


Abstract
Predictive toxicology using stem cells or their derived tissues has gained increasing importance in biomedical and pharmaceutical research. Here, we show that toxicity category prediction by support vector machines (SVMs), which uses qRT-PCR data from 20 categorized chemicals based on a human embryonic stem cell (hESC) system, is improved by the adoption of gene networks, in which network edge weights are added as feature vectors when noisy qRT-PCR data fail to make accurate predictions. The accuracies of our system were 97.5–100% for three toxicity categories: neurotoxins (NTs), genotoxic carcinogens (GCs) and non-genotoxic carcinogens (NGCs). For two uncategorized chemicals, bisphenol-A and permethrin, our system yielded reasonable results: bisphenol-A was categorized as an NGC, and permethrin was categorized as an NT; both predictions were supported by recently published papers. Our study has two important features: (i) as the first study to employ gene networks without using conventional quantitative structure-activity relationships (QSARs) as input data for SVMs to analyze toxicogenomics data in an hESC validation system, it uses additional information of gene-to-gene interactions to significantly increase prediction accuracies for noisy gene expression data; and (ii) using only undifferentiated hESCs, our study has considerable potential to predict late-onset chemical toxicities, including abnormalities that occur during embryonic development.
INTRODUCTION
In recent years, several massive toxicological analyses have yielded large volumes of microarray data sets. Such undertakings as the chemical effects in biological systems (CEBS) project (1), a study of the effects of 2,4,6-trinitrotoluene (TNT) on the liver (2) and the TGP Consortium in Japan (3) have taken on the challenge of systematically measuring, analyzing and understanding the toxic effects of chemicals on the human body, as well as developing new bioinformatics methods for mining large-scale toxicology data (4,5). However, due to legal, ethical and/or religious concerns that prohibit direct testing on the human body, chemical toxicity studies performed over the last few decades have had to use animals as human models. Unfortunately, animal models are not equivalent to the human system, and some drugs that have been approved for release after successful animal studies have yielded tragic results in humans, including the delayed occurrence of mental deficiencies or cancers. For example, thalidomide [3-(N-phthalimido) glutarimide], a well-known versatile drug used to treat such serious diseases as red nodules in Hansen's disease, multiple myeloma and HIV, was responsible for various forms of birth defects when it was administered to pregnant women, via mechanisms that remain unknown to this day (6). Methylmercury, a well-known chemical pollutant used since ancient times, has poisoned tens of thousands of individuals in Japan (7). The toxicities of those two chemicals were very difficult to assess because they exerted weaker effects in mice than in humans, or different effects altogether. Therefore, in order to overcome the problems of animal test systems, we urgently need to develop a novel ‘pseudo-human’ system for drug testing.
In this regard, it is likely that toxicological studies based on various types of stem cells, including embryonic, induced pluripotent and mesenchymal stem cells, as well as induced differentiated cells, will facilitate the development of novel drugs that act on various diseases with improved efficiencies. A recent report showed that human and mouse embryonic stem (ES) cell-derived neural cells exhibited completely different responses to methylmercury due to differences in the underlying gene networks in the two species (8). Although the authors of that study clearly demonstrated the existence of chemical effects that were specific to the artificially but fully differentiated neural cells, it was not straightforward to reproduce the entire process of neuronal development, during which the effects of the chemicals were expected to be observed.
In this study, we developed an alternative approach that uses human ES cells (hESCs) without further differentiation. Our aim was to predict the ‘late-onset’ effects of toxic chemicals whose clinical symptoms appear months or years after exposure. Using only 10 genes pre-selected by multivariate statistical analysis of limited microarray data, we performed 9768 qRT-PCR measurements and extensive bioinformatics studies to predict the toxic effects of 20 categorized and two uncategorized chemicals. Using this system, we achieved as high as 97.5–100% prediction accuracies for three representative toxicity categories: neurotoxins (NTs), genotoxic carcinogens (GCs) and non-genotoxic carcinogens (NGCs). We also found that gene network information reinforced prediction accuracy when qRT-PCR data alone had little predictive power. The resultant gene networks exhibited some conserved patterns as well as variations among chemicals belonging to the same toxicity category, indicating that sub-categorical information that is not extracted from raw qRT-PCR data should be included in the gene network architecture.
MATERIALS AND METHODS
Toxicity tests on the hESC system
Ethics statement
The hESC line KhES-3 (XY karyotype) was provided by Dr Hirofumi Suemori, Research Center of Stem Cells, Institute for Frontier Medical Science, Kyoto University (9). All experiments using hESCs were approved by the ethics committees of the National Institute for Environmental Studies and The University of Tokyo, in accordance with the guidelines of the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Selection of toxic chemicals
Twenty two chemicals were carefully selected according to the literature (10,11) from among three representative toxicity categories: 9 neurotoxins (NTs), 5 genotoxic carcinogens (GCs) and 6 non-genotoxic carcinogens (NGCs) and two uncategorized toxins. The NTs were valproic acid, cyclopamine, phenytoin, methylmercury, acrylamide, 4-OH-2′,3,3′,4′,5′-PCB, 2,5-hexanedione, warfarin and thalidomide; the GCs were benzo[a]anthracene, 3-methylcholanthrene, benzo[a]pyrene, diethylnitrosamine and diethylstilbestrol; the NGCs were 2,3,7,8-Tetrachlorodibenzodioxin (TCDD), lithocholic acid, thioacetamide, butylated hydroxyanisole, methapyrilene hydrochloride and phenobarbital; and the uncategorized toxins were bisphenol-A and permethrin. To guide our selection process, we chose NTs, GCs and NGCs with well-established toxicities in humans (NTs: valproic acid, phenytoin, warfarin and thalidomide; GCs: benzo[a]pyrene, diethylnitrosamine and diethylstilbestrol; NGCs: TCDD) and/or well-established mechanisms (NTs: cyclopamine; GCs: benzo[a]pyrene and diethylstilbestrol; NGCs: TCDD). Other NTs were selected based on data from animal experiments and human epidemiology data (12,13), and other GCs and NGCs were selected based on U.S. and European international projects (14–16) and the International Agency for Research on Cancer monograph on human carcinogens (http://monographs.iarc.fr/ENG/Monographs/PDFs/index.php). Uncategorized chemicals were selected from among widely used representative materials found in plastics and pesticides. All of the selected chemicals could be handled using normal laboratory equipment with sufficient care for safety. Detailed information of the 22 chemicals, including chemical abstract services registry numbers (CASRNs), suppliers and references, is shown in Supplementary Table S1.
Exposure system
Human KhES-3 ES cells were cultured on MatrigelTM in mTeSR1 (a feeder-free maintenance medium for hESCs). As in conventional toxicology experiments (17–19), to avoid drawing only over-dosed responses, such as apoptosis, and to enable the detection of subtle gene expression changes that may happen in human embryos, hESCs were exposed to each of the above 22 chemicals (9 NTs, 5 GCs, 6 NGCs and two uncategorized toxins) at five doses (1, 1/2, 1/4, 1/8 and 1/16, where 1 indicates the highest dose that did not impair cell viability), and mRNA samples were taken at four time points (24, 48, 72 and 96 h after initiation of exposure), to yield 11 genes × 22 chemicals × 5 doses × 4 time points = 4840 measurements. As the negative control, mRNA levels were also measured at the same four time points in cells not exposed to any chemical, thus yielding 11 genes × 4 time points = 44 measurements. Then, we repeated all the measurements, thereby performing (4840 + 44) × 2 = 9768 measurements in total.
Correspondence analysis (CA) to narrow down candidate feature genes
Microarray analysis of 10 chemicals (valproic acid, cyclopamine, phenytoin, methylmercury, acrylamide, benzo[a]anthracene, 3-methylcholanthrene, benzo[a]pyrene, diethylnitrosamine and diethylstilbestrol) and one control (no exposure), for a total of 11 experimental conditions, was performed to select marker or feature genes for the gene network and toxicity prediction analysis. Feature genes constitute feature vectors in machine learning approaches; such genes are expected to both characterize and predict toxicity effects. Because we assumed that the mechanisms of normal development are primarily controlled by transcription factors, the feature genes were selected from genes encoding transcription factors. In order to simply detect genes sensitive to chemicals, or candidate feature genes at the stem cell stage, we performed microarray analysis of undifferentiated KhES-3 ES cells rather than differentiated cells or embryos. The raw data obtained 48 h after exposure to 1/2 dose were log2-transformed and quantile-normalized using the BeadArray (20) library of the Bioconductor (http://bioconductor.org/) package in the R (http://www.r-project.org/) statistical computing language. Among the 48 206 gene probes, 30 990 probes with signal detection P-value < 0.05 in at least one of the 11 experiments were analyzed further. Next, we performed a classical unsupervised multivariate statistical method called correspondence analysis (21) using the corresp function from the MASS (22) library in R on the matrix data of 11 samples, adopting 3492 probes, which correspond to genes encoding transcription factors. Importantly, this process does not use any toxicity categorical information and thus, it should not affect the subsequent toxicity predictions. We conditionally selected 10 candidate marker or feature genes that satisfied the following three criteria: (i) signal detection P-value < 0.1 for three or more samples; (ii) the encoded protein is a transcription factor; and (iii) within the top 20 highest (or lowest for negative) weights in each principal component. Using these three criteria, we narrowed down the initial 48 206 genes to 167 non-redundant candidate genes. However, the number was still large and thus might include spurious genes. Therefore, we added a fourth criterion that we considered to be the most important: (iv) abundant expression in hESCs, as determined by qRT-PCR analysis. Ultimately, we selected the following 10 genes: NANOG, SOX2, DMTF1, ZNF208, ADRM1, TRIB1, CRY1, SMAD7, SMAD6 and VHL1. The top 20 genes with the highest (or lowest) weights in each principal component are shown in Supplementary Table S2. The microarray data are available on our website (http://stemcellinformatics.org/toxicology/) or the Gene Expression Omnibus (GSE60154).
qRT-PCR analysis of 22 chemicals
Using the 10 marker genes listed above, we performed qRT-PCR analysis of mRNAs in hESCs exposed to the 22 toxic chemicals. Details of the exposure doses and the toxicity categories are provided in Table 1. qRT-PCR experiments were performed according to a previously described protocol (8), and the primer sequences used in our analysis are shown in Supplementary Table S3. To calibrate gene expression levels from qRT-PCR measurements, we also measured the levels of β-actin (ACTB) mRNA as the internal standard; thus, a total of 11 genes were used in the qRT-PCR analysis, yielding 9768 measurements. Raw data from the qRT-PCR experiments and the sample preparation information are available at our website (http://stemcellinformatics.org/toxicology/).
Table 1. Complete list of the 22 toxic chemicals and doses used in the exposure experiments.
| Compound Name | Dose (nM) | Toxicity Category | |||||
|---|---|---|---|---|---|---|---|
| Valproic acid | 1000 | 500 | 250 | 125 | 62.5 | 0 | Neurotoxin |
| Cyclopamine | 1000 | 500 | 250 | 125 | 62.5 | 0 | (NT) |
| Phenytoin | 1000 | 500 | 250 | 125 | 62.5 | 0 | |
| Methylmercury | 50 | 25 | 12.5 | 6.25 | 3.125 | 0 | |
| Acrylamide | 5000 | 2500 | 1250 | 625 | 312.5 | 0 | |
| 4-OH-2′,3,3′,4′,5′-PCB107 | 10 | 5 | 2.5 | 1.25 | 0.625 | 0 | |
| 2,5-Hexanedione | 1000 | 500 | 250 | 125 | 62.5 | 0 | |
| Warfarin | 1000 | 500 | 250 | 125 | 62.5 | 0 | |
| Thalidomide | 500 | 250 | 125 | 62.5 | 31.25 | 0 | |
| Benzo[a]anthracene | 50 | 25 | 12.5 | 6.25 | 3.125 | 0 | Genotoxic carcinogen |
| 3-Methylcholanthrene | 500 | 250 | 125 | 62.5 | 31.25 | 0 | (GC) |
| Benzo[a]pyrene | 500 | 250 | 125 | 62.5 | 31.25 | 0 | |
| Diethylnitrosamine | 500 | 250 | 125 | 62.5 | 31.25 | 0 | |
| Diethylstilbestrol | 500 | 250 | 125 | 62.5 | 31.25 | 0 | |
| 2,3,7,8-TCDD | 10 | 5 | 2.5 | 1.25 | 0.625 | 0 | Non-genotoxic carcinogen |
| Lithocholic acid | 1000 | 500 | 250 | 125 | 62.5 | 0 | (NGC) |
| Thioacetamide | 1000 | 500 | 250 | 125 | 62.5 | 0 | |
| Butylated hydroxyanisole | 1000 | 500 | 250 | 125 | 62.5 | 0 | |
| Methapyrilene hydrochloride | 1000 | 500 | 250 | 125 | 62.5 | 0 | |
| Phenobarbital | 1000 | 500 | 250 | 125 | 62.5 | 0 | |
| Bisphenol-A | 1000 | 500 | 250 | 125 | 62.5 | 0 | Uncategorized |
| Permethrin | 1000 | 500 | 250 | 125 | 62.5 | 0 | |
Preprocessing and data normalization
The raw qRT-PCR data were processed using the limma (23) library of the Bioconductor package. Normalization was performed in two steps. First, we estimated batch effects in standard 96-well plates using the empirical Bayesian linear model provided in the limma library based on the following equation:
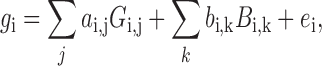 |
(1) |
where i, gi, Gi, j, Bi, k and 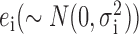 indicate gene, expression value, chemical effect, batch effect and noise, respectively. For each of the 11 genes at four different time points (thus, i ranges from 1 to 44), the function gi consists of j (22 chemicals + 1 control) and k (10 plates) terms, whereas only one each of ai, j and bi, k has non-zero values. Next, we estimated the linear function gi by empirical Bayesian linear fitting (the lmfit function in limma) over the 222 data points, which consisted of two biological replicates of 22 chemicals multiplied by 5 doses and 1 control (2 × (22 × 5 + 1) = 222). Once ai, j and bi, k were estimated, we subtracted the batch effect terms to leave only the chemical effects. Finally, the batch-effect-adjusted mRNA levels were calculated by dividing them by the level of the internal standard (ACTB), and the adjusted values were used for further analysis.
indicate gene, expression value, chemical effect, batch effect and noise, respectively. For each of the 11 genes at four different time points (thus, i ranges from 1 to 44), the function gi consists of j (22 chemicals + 1 control) and k (10 plates) terms, whereas only one each of ai, j and bi, k has non-zero values. Next, we estimated the linear function gi by empirical Bayesian linear fitting (the lmfit function in limma) over the 222 data points, which consisted of two biological replicates of 22 chemicals multiplied by 5 doses and 1 control (2 × (22 × 5 + 1) = 222). Once ai, j and bi, k were estimated, we subtracted the batch effect terms to leave only the chemical effects. Finally, the batch-effect-adjusted mRNA levels were calculated by dividing them by the level of the internal standard (ACTB), and the adjusted values were used for further analysis.
Bayesian gene network analysis
Many gene network reconstruction methods are available. We tested structure equation modelings (SEMs), graphical Gaussian modelings (GGMs), cross correlation coefficients (CCCs) and Bayesian networks (BNs) in our previous study (24), and found no large differences. Thus, we simply used BNs as they are one of the sophisticated network inference methods used for analyzing biological data (25).
We reconstructed BNs from the qRT-PCR data of each of the 22 chemical exposure experiments. By ‘reconstruction of gene networks,’ we mean inference of the directed edge weights for a complete graph of q × q edges connecting q nodes, or genes; in this study, q = 10. Due to statistical limitations, BNs are characterized by the well-known constraint that they generate only non-cyclic (acyclic) graphs. For BN reconstruction, we used the previously described inference algorithm TAO-Gen (26,27), which was developed using the Gibbs sampling method. As the details of the original TAO-Gen algorithm can be downloaded from our website (http://stemcellinformatics.org/toxicology/), we describe only the additional improved algorithm here. In order to achieve more efficient and reliable sampling, we further improved this algorithm by creating a version optimized for parallel computation, based on standard ensemble Markov chain Monte Carlo (MCMC) sampling or replica-exchange MCMC sampling (28,29). The improved algorithm, called ‘RX-TAOgen,’ can run multiple Gibbs sampling series and exchange sampling parameters between neighbor series as follows: It exchanges the l and l + 1 series of different parameters Cl, only if the ratio of the posterior probabilities of BNs, which are calculated by function f in the original TAO-Gen, is greater than some threshold. The ratio is given by the following equation:
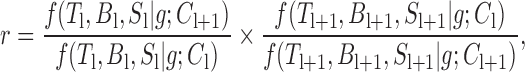 |
(2) |
where Tl and Bl are q × q transition and weight matrices, respectively, Sl is a vector of the standard deviations of gene expression values for q genes, and g is the observed gene expression data. For the threshold, we used random values in [0, 1]. A total of 10 × 10 = 100 gene-to-gene weights ( i, j in Figure 1) on network edges were obtained and used as additional features for the support vector machines (SVM). Once the 10 × 10 edge weights for each chemical were obtained, they were added to the qRT-PCR data for the corresponding chemicals; thus, the input data were increased from 10 genes × 5 doses × 4 time points = 200 values to 200 + (10 × 10) genes = 300 values in the toxicity prediction system. The binary program and a detailed description of RX-TAOgen can be downloaded from our website (http://stemcellinformatics.org/toxicology/).
i, j in Figure 1) on network edges were obtained and used as additional features for the support vector machines (SVM). Once the 10 × 10 edge weights for each chemical were obtained, they were added to the qRT-PCR data for the corresponding chemicals; thus, the input data were increased from 10 genes × 5 doses × 4 time points = 200 values to 200 + (10 × 10) genes = 300 values in the toxicity prediction system. The binary program and a detailed description of RX-TAOgen can be downloaded from our website (http://stemcellinformatics.org/toxicology/).
Figure 1.

Schematic view of processes from data generation to SVM prediction. For each of the 22 toxic chemicals, 200 qRT-PCR measurements of 10 genes were conducted on cells treated with five doses at four time points. In addition, 100 BN data from 10 × 10 genes were generated. qRT-PCR data, BN data or both data types were used to train SVMs and predict each of the three toxicity categories.
Toxicity prediction by SVM
There exist a variety of statistical or machine learning methods, such as neural networks, partial least squares, random forests, etc., which can be applied to supervised toxicity prediction. In this study, we did not focus on the comparison of existing methods; rather, we focused on whether or not the pseudo-human system based on hESCs has sufficient potential in general to predict late-onset chemical toxicities. Thus, we simply used SVMs (30) as one of the state-of-the-art learning methods (31). SVMs are widely used for the classification of samples on the basis of their input (gene expression) values (32), and it has been reported that their classification performance is not significantly different from random forests (33). In its basic form, an SVM is a binary classifier based on a hyper-plane that distinguishes two distributions of M-dimensional vectors or samples from different classes with a linear or non-linear similarity function called kernel matrix. As the principles of SVMs are found in many papers and books, we omit the details in this paper.
Kernel types and leave-one-chemical-out-prediction (LOCOP)
According to our previous study (34), here we used six (linear, polynomial, RBF, EKM, Saigo and ME) kernels. Three of these (linear, polynomial and RBF) constitute the widely used vectorial kernel family, and the three others (EKM, Saigo and ME) constitute the structured kernel family (35). In the prediction of complex data, the structured kernel family often outperforms the vectorial kernel family. For each of the 22 chemicals, a pair of repeat qRT-PCR measurements was performed; consequently, a total of 44 data points were generated. To carefully evaluate the prediction accuracy for new chemicals, we performed not conventional leave-one-out-prediction (LOOP) but the leave-one-chemical-out-prediction (LOCOP) test, in which a pair of repeat data for each of the 22 chemicals was eliminated during the learning process, and then used to test the prediction.
Feature selection and addition of gene network edge weights
As shown in Figure 1, we used two different types of features for SVM predictions: feature 1, which denotes the raw qRT-PCR data consisting of 200 measurements, and feature 2, which denotes BN edge data consisting of 100 weight values. Note that feature 1 is raw gene expression data, whereas feature 2 is computer-inferred gene-to-gene interaction data derived from the raw gene expression data. To optimize the number of features (qRT-PCR data, BN edge weights or both) for input into the SVM, we used standard two-sample t-statistics in each iteration of the LOCOP cross-validation tests in order to rank the features according to their discriminative potential or P-values. Thus, the six kernels were tested with SVMs in order to analyze their classification power with various numbers of features, and their prediction accuracies were recorded for each number of features. More specifically, we initially ranked 200, 100 and 300 features for qRT-PCRs, BNs and qRT-PCRs + BNs, respectively and then trained the SVM with 21 chemicals and tested the 22nd chemical (two repeats for each) with various numbers of the top n features with n ranging from 1 to the maximum number: 200 (qRT-PCRs), 100 (BNs) and 300 (qRT-PCRs + BNs).
Performance measure
For all the six kernels tested, seven SVM parameter values (C = 10−3, 10−2, 10−1, 1, 10, 102, 103) were tested. Additionally, for the polynomial kernel, 10 values (D = 1, 2, 3, 4, 5, 6, 7, 8, 9, 10) were tested, and for the RBF, EKM and Saigo kernels, 11 values (σ = 10−10, 10−9, 10−8, 10−7, 10−6, 10−5, 10−4, 10−3, 10−2, 10−1, 1) were tested. For the ME kernel, we tested parameters in the range G = 2−5, 2−4, 2−3, 2−2, 2−1, 1, 2, 22, 23, 24, 25. Note that the number of parameter combinations in the ME kernel is equal to those in the RBF, EKM and Saigo kernels used in this study. For convenience, all accuracies reported in this paper were calculated using the following formula:
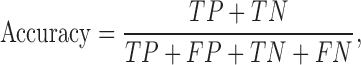 |
(3) |
where TP, FP, TN and FN are true positive, false positive, true negative and false negative frequencies, respectively. We also performed area under the curve (AUC) calculations using the pROC library (36) in R. No smoothing method was applied to the receiver operator characteristic (ROC) curve.
RESULTS
Previously, we used neural cells differentiated from hESCs (24). In that study, we showed that when qRT-PCR data and BN edge weights were given as the input data of SVMs, we were able to classify 15 chemicals as NTs or carcinogens (including GCs and NGCs) with prediction accuracies of 93.3% and 100.0%, respectively. In that case, the marker genes used as features for SVM predictions were pre-selected by an expert.
Here, we investigated difficult-to-predict late-onset chemical toxicities using only hESCs not subjected to further differentiation. In the experiments, the number of chemicals was increased from 15 to 22. In addition, the marker genes were carefully selected from microarray data obtained from cells treated with 10 toxic chemicals and one control, using classical unsupervised multivariate statistics (correspondence analysis, CA) and a filter based on four criteria. Ultimately, we selected 10 genes as qRT-PCR targets: NANOG, SOX2, DMTF1, ZNF208, ADRM1, TRIB1, CRY1, SMAD7, SMAD6 and VHL1. The CA weights and the gene descriptions are shown in Table 2. The full list of chemicals and the doses used in the exposure experiments are shown in Table 1. The prediction scheme is shown in Figure 1.
Table 2. Ten genes selected by correspondence analysis (CA) for 9768 qRT-PCR data.
| Gene Symbol | CA Component (Weight) | Definition* | Synonym |
|---|---|---|---|
| ADRM1 | 1 (1.70) | Homo sapiens adhesion regulating molecule 1 (ADRM1), transcript variant 2, mRNA. | Rpn13; GP110; MGC29536 |
| CRY1 | 4 (2.47) | Homo sapiens cryptochrome 1 (photolyase-like) (CRY1), mRNA. | PHLL1 |
| DMTF1 | 6 (−2.44) | Homo sapiens cyclin D binding myb-like transcription factor 1 (DMTF1), mRNA. | DMP1; DMTF; hDMP1; FLJ41265 |
| NANOG | 5 (3.03) | Homo sapiens Nanog homeobox (NANOG), mRNA. | – |
| SMAD6 | 6 (3.68), 7 (−2.88), 8 (2.72), 9 (−2.49) | Homo sapiens SMAD family member 6 (SMAD6), transcript variant 1, mRNA. | MADH6; HsT17432; MADH7 |
| SMAD7 | 1 (1.66) | Homo sapiens SMAD family member 7 (SMAD7), mRNA. | FLJ16482; MADH8; MADH7 |
| SOX2 | 1 (1.64), 4 (2.19) | Homo sapiens SRY (sex determining region Y)-box 2 (SOX2), mRNA. | ANOP3; MGC2413; MCOPS3 |
| TRIB1 | 1 (−3.25), 2 (−2.77) | Homo sapiens tribbles homolog 1 (Drosophila) (TRIB1), mRNA. | GIG2; SKIP1; C8FW |
| VHL1 | 1(1.77), 2 (−2.72), 4 (2.49) | Homo sapiens von Hippel-Lindau tumor suppressor (VHL), transcript variant 2, mRNA. | HRCA1; RCA1; VHL1 |
| ZNF208 | 6 (−2.54), 10 (−3.71) | Homo sapiens zinc finger protein 208 (ZNF208), mRNA. | ZNF95; PMIDP |
*The information in the ‘Definition” column is an exact copy of data in the microarray descriptions published by Illumina.
qRT-PCR results revealed little dose dependence
The 9768 qRT-PCR measurements gave important general features of genes with regard to dose responses. When we checked the general tendencies of mRNA abundances, among the selected 10 marker genes, two genes, DMTF1 and SMAD6, were undetectable in a large number of experiments, probably due to their low expressions. SMAD7 was also undetectable in several experiments. Thus, most of the results of our statistical analysis could be, in principle, a reflection of the remaining seven genes. To investigate the global trends in gene expression in response to the 22 chemicals, we constructed a heat map of the Pearson correlation coefficients of the qRT-PCR data (Figure 2). For each of the 22 chemicals, the Pearson correlations between two chemicals administered at five doses were distinctively plotted on a color scale. Time-dependent values from two repeat experiments were averaged within the repeats and plotted as a single correlation coefficient. This plot revealed two important observations: first, there were no remarkable changes among the five doses used for each chemical, although the 1/16 dose was sometimes weakly correlated with the other doses; and second, the Pearson correlations (ranging from r = 0.778 to 0.878) did not differ significantly between chemicals of the same category and those of different categories. It should be noted, however, that for each of the three categories, the average correlations within the same category were always stronger than those among different categories.
Figure 2.

Pearson correlation coefficients in 100 toxic chemical experiments, constructed based on batch-adjusted qRT-PCR data. Pairwise Pearson correlation coefficients were calculated for 20 toxic chemicals administered at five doses (100 experiments).
Analysis of gene network patterns inferred from chemical exposure experiments
We inferred BNs consisting of 10 genes from the qRT-PCR data for all 20 chemicals. Each chemical exposure experiment generated only 40 data points (4 time points × 5 doses × 2 repeats = 40). Due to the limited amount of data, RX-TAOgen could infer only one BN for two repeat experiments. For each chemical, we ran RX-TAOgen in eight parallel settings with 20 000 exchanges, each executed at 10 Gibbs samplings, and took only the best network result (i.e. the one that yielded the highest likelihood) for further SVM predictions. For each chemical, this calculation took 10–15 min on a 2.93 GHz Intel X5570 CPU. The reconstructed BN structures for the 20 chemicals are shown in Supplementary Figure S1. To characterize those networks, we first performed topological analysis of the network structures. Figure 3 shows the summarized circular network diagrams displaying all the network edges that are high-weighted by any of the toxic chemicals in each of the three toxicity categories. Red edges with triangular tips indicate positive influences, whereas blue edges with diamond tips indicate negative ones. The number of colored edges between the same two nodes corresponds to the number of supported chemicals. Whereas the diagram of NTs contained a large number of colored edges representing high weights, that of NGCs exhibited a small number of edges, since NTs had higher variations than NGCs in the gene network patterns among the chemicals. It is noteworthy that GCs contained a smaller number of chemicals but showed more variations in the gene network patterns than NGCs.
Figure 3.

Category-dependent high-weighted edges of BN structures in circular network diagrams. (A) Neurotoxins (9 data), (B) genotoxic carcinogens (5 data) and (C) non-genotoxic carcinogens (6 data). The network for non-genotoxic carcinogens represents limited edges, indicating that the network may be specific and similar among different chemicals and thus easy to predict, whereas that for neurotoxins contains almost all edges, indicating that the network is not easy to characterize and thus difficult to predict.
BN information differs from raw qRT-PCR data
The gene network variations were revealed more clearly in the Pearson correlation analysis. Figure 4 shows heat maps of the Pearson correlation coefficients of (i) dose-averaged qRT-PCR data (Figure 4A) and (ii) the 10 × 10 = 100 edge weights in the reconstructed BNs for the 20 chemicals (Figure 4B). Comparing the heat maps of the qRT-PCRs and the BNs, we observed differences between the two Pearson correlations, indicating that the BNs provide different information from qRT-PCR data. To clarify which data contributed more, we generated a subtractive heat map, in which Pearson correlations of BNs subtracted by those of qRT-PCR for each pair of chemicals after normalization are shown in red-blue colors. In Figure 4C, Pearson correlations where BNs are more positive than qRT-PCRs are indicated by red, and those where qRT-PCRs are more positive than BNs are indicated by blue. For example, whereas the Pearson correlations for methylmercury, warfarin and thalidomide against other chemicals are more positive in qRT-PCRs, those for valproic acid and lithocholic acid are more positive in BNs. This analysis indicates the importance of using both BN and qRT-PCR data to boost the toxicity predictions, because together they could compensate for the lack of information by either method alone.
Figure 4.

Analysis of BN information by Pearson correlation coefficients. Heat maps of pairwise Pearson correlation coefficients of the 20 toxic chemicals constructed based on (A) five dose-averaged qRT-PCR Pearson correlations and (B) 10 × 10 BN edge weights, where two experimental repeats were averaged for use in the calculation, are shown. (C) The qRT-PCR and BN contributions of the Pearson correlation coefficients calculated by, 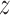 (|BN|) −
(|BN|) − 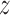 (|qRT-PCR|), where z is Gaussian normalization, are indicated by colors: red for BN and blue for qRT-PCR contributions.
(|qRT-PCR|), where z is Gaussian normalization, are indicated by colors: red for BN and blue for qRT-PCR contributions.
Predictions of chemical toxicity using embryonic stem cells
Using the above 10 genes, we trained SVMs for the 3 toxicity categories by supervised learning except for the ME kernel, using all the chemicals except for a set of test chemicals. The ME kernel requires the generation of kernel matrix by only a semi-supervised setting (37), i.e. learning all the chemicals including test chemicals that have no class labels, due to its intrinsic mechanisms. The two uncategorized chemicals, bisphenol-A and permethrin, were also included during the training iterations in the ME kernel. All data were assigned a class label of +1 or −1 for positive and negative data, respectively or 0 for uncategorized and test data. The uncategorized chemicals were not included in the accuracy and AUC calculations in the LOCOP tests. A total of 200, 100 and 300 feature values (see Materials and Methods) for qRT-PCR, BN and qRT-PCR+BN data, respectively, were re-ranked by P-values derived from the standard two-sample t-tests performed on the training data in each iteration of the LOCOP cross-validation tests, and the number of features from the top of the list was gradually increased. Table 3 summarizes the highest SVM prediction accuracies and their AUCs for various combinations of parameters and numbers of features for qRT-PCR, BN and qRT-PCR+BN input data in each category. The upper table is for non-adjusted data, and the lower table is for batch-adjusted qRT-PCR data. Prediction accuracies by vectorial and structured kernels are separately shown in two lines. ROC curves with AUCs corresponding to the Table are shown in Supplementary Figure S2.
Table 3. Summary of toxicity category predictions.
| Toxicity Category | #Data* | Random** | qRT-PCR*** | BN*** | qRT-PCR+BN*** |
|---|---|---|---|---|---|
| Upper: Vectorial Kernel (linear, polynomial and RBF)/Lower: Structured Kernel (EKM, Saigo and ME) | |||||
| Non-adjusted (Raw) qRT-PCR | |||||
| Neurotoxin | 18/40 | 55.00 | 70.0 (0.664) | 80.0 (0.758) | 70.0 (0.697) |
| 77.5 (0.773) | 95.0 (0.889) | 82.5 (0.881) | |||
| Genotoxic carcinogen | 10/40 | 75.00 | 82.5 (0.553) | 90.0 (0.893) | 82.5 (0.863) |
| 82.5 (0.777) | 100.0 (1.000) | 97.5 (0.980) | |||
| Non-genotoxic carcinogen | 12/40 | 70.00 | 100.0 (1.000) | 90.0 (0.929) | 100.0 (1.000) |
| 100.0 (1.000) | 95.0 (0.964) | 100.0 (1.000) | |||
| Batch-adjusted qRT-PCR | |||||
| Neurotoxin | 18/40 | 55.00 | 70.0 (0.705) | 90.0 (0.859) | 87.5 (0.919) |
| 80.0 (0.874) | 95.0 (0.980) | 97.5 (0.998) | |||
| Genotoxic carcinogen | 10/40 | 75.00 | 85.0 (0.800) | 85.0 (0.640) | 82.5 (0.793) |
| 85.0 (0.830) | 100.0 (1.000) | 92.5 (0.940) | |||
| Non-genotoxic carcinogen | 12/40 | 70.00 | 100.0 (1.000) | 90.0 (0.964) | 97.5 (1.000) |
| 100.0 (1.000) | 95.0 (0.929) | 97.5 (1.000) | |||
*#Data: X/Y, where X = number of samples in the indicated category, and Y = total number of samples.
**Random (prior) = max {Y/X, (40 − Y)/X} × 100, with X and Y defined as in ‘#Data”
***Values for qRT-PCR, BN and qRT-PCR+BN represent maximum accuracy and AUC (in parentheses) in each toxicity category.
Even with qRT-PCR data alone, the prediction was generally successful due to the high performance of the kernel SVM method itself; the accuracies ranged from 77.5 to 100.0%, and were significantly higher than those of random predictions, i.e. 55.0–75.0%. Surprisingly, the use of BN data as SVM input tended to increase the prediction accuracies to as high as 95.0–100.0%. For NT prediction using batch-adjusted data, the accuracies were improved even more when qRT-PCR data and BN data were combined for use as SVM input: the resultant accuracies were 80.0% (qRT-PCR), 95.0% (BN) and 97.5% (qRT-PCR+BN). Furthermore, BN data alone achieved perfect accuracy (100.0%) in GC prediction. In NT and GC predictions using qRT-PCR data alone, accuracies were higher when the analyses were performed on batch-adjusted data; by contrast, BN predictions often yielded similar accuracies in both batch-adjusted and non-adjusted data. These results indicate that BN predictions may be more robust than qRT-PCR predictions, perhaps because the gene-to-gene interaction information is not significantly affected by noise caused by batch effects.
To summarize, if qRT-PCR data yield accuracies lower than 100.0%, BN (as well as a combination of qRT-PCR+BN in some cases) can increase accuracy in all predictions; therefore, it is advantageous to add BN to qRT-PCR as input data. Conversely, if BN alone achieves high predictive power (such as 100.0% in the GC case), the prediction accuracy is sometimes decreased when BN is used in combination with qRT-PCR, probably because the latter method provides noisy and low-level information that prevents SVMs from producing better discriminant functions.
Effective genes, hours and doses for SVM prediction
Figure 5 shows the effects of the number of features on the prediction accuracy. Peaks were observed for the combined data of NTs and GCs. For NTs, the highest accuracies were obtained at around 170 features, whereas for GCs, two small peaks were observed at around 50 and 250 features. For NGCs, no distinct peak was observed; nevertheless, the accuracy was consistently high for all numbers of features. When we examined the top 100 of 300 features (ranked by the two-sample t-test) in LOCOP iterations performed on the combined data, the choice frequencies of features at 96 h were the lowest among the four time points (50 features for each): 12.14, 13.95 and 9.82 times on average for NTs, GCs and NGCs, respectively, whereas the randomly expected choice frequencies should be 50/300 × 100 = 16.67. This result indicates that the 96-h data were segregated from the top candidate features, probably due to low data quality caused by sample degradation resulting from long-term toxic chemical exposure. Furthermore, no choice frequency preference was observed for the rest of the time points (24-, 48- or 72-h) in the top 100 features among the three toxicity categories.
Figure 5.

Maximum accuracies for various numbers of features. (A) Neurotoxins (9 data), (B) genotoxic carcinogens (5 data) and (C) non-genotoxic carcinogens (6 data). qRT-PCR only (200 features), BN only (100 features) and qRT-PCR + BN (300 features) are shown by different lines. In the qRT-PCR + BN lines, NTs and GCs exhibit peak accuracies at specific feature numbers, whereas NGCs constantly exhibit 90.0% or higher accuracy.
Typing of gene networks inferred from toxic chemical experiments
Detailed analysis of the SVM predictions in each toxicity category may give us a better understanding of the characteristics of the chemicals used in this study. Therefore, we determined which chemicals were difficult to predict, and which chemicals exhibited improved predictions in combination with gene networks. In the case of NT prediction, 11 chemicals for which qRT-PCR data only prediction failed (accuracy: 80.0%) were noted: cyclopamine (NT), benzo[a]anthracene (GC), 3-methylcholanthrene (GC), methylmercury (NT), benzo[a]pyrene (GC), diethylstilbestrol (GC), 4-OH-2′,3,3′,4′,5′-PCB (NT), 2,5-hexanedione (NT), warfarin (NT), 2,3,7,8-TCDD (NGC) and phenobarbital (NGC). After BNs were incorporated, the predictions were dramaticaly improved and only one chemical, acrylamide (NT), failed to be predicted (accuracy: 97.5%), representing a new failure of the combined prediction method.
In the case of GC prediciton, qRT-PCR data only prediction (accuracy: 85.0%) failed for as many as 10 types of chemicals, including valproic acid (NT), benzo[a]anthracene (GC), 3-methylcholanthrene (GC), methylmercury (NT), acrylamide (NT), benzo[a]pyrene (GC), diethylnitrosamine (GC), diethylstilbestrol (GC), 2,5-hexanedione (NT) and methapyrilene hydrochloride (NGC). That number was reduced to two in qRT-PCR+BN prediction (accuracy: 92.5%): 3-methylcholanthrene (GC) and diethylnitrosamine (GC). Thus, we conclude that these two GC chemicals are difficult to predict as GCs, probably due to their unique gene response and BN architecture (see Supplementary Figure S1).
DISCUSSION
In this study, we attempted to predict late-onset chemical toxicities using only an undifferentiated hESC system. The results indicated that this system is sufficiently powerful to predict what will happen during the later stages of development when using SVM with gene networks in which network edge weights are given as input feature vectors.
Comparisons with QSAR
As the most popular toxic chemical prediction method, the quantitative structure-activity relationship (QSAR) model is often used, assuming that chemical structures are intrinsically related to specific biological activities (38–40). However, the QSAR model is plagued by several problems, including: (i) the inability to directly measure cellular activities, (ii) difficulty in predicting racemic chemicals, such as thalidomide, (iii) dependence of prediction accuracies on how molecular descriptors are set, and so on (41). Recent QSAR-based approaches that use such molecular descriptors have been improved with the introduction of machine learning techniques (42–45). However, even the most recent approaches can handle only analogous chemicals or their derivatives due to intrinsic difficulties in the prediction of active chemicals that have no or little commonality of the structural framework or functional groups to the learning chemicals. This is exactly the case in our selected chemicals, as shown in Supplementary Table S1.
Alternatively, our gene expression-based approach directly measures biological activities and is thus expected to have great predictive power for various chemicals, even if learning data for similar transcriptional (network) reactions are available. Moreover, the QSAR model basically does not consider conditional cellular activities, such as developmental-stage-specific reactions. Thus, our qRT-PCR measurements based on the hESC system should be more accurate because they reflect initial as well as complex molecular reactions in actual developmental processes.
As a simple proof of study, we generated 1665 molecular descriptors for our selected chemicals using E-Dragon (46) website (http://www.vcclab.org/lab/edragon/) and predicted chemical toxicity categories with our SVM using the same scheme as BNs. We exhaustively tested the number of feature descriptors ranked by two-sample t-test (see Materials and Methods) from 1 to the full range of 1665, which is more than 16 times larger than the range of BNs that have only 100 edge weight parameters. As a result, the highest accuracies for NTs, GCs, and NGCs were as low as 75.0%, 95.0% and 80.0%, respectively, whereas those were 95.0%, 100.0% and 95.0%, respectively, for BN only predictions, as shown in Table 3.
Prediction of uncategorized chemicals
One of the most intriguing issues surrounding this high-accuracy prediction method is whether it is applicable to new toxic chemicals. We were limited by cost and time due to the large number (9768) of qRT-PCR experiments performed on hESCs. Nonetheless, we incorporated two uncategorized chemicals, bisphenol-A and permethrin, into our SVM system with an eye to predicting their potential toxic effects. Bisphenol-A is a weak environmental hormone, and is suspected to be potentially toxic as an NT and/or an NGC. Several studies have shown that bisphenol-A prevents synaptogenic response to estradiol (47). On the other hand, due to its estrogenicity, bisphenol-A can also cause neural disorders or early maturation in animal experiments (48). In addition, developmental exposure to bisphenol-A increases susceptibility to prostate carcinogenesis via unknown epigenetic mechanisms (49). Permethrin is a pyrethroid that is widely used as an insecticide; it also functions as an NT in mice by decreasing Ca2 + influx into neurons and repressing mRNA expression of activity-dependent c-fos and brain-derived neurotrophic factor (BDNF) (50). Neonatal exposure to permethrin leads to long-lasting effects, causing changes in behaviors and striatal monoamine levels and increasing oxidative stress (51).
Figure 6 shows chemicals with the highest similarity to bisphenol-A and permethrin at the BN-structure level. Interestingly, bisphenol-A has marked similarity to 2,3,7,8-TCDD (an NGC; r = 0.713), whereas permethrin has moderate similarity to both 4-OH-2′,3,3′,4′,5′-PCB (an NT; r = 0.505) and methapyrilene hydrochloride (an NGC; r = 0.456). When we investigated the SVM prediction results using both qRT-PCR data and BNs at their maximum accuracies, bisphenol-A was predicted to have an NGC effect, and permethrin was predicted to have both NT and NGC effects. GC effects were not predicted for either chemical.
Figure 6.

Pearson correlations of bisphenol-A and permethrin against toxic chemicals based on qRT-PCR and BN edge weights. (A) Pearson correlation coefficients calculated by qRT-PCR and BN edge weights. (B) Bisphenol-A BN exhibits the highest similarity to 2,3,7,8-TCDD (an NGC), with a relatively high Pearson correlation coefficient (r = 0.713), whereas (C) permethrin BN is moderately similar to both 4-OH-2′,3,3′,4′,5′-PCB (an NT; r = 0.505) and methapyrilene hydrochloride (an NGC; r = 0.456).
Several recent papers have described the relationship between bisphenol-A and its carcinogenicity: specifically, fetal exposure to bisphenol-A induces preneoplastic and neoplastic lesions in adult rat mammary gland (52), and bisphenol-A induces human ovarian cancer growth (53). Other reports have described the relationships between permethrin and neural disorders. For example, prenatal exposure to permethrin influences vascular development in mouse fetal brain (54). To our knowledge, however, no previous report has provided solid evidence of a relationship between permethrin and NGC toxicity. Because the underlying mechanisms of the suspected toxicities of these two uncategorized chemicals remain unknown, additional investigations are necessary to determine their toxicity types.
Further improvement and implications for kernel methods
In this study, we used the two-sample t-test to rank 300 features and observed the tendency for the 96-h qRT-PCR data to be segregated from the top 100 features. Our method of selecting features is often called the ‘filters method.’ There also exist other selection methods, such as wrappers, embedded selection and others (55). Although not the primary focus of this study, the use of recursive feature elimination (RFE) might have enabled us to effectively eliminate non-informative features (56).
It is also notable that the method described here, which provides BN structured data for input as features, offers new insight into research fields that use kernel methods. The use of gene networks, or any other data reconstructed from original raw data, as the input data for an SVM may be considered an external kernel, which sometimes yields explicit models and thus provides more discriminant power in non-linear and/or conceptualized data space than raw data in the original data space. The concept of an external kernel is expected to provide a new arena for SVMs and other machine-learning methodologies, as well as multivariate statistical analyses, which may yield better mathematical models for real problems, leading in turn to better solutions.
CONCLUSION
We demonstrated that a human stem cell-based system that uses only hESCs without further differentiation shows considerable promise for predicting late-onset chemical toxicities, such as abnormalities that occur during embryonic development. Particularly, we show that toxicity category prediction was dramatically improved by SVMs when adopting gene networks in which the network edge weights are added as feature vectors. In recent years, in order to circumvent ethical problems, methods based on human induced pluripotent stem (iPS) cells rather than hESCs have attracted a great deal of attention in the field of stem cell research. As proposed by Anson et al. (57), human iPS cells and their derivatives, such as tissues and organs, are a promising resource for toxicity assessment.
Supplementary Material
Acknowledgments
The authors are deeply grateful to Dr Yuki Kato and Dr Daisuke Nakajima for helpful discussions.
Footnotes
Present addresses:
Sachiyo Aburatani, Biotechnology Research Institute for Drug Discovery, Department of Life Science and Biotechnology, Advanced Industrial Science and Technology (AIST), Central 2, 1-1-1 Umezono, Tsukuba, Ibaraki 305-8568, Japan.
Tsuyoshi Kato, Division of Electronics and Informatics, Faculty of Science and Technology, Gunma University, 1-5-1 Tenjin-cho, Kiryu, Gunma 376-8515, Japan.
Hideko Sone, Center for Health and Environmental Risk Research, National Institute for Environmental Studies, 16-2 Onogawa, Tsukuba, Ibraki 305-8506, Japan.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Health and Labor Sciences Research Grant from the Ministry of Health, Labour and Welfare, Japan [KAKENHI Grant Numbers H21-Kagaku·Ippan-003 to S.O.]; a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Culture and Sports of Japan [15H01749] to H.S.; Core Center for iPS Cell Research, Research Center Network for Realization of Regenerative Medicine from JST. Funding for open access charge: Kyoto University.
Conflict of interest statement. None declared.
REFERENCES
- 1.Waters M., Stasiewicz S., Merrick B.A., Tomer K., Bushel P., Paules R., Stegman N., Nehls G., Yost K.J., Johnson C.H., et al. CEBS–Chemical Effects in Biological Systems: a public data repository integrating study design and toxicity data with microarray and proteomics data. Nucleic Acids Res. 2008;36(Suppl 1):D892–D900. doi: 10.1093/nar/gkm755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deng Y., Johnson D.R., Guan X., Ang C.Y., Ai J., Perkins E.J. In vitro gene regulatory networks predict in vivo function of liver. BMC Syst. Biol. 2010;4:153. doi: 10.1186/1752-0509-4-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noriyuki N., Igarashi Y., Ono A., Yamada H., Ohno Y., Urushidani T. Evaluation of DNA microarray results in the Toxicogenomics Project (TGP) consortium in Japan. J. Toxicol. Sci. 2011;37:791–801. doi: 10.2131/jts.37.791. [DOI] [PubMed] [Google Scholar]
- 4.Low Y., Uehara T., Minowa Y., Yamada H., Ohno Y., Urushidani T., Sedykh A., Muratov E., Kuz'min V., Fourches D., et al. Predicting drug-induced hepatotoxicity using QSAR and toxicogenomics approaches. Chem. Res. Toxicol. 2011;24:1251–1262. doi: 10.1021/tx200148a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirai T., Kiyosawa N. Computational Toxicology. Humana Press; 2013. Developing a practical toxicogenomics data analysis system utilizing open-source software; pp. 357–374. [DOI] [PubMed] [Google Scholar]
- 6.Ito T., Ando H., Suzuki T., Ogura T., Hotta K., Imamura Y., Yamaguchi Y., Handa H. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327:1345–1350. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 7.Davidson P.W., Myers G.J., Weiss B. Mercury exposure and child development outcomes. Pediatrics. 2004;113(Suppl 3):1023–1029. [PubMed] [Google Scholar]
- 8.He X., Imanishi S., Sone H., Nagano R., Qin X.-Y., Yoshinaga J., Akanuma H., Yamane J., Fujibuchi W., Ohsako S. Effects of methylmercury exposure on neuronal differentiation of mouse and human embryonic stem cells. Toxicol. Lett. 2012;212:1–10. doi: 10.1016/j.toxlet.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 9.Suemori H., Yasuchika K., Hasegawa K., Fujioka T., Tsuneyoshi N., Nakatsuji N. Efficient establishment of human embryonic stem cell lines and long-term maintenance with stable karyotype by enzymatic bulk passage. Biochem. Biophysic. Res. Commun. 2006;345:926–932. doi: 10.1016/j.bbrc.2006.04.135. [DOI] [PubMed] [Google Scholar]
- 10.Judson R., Richard A., Dix D.J., Houck K., Martin M., Kavlock R., Dellarco V., Henry T., Holderman T., Sayre P., et al. The toxicity data landscape for environmental chemicals. Environ. Health Perspect. 2009;117:685–695. doi: 10.1289/ehp.0800168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakuratani Y., Zhang H., Nishikawa S., Yamazaki K., Yamada T., Yamada J., Gerova K., Chankov G., Mekenyan O., Hayashi M. Hazard Evaluation Support System (HESS) for predicting repeated dose toxicity using toxicological categories. SAR QSAR Environ. Res. 2013;24:351–363. doi: 10.1080/1062936X.2013.773375. [DOI] [PubMed] [Google Scholar]
- 12.Kadereit S., Zimmer B., van Thriel C., Hengstler J., Leist M. Compound selection for in vitro modeling of developmental neurotoxicity. Front. Biosci. 2012;17:2442–2460. doi: 10.2741/4064. [DOI] [PubMed] [Google Scholar]
- 13.Theunissen P., Robinson J., Pennings J., de Jong E., Claessen S., Kleinjans J., Piersma A. Transcriptomic concentration-response evaluation of valproic acid, cyproconazole, and hexaconazole in the neural embryonic stem cell test (ESTn) Toxicol. Sci. 2012;125:430–438. doi: 10.1093/toxsci/kfr293. [DOI] [PubMed] [Google Scholar]
- 14.Magkoufopoulou C., Claessen S., Tsamou M., Jennen D., Kleinjans J., van Delft J. A transcriptomics-based in vitro assay for predicting chemical genotoxicity in vivo. Carcinogenesis. 2012;33:1421–1429. doi: 10.1093/carcin/bgs182. [DOI] [PubMed] [Google Scholar]
- 15.Gómez-Lechón M., Tolosa L., Castell J., Donato M. Mechanism-based selection of compounds for the development of innovative in vitro approaches to hepatotoxicity studies in the LIINTOP project. Toxicol. In Vitro. 2010;24:1879–1889. doi: 10.1016/j.tiv.2010.07.018. [DOI] [PubMed] [Google Scholar]
- 16.Vinken M., Doktorova T., Ellinger-Ziegelbauer H., Ahr H., Lock E., Carmichael P., Roggen E., van Delft J., Kleinjans J., Castell J., et al. The carcinoGENOMICS project: critical selection of model compounds for the development of omics-based in vitro carcinogenicity screening assays. Toxicol. In Vitro. 2008;659:202–210. doi: 10.1016/j.mrrev.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 17.Do J.H. Neurotoxin-induced pathway perturbation in human neuroblastoma SH-EP cells. Mol. Cells. 2014;37:672–684. doi: 10.14348/molcells.2014.0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pisani C., Gaillard J.-C., Nouvel V., Odorico M., Armengaud J., Prat O. High-throughput, quantitative assessment of the effects of low-dose silica nanoparticles on lung cells: grasping complex toxicity with a great depth of field. BMC Genomics. 2015;16:315. doi: 10.1186/s12864-015-1521-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lan J., Gou N., Rahman S.M., Gao C., He M., Gu A.Z. A quantitative toxicogenomics assay for high-throughput and mechanistic genotoxicity assessment and screening of environmental pollutants. Environ. Sci. Technol. 2016;50:3202–3214. doi: 10.1021/acs.est.5b05097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ritchie M.E., Dunning M.J., Smith M.L., Shi W., Lynch A.G. BeadArray expression analysis using bioconductor. PLoS Comp. Biol. 2011;7:e1002276. doi: 10.1371/journal.pcbi.1002276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benzecri J. Analyse des Données: l'Analyse des Correspondances (Tome 2, Dunod) 1973.
- 22.Ripley B. MASS: support functions and datasets for Venables and Ripley's MASS. 2011.
- 23.Smyth G.K. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. NY: Springer; 2005. limma: Linear Models for Microarray Data; pp. 397–420. [Google Scholar]
- 24.Fujibuchi W., Aburatani S., Yamane J., Imanishi S., Akanuma H., Sone H., Ohsako S. Proc. of the 2011 Joint Conference of CBI-Society and JSBi. Japanese Society for Bioinformatics; 2011. Prediction of chemical toxicity by network-based SVM on ES-cell validation system. [Google Scholar]
- 25.Friedman N., Linial M., Nachman I., Pe'er D. Using Bayesian networks to analyze expression data. J. Comp. Biol. 2000;7:601–620. doi: 10.1089/106652700750050961. [DOI] [PubMed] [Google Scholar]
- 26.Yamanaka T., Toyoshiba H., Sone H., Parham F.M., Portier C.J. The TAO-Gen algorithm for identifying gene interaction networks with application to SOS repair in E. coli. Environ. Health Perspect. 2004;112:1614–1621. doi: 10.1289/txg.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagano R., Akanuma H., Qin X.-Y., Imanishi S., Toyoshiba H., Yoshinaga J., Ohsako S., Sone H. Multi-parametric profiling network based on gene expression and phenotype data: a novel approach to developmental neurotoxicity testing. Int. J. Mol. Sci. 2011;13:187–207. doi: 10.3390/ijms13010187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swendsen R.H., Wang J.-S. Replica Monte Carlo simulation of spin-glasses. Phys. Rev. Lett. 1986;57:2607. doi: 10.1103/PhysRevLett.57.2607. [DOI] [PubMed] [Google Scholar]
- 29.Earl D.J., Deem M.W. Parallel tempering: Theory, applications, and new perspectives. Phys. Chem. Chem. Phys. 2005;7:3910–3916. doi: 10.1039/b509983h. [DOI] [PubMed] [Google Scholar]
- 30.Vapnik V.N. The Nature of Statistical Learning Theory. NY: Springer-Verlag; 1995. [Google Scholar]
- 31.Vapnik V.N. Statistical Learning Theory. Vol. 1. NY: John Wiley & Sons; 1998. [Google Scholar]
- 32.Ramaswamy S., Tamayo P., Rifkin R., Mukherjee S., Yeang C.-H., Angelo M., Ladd C., Reich M., Latulippe E., Mesirov J.P., et al. Multiclass cancer diagnosis using tumor gene expression signatures. Proc. Natl. Acad. Sci. U.S.A. 2001;98:15149–15154. doi: 10.1073/pnas.211566398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fernández-Delgado M., Cernadas E., Barro S., Amorim D. Do we need hundreds of classifiers to solve real world classification problems? J. Mach. Learn. Res. 2014;15:3133–3181. [Google Scholar]
- 34.Fujibuchi W., Kato T. Classification of heterogeneous microarray data by maximum entropy kernel. BMC Bioinformatics. 2007;8:267. doi: 10.1186/1471-2105-8-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kato T., Fujibuchi W. Kernel Classification Methods for Cancer Microarray Data. In: Emmert-Streib F, Dehmer M, editors. Medical biostatistics for complex diseases. John Wiley & Sons; 2010. pp. 279–303. [Google Scholar]
- 36.Robin X., Turck N., Hainard A., Tiberti N., Lisacek F., Sanchez J.-C., Müller M. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bennett K.P., Demiriz A. Advances in neural information processing systems. MIT Press; 1998. Semi-supervised support vector machines; pp. 368–374. [Google Scholar]
- 38.Schultz T.W. Structure-toxicity relationships for benzenes evaluated with Tetrahymena pyriformis. Chem. Res. Toxicol. 1999;12:1262–1267. doi: 10.1021/tx9900730. [DOI] [PubMed] [Google Scholar]
- 39.Jalali-Heravi M., Mani-Varnosfaderani A. QSAR modelling of integrin antagonists using enhanced Bayesian regularised genetic neural networks. SAR QSAR Environ. Res. 2011;22:293–314. doi: 10.1080/1062936X.2011.569758. [DOI] [PubMed] [Google Scholar]
- 40.Cumming J.G., Davis A.M., Muresan S., Haeberlein M., Chen H. Chemical predictive modelling to improve compound quality. Nat. Rev. Drug Discov. 2013;12:948–962. doi: 10.1038/nrd4128. [DOI] [PubMed] [Google Scholar]
- 41.Koutsoukas A., Paricharak S., Galloway W.R., Spring D.R., IJzerman A.P., Glen R.C., Marcus D., Bender A. How diverse are diversity assessment methods? A comparative analysis and benchmarking of molecular descriptor space. J Chem. Inf. Model. 2013;54:230–242. doi: 10.1021/ci400469u. [DOI] [PubMed] [Google Scholar]
- 42.Su B.-H., Tu Y.-S., Esposito E.X., Tseng Y.J. Predictive toxicology modeling: protocols for exploring hERG classification and Tetrahymena pyriformis end point predictions. J. Chem. Inf. Model. 2012;52:1660–1673. doi: 10.1021/ci300060b. [DOI] [PubMed] [Google Scholar]
- 43.Cao D.-S., Zhao J.-C., Yang Y.-N., Zhao C.-X., Yan J., Liu S., Hu Q.-N., Xu Q.-S., Liang Y.-Z. In silico toxicity prediction by support vector machine and SMILES representation-based string kernel. SAR QSAR Environ. Res. 2012;23:141–153. doi: 10.1080/1062936X.2011.645874. [DOI] [PubMed] [Google Scholar]
- 44.Jolly R., Ahmed K. B.R., Zwickl C., Watson I., Gombar V. An evaluation of in-house and off-the-shelf in silico models: Implications on guidance for mutagenicity assessment. Regul. Toxicol. Pharmacol. 2015;71:388–397. doi: 10.1016/j.yrtph.2015.01.010. [DOI] [PubMed] [Google Scholar]
- 45.Omer A., Singh P., Yadav N., Singh R. An overview of data mining algorithms in drug induced toxicity prediction. Mini Rev. Med. Chem. 2014;14:345–354. doi: 10.2174/1389557514666140219110244. [DOI] [PubMed] [Google Scholar]
- 46.Tetko I.V., Gasteiger J., Todeschini R., Mauri A., Livingstone D., Ertl P., Palyulin V.A., Radchenko E.V., Zefirov N.S., Makarenko A.S., et al. Virtual computational chemistry laboratory–design and description. J. Comput. Aid. Mol. Des. 2005;19:453–463. doi: 10.1007/s10822-005-8694-y. [DOI] [PubMed] [Google Scholar]
- 47.Leranth C., Hajszan T., Szigeti-Buck K., Bober J., MacLusky N.J. Bisphenol A prevents the synaptogenic response to estradiol in hippocampus and prefrontal cortex of ovariectomized nonhuman primates. Proc. Natl. Acad. Sci. U.S.A. 2008;105:14187–14191. doi: 10.1073/pnas.0806139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beronius A., Johansson N., Rudén C., Hanberg A. The influence of study design and sex-differences on results from developmental neurotoxicity studies of bisphenol A, implications for toxicity testing. Toxicology. 2013;311:13–26. doi: 10.1016/j.tox.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 49.Ho S.-M., Tang W.-Y., de Frausto J.B., Prins G.S. Developmental exposure to estradiol and bisphenol A increases susceptibility to prostate carcinogenesis and epigenetically regulates phosphodiesterase type 4 variant 4. Cancer Res. 2006;66:5624–5632. doi: 10.1158/0008-5472.CAN-06-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Imamura L., Hasegawa H., Kurashina K., Hamanishi A., Tabuchi A., Tsuda M. Repression of activity-dependent c-fos and brain-derived neurotrophic factor mRNA expression by pyrethroid insecticides accompanying a decrease in Ca2+ influx into neurons. J. Pharmacol. Exp. Ther. 2000;295:1175–1182. [PubMed] [Google Scholar]
- 51.Nasuti C., Gabbianelli R., Falcioni M.L., Di Stefano A., Sozio P., Cantalamessa F. Dopaminergic system modulation, behavioral changes, and oxidative stress after neonatal administration of pyrethroids. Toxicology. 2007;229:194–205. doi: 10.1016/j.tox.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 52.Ptak A., Hoffman M., Gruca I., Barc J. Bisphenol A induce ovarian cancer cell migration via the MAPK and PI3K/Akt signalling pathways. Toxicol. Lett. 2014;229:357–369. doi: 10.1016/j.toxlet.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 53.Dhimolea E., Wadia P.R., Murray T.J., Settles M.L., Treitman J.D., Sonnenschein C., Shioda T., Soto A.M. Prenatal exposure to BPA alters the epigenome of the rat mammary gland and increases the propensity to neoplastic development. PLoS One. 2014;9:e99800. doi: 10.1371/journal.pone.0099800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Imanishi S., Okura M., Zaha H., Yamamoto T., Akanuma H., Nagano R., Shiraishi H., Fujimaki H., Sone H. Prenatal exposure to permethrin influences vascular development of fetal brain and adult behavior in mice offspring. Environ. Toxicol. 2013;28:617–629. doi: 10.1002/tox.20758. [DOI] [PubMed] [Google Scholar]
- 55.Guyon I., Elisseeff A. An introduction to variable and feature selection. J. Mach. Learn. Res. 2003;3:1157–1182. [Google Scholar]
- 56.Guyon I., Weston J., Barnhill S., Vapnik V. Gene selection for cancer classification using support vector machines. Mach. Learn. 2002;46:389–422. [Google Scholar]
- 57.Anson B.D., Kolaja K., Kamp T.J. Opportunities for human iPS cells in predictive toxicology. Clin. Pharmacol. Ther. 2011;89:754–758. doi: 10.1038/clpt.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.