1016/j.molcel.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 97.Lu G., Duan J., Shu S., Wang X., Gao L., Guo J., Zhang Y. Ligase I and ligase III mediate the DNA double-strand break ligation in alternative end-joining. Proc. Natl. Acad. Sci. U.S.A. 2016;113:1256–1260. doi: 10.1073/pnas.1521597113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kitagawa Y., Inoue K., Sasaki S., Hayashi Y., Matsuo Y., Lieber M.R., Mizoguchi H., Yokota J., Kohno T. Prevalent involvement of illegitimate V(D)J recombination in chromosome 9p21 deletions in lymphoid leukemia. J. Biol. Chem. 2002;277:46289–46297. doi: 10.1074/jbc.M208353200. [DOI] [PubMed] [Google Scholar]
- 99.Cayuela J.M., Gardie B., Sigaux F. Disruption of the multiple tumor suppressor gene MTS1/p16(INK4a)/CDKN2 by illegitimate V(D)J recombinase activity in T-cell acute lymphoblastic leukemias. Blood. 1997;90:3720–3726. [PubMed] [Google Scholar]
- 100.Burman B., Zhang Z.Z., Pegoraro G., Lieb J.D., Misteli T. Histone modifications predispose genome regions to breakage and translocation. Genes Dev. 2015;29:1393–1402. doi: 10.1101/gad.262170.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang Y., Shishkin A.A., Nishida Y., Marcinkowski-Desmond D., Saini N., Volkov K.V., Mirkin S.M., Lobachev K.S. Genome-wide screen identifies pathways that govern GAA/TTC repeat fragility and expansions in dividing and nondividing yeast cells. Mol. Cell. 2012;48:254–265. doi: 10.1016/j.molcel.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang G., Vasquez K.M. Impact of alternative DNA structures on DNA damage, DNA repair, and genetic instability. DNA Repair. 2014;19:143–151. doi: 10.1016/j.dnarep.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lin Y., Wilson J.H. Transcription-induced DNA toxicity at trinucleotide repeats: double bubble is trouble. Cell Cycle. 2011;10:611–618. doi: 10.4161/cc.10.4.14729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chan Y.A., Hieter P., Stirling P.C. Mechanisms of genome instability induced by RNA-processing defects. Trends Genet. 2014;30:245–253. doi: 10.1016/j.tig.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hamperl S., Cimprich K.A. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair. 2014;19:84–94. doi: 10.1016/j.dnarep.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nelson L.D., Bender C., Mannsperger H., Buergy D., Kambakamba P., Mudduluru G., Korf U., Hughes D., Van Dyke M.W., Allgayer H. Triplex DNA-binding proteins are associated with clinical outcomes revealed by proteomic measurements in patients with colorectal cancer. Mol. Cancer. 2012;11:38. doi: 10.1186/1476-4598-11-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kostyuk S.V., Konkova M.S., Ershova E.S., Alekseeva A.J., Smirnova T.D., Stukalov S.V., Kozhina E.A., Shilova N.V., Zolotukhina T.V., Markova Z.G., et al. An exposure to the oxidized DNA enhances both instability of genome and survival in cancer cells. PLoS One. 2013;8:e77469. doi: 10.1371/journal.pone.0077469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lee Y.A., Durandin A., Dedon P.C., Geacintov N.E., Shafirovich V. Oxidation of guanine in G, GG, and GGG sequence contexts by aromatic pyrenyl radical cations and carbonate radical anions: relationship between kinetics and distribution of alkali-labile lesions. J. Phys. Chem. B. 2008;112:1834–1844. doi: 10.1021/jp076777x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Adhikary A., Khanduri D., Sevilla M.D. Direct observation of the hole protonation state and hole localization site in DNA-oligomers. J. Am. Chem. Soc. 2009;131:8614–8619. doi: 10.1021/ja9014869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lee Y.M., Kim H.E., Park C.J., Lee A.R., Ahn H.C., Cho S.J., Choi K.H., Choi B.S., Lee J.H. NMR study on the B-Z junction formation of DNA duplexes induced by Z-DNA binding domain of human ADAR1. J. Am. Chem. Soc. 2012;134:5276–5283. doi: 10.1021/ja211581b. [DOI] [PubMed] [Google Scholar]
- 111.de Rosa M., de Sanctis D., Rosario A.L., Archer M., Rich A., Athanasiadis A., Carrondo M.A. Crystal structure of a junction between two Z-DNA helices. Proc. Natl. Acad. Sci. U.S.A. 2010;107:9088–9092. doi: 10.1073/pnas.1003182107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Schlacher K., Wu H., Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22:106–116. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.