


Abstract
Hepatocellular carcinoma (HCC) has a high mortality rate and early detection of HCC is crucial for the application of effective treatment strategies. HCC is typically caused by either viral hepatitis infection or by fatty liver disease. To diagnose and treat HCC it is necessary to elucidate the underlying molecular mechanisms. As a major cause for development of HCC is fatty liver disease, we here investigated anomalies in regulation of lipid metabolism in the liver. We applied a tailored network-based approach to identify signaling hubs associated with regulation of this part of metabolism. Using transcriptomics data of HCC patients, we identified significant dysregulated expressions of lipid-regulated genes, across many different lipid metabolic pathways. Our findings, however, show that viral hepatitis causes HCC by a distinct mechanism, less likely involving lipid anomalies. Based on our analysis we suggest signaling hub genes governing overall catabolic or anabolic pathways, as novel drug targets for treatment of HCC that involves lipid anomalies.
INTRODUCTION
The incidence of hepatocellular carcinoma (HCC) continues to increase worldwide, and lack of early detection and treatment causes a major burden for the health care sector (1). This highly lethal cancer rarely shows symptoms at an initial stage, and it is therefore difficult to detect at an early stage (2). Moreover, there is no effective means of treatment (3) due to its unknown pathogenic mechanism. Furthermore, HCC is a highly recurring cancer even after liver resection (2).
Several lines of evidence imply that lipid anomalies underlie HCC pathogenesis: increasing risks in patients with obesity (4), diabetes (5) and hepatic steatosis (6). Also, increased de novo lipogenesis in tumor samples (7) substantiates lipid anomaly underlying its pathogenesis. This has prompted to develop genetic or metabolic markers relevant to lipid metabolism for detection of HCC (8–10). However, the complex manner of lipid regulation impedes the identification of genes responsible for lipid anomalies in HCC.
The liver has a key role in lipid metabolism and in maintaining plasma lipoprotein homeostasis by lipid-sensing regulators and lipid-regulating enzymes. Peroxisome proliferator-activated receptors (PPARs), members of the nuclear receptor superfamily, serve as hepatic lipid sensors and govern intrahepatic lipid metabolism by controlling enzymes involved in lipid metabolism through signaling events (11,12). Enzymes of lipid metabolism catalyze reactions in the beta-oxidation of fatty acids, de novo lipogenesis, lipid droplet formation and lipid uptake or secretion (12). Signal transductions enable controlling the level of enzymes in lipid metabolism based on sensed intracellular lipid level and hereby produce lipids required by the cell or degradation of excess fatty acids. However, signaling proteins mediating the communication between sensors and transcription factors controlling expression of enzyme encoding genes remain to be elucidated despite their mechanistic importance. This lack of knowledge is due to the complex and massive nature of signaling networks.
Despite much knowledge about signaling networks, a lack of a tailored network approach delays detection of implicated signaling genes, especially involved in lipid metabolism. Unlike well-characterized genome-scale metabolic models (GEMs) (13,14), analysis and simulation of signaling networks poses a daunting challenge. Some well-curated databases of signaling pathways provide only limited signaling maps (15–17). However, recent large-scale interactome data allow uncovering signaling events implicated in various conditions (18–21), but underlying algorithms have not accounted for the context of metabolic regulation. Thus, signaling genes involved in lipid anomalies have not been examined in a comprehensive manner. Recently, taking metabolic context into account, a novel hubness, called bridgeness (22), allowed identification of signaling hub genes linking bile acid sensors and bile acid enzymes. Unlike other hubness measures that count overall connectivity in a network, bridgeness focuses on genes having connectivity within designated signaling paths, such as paths between metabolic regulators and enzymes. Its formula also showed the possibility to identify important signaling genes implicated in other parts of metabolism.
Here, we applied a tailored network-based approach to investigate lipid-regulating genes, signaling hub genes and enzymes, in HCC. Based on an integrated signaling network, which combines both curated signaling pathway data and large-scale interactome data, we first identified signaling hub genes involved in various lipid metabolic pathways in the liver. Investigating RNA-seq data of HCC patients, from a large genomic data repository of cancer patients, The Cancer Genome Atlas (TCGA), we examined dysregulated expressions of lipid-regulated genes, across all lipid pathways. Also, comparing HCC patients having a viral hepatitis factor, we attempted to distinguish pathogenic mechanism of viral hepatitis from lipid anomalies. Finally, harnessing the power to identify implicated signaling hub genes, we proposed signaling hub genes governing overall catabolic or anabolic pathways that could be new potential drug targets for treatment of HCC.
MATERIALS AND METHODS
Identifying signaling hubs, based on an integrated network and a curated metabolic model
In order to establish a comprehensive signaling network, six BioPAX (23)-formatted databases were integrated by using the Java library Paxtools (24) (the first step in Supplementary Figure S1). By Paxtools, all databases were merged and converted into ‘Single Interaction Format (SIF)’, suited for topological analysis, by ‘Level 3’ rules, which are sets of terms describing types of biomolecular interactions, without indirect interaction rules like ‘Consecutive Catalysis Rule’, ‘Controls Together Rule’ and ‘Metabolic Catalysis’ of ‘Control Rule’. During integration, we excluded interactions containing small molecules in order to find only macromolecular hubs, i.e. genes. Also, ‘generic’ molecules, which are defined by generic name and thus not mapped with unique molecule IDs, were excluded during integration. Basically, this network is a directed network, combined with unidirectional interactions (e.g. transcriptional regulation) and bidirectional interactions (e.g. protein–protein interaction). An integrated network with SIF-format was imported into a network object of the igraph R package (25), with annotations of lipid sensors and enzymes in the network and through this object its network characteristics such as shortest path length were analyzed (the third step in Supplementary Figure S1). To quantify how a given protein links between sensors and enzymes we used a hubness score that was formalized using a previously developed measure, bridgeness (22). We applied a bridgeness measure on all genes in the integrated network, using a shortest path information generated by igraph. Finally, based on a high bridgeness, we selected top-100 hub genes between given pathway enzymes and sensors, regarding them as signaling hubs of a given lipid pathway (the fourth step in Supplementary Figure S1). In addition, we excluded genes not-mapped having an Entrez gene ID from signaling hub candidates because of difficulties with follow up studies when the gene does not have a unique ID (e.g. gene expression analysis).
Taking only free fatty acids (FFAs) and eicosanoids into consideration, we selected (or reconstructed) 37 lipid metabolic pathways (i.e. subsystems) having those lipids as substrates of their reactions from a curated liver GEM, iHepatocyte2322 (13) (the second step in Supplementary Figure S1). Enzymes with their corresponding lipid metabolic pathways were obtained from iHepatocyte2322 by a Java library, JSBML (26). Focusing on only lipid metabolism, we reconstructed a new lipid-specific ‘Pool reactions’ pathway, by selecting enzymes of reactions pooling FFAs: a list of FFAs was obtained from fatty acids contained in ‘NEFA blood pool’ and ‘SMCFA blood pool’ in the model. Likewise, we reconstructed a ‘Transport, FFA’ pathway, by selecting enzymes transporting FFAs. In case of reconstructing a ‘Transport, Pool’ pathway, enzymes transporting FFA-containing pool metabolites, shown in ‘Pool reactions’ pathway, were used. These three lumped pathways were also included in the analysis.
Comparison to known signaling genes in existing curated pathway databases
We collected information about known signaling genes of PPAR or nuclear receptor pathways from the Reactome and BioCarta databases through the MSigDB repository (27). We aggregated the top-100 signaling hubs for the 37 lipid metabolic pathways described above and compared those hubs with signaling genes of PPAR or nuclear receptor pathways by hypergeometric tests. We also identified the top-100 hubs in the integrated network by degree, closeness and betweenness, and compared them with the signaling genes by hypergeometric tests.
Comparison to lipid anomaly genes
We first collected lipid anomaly genes from a human phenotype ontology (HPO) database (28). From abnormal phenotype-to-gene mappings provided in the database, genes that are mapped to ‘Abnormality of lipid metabolism’ (HP: 0003119) were selected as lipid anomaly genes (Supplementary Table S1). To test the statistical significance of overlap between lipid anomaly genes and signaling hubs or enzymes, we only used lipid anomaly genes that are shown in the integrated network. Considering all genes in the network as background, we examined the statistical significance of overlap using a hypergeometric test. Also, we used enzymes that are shown in the integrated network, for comparison between lipid anomaly genes and enzymes.
Comparison to genes associated with tissue-specific phenotypes
For comparison, we collected genes associated with tissue-specific phenotypes from the phenotype-to-gene mapping table of HPO database (28). For liver tissue, genes associated with ‘Abnormality of the liver’ (HP:0001392), for bone marrow tissue, those with ‘Abnormality of bone marrow cell morphology’ (HP:0005561), and for kidney tissue, those with ‘Abnormality of the kidney’ (HP:0000077) were selected and compared with tissue-specific signaling hubs. To select tissue-specific signaling hubs, we first identified signaling hubs of 37 lipid metabolic pathways from the generic human GEM (HMR v2.0) and tissue-specific GEMs for liver, bone marrow and kidney (13,29) over the same integrated network. Then, across lipid metabolic pathways for each tissue-specific GEM, we obtained corresponding tissue-specific signaling hubs by excluding signaling hubs that can be identified by tissue-specific GEM as well as by generic human GEM. After identifying tissue-specific signaling hubs by each metabolic pathway, we aggregated those hubs and compared to genes associated with their corresponding tissue phenotypes by hypergeometric tests.
Pathway-level analysis of gene expressions
RNA-seq data of HCC patients were collected from the TCGA database using the TCGA-Assembler (30): by giving patient TCGA IDs as an input to TCGA-Assembler, we obtained gene-level RNA-seq data of corresponding patients (expression levels in terms of transcripts per million (TPM)). After rounding off normalized values to integers, we identified the significance of differential gene expressions, as gene-level statistics, between tumor and matched control samples by a negative binomial test using the DESeq R package (31) (the fifth step of Supplementary Figure S1). Additionally, enriched gene ontology (GO) terms of differentially expressed genes having adjusted P-value < 0.01 were examined by the BiNGO package (32). After calculating P-values of all genes from negative binomial tests we have compared log P-value distributions between background genes (i.e. all genes detected in RNA-seq data) and signaling hubs or enzymes in a given lipid pathway (the sixth step of Supplementary Figure S1). To compare P-value distributions, we conducted a one-sided Kolmogorov–Smirnov (KS) test, using the built-in ‘stats’ package in R.
Based on the clinical information of TCGA RNA-seq data, we classified RNA-seq data of HCC patients into two groups, a group of HCC patients with viral hepatitis and a group of HCC patients without viral hepatitis. RNA-seq samples having no clinical information about their patient status or medical history about viral hepatitis were excluded in this stage. We examined differential expressions of tumors between the two patient groups (HCC patients with viral hepatitis and HCC patients without viral hepatitis). Limma R statistical package (33) was used to normalize corresponding TCGA RNA-seq data (Voom function (34)) and to calculate gene-level statistics of a factorial model via design and contrast matrices. Making a factorial model by the design and contrast matrices, with factoring sample conditions (i.e. tumors and controls) and sample subgroups (patients with viral hepatitis and patients without viral hepatitis) in the model, we calculated Limma's moderated t-statistics for the difference of differential tumor expressions (i.e. differential gene expressions between tumors and matched controls) between HCC patients with viral hepatitis and HCC patients without viral hepatitis. From P-values of all genes calculated from the moderated t-statistics, we compared log P-value distributions between the background genes and lipid regulating genes, signaling hubs or enzymes, of given lipid pathway by KS one-sided tests. Next, we classified RNA-seq samples into three subgroups based on Pearson's correlation coefficients of gene expressions of samples by hierarchical clustering. By each subgroup of RNA-seq samples, we re-examined differential expressions of tumors between the two patient groups at the gene- and pathway-level, like described above.
In-group specificity
Catabolic or anabolic signaling hubs were identified by in-group specificity. On the basis of cosine similarity, hubness vector (i.e. hubness scores across pathways) and specificity vector of a given gene were used to formalize in-group specificity like below:
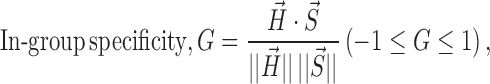 |
where hubness vector, 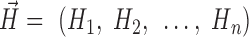 , Hi = a bridgeness in a given pathway i, n = a number of all pathways and specificity vector,
, Hi = a bridgeness in a given pathway i, n = a number of all pathways and specificity vector, 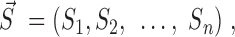 Si =
Si = 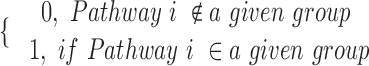
If the hubness vector of a given gene resembles the specificity vector of a certain group, thus showing high hubness scores exclusively in that group, its in-group specificity tends to be high. We analyzed in-group specificities on seed hub genes, which were selected by averaged hubness scores in a given group of pathway. We took 10% as criterion of selecting seed hub genes over genes shown in the integrated network, except sensors, enzymes and genes not-mapped with Entrez IDs.
RESULTS
Identification of signaling hubs of liver lipid metabolism
We identified signaling hub genes of liver lipid metabolism that provide links between lipid-sensing regulators (PPAR-α, β/δ and γ) and lipid-regulated enzymes in liver (Figure 1A). We first established a comprehensive signaling network by integrating databases of curated signaling pathways (PID (17), Panther (15) and Reactome (16)) and large-scale interactomes (PhosphositePlus (35), HPRD (36) and CORUM (37)), resulting in a network with 15 915 genes and 575 634 interactions (see network characteristics in Supplementary Figure S2). Information about lipid enzymes in the liver was taken from a well-curated GEM for liver cells (iHepatocyte2322) (13): we chose lipid enzymes from 37 lipid metabolic pathways regulating free fatty acids or eicosanoids, which are major lipids involved in PPAR signaling (11,12,38). By each lipid metabolic pathway given we separately identified signaling hub genes to corresponding enzymes of the pathway. Based on a hubness tailored for signaling hubs of metabolism called bridgeness (22), we selected the top-100 genes from the integrated network, and considered them as signaling hub genes of a given lipid pathway (signaling hubs shown in Supplementary Table S2 and overall pipeline in Supplementary Figure S1). Unlike other general hubness (e.g. betweenness), bridgeness is designed to quantify a hubness within specific paths between given sensors and enzymes and gives different scores to same genes in the network according to given sensors and enzymes of interest. For that reason, signaling hub genes that we identified did not show high hubness in overall network and thus mostly differ from general hub genes of the network (ranks of the signaling hub genes in other general hubness measures shown in Supplementary Figure S3). Intriguingly, we found that known signaling genes of PPAR or nuclear receptor pathways, which are known as actively signaling in the liver (39), from canonical signaling pathway databases (Reactome and BioCarta) were significantly overlapping with hub genes identified by bridgeness (hypergeometric test P < 1 × 10−12), even more than hub genes identified by other general hubness measures (Table 1). Thus, we confirmed that a tailored hubness, bridgeness was suitable to identify signaling genes within specific paths, like paths between lipid-sensing regulators, PPARs and lipid enzymes.
Figure 1.

Signaling hubs of liver lipid metabolism. (A) Hub genes of signaling between lipid sensors (PPAR-α, β/δ and γ) and lipid enzymes were identified from an integrated signaling network. Hepatic lipid enzymes were chosen from a curated genome-scale metabolic model for liver cells, iHepatocyte2322 and classified into 37 lipid metabolic pathways. For every lipid pathway, we identified top-100 signaling hub genes separately. (B) Hub genes overlapping across lipid pathways were shown in a heatmap. Each row indicates hub genes involved in a certain lipid pathway (if involved, they were colored as black). Most of the largely overlapping genes, referred to as global hubs, were genes supporting nuclear receptor, including PPAR, signaling, such as RXRs, HDACs and nuclear receptor (NR) co-activators/-repressors. Known lipid regulators, SREBPs, LXRα and HNF-4α were less overlapping and therefore referred to as local hubs. Also, based on the HPO database, we examined whether hub genes (C) or sensors/enzymes (D) are associated with a lipid anomaly phenotype and both hub genes and sensors/enzymes were significantly overlapped with known lipid anomaly genes in the database. Next, we examined if (E) identified hub genes of liver lipid metabolism were specifically overlapping with gens associated with liver tissue phenotype. Likewise, we identified signaling hubs of lipid metabolism in other tissues, such as (F) bone marrow and (G) kidney and compared them with genes associated with their tissue phenotypes. Interestingly, those signaling hubs were significantly overlapping with their tissue phenotypic genes. Abbreviations: FA, fatty acid; FFA, free fatty acid; UFA, unsaturated fatty acid; PUFA, poly-unsaturated fatty acid.
Table 1. Overlap between signaling hubs we found and known signaling genes of existing signaling pathway databases. Statistical significances of overlaps were calculated by hypergeometric tests.
| Source | Pathway | Overlap to signaling hubs identified by bridgeness (438 genes): number (P-value) | Overlap to top-100 hubs identified by degree: number (P-value) | Overlap to top-100 hubs identified by closeness: number (P-value) | Overlap to top-100 hubs identified by betweenness: number (P-value) |
|---|---|---|---|---|---|
| REACTOME | Nuclear receptor transcription pathway (51 genes) | 18 (6.7 × 10−16)* | 0 (1) | 4 (2.9 × 10−4)* | 3 (4.0 × 10−3)* |
| REACTOME | Regulation of lipid metabolism by PPAR alpha (115 genes) | 67 (<1 × 10−20)* | 3 (0.036) | 6 (8.6 × 10−5)* | 3 (0.036) |
| BIOCARTA | PPARA pathway (57 genes) | 34 (<1 × 10−20)* | 4 (4.5 × 10−4)* | 18 (<1 × 10−20)* | 14 (<1 × 10−20)* |
| BIOCARTA | RAR RXR pathway (15 genes) | 10 (6.0 × 10−13)* | 1 (0.09) | 2 (3.9 × 10−3)* | 1 (0.09) |
*P-value <0.01.
Some signaling hub genes we found were linked to many lipid metabolic pathways extensively (Figure 1B). Based on the number of involved lipid pathways, hub genes can be classified into two kinds of hubs: global hubs and local hubs. A total of 47 hub genes showed up in an average of 33.9 of 37 lipid pathways (91.6%), and they therefore represent global hubs (a black bracket region in Figure 1B). Essential supporters of PPAR signaling—a sort of nuclear receptor signaling—were most of global hubs, including retinoid X receptors (PPAR heterodimers (11)), histone deacetylases (40), and nuclear receptor co-activators/-repressors (40). In accordance with the PPAR-supporting molecular function, global hubs are enriched in GO terms like ‘nucleoplasm’, ‘transcription regulator activity’ and ‘nuclear hormone receptor binding’ (adjusted p-value: 6.0×10−27, 6.1×10−28 and 1.7×10−20, respectively). On the other hand, local hub genes (total 391 genes) appeared in an average of 5.38 lipid pathways (14.6%), with showing the diverse spectrum of molecular functions. Interestingly, among local hubs are well-known lipid regulators such as, sterol regulatory element-binding proteins (SREBPs) (41), liver X receptor alpha (LXRα) (42) and hepatocyte nuclear factor 4 alpha (HNF-4α) (43).
More intriguingly, we found that many hub genes are associated with abnormal lipid phenotype (Figure 1C and D). We collected 94 genes involved in lipid anomalies (Supplementary Table S1) from a HPO database, which comprehensively displays genes involved in abnormal human phenotypes, based on literature evidence (28). Hypergeometric tests showed that lipid anomaly genes are significantly overlapping with signaling hubs (Figure 1C, P = 2.6 × 10−4) as well as with lipid sensors (PPARs)/enzymes (Figure 1D, P = 1.5 × 10−4). Thus, both lipid sensors/enzymes and signaling hub genes are likely involved in lipid anomalies and may also be involved in causing lipid anomalies in liver tissue.
In addition, we examined if the signaling hubs we identified from the integrated network, by bridgeness, were specifically associated with liver tissue phenotype (Figure 1E and Table 2). From HPO database we collected genes associated with tissue-specific phenotypes, such as liver, bone marrow, and kidney phenotypes, and compared them with identified signaling hubs. We chose bone marrow and kidney tissues for comparison because they are metabolically distinct from liver metabolism. For more specific comparison, we selected liver-specific signaling hubs of lipid pathways that can be identified by liver metabolism (i.e. iHepatocyte2322), not by generic human metabolism (i.e. HMR v2.0) (Supplementary Table S3). Interestingly, we observed that liver-specific signaling hubs were significantly overlapping with genes associated with liver phenotype (P-value < 0.05, Figure 1E), not with genes associated with bone marrow and kidney phenotypes (P-values, 0.27 and 0.074, respectively, see Table 2). For benchmarking, we also identified tissue-specific signaling hubs of bone marrow and kidney based on tissue-specific GEMs (29) (Supplementary Tables S4 and S5), in a similar way that the liver-specific signaling hubs were identified. We found that those tissue-specific signaling hubs were significantly and specifically overlapping with genes of the corresponding tissue phenotypes (Figure 1F and G, Table 2), thus confirming that signaling hubs of liver lipid metabolism identified by bridgeness were highly liver-specific.
Table 2. Overlap between tissue-specific signaling hubs of liver, bone marrow and kidney and genes associated with their tissue-specific anomaly phenotypes. Each row indicates a set of tissue-specific signaling hubs and each column indicates a set of tissue-specific phenotypic genes that were compared with tissue-specific signaling hubs. Statistical significances of overlaps were calculated by hypergeometric tests. Bolded was the most significant among overlaps of tissue-specific signaling hubs to genes associated with tissue-specific phenotypes of liver, bone marrow and kidney.
| Tissue-specific signaling hubs | Overlap to liver phenotype genes (493 genes): number (P-value) | Overlap to bone marrow phenotype genes (289 genes): number (P-value) | Overlap to kidney phenotype genes (517 genes): number (P-value) |
|---|---|---|---|
| Liver-specific signaling hubs (98 genes) | 8 (0.011)* | 3 (0.26) | 6 (0.099) |
| Bone marrow-specific signaling hubs (198 genes) | 8 (0.27) | 10 (0.0034)** | 13 (0.013)* |
| Kidney-specific signaling hubs (166 genes) | 9 (0.074) | 5 (0.18) | 12 (0.0081)** |
*P-value < 0.05.
**P-value < 0.01.
Dysregulated signaling hubs and enzymes in patients with HCC
Aiming to investigate dysregulated lipid metabolism in HCC, we analyzed gene expressions of lipid-regulating genes, both signaling hub genes and lipid enzymes, in patients with HCC (Figure 2). We first gathered RNA-seq data of 207 patients with HCC, from TCGA. Applying a negative binomial test on the RNA-seq data using the DESeq analysis package (31), we examined differential expressions between 207 tumor samples and 49 matched control samples from patients (see differentially expressed genes (DEGs) (adjusted P-value < 0.01) in Supplementary Table S6). Interestingly, among all the DEGs, the most highly enriched GO terms (Supplementary Table S7) were lipid metabolism-related terms (e.g. ‘carboxylic acid metabolic process’), thereby reinforcing that lipid anomaly is a key part of HCC pathogenesis.
Figure 2.

Dysregulated expressions of signaling hubs and enzymes across lipid pathways, in patients with HCC. For each lipid pathway, we examined differential expressions of enzymes (top) and signaling hubs (bottom), between tumor samples and matched control samples. Lipid pathways were classified into four major categories: catabolism, anabolism, pooling and transport. Many enzymes in catabolic and transport pathways were dysregulated at the gene expression level. On the other hand, many hub genes in anabolic and pooling pathways were dysregulated at the gene expression level. Abbreviations: same as Figure 1.
We also examined expressions of lipid-regulating genes at the pathway-level. For each lipid pathway we estimated the statistical significance of differential expressions of signaling hubs or enzymes. For the estimation, P-values of signaling hub genes or enzymes in a given lipid pathway were compared with P-values of background genes (i.e. all detected genes in RNA-seq data) using a Kolmogorov–Smirnov (KS) test. Throughout many lipid pathways, expressions of signaling hubs or enzymes were dysregulated in patients with HCC (Figure 2). Among metabolic pathways being highly dysregulated (i.e. pathways having P-values under 0.05) are catabolic pathways (60%) and transport pathways (100%) (Figure 2, top), whereas in case of signaling hubs anabolic pathways (46.7%) and pooling pathways (60%) (lipid pathway classification shown in Supplementary Table S8) were the most significant (Figure 2, bottom). As a whole, most lipid pathways were disturbed by dysregulated expressions of their lipid-regulating genes, signaling hub genes and enzymes.
Among the dysregulated lipid pathways, noteworthy are eicosanoid-related pathways (Figure 2). Eicosanoids are signaling lipids having impact on inflammatory and immune response and even cancer development (44). Unlike other anabolic pathways, most enzymes of eicosanoid-related pathways (arachidonic acid, leukotriene and prostaglandin pathways) were dysregulated. Even in an ‘eicosanoid metabolism’ pathway with many non-dysregulated enzymes, signaling hubs and key synthases of cancer-promoting lipid, PGE2 were significantly dysregulated (prostaglandin E synthase (PTGES) and prostaglandin E synthase 2 (PTGES2): adjusted P-value = 1.9 × 10−4 and 3.5 × 10−2; fold change = 7.5 and 1.5, respectively). Thus, by investigating both signaling hubs and enzymes we identified lipid anomalies to be associated with HCC.
HCC patients with viral hepatitis reveal a distinct mechanism, less likely involving lipid anomalies
We next sought to further investigate the dysregulation of lipid-regulating genes associated with a major risk factor of HCC, viral hepatitis (B and C) infection. Despite obvious hepatocellular carcinogenesis of viral hepatitis, the underlying mechanisms still remain obscure (45). Here, we attempted to identify if lipid anomalies are also involved in HCC caused by viral hepatitis, by analyzing expressions of signaling hubs and enzymes of lipid metabolism. First, based on patients’ clinical information in TCGA, we collected RNA-seq data of HCC patients by classifying them into two groups: HCC patients with viral hepatitis, both B and C (53 patients, with samples of 53 tumors and 12 matched controls) and HCC patients without viral hepatitis (124 patients, with samples of 124 tumors and 34 matched controls). Noticeably, we found that between the two patients groups, matched control samples are highly correlated (mean ρ = 0.93), but HCC tumor samples are not (mean ρ = 0.67) (Supplementary Figure S4A). We then examined differential expressions between tumors and matched controls by each patient group, and lastly, estimated the statistical significance of differential expressions of signaling hubs and enzymes at the pathway-level (Figure 3), in a similar way of Figure 2.
Figure 3.

Distinct expressions of signaling hubs and enzymes revealed in HCC patients with viral hepatitis. In two patient groups of HCC, (A and B) a patient group with viral hepatitis B and C and (C and D) a patient group without viral hepatitis, we separately examined differential expressions of signaling hubs and enzymes. Interestingly, in patients with viral hepatitis, (A) most pathways’ enzymes and (B) signaling hubs were less dysregulated. Of note, fatty acid biosynthesis pathways (a red bracket), called de novo lipogenesis pathways, did not reach the statistical significance of P-value = 0.05, suggesting that viral hepatitis leads to HCC, irrespective of lipid anomalies. Abbreviations: same as Figure 1.
From pathway-level P-values (KS test P-values) of the two patient groups (Figure 3), we found that expressions of lipid-regulating genes between tumors and controls were markedly distinct in a group of patients with viral hepatitis, compared to a group of patients without viral hepatitis. In case of enzymes (Figure 3A and C), many lipid pathways tend to be less significant in patients with viral hepatitis. In addition, in case of signaling hubs (Figure 3B and D), many lipid pathways, even tend to be non-significant in patients with viral hepatitis. In particular, anabolic pathways’ signaling hubs, which were mostly dysregulated in overall HCC patients, showed substantial differences. Only 2 of 15 anabolic pathways were dysregulated in patients with viral hepatitis. Moreover, pathways involved in de novo lipogenesis, a prominent feature of HCC, were not dysregulated (a red bracket in Figure 3B and D).
We further examined differential expressions in tumors between the two patient groups by additional statistical tests. First, from the factorial model of Limma R statistical package, we investigated individual genes if their differential expressions between tumors and matched controls were distinct in a specific patient group (Supplementary Table S9). From the gene-level statistics (i.e. Limma's moderated t-statistics) of the factorial model, we examined pathway-level differential expressions of lipid-regulating genes in tumors between the two patient groups (Supplementary Figure S5A), in a similar way of Figure 2. Here, we observed weak differential expressions of signaling hubs in tumors between the two patient groups because of tumor heterogeneity, as revealed in sample correlations. In order to control tumor heterogeneity while comparing the two patient groups, we clustered tumor samples into three subgroups based on gene expression similarity (Supplementary Figure S4) and within each subgroup we compared tumor gene expressions between the two patient groups again, through the factorial model like above. A subgroup having most low-grade tumors, class 1 (Supplementary Figure S4B), has significantly distinct tumor expressions in signaling hubs, in particular those of de novo lipogenesis, between the two patient groups (Supplementary Figure S5B), implying that even before tumors became aggressive, viral hepatitis showed a distinct pathogenesis. On the other hand, a subgroup having more high-grade tumors, class 3, has significantly distinct tumor expressions in signaling hubs of most lipid pathways (48.6%) between the two patient groups (Supplementary Figure S5C). For class 2 subgroup, which has most aggressive tumors among subgroups, we could not perform the comparison as there were only data for tumors from patients with viral hepatitis. Based on those findings, we concluded that viral hepatitis leads to HCC by a distinct mechanism, less likely involving lipid anomalies.
Catabolic or anabolic signaling hubs
We next attempted to find signaling hub genes governing overall catabolic or anabolic pathways. Applying an in-group specificity, which measures hubness elevated exclusively in a given group of pathways, we examined the catabolic or the anabolic specificity of signaling hubs (Figure 4). Interestingly, signaling hubs only present in catabolic pathways tend to have higher catabolic specificity and lower anabolic specificity and signaling hubs only found in anabolic pathways showed opposite tendencies. By t-tests, we found that these differences were statistically significant (between two signaling hub groups, the difference of catabolic specificities: t-test P-value = 3.74 × 10−11; the difference of anabolic specificities: t-test P-value = 3.72 × 10−10). Thus, taking advantage of in-group specificity, we applied in-group specificities on seed hub genes, which are the 10% genes with highest mean hubness in catabolic or anabolic pathways. Among the seed hub genes, we selected the top-10 genes with highest in-group specificities as group-specific hubs: catabolic or anabolic signaling hubs (Table 3).
Figure 4.

In-group specificity of signaling hubs. In order to distinguish catabolism- and anabolism-specific hub genes, we measured the in-group specificity of hub genes in catabolic or anabolic pathways. Notably, hubs shown only in anabolic pathways (blue) tend to be located in top-left region (high anabolic specificity and low catabolic specificity). On the other hand, hubs shown only in catabolic pathways (red) tend to be in bottom-right region. We also found significant differences of catabolic and anabolic specificity between two hub groups (red and blue) by t-tests (P-values, 3.74 × 10−11 and 3.72 × 10−10, respectively). Based on in-group specificities, we identified catabolic or anabolic signaling hubs.
Table 3. Catabolic or anabolic signaling hubs. We showed the characteristics of catabolic or anabolic signaling hubs, with in-group specificity (with rank) and representative ranks of hubness scores (averaged-rank and top-rank) across a given group of pathways.
| Hub type | Name (Entrez ID) | In-group specificity (rank) | Hubness rank in catabolic pathways: average (top) | Hubness rank in anabolic pathways: average (top) | Hubness rank in pooling pathways: average (top) | Hubness rank in transport pathways: average (top) |
|---|---|---|---|---|---|---|
| Catabolic signaling hubs | GSC (145258) | 0.6673 (1) | 1039.8 (79) | 3073.9 (2219) | 2824.0 (1595) | 3449.5 (2427) |
| FOXA2 (3170) | 0.6663 (2) | 754.9 (4) | 1323.9 (816) | 1671.4 (1419) | 1393.0 (1391) | |
| GLI1 (2735) | 0.6587 (3) | 408.5 (14) | 1005.7 (671) | 1058.6 (888) | 669.5 (580) | |
| RUNX3 (864) | 0.6555 (4) | 351.5 (108) | 1054.8 (462) | 1108.0 (789) | 762.0 (681) | |
| FOXH1 (8928) | 0.6492 (5) | 519.4 (191) | 1093.4 (566) | 1139.0 (900) | 1628.0 (1610) | |
| FOXG1 (2290) | 0.6489 (6) | 709.3 (300) | 1492.8 (816) | 1582.6 (1245) | 1549.5 (1476) | |
| SMAD9 (4093) | 0.6485 (7) | 70.9 (13) | 225.9 (24) | 161.8 (27) | 107.0 (46) | |
| SKP1 (6500) | 0.6483 (8) | 773.1 (416) | 1645.1 (465) | 1697.8 (1129) | 1257.0 (1077) | |
| SLC2A4 (6517) | 0.6478 (9) | 86.3 (9) | 203.5 (9) | 284.8 (142) | 115.5 (59) | |
| NCOA2 (10499) | 0.6450 (10) | 13.7 (11) | 55.2 (13) | 86.0 (19) | 23.5 (19) | |
| Anabolic signaling hubs | SREBF1 (6720) | 0.6758 (1) | 836.1 (678) | 361.7 (59) | 722.6 (191) | 645.5 (540) |
| PNO1 (56902) | 0.6707 (2) | 2038.8 (1322) | 890.5 (173) | 1854.4 (988) | 1459.5 (1266) | |
| ZBTB17 (7709) | 0.6702 (3) | 567.9 (374) | 262.4 (120) | 655.0 (322) | 529.5 (249) | |
| EWSR1 (2130) | 0.6698 (4) | 469.9 (50) | 202.7 (7) | 326.0 (142) | 530.5 (391) | |
| SP1 (6667) | 0.6668 (5) | 194.0 (63) | 91.1 (6) | 115.2 (52) | 114.5 (97) | |
| USF2 (7392) | 0.6655 (6) | 1250.5 (564) | 763.7 (221) | 1126.2 (807) | 921.0 (715) | |
| TP63 (8626) | 0.6650 (7) | 351.9 (122) | 225.3 (12) | 484.0 (225) | 507.5 (284) | |
| C1D (10438) | 0.6640 (8) | 1144.9 (928) | 986.3 (264) | 2219.4 (1179) | 994.5 (874) | |
| RPS3A (6189) | 0.6632 (9) | 1609.9 (1240) | 1166.9 (35) | 1497.6 (925) | 1944.5 (1611) | |
| NDRG1 (10397) | 0.6627 (10) | 1161.1 (212) | 924.7 (24) | 1200.0 (772) | 1226.5 (948) |
Some hub genes were found to regulate de novo lipogenesis (anabolism) or fatty acid oxidation (catabolism). The top-scoring anabolic hub gene, the sterol regulatory element-binding protein 1 (SREBP1, synonym: SREBF1), is a known master regulator of de novo lipogenesis (41). By regulating genes for fatty acid synthesis, SREBP-1c (hepatic major isoform of SREBP1) governs overall lipid synthesis in the liver. The second highest scoring catabolic hub gene, forkhead box protein A2 (FOXA2) also controls lipid metabolism, by activating genes involved in beta-oxidation (46,47). Mice with haplo-insufficient FOXA2 have a decreased beta-oxidation index in liver, thus substantiating its role as a catabolic regulator. Other genes also revealed their association with lipid metabolism: p63 and the glucose transporter type 4 (GLUT4). p63 (synonym: TP63), a homolog of the metabolic regulator p53, also regulates lipid metabolism. Recent studies revealed that loss of TAp63 (p63 isoform) leads to deregulation of lipid metabolism, with increasing fatty acid synthesis and decreased fatty acid oxidation (48). Also, GLUT4 (synonym: SLC2A4) is related to lipid metabolism: GLUT4-null mice show increased hepatic de novo lipogenesis (49) which underpins its association to lipid metabolism. In addition, an enriched GO term ‘transcription factor activity’ (adjusted P-value = 1.65 × 10−7) in both catabolic and anabolic hubs implies that even other signaling hubs with unknown association with lipid metabolism have a pivotal role in regulating certain biological processes, including lipid metabolism.
DISCUSSION
Here, we identified signaling hub genes of liver lipid metabolism in a systematic manner, and examined their co-expression with lipid enzymes in HCC. These signaling hub genes have not earlier been linked to lipid anomalies in HCC, but using our tailored network-based approach we could identify these genes. Our analysis revealed that not only lipid enzymes, but also signaling hub genes showed dysregulated expressions, and even synergistically leading to lipid anomalies in HCC. We further found that there are distinct signaling hub genes and enzymes associated with HCC pathogenesis caused by viral hepatitis. Lastly, our tailored network-based approach uncovered catabolic and anabolic signaling hub genes that could serve as novel drug targets for treatment of HCC, by alleviating lipid anomalies during its pathogenesis.
Until now, many studies have related lipid-sensing regulators or lipid-regulating enzymes to lipid anomalies and considered them as therapeutic drug targets (50–52). Fibrates or statins are well-known lipid-regulating drugs by targeting PPARs (lipid sensor) or HMG-CoA reductase (cholesterol biosynthetic enzyme), and they have also been found to have potentials to reduce HCC risks (53). However, these drugs have been used for improving systemic lipid metabolism, not targeted for lipid anomalies in liver tissues. Instead, a drug targeting signaling hubs of liver lipid metabolism might be a new therapeutic option for alleviating lipid abnormality only in the liver tissue. In the present study, we found significant abnormalities of lipid anabolic pathways in HCC patients, and these anabolic signaling hubs would be promising selective drug targets of lipid dysfunctional metabolism associated with HCC tumors.
Notably, beside an abnormal lipid, HCC pathogenesis involves abnormal immune response, called cirrhosis (54): almost 80% of patients with HCC have cirrhotic livers (2). Hence, till now, lipid anomaly has been considered as a first alteration and cirrhosis as a second alteration to cause HCC (55). Interestingly, we found that eicosanoids, important signaling lipids associated with inflammation, were dysregulated, thus possibly indicating abnormal inflammation. Therefore, the dysregulation of lipid-regulating genes we identified might enable diagnosis of both the first and second alteration causing HCC. Comparing eicosanoid anomalies between fatty liver disease (without inflammation) and HCC (with inflammation) might allow us to understand the gap between these two pathological conditions, and furthermore to diagnose the progression of fatty liver disease into HCC before the second alteration.
The relationships identified here between the signaling and other metabolic pathways also extend our knowledge about the occurrence of HCC. In a cell, the regulation of energy-controlling metabolites, such as glucose, lipid, cholesterol and bile acid, is strongly coordinated with each other. Intriguingly, from our signaling hub genes, we found both nuclear receptors and AMP-activated protein kinase (AMPK), which regulate other metabolic pathways: LXRα (synonym: NR1H3) that regulates cholesterols and lipids (42,56), small heterodimer partner (SHP) (synonym: NR0B2) regulating bile acids (57), and AMPK1 and AMPK2 (synonym: PRKAA1 and PRKAA2) that regulates the overall energy metabolism (58). Therefore, developing more comprehensive methods for investigating overall energy metabolism will give more insights to our understanding of inter-regulations within overall energy metabolism, eventually mechanisms of all metabolic anomalies underlying several diseases, including HCC.
In summary, to identify lipid anomalies shown in HCC, we identified signaling hub genes by a tailored network-based approach. Based on RNA-seq data of patients with HCC, we examined lipid-regulating genes, both signaling hub genes and lipid enzymes, and found significant dysregulated expressions among these genes. We also found that viral hepatitis causes HCC in a distinct mechanism, less likely involving lipid anomalies. Finally we identified catabolic or anabolic signaling hubs also that might be associated with HCC pathogenesis and be possible drug targets for treatment of HCC.
Supplementary Material
Acknowledgments
The authors thank The Cancer Genome Atlas (TCGA) to provide access of restricted data. The authors also thank Elias Björnson for valuable discussions and comments on the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Knut and Alice Wallenberg Foundation; Bio-Synergy Research Project of the Ministry of Science, ICT; Future Planning through the National Research Foundation of Korea [NRF- 2012M3A9C4048758 to D.L.]. Funding for open access charge: Knut and Alice Wallenberg Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1.Siegel R., Ma J., Zou Z., Jemal A. Cancer statistics, 2014. CA Cancer J. Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 3.Michelotti G.A., Machado M.V., Diehl A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013;10:656–665. doi: 10.1038/nrgastro.2013.183. [DOI] [PubMed] [Google Scholar]
- 4.Calle E.E., Rodriguez C., Walker-Thurmond K., Thun M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 5.Mittal S., El-Serag H.B. Epidemiology of hepatocellular carcinoma: consider the population. J. Clin. Gastroenterol. 2013;47(Suppl):S2–S6. doi: 10.1097/MCG.0b013e3182872f29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pekow J.R., Bhan A.K., Zheng H., Chung R.T. Hepatic steatosis is associated with increased frequency of hepatocellular carcinoma in patients with hepatitis C-related cirrhosis. Cancer. 2007;109:2490–2496. doi: 10.1002/cncr.22701. [DOI] [PubMed] [Google Scholar]
- 7.Calvisi D.F., Wang C., Ho C., Ladu S., Lee S.A., Mattu S., Destefanis G., Delogu S., Zimmermann A., Ericsson J., et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology. 2011;140:1071–1083. doi: 10.1053/j.gastro.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uccello M., Malaguarnera G., Pelligra E.M., Biondi A., Basile F., Motta M. Lipoprotein(a) as a potential marker of residual liver function in hepatocellular carcinoma. Indian J. Med. Paediatr. Oncol. 2011;32:71–75. doi: 10.4103/0971-5851.89775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin H.J., Wu P.C., Ho J.C. The ether lipid tumour marker in human liver with hepatocellular carcinoma. Br. J. Cancer. 1980;41:320–324. doi: 10.1038/bjc.1980.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patterson A.D., Maurhofer O., Beyoglu D., Lanz C., Krausz K.W., Pabst T., Gonzalez F.J., Dufour J.F., Idle J.R. Aberrant lipid metabolism in hepatocellular carcinoma revealed by plasma metabolomics and lipid profiling. Cancer Res. 2011;71:6590–6600. doi: 10.1158/0008-5472.CAN-11-0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahmadian M., Suh J.M., Hah N., Liddle C., Atkins A.R., Downes M., Evans R.M. PPARgamma signaling and metabolism: the good, the bad and the future. Nat. Med. 2013;19:557–566. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bechmann L.P., Hannivoort R.A., Gerken G., Hotamisligil G.S., Trauner M., Canbay A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012;56:952–964. doi: 10.1016/j.jhep.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 13.Mardinoglu A., Agren R., Kampf C., Asplund A., Uhlen M., Nielsen J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014;5:3083. doi: 10.1038/ncomms4083. [DOI] [PubMed] [Google Scholar]
- 14.Mardinoglu A., Agren R., Kampf C., Asplund A., Nookaew I., Jacobson P., Walley A.J., Froguel P., Carlsson L.M., Uhlen M., et al. Integration of clinical data with a genome-scale metabolic model of the human adipocyte. Mol. Syst. Biol. 2013;9:649. doi: 10.1038/msb.2013.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mi H., Muruganujan A., Thomas P.D. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–D386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Croft D., Mundo A.F., Haw R., Milacic M., Weiser J., Wu G., Caudy M., Garapati P., Gillespie M., Kamdar M.R., et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014;42:D472–D477. doi: 10.1093/nar/gkt1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaefer C.F., Anthony K., Krupa S., Buchoff J., Day M., Hannay T., Buetow K.H. PID: the Pathway Interaction Database. Nucleic Acids Res. 2009;37:D674–D679. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gitter A., Carmi M., Barkai N., Bar-Joseph Z. Linking the signaling cascades and dynamic regulatory networks controlling stress responses. Genome Res. 2013;23:365–376. doi: 10.1101/gr.138628.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui Q., Ma Y., Jaramillo M., Bari H., Awan A., Yang S., Zhang S., Liu L., Lu M., O'Connor-McCourt M., et al. A map of human cancer signaling. Mol. Syst. Biol. 2007;3:152. doi: 10.1038/msb4100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang R.S., Albert R. Elementary signaling modes predict the essentiality of signal transduction network components. BMC Syst. Biol. 2011;5:44. doi: 10.1186/1752-0509-5-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chasman D., Ho Y.H., Berry D.B., Nemec C.M., MacGilvray M.E., Hose J., Merrill A.E., Lee M.V., Will J.L., Coon J.J., et al. Pathway connectivity and signaling coordination in the yeast stress-activated signaling network. Mol. Syst. Biol. 2014;10:759. doi: 10.15252/msb.20145120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee S., Lee K., Yoon S., Lee J.W., Lee D. Anomalies in network bridges involved in bile Acid metabolism predict outcomes of colorectal cancer patients. PLoS One. 2014;9:e107925. doi: 10.1371/journal.pone.0107925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demir E., Cary M.P., Paley S., Fukuda K., Lemer C., Vastrik I., Wu G., D'Eustachio P., Schaefer C., Luciano J., et al. The BioPAX community standard for pathway data sharing. Nat. Biotechnol. 2010;28:935–942. doi: 10.1038/nbt.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Demir E., Babur O., Rodchenkov I., Aksoy B.A., Fukuda K.I., Gross B., Sumer O.S., Bader G.D., Sander C. Using biological pathway data with paxtools. PLoS Comput. Biol. 2013;9:e1003194. doi: 10.1371/journal.pcbi.1003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Csardi G., Nepusz T. The igraph software package for complex network research. Int. J. Comp. Syst. 2006;1695:1–5. [Google Scholar]
- 26.Drager A., Rodriguez N., Dumousseau M., Dorr A., Wrzodek C., Le Novere N., Zell A., Hucka M. JSBML: a flexible Java library for working with SBML. Bioinformatics. 2011;27:2167–2168. doi: 10.1093/bioinformatics/btr361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kohler S., Doelken S.C., Mungall C.J., Bauer S., Firth H.V., Bailleul-Forestier I., Black G.C., Brown D.L., Brudno M., Campbell J., et al. The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data. Nucleic Acids Res. 2014;42:D966–D974. doi: 10.1093/nar/gkt1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uhlen M., Fagerberg L., Hallstrom B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A., et al. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 30.Zhu Y., Qiu P., Ji Y. TCGA-assembler: open-source software for retrieving and processing TCGA data. Nat. Methods. 2014;11:599–600. doi: 10.1038/nmeth.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maere S., Heymans K., Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 33.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Law C.W., Chen Y., Shi W., Smyth G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hornbeck P.V., Zhang B., Murray B., Kornhauser J.M., Latham V., Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–D520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keshava Prasad T.S., Goel R., Kandasamy K., Keerthikumar S., Kumar S., Mathivanan S., Telikicherla D., Raju R., Shafreen B., Venugopal A., et al. Human Protein Reference Database–2009 update. Nucleic Acids Res. 2009;37:D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruepp A., Waegele B., Lechner M., Brauner B., Dunger-Kaltenbach I., Fobo G., Frishman G., Montrone C., Mewes H.W. CORUM: the comprehensive resource of mammalian protein complexes–2009. Nucleic Acids Res. 2010;38:D497–D501. doi: 10.1093/nar/gkp914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wahli W., Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012;23:351–363. doi: 10.1016/j.tem.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 39.Rando G., Wahli W. Sex differences in nuclear receptor-regulated liver metabolic pathways. Biochim Biophys. Acta. 2011;1812:964–973. doi: 10.1016/j.bbadis.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 40.Perissi V., Rosenfeld M.G. Controlling nuclear receptors: the circular logic of cofactor cycles. Nat. Rev. Mol. Cell Biol. 2005;6:542–554. doi: 10.1038/nrm1680. [DOI] [PubMed] [Google Scholar]
- 41.Horton J.D., Goldstein J.L., Brown M.S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hong C., Tontonoz P. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat. Rev. Drug Discov. 2014;13:433–444. doi: 10.1038/nrd4280. [DOI] [PubMed] [Google Scholar]
- 43.Hayhurst G.P., Lee Y.H., Lambert G., Ward J.M., Gonzalez F.J. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol. Cell. Biol. 2001;21:1393–1403. doi: 10.1128/MCB.21.4.1393-1403.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang D., Dubois R.N. Eicosanoids and cancer. Nat. Rev. Cancer. 2010;10:181–193. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arzumanyan A., Reis H.M., Feitelson M.A. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat. Rev. Cancer. 2013;13:123–135. doi: 10.1038/nrc3449. [DOI] [PubMed] [Google Scholar]
- 46.Wolfrum C., Asilmaz E., Luca E., Friedman J.M., Stoffel M. Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature. 2004;432:1027–1032. doi: 10.1038/nature03047. [DOI] [PubMed] [Google Scholar]
- 47.Wolfrum C., Stoffel M. Coactivation of Foxa2 through Pgc-1beta promotes liver fatty acid oxidation and triglyceride/VLDL secretion. Cell Metab. 2006;3:99–110. doi: 10.1016/j.cmet.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 48.Su X., Gi Y.J., Chakravarti D., Chan I.L., Zhang A., Xia X., Tsai K.Y., Flores E.R. TAp63 is a master transcriptional regulator of lipid and glucose metabolism. Cell Metab. 2012;16:511–525. doi: 10.1016/j.cmet.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kotani K., Peroni O.D., Minokoshi Y., Boss O., Kahn B.B. GLUT4 glucose transporter deficiency increases hepatic lipid production and peripheral lipid utilization. J. Clin. Invest. 2004;114:1666–1675. doi: 10.1172/JCI21341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dobrzyn A., Ntambi J.M. Stearoyl-CoA desaturase as a new drug target for obesity treatment. Obes. Rev. 2005;6:169–174. doi: 10.1111/j.1467-789X.2005.00177.x. [DOI] [PubMed] [Google Scholar]
- 51.Wu M., Singh S.B., Wang J., Chung C.C., Salituro G., Karanam B.V., Lee S.H., Powles M., Ellsworth K.P., Lassman M.E., et al. Antidiabetic and antisteatotic effects of the selective fatty acid synthase (FAS) inhibitor platensimycin in mouse models of diabetes. Proc. Natl. Acad. Sci. U.S.A. 2011;108:5378–5383. doi: 10.1073/pnas.1002588108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pahan K. Lipid-lowering drugs. Cell Mol. Life Sci. 2006;63:1165–1178. doi: 10.1007/s00018-005-5406-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Singh S., Singh P.P., Singh A.G., Murad M.H., Sanchez W. Statins are associated with a reduced risk of hepatocellular cancer: a systematic review and meta-analysis. Gastroenterology. 2013;144:323–332. doi: 10.1053/j.gastro.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 54.Farazi P.A., DePinho R.A. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat. Rev Cancer. 2006;6:674–687. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 55.Byrne C.D. Fatty liver: role of inflammation and fatty acid nutrition. Prostaglandins Leukot Essent Fatty Acids. 2010;82:265–271. doi: 10.1016/j.plefa.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 56.Zhao C., Dahlman-Wright K. Liver X receptor in cholesterol metabolism. J. Endocrinol. 2010;204:233–240. doi: 10.1677/JOE-09-0271. [DOI] [PubMed] [Google Scholar]
- 57.Thomas C., Pellicciari R., Pruzanski M., Auwerx J., Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 2008;7:678–693. doi: 10.1038/nrd2619. [DOI] [PubMed] [Google Scholar]
- 58.Hardie D.G., Ross F.A., Hawley S.A. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.