


Abstract
The template-directed incorporation of nucleotides at the terminus of a growing primer is the basis of the transmission of genetic information. Nature uses polymerases-catalyzed reactions, but enzyme-free versions exist that employ nucleotides with organic leaving groups. The leaving group affects yields, but it was not clear whether inefficient extensions are due to poor binding, low reactivity toward the primer, or rapid hydrolysis. We have measured the binding of a total of 15 different activated nucleotides to DNA or RNA sequences. Further, we determined rate constants for the chemical step of primer extension involving methylimidazolides or oxyazabenzotriazolides of deoxynucleotides or ribonucleotides. Binding constants range from 10 to >500 mM and rate constants from 0.1 to 370 M−1 h−1. For aminoterminal primers, a fast covalent step and slow hydrolysis are the main factors leading to high yields. For monomers with weakly pairing bases, the leaving group can improve binding significantly. A detailed mechanistic picture emerges that explains why some enzyme-free primer extensions occur in high yield, while others remain recalcitrant to copying without enzymatic catalysis. A combination of tight binding and rapid extension, coupled with slow hydrolysis induces efficient enzyme-free copying.
INTRODUCTION
The incorporation of nucleotides complementary to a template base at the terminus of a growing primer is the molecular basis of replication and transcription (1). Best known are polymerase-catalyzed forms of this reaction, but the most pristine version of it is enzyme-free primer extension (2,3). Enzyme-free primer extension is solely driven by molecular recognition between nucleotides and naked DNA or RNA and the intrinsic reactivity of activated monomers and primer terminus. The reaction has a special aura, as it has the potential to explain how early versions of replication may have occurred when life first arose from inanimate material (4). Almost fifty years ago, pioneering work in the field involving long homopolymer templates and non site-specific oligomerization of activated nucleotides showed the feasibility of the reaction (5,6) but yields were low. The advent of automated oligonucleotide synthesis (7) made it possible to study extension reactions at a specific site of a primer-template complex or hairpin (8,9) but studies of broader ranges of sequence space gave mixed results (10,11). Unless non-natural aminoterminal primers are used, incomplete conversion is observed on many sequences (12).
The activation of the nucleotide is critically important for the yield of primer extensions. Polymerase-catalyzed reactions use triphosphates, but triphosphates are not reactive enough in enzyme-free versions. Nucleotides with amino acids as leaving groups are accepted by some polymerases (13,14), but no enzyme-free version is known. Instead, nucleotides with aromatic heterocycles as leaving groups are used for copying DNA or RNA templates in the absence of enzymes. The list of leaving groups known to provide sufficient reactivity includes 2-methylimidazole (MeIm) (15), imidazolide (Im) (15,16), and oxyazabenzotriazole (OAt) (17). Alkylated adenines as leaving groups lead to oligomerization on mineral surfaces (18), a reaction that is related mechanistically to chemical primer extension. Recently, in situ activation by a water-soluble carbodiimide, combined with an organocatalyst has been found to induce enzyme-free extension of aminoterminal (19) or ribonucleotide primers (20).
The yield of enzyme-free chain extension is also dependent on the nucleophile found at the primer terminus. For straight DNA primers, no broadly applicable method exists for extension (21), but RNA primers with their terminal 2′/3′-diol are sufficiently reactive for extension to occur when allowed to react with monomers activated by one of the methods producing nucleotides with heterocyclic leaving groups. Still, the low reactivity of the ribose terminus often causes hydrolysis to compete successfully with extension, resulting in significant concentrations of free nucleotides that act as competitive inhibitors (22). The most common type of primer used for high-yielding extension features an amino group at the 3′- or 2′-position of the 3′-terminal residue, and numerous examples exist for this type of chain extension that produces phosphoramidate linkages (12,23–27). Because it is unclear whether entirely ribonucleotide-based systems have the ability to undergo replication, phosphoramidate-producing reactions continue to be studied in enzyme-free systems. But for either chemistry, full replication of RNA or DNA sequences from monomers has remained an elusive goal (28).
It is important to understand why many primer extensions stall after incomplete conversion. Why does chemical primer extension not behave like a conventional bimolecular reaction that can be driven to completion by increasing the concentration of the reactants, increasing the temperature, adding a catalyst, or a combination of such measures? A quantitative understanding of the kinetic and thermodynamic factors governing this reaction should provide answers and pave the way for improved versions of enzyme-free copying, both in fundamental research and for practical applications, such as the read-out of genetic information in analytical samples (29).
Chemical primer extension reactions are mechanistically complex. At the very least, they involve a binding step, during which the nucleotide monomer pairs with the primer-template duplex, and one or several chemical steps leading to the formation of the new covalent bond and the release of the leaving group (Figure 1). At the same time, background hydrolysis converts monomers into free nucleotides that can still bind, but in doing so block extension sites, acting as competitive inhibitors (22). Dissecting the overall process kinetically and thermodynamically requires detailed experimental work. We have recently reported methodologies for measuring the binding of nucleotides to primer-template complexes, using two complementary techniques, namely NMR-monitored titration and inhibitor assays (30). Dissociation constants (Kd's) of 2–280 mM were measured for the four different free deoxynucleotides (dAMP, dCMP, dGMP and dTMP) and a Kd value of 15 mM was found for GMP binding to an RNA hairpin. Subsequently, Szostak and coworkers measured binding constants for complexes of three of the four unactivated ribonucleotides (A, C and G) for self-complementary ribonucleotide duplexes, using NMR (31). As part of our mechanistic work, we had developed a quantitative mathematical description of primer extension (30), but the ability to model and predict yields was limited because the effect of the leaving group on binding was unknown, so that it had to be assumed that the activated monomers bind with the same strength as the free nucleotides. Further, rate constants for the covalent step had been measured for oxyazabenzotriazolides only, and data for the more popular methylimidazolides was missing. In order to tackle the problem of incomplete conversion systematically, quantitative data was needed on how leaving groups affect binding, the covalent step of primer extension, as well as hydrolysis, both for DNA and RNA templates.
Figure 1.

Mechanistic pathways for a monomer (M) in the context of enzyme-free extension of a primer (P) on a template (T). A quantitative model requires rate constants (k) and dissociation constants (K). The expansion in the upper part of the shows molecular details of the covalent step.
Here we report the dissociation constants of complexes of activated deoxynucleotide and ribonucleotide monomers to hairpins, as well as rates of hydrolysis that were hitherto unknown. Further, we have extracted rate constants for the covalent step of primer extension from kinetic assays with primer:template duplexes. Dissociation constants for nucleotides with 2-methylimidazole (MeIm) or oxyazabenzotriazole (OAt) as leaving groups were found to be in the range of 20–240 mM for DNA and 11 to >500 mM for RNA. The rates and yields of extension reactions were determined for each of the four nucleobases, both for DNA templates and RNA templates. Using the refined mathematical model we were able to simulate the time-yield relationships for primer extension reactions.
MATERIALS AND METHODS
Activated nucleotides
The 2′-deoxynucleotides (dNMPs, 1a–t) were activated to oxyazabenzotriazolides (OAt esters) 2a–t or 2-methylimidazolides (MeIm-dNMPs) 3a–t. Monomers for enzyme-free primer extension on RNA templates were synthesized from ribonucleotides (NMPs) 4a–u and gave OAt esters 5a–u or methylimidazolides 6a–u. In either case, slightly modified versions of known protocols (6,33) were employed. Brief, representative protocols for the preparation of MeIm-dGMP (3g) and OAt-GMP (5g) are given below. Prior to activation, sodium salts of commercial ribonucleotides were treated with cation exchange resin (Dowex 50WX8 in triethylammonium form) to improve solubility.
The methylimidazolide of dGMP (3g) was prepared by treating a solution of its sodium salt (50 mg, 135 μmol) with 2-methylimidazole (111 mg, 1.4 mmol), 2,2′-dipyridyldisulfide (89 mg, 405 μmol) and triphenylphosphine (71 mg, 270 μmol) in dry DMF/DMSO (2 mL, 1:1, v/v) (14). The solution was stirred for 1.5 h at 22°C under argon, followed by precipitation with a solution of sodium perchlorate (33 mg, 270 μmol) in cold acetone/diethylether (35 mL, 4:1, v/v). The pellet was washed with acetone/diethylether (4 × 15 mL), and the resulting crude was dried at < 10−3 mbar, followed by cartridge purification in two portions on Sep Pak RP18 cartridges (Waters, 12 cc). The cartridges had previously been washed with acetonitrile and then with demineralized water. An aqueous solution of one half of the crude product in water (500 μL for 15 mg crude product) was loaded onto the cartridge, followed by washing with aqueous NaCl (1 M, 20 mL) and elution of the activated nucleotide with a gradient of acetonitrile (0 to 10%) in H2O. Methylimidazolide 3g eluted at 2% CH3CN. The combined product fractions from the two cartridges were immediately frozen and then lyophilized to dryness, yielding 23 mg yield (53 μmol, 39%) of 3g. If needed, purified monomers were stored in dry form at −20°C.
To prepare OAt-GMP (5g), the triethylammonium salt of GMP (4g, 50 mg, 123 μmol) was treated with O-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate (HATU, 140 mg, 369 μmol), 1-hydroxy-7-azabenzotriazole (HOAt, 33 mg, 246 μmol) and triethylamine (14 μl, 185 μmol) in dry DMF (2 mL) (33). The reaction mixture was stirred for 1.5 h at 22°C under argon, followed by precipitation of the product by adding the mixture to a solution of sodium perchlorate (30 mg, 246 μmol) in cold acetone/diethylether (35 mL, 4:1, v/v). After washing with acetone/diethylether (4 × 15 mL) and drying at <10−3 mbar, the same cartridge purification was used as for 3g, above, with 5g eluting at 4% CH3CN, yielding 13 mg (27 μmol, 22%) of pure active ester. Further details of synthesis and purification protocols can be found in the Supporting Data.
NMR experiments
The NMR spectra were measured on a Bruker AVANCE 500 MHz spectrometer at 20°C in D2O containing phosphate buffer (200 mM, NaCl 400 mM and MgCl2 80 mM). For OAt esters, a pH of 8.9 was used, and for methylimidazolides the pH was 7.0 (either value uncorrected for deuterium effect). A method for determining dissociation constants described earlier was used (30). Briefly, a solution (200 μL) of the hairpin (1 mM for 7a–t or 0.5 mM for 8a–u) was treated with aliquots of a stock solution of the monomer in the same buffer. After brief mixing and centrifugation, 1H NMR spectra of the resulting solution were recorded immediately. Each set of titration experiments was performed in less than 90 min to minimize reactions. Data analysis was performed as previously described (30). Hydrolysis rates for activated monomers were determined from series of 31P NMR spectra of freshly prepared solutions of the respective monomer (25 mM), using a fit procedure as described earlier (12,22,32). Further information can be found in the Supporting Data.
Kinetic assays
The methodology employed for primer extension assays was similar to that described previously (12,33). A representative assay was performed as follows. A solution (10 μL final volume) containing DNA template 9n (54 μM) and aminoterminal primer 10 (36 μM), prepared as described (34), in buffer (HEPES 200 mM, NaCl 400 mM, and MgCl2 80 mM) at pH 8.9 for reaction with OAt esters or pH 7.0 for methylimidazolides, at 20°C was treated with aliquots of a stock solution of the activated monomer (2a–t or 3a–t). The same procedure was used for RNA primer extension assays and monomers 5a–u or 6a–u, except that primer 13 and template 12a–u were used at 36 μM each, and the pH adjusted to 7.7 for methylimidazolides. The extension of the primer was measured via MALDI-ToF mass spectrometry of samples drawn at stated intervals under conditions that allow for quantitative detection (35,36).
Mathematical model
Calculations with the binding constants and rate constants listed in Tables 1–5 were performed with the solver tool of Excel 2010 (Microsoft). Simulation of the extension of a primer through reaction with a given activated monomer were performed using an expanded mathematical model presented in the Supplementary Information of reference (30). For each simulation, the inhibition caused by hydrolyzed monomer was studied by calculating yield versus time curves for hypothetical reactions without hydrolysis. Additional plots of calculated and experimental data are shown in the Supporting Data.
Table 1. Rate constants of hydrolysis for activated nucleotides as determined by 31P NMRa.
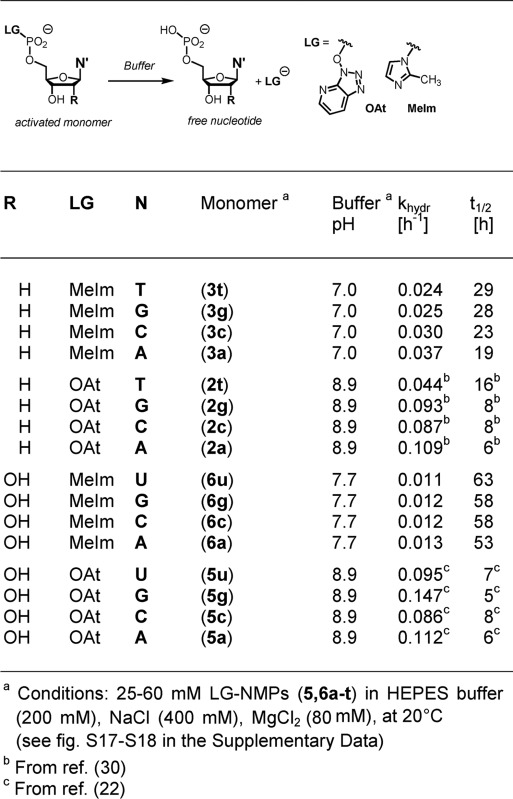
|
Table 5. Kinetic constants and conversion maxima for extension of RNA primers with ribonucleotide monomers, as determined by MALDI-MSa.
| Nucleotide | Conc. [mM] | Template | pH | k' [h−1 M−1] | Conversionb | kcovc [h−1] | |
|---|---|---|---|---|---|---|---|
| OAt-UMP | (5u) | 22 | 12a | 8.9 | 1.1 | 18 | 0.07 |
| OAt-GMP | (5g) | 14 | 12c | 8.9 | 3.8 | 28 | 0.10 |
| OAt-CMP | (5c) | 7.2 | 12g | 8.9 | 4.6 | 29 | n.d.d |
| OAt-AMP | (5a) | 7.2 | 12u | 8.9 | 2.9 | 16 | 0.06 |
| MeIm-UMP | (6u) | 60 | 12a | 7.7 | 0.1 | 24 | n.d.d |
| MeIm-GMP | (6g) | 36 | 12c | 7.7 | 0.3 | 48 | 0.02 |
| MeIm-CMP | (6c) | 30 | 12g | 7.7 | 0.5 | 59 | (0.03)e |
| MeIm-AMP | (6a) | 60 | 12u | 7.7 | 0.1 | 29 | 0.01 |
aConditions: HEPES buffer (200 mM), NaCl (400 mM), MgCl2 (80 mM) 20°C, and pH 8.9 (OAt) or 7.7 (MeIm).
bCalculated conversion at infinite reaction time.
cDetermined from initial rates and calculated occupancy of extension site.
dNot determined because numerical value of dissociation constant unknown.
eBased on dissociation constant determined in salt-free solution (compare Table 3).
RESULTS
To quantitatively describe primer extension (Figure 1), it is important to understand the fate of the nucleotide monomer, which can enter several reaction channels. It may bind to the template-primer complex, as quantitatively described by the dissociation constant of the complex, and then react to give the extended primer with the rate of the covalent step. Alternatively, the monomer may hydrolyze, a process quantitatively described by the rate constant khydr, producing the free nucleotide (Mh). The hydrolyzed monomer can also bind to the primer-template complex, thereby acting as an inhibitor that blocks the extension site. The inhibitory effect can be understood on the basis of the dissociation constant Kdh. The reactivity of a bound monomer toward the primer terminus manifests itself in the rate constant for the covalent step (kcov). The yield of the reaction thus depends on the concentrations of the reactants and four constants that have to be determined: two dissociation constants, and two rate constants. The yield of extended primer (Yp+1) over time may then be calculated using Equation (1), which was elaborated as part of our earlier work on quantitatively modeling enzyme-free primer extension (30).
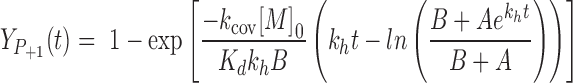 |
(1) |
With the definitions:
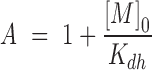 |
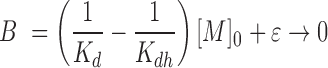 |
In our current work, we wished to quantitatively understand reactions of OAt esters and methylimidazolides of nucleotides with aminoterminal DNA primers and RNA primers, respectively. This meant having to access the binding constants, rates of hydrolysis and rate constants of the covalent step of primer extension for nucleotides with either of these two common types of leaving groups, both for ribonucleotides and 2′-deoxynucleotides. Dissociation constants for complexes of free nucleotides under our reaction conditions were known for the four deoxynucleotides (dAMP, dCMP, dGMP and TMP) and GMP as ribonucleotide (30). For the latter and the remaining three unactivated ribonucleotides, binding constants were also reported by Szostak and coworkers in a different sequence context during the course of our experimental work (31). The kcov values for the OAt esters reacting with aminoprimers were also known (30), but the corresponding values for reactions of methylimidazolides were lacking.
First, we determined rate constants of hydrolysis (khydr) under reaction conditions for extension of aminoprimers (12) or RNA primers (22) for all cases for which such data was not available. The activated monomers were dissolved in extension buffer and their hydrolysis was monitored by 31P NMR. Table 1 lists the rate constants for the entire set of 16 monomers, namely OAt esters and methylimidazolides of deoxy- or ribonucleotides with any of the four bases (A, C, G or T/U). Under the basic reaction conditions, OAt esters showed a half-life time of approx. 8 hours, except for OAt-TMP (2t), which gave twice this value. The slower hydrolysis of 2t may be due to the steric shielding of the phosphodiester by the methyl group of the nucleobase. A conformational search by molecular modeling corroborated this hypothesis. It led to a number of structures where the substituent at position 5 of the pyrimidine blocks possible trajectories of attack of water or hydroxide ions on the active ester (Supplementary Figure S35, Supporting Data).
The half-life time of methylimidazolides in the reaction buffer was found to be approximately one day at pH 7.0 (deoxynucleotide monomers) and two-and-a half days at pH 7.7 (ribonucleotide monomers). The half-life time of MeIm-GMP 6g at a higher Mg2+ concentration (200 mM MgCl2) and 37°C had previously been measured to be approx. 27 h at pH 8 (37), which is close, given the differences in conditions. Overall, the nucleobases were found to have a modest effect on hydrolysis, with slightly higher rates of hydrolysis for the purines and the slowest rate of hydrolysis for T.
Next, we measured dissociation constants for complexes of activated nucleotides. For this, we chose hairpins with an unreactive natural deoxynucleotide at the 3′-terminus (30), with a sequence studied previously (38,30). Binding data from hairpins have previously been cross-validated on long templates, using a complementary technique and free nucleotides (30). The activated nucleotides were obtained in pure form by a combination of precipitation and cartridge purification and were added to solutions containing hairpins 7a–t (Figure 2). Hairpins with stem sequences analogous to those of 7a–t gave melting points >40°C in temperature-dependent NMR studies (30). Because NMR measurements can be performed within minutes and the half-life times of the active monomers are all ≥6 h, conventional NMR experiments were performed. Because hydrolysis manifests itself in NMR spectra, every titration experiment produced data that confirmed that no more than minimal quantities of free nucleotides were present. Figure 3 shows representative NMR data, measured upon addition of 2t to a solution of 7a, together with a plot of the chemical shift of the H2 proton of the pairing nucleotide in the hairpin versus concentration of the monomer. Even for this weakly pairing base, the OAt ester bound tightly enough to yield a well-defined dissociation constant (71 mM). Table 2 lists the dissociation constants obtained.
Figure 2.

Hairpins and monomers used for determining dissociation constants via NMR titration. Loops are hexaethylene glycol linkers (HEG).
Figure 3.

Representative results from an NMR titration. (A) Overlay of spectra showing the 1H NMR signals of nucleobase protons of hairpin 5′-ACAG(HEG)CTG (7a) (0.5 mM) at increasing concentration of OAt-TMP (2t). (B) Chemical shift displacement of the H2 proton of the 5′-terminal A residue. The line is the fit to the experimental data points (open circles).
Table 2. Dissociation constants for complexes of deoxynucleotides binding to the termini of DNA hairpins, as determined by NMR titrationa.
| Nucleotide | Templating base | Hairpin | pH | Kd [mM]b | |
|---|---|---|---|---|---|
| dTMP | (1t) | A | (7a) | 8.9 | 241c |
| OAt-dTMP | (2t) | A | (7a) | 8.9 | 71 |
| MeIm-dTMP | (3t) | A | (7a) | 7.0 | 236 |
| dGMP | (1g) | C | (7c) | 8.9 | 27 |
| OAt-dGMP | (2g) | C | (7c) | 8.9 | 26 |
| MeIm-dGMP | (3g) | C | (7c) | 7.0 | 25 |
| dCMP | (1c) | G | (7g) | 8.9 | 34d |
| OAt-dCMP | (2c) | G | (7g) | 8.9 | 25 |
| MeIm-dCMP | (3c) | G | (7g) | 7.0 | 26 |
| dAMP | (1a) | T | (7t) | 8.9 | 38d |
| OAt-dAMP | (2a) | T | (7t) | 8.9 | 20 |
| MeIm-dAMP | (3a) | T | (7t) | 7.0 | 37 |
aConditions: 0.5 to 1 mM hairpin in D2O (99.9%) and phosphate buffer (200 mM), NaCl (400 mM), MgCl2 (80 mM), pH 8.9 or 7.0 (uncorrected for deuterium effect), 20°C.
bDetermined by fit using law of mass action.
cAverage value from two titration experiments.
dFrom (30).
It can be discerned that for all deoxynucleotides except dGMP, the OAt leaving group improves binding. For dGMP, neither the methylimidazole group nor the oxyazabenzotriazolide changed the Kd value significantly over that of the free nucleotide. For dCMP, a modest increase in binding strength was found for both the MeIm and the OAt group. For dAMP, the most lipophilic of all nucleotides, little increase in binding strength was detectable for the methylimidazolide, whereas the OAt ester gave the lowest Kd value of all deoxynucleotide-hairpin combinations (20 mM). The largest effect of a leaving group on binding by far was found for dTMP, with an approx. four-fold increase in binding strength upon furnishing it with the OAt leaving group.
When the titration was performed with ribonucleotides and RNA hairpins 8a–u, the results shown in Table 3 were obtained. The titrations were again performed at 20°C, i.e. at a temperature 16°C below the melting point of the RNA hairpins (Supplementary Figure S19, Supporting Data). Binding of both UMP itself and its methylimidazolide 6u was too weak to calculate a dissociation constant. The OAt ester of this weakly stacking ribonucleotide gave a Kd value of 43 mM, though, again suggesting that for weakly pairing nucleotides this leaving group can significantly improve binding. For hairpin 8g, addition of the primer extension buffer led to broadening of signals, possibly because the dangling G residues engaged in G quartet formation at NMR concentrations. This prevented the determination of Kd's in extension buffer. For free CMP 4c and its methylimidazolide 6c, dissociation constants could be measured under low salt conditions. For 5c, even those conditions gave signals too broad for proper analysis. Under the (electrostatically unfavorable) low salt conditions, CMP (4c) gave a Kd for the complex with 8g that was lower than those of the complexes of the other three ribonucleotides and their complementary hairpins. This corroborates the finding of Zhang, Szostak and colleagues, who had recently identified CMP as the most strongly pairing nucleotide among the four free NMPs (31).
Table 3. Dissociation constants for complexes of ribonucleotides binding to the terminus of RNA hairpins, as determined by fitting NMR dataa.
| Ribonucleotide | Templating base | Hairpin | pH | Kd [mM] | |
|---|---|---|---|---|---|
| UMP | (4u) | A | (8a) | 7.0 | > 500 |
| OAt-UMP | (5u) | A | (8a) | 7.0 | 43b |
| MeIm-UMP | (6u) | A | (8a) | 7.0 | > 500 |
| GMP | (4g) | C | (8c) | 7.0 | 27 (± 9)d |
| OAt-GMP | (5g) | C | (8c) | 7.0 | 11b |
| MeIm-GMP | (6g) | C | (8c) | 7.0 | 23 |
| CMP | (4c) | G | (8g) | 7.0 | 19c |
| OAt-CMP | (5c) | G | (8g) | 7.0 | n.d. |
| MeIm-CMP | (6c) | G | (8g) | 7.0 | (27)c |
| AMP | (4a) | U | (8u) | 7.0 | 90 |
| OAt-AMP | (5a) | U | (8u) | 7.0 | 14 |
| MeIm-AMP | (6a) | U | (8u) | 7.0 | 42 |
aConditions: 0.5 mM RNA hairpin in D2O and phosphate buffer (200 mM), NaCl (400 mM), MgCl2 (80 mM) at pH 7.0 (uncorrected values for deuterium effect) and 20°C.
bAverage value from 2 titrations.
cTitrations with CMP (4c) and MeIm-CMP (6c) were performed in D2O, without buffer salts, to avoid broadening of hairpin signals.
dAverage of two measurements done in this work and the value reported in (30), with standard deviation in parentheses.
Our RNA hairpin displaying uracil as templating base (8u) gave dissociation constants in the range of 14–90 mM, depending on the base and the leaving group. In either case, leaving groups improved binding. For the methylimidazolides of ribonucleotides, the GMP derivative formed the most stable complex, with a Kd of 23 mM. This monomer (6g) is the most popular monomer for enzyme-free primer extension in the literature (39,11,37). When comparing the pairing strength of the MeIm derivatives, the order found was G > C > A >> U. The lowest Kd values of all monomers tested in our study were those of OAt esters of purine ribonucleotides, with Kd values of 11 and 14 mM for 5g and 5a, respectively.
We then proceeded to measuring rate constants for the covalent step of the enzyme-free primer extension reaction. Using the primer-template combinations presented in Figure 4, we measured extension rates at different monomer concentrations, calculated the concentration of the active complex M…PT (Figure 1) and determined how rapidly it reacts to give the extended primer. The resulting kcov values that show how reactive a monomer with a specific leaving group is are compiled in Tables 4 and 5 for deoxynucleotides and ribonucleotides, respectively. The data shows that the OAt esters can not only improve binding, but also provide the monomers with increased reactivity in the covalent step. In every case studied, the OAt ester of a deoxynucleotide underwent the covalent step faster than its MeIm analog. On average, the increase in rate is approx. one order of magnitude. In either group of monomers, the most weakly binding base (T/U) also gives the slowest rate in the covalent step and the strongly pairing bases (G/C) give the fastest chemical step. The difference between the most reactive and the least reactive nucleotide in one group is approx. a factor of four, which is significant, but considerably less than the effect of sequence context on the extension of aminoterminal primers with deoxynucleotides (12).
Figure 4.

Oligonucleotide sequences and 2′-deoxynucleotides used for template-directed primer extension reaction. Assays at increasing concentrations of activated monomers were performed using the following conditions: 36 μM template: 3′-aminoprimer complex, 0.18 to 7.2 mM matching activated monomer (2a–t or 3a–t) in primer extension buffer (HEPES 200 mM, NaCl 400 mM, MgCl2 80 mM), pH 8.9 (for OAt-dNMP) or 7.0 (for MeIm-dNMP) at 20°C.
Table 4. Kinetic constants and conversion for nucleotides extending primers in DNA template-directed reaction, as determined by MALDI-MSa.
| Nucleotide | Template | pH | k' [h−1 M−1]b | Conversion c | kcov [h−1]d | |
|---|---|---|---|---|---|---|
| OAt-dTMP | (2t) | 9a | 8.9 | 47 | 74 | 1.7 |
| OAt-dGMP | (2g) | 9c | 8.9 | 280 | 99 | 8.6 |
| OAt-dCMP | (2c) | 9g | 8.9 | 370 | 99 | 9.9 |
| OAt-dAMP | (2a) | 9t | 8.9 | 140 | 98 | 3.2 |
| MeIm-dTMP | (3t) | 9a | 7.0 | 8 | 15 | 0.4 |
| MeIm-dGMP | (3g) | 9c | 7.0 | 51 | 99 | 1.4 |
| MeIm-dCMP | (3c) | 9g | 7.0 | 22 | 96 | 0.4 |
| MeIm-dAMP | (3a) | 9t | 7.0 | 13 | 67 | 0.3 |
aConditions: 3.6 mM LG-dNMPs (2a–t or 3a–t) in HEPES buffer (200 mM), NaCl (400 mM), MgCl2 (80 mM), pH 8.9 for LG = OAt or 7.0 for LG = MeIm, at 20°C.
bSecond order rate constant, as determined by fitting using equation S3.
cMaximum conversion at infinite reaction time, as obtained by mathematical fit.
dDetermined from initial rates kexp and calculated occupancy of extension site.
Assays with ribonucleotide monomers and RNA primer were performed using the sequences shown in Figure 5. Reactions did not lead to full conversion of the primers. Monomers with weakly pairing nucleobases (A/U) showed particularly low yields, so that the kcov values obtained by fitting are fraught with more uncertainty than those obtained with aminoterminal primers. Also, it was in early assays with ribonucleotides that we first noticed a side product in the crudes of methylimidazolides that initially complicated our kinetic analysis. This side product, which can also be found in unpurified methylimidazolides of deoxynucleotides, gives a peak in the 31P spectrum at −10.8 ppm. It was tentatively assigned to an imidazoliumbisphosphate, where one leaving group is covalently linked to two nucleotides. Supplementary Figure S32 of the Supporting Data shows data for unpurified monomer 3g. Unless the side product, which appears to be formed to a significant extent under the traditional activation conditions (6), was rigorously removed by purification, an initial burst phase was observed in the kinetics of primer extension (see Supplementary Figures S33 and S34). With pure methylimidazolides 6a–u, smooth, but slower kinetics were observed. Still, methylimidazolides of ribonucleotides required higher concentrations (up to 60 mM of the monomer) than the corresponding OAt esters, to obtain the values for primer conversion listed in Table 5.
Figure 5.

Oligoribonucleotide sequences and ribonucleotide monomers used for template-directed primer extension assays. Conditions: 36 μM template:primer complex, 1.8 to 60 mM complementary monomer (5a–u or 6a–u) in primer extension buffer (200 mM HEPES, 80 mM MgCl2, 400 mM NaCl, pH 8.9 (for OAt-NMP) or 7.7 (for MeIm-NMP) at 20°C.
We then used the kinetic data to calculate kcov values from the initial rate and the occupancy of the extension site, accessible via the Kd values. These kcov values that were calculated from sets of kinetic data at different monomer concentration provide a quantitative picture of the intrinsic reactivity of the complex of monomers and primer-template duplexes (last column of Tables 4 and 5). For example, for aminoterminal primers reacting with deoxynucleotides, the chemical step of the mechanism is less than one order of magnitude faster for OAt esters than for methylimidazolides when the nucleobase of the monomer is T or G. The only monomer for which the kcov value differs by significantly more than one order of magnitude is dCMP, not because of an unusual value for the MeIm monomer, but because of the high value of the rate constant for the OAt ester. For the ribonucleotides reacting with oligoribonucleotide primers, the situation is similar, except that the absolute values of kcov are considerably smaller, both for OAt esters and for methylimidazolides, compared to the reactions with aminoprimers. Among the OAt esters for which data is available, the GMP derivative gives the fastest reaction and the reactivity differences are small. Among the methylimidazolides, all available data point to more significant differences in reactivity and a molecular situation that favors CMP.
With the Kd values for activated nucleotides and a selection of kcov values in hand, we were in a position to simulate primer extension on a new level. Figures 6 and 7 show calculated time-conversion curves for either type of primer and each of the two different types of leaving groups. Additional plots of simulated and experimental data are shown in the Supporting Data (Supplementary Figures S28–S31). For the aminoterminal primer, the agreement of experimental and theoretical values (Figure 6), suggests that the process is well described by the theory. Inspection of the calculated yield curves for hypothetical reactions without inhibition by spent monomer (broken lines in Figure 6) highlights differences between the two leaving groups. Whereas the OAt ester 2a reacts so fast and efficiently that inhibition does not develop into a significant problem, the incomplete conversion of aminoterminal primer 10 when reacting with MeIm monomer 3a could be all but avoided, if the spent monomer was not inhibiting the extension after the initial phase of the assay (Figure 6B).
Figure 6.

Time-conversion curves for extension of an aminoterminal primer: experimental data points (symbols) and simulated time course (lines). (A) Primer 10, template 9t, and monomer OAt-dAMP (2a) at 3.6 mM (red), 1.8 mM (blue), 0.36 mM (green) or 0.18 mM (purple) concentration, simulation with Kd = 20 mM, Kdh = 38 mM; kcov = 3.2 h−1 and khydr = 0.109 h−1. The broken black line shows hypothetical kinetics without the formation of an inhibitor through hydrolysis of 3.6 mM 2a. (B) Primer 10, template 9t and MeIm-dAMP (3a) at 5.0 mM (red), 2.5 mM (blue) or 1.3 mM (green) concentration; simulation with Kd = 37 mM, Kdh = 38 mM; kcov = 0.31 h−1, khydr = 0.037 h−1. Again, the broken black line is hypothetical kinetics without hydrolysis/inhibition at 5 mM monomer concentration.
Figure 7.

Simulated extension of an RNA primer according to Equation (1) (lines) with experimental data points (symbols) of the corresponding reactions. (A) Primer 13, template 12c (36 μM and MeIm-GMP (6g) at 36 mM (red), 24 mM (blue) or 12 mM (green); simulation with Kd = 23 mM, Kdh = 27 mM; kcov = 0.020 h−1, khydr = 0.012 h−1. The broken black line shows hypothetical kinetics without hydrolysis at 36 mM monomer. (B) Primer 13, template 12c and OAt-GMP 5g at 7.2 mM (green), 3.6 mM (blue), 1.8 mM (red) concentration, simulated with Kd = 11 mM; Kdh = 35 mM; khydr = 0.147 h−1, kcov = 0.095 h−1 and with the broken black line showing hypothetical kinetics without hydrolysis/inhibition at 7.2 mM nucleotide. Note that the dissociation constants are lower limits of the true value (compare Table 3).
For ribonucleotides reacting with oligoribonucleotide primers, the quantitative picture based on the Kd values for activated nucleotides, the hydrolysis rates and the kcov values extracted from extension assays is shown in Figure 7. The numerical agreement is poorer than for the reactions of aminoterminal primers, but it is clear why more quantitative extensions could be achieved when the effects of inhibition were to be avoided. In either case, the hypothetical reactions without inhibition by spent monomer are significantly higher yielding than those found experimentally, explaining why immobilizing primer and template and periodically replacing the supernatant helps (22).
DISCUSSION
Oxyazabenzotriazole was introduced as a leaving group for high-yielding enzyme-free primer extensions more than a decade ago (17,33), but its effect on primer extensions had remained unexplained on a mechanistic level. Our results now show that the increase in rate and yield with OAt leaving groups can be traced back to two critical factors. The oxyazabenzotriazolide can improve binding to primer-template complexes, and it can accelerate extension without accelerating hydrolysis to the same extent. The first is a phenomenon that has its roots in ground-state binding, whereas the latter is due to differences in transition state energies for the desired pathway and the undesired pathway of hydrolysis.
Binding is induced by intermolecular forces that stabilize the bound form, including, but not limited to hydrogen bonding, van der Waals interactions, hydrophobic effect, and Coulombic interactions. Either of the two leaving groups has an imino nitrogen that can be protonated, but the OAt ester has a larger surface area than methylimidazole, and thus offers more sites for hydrogen bonding, van der Waals interactions, and favorable dipole-dipole interactions. This may explain why the OAt ester strengthens binding more than MeIm in most cases (Tables 2 and 3). Because of the position of the leaving group in the complex of the monomer with the primer-template duplex and the good sequence selectivity observed with this leaving group (12), it is unlikely that the interactions stabilizing the complex involve binding of the oxyazabenzotriazolide in an intercalative mode. More likely, the additional interactions occur with the backbone or in one of the grooves. To unravel the contributions of each of these interactions, both for correctly paired and mismatched nucleotides, is a challenge that remains to be addressed.
In order to explain why OAt esters increase the yield of extension of aminoterminal primers, it is instructive to plot the ratio of the rates of the covalent step of extension (kcov) against that of hydrolysis (khydr). The more extension is accelerated over hydrolysis, the higher the yield of the reaction. So, the higher the ratio kcov/khydr, the more productive is the activated monomer in enzyme-free primer extension. Figure 8 shows that for deoxynucleotides reacting with aminoprimers, the ratio is more favorable for OAt ester than for methylimidazolides in each case (nucleobases A, C, G or T). The effect is most pronounced for dCMP (8.1-fold better ratio) and least significant for dGMP (1.6-fold better ratio). The OAt ester also gives a more even distribution of absolute reactivities over the four different bases, with kcov/khydr ratios differing by no more than a factor of 4.4 between the best and poorest base. On the other hand, the kcov/khydr ratio is less uniform for methylimidazolides, differing by up to a factor of 7, a finding that helps to explain why some combinations of template bases/monomers give poorer results with this leaving group (25, 40).
Figure 8.

Ratios of rate constants for the covalent step of extension (kcov) and hydrolysis (khydr) for different nucleotides and OAt or MeIm as leaving group. The larger the value, the more likely that a bound monomer will react with the primer, whereas a low ratio means that the given monomer is more likely to suffer hydrolysis. (A) kcov/khydr ratios for deoxynucleotide monomers reacting with aminoterminal primers, (B) kcov/khydr ratios for those ribonucleotides reacting with RNA primers for which reliable data is available. Note the much smaller absolute values compared to A).
Not surprisingly, the ratio kcov/khydr is less favorable in the case of ribonucleotides reacting with RNA primers (Figure 8B), as the nucleophile at the terminus of the primer is less reactive than in the case of the 3′-amines. The available values are close to unity, suggesting that the 2′,3′-diol is intrinsically not very different in its reactivity than the water nucleophiles at the given pH values, at least for the trajectories for nucleophilic attack that are accessible in the monomer-primer-template complexes. Interestingly, the differences in the kcov/khydr values for the different leaving groups and bases are also smaller than for deoxynucleotides and aminoterminal primers. Apparently, the OAt group gains over the methylimidazole mostly by improving binding (Tables 2 and 3), and not by reacting selectively with the primer terminus rather than the solvent.
How, then, does the OAt ester improve chemoselectivity for the amino group of primers, without accelerating the rate of the reaction with an oxygen nucleophile (hydrolysis) to the same extent? The pyridinic nitrogen of the six-membered ring of the benzotriazole may have a role as acid/base catalyst, as first proposed by Carpino in peptide couplings with OAt-activated amino acid building blocks (41). Figure 9 shows how a deprotonation of the ammonium form of the primer terminus can produce the amino group, thus making the primer competent as a nucleophile. The same mechanistic path will most probably not be accessible to a methylimidazolide, as the imidazole nitrogen is not positioned well for deprotonating the primer terminus. In either case, the protonated form can depart as a leaving group more readily than the neutral form that would have existed without protonation.
Figure 9.
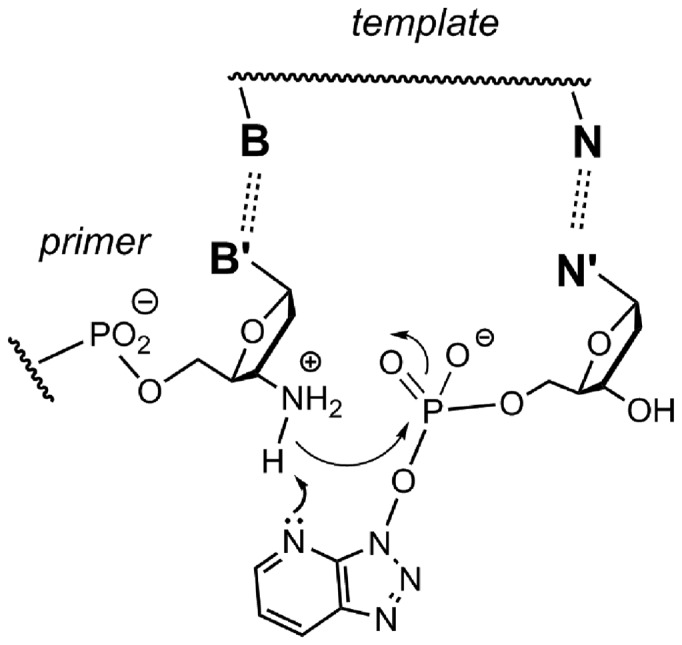
Mechanistic proposal for the role of the pyridinic nitrogen in promoting the formation of the kinetically competent form of the amino group at the terminus of a primer through deprotonation of the ammonium form.
The subtle differences in binding and reactivity between individual nucleobases may be a consequence of structural details of the monomer-primer-template complex. For example, the size and position of the nucleobase and the tightness of the base pair with the templating base may either position the phosphate of the monomer correctly for the nucleophilic attack of the primer, or may hold it in a less productive state. Once the nucleophilic attack has occurred, producing a pentavalent intermediate, some complexes may undergo pseudorotation more readily to place the leaving group in an apical position than others. Taken together, this may explain the differences in rate and yield of reactions induced by the four different base pairs and either of the two different leaving groups. The differences between reactions of deoxynucleotide monomers with aminoterminal primers and ribonucleotide monomers and RNA primers can be rationalized, if one remembers that in the latter case chemoselectivity will operate to a much smaller extent. Here, both the nucleophile at the primer terminus and the nucleophile causing hydrolysis are oxygen nucleophiles, though the 2′,3′-diol of the 3′-terminal residue is easier to deprotonate than water. So, the small differences in kcov/khydr compared to those of reactions with aminoprimers (Figure 8) are not unexpected.
With this quantitative picture of enzyme-free primer extension, it is interesting to step back and ask larger questions, such as what the likelihood is that enzyme-free copying may have driven replication in a prebiotic setting. Figure 10 shows plots of simulated time-yield relationships for increasing concentrations of monomers, calculated after 10 days, i.e. 8–15 times the half-life time of hydrolysis for OAt and MeIm monomers. To reach full conversion of the primer (≥99%), only 1.5 mM OAt-dGMP is needed, whereas 10 mM OAt-dTMP is required. These values are to be compared with the 150 mM concentration of MeIm-dTMP that is needed for achieving the same yield, according to this simulation. The corresponding calculation for ribonucleotides reacting with an RNA primer (Figure 10B) shows that an unrealistic concentration of LG-GMP would be required in the current sequence context to induce full conversion. No more than 84% conversion is reached at the end of the assay, even at 500 mM concentration of MeIm-GMP. For OAt-GMP a similar level of conversion is calculated within 10 days under the chosen reaction conditions. It should be remembered, though, that the kcov values for ribonucleotides are the result of fits to noisy data from low-yielding reactions. Still, it is clear that periodic removal of spent monomers (22) or the presence of microhelper strands (42) is required to induce quantitative conversion of RNA primers in unfavourable sequence contexts, even if very high monomer concentrations are used.
Figure 10.

Concentration dependence of yield. Simulated yields of primer extension after 10 days reaction time versus monomer concentration. (A) Reaction of OAt-dGMP on template 9c or OAt-dTMP or MeIm-dTMP on template 9a. The following values were used for the simulation: Kd (OAt-dGMP) 26 mM, Kdh (dGMP) 27 mM; kcov = 8.6 h−1, khydr = 0.093 h−1; Kd (OAt-dTMP) 71 mM, Kdh (TMP) 241 mM; kcov = 1.69 h−1, khydr = 0.044 h−1 and Kd (MeIm-TMP) 236 mM, Kdh (TMP) 241 mM; kcov = 0.35 h−1, khydr = 0.024 h−1. (B) Extension of RNA primer 13 with MeIm-GMP on template 12c, or OAt-GMP on template 12c. The values used for the simulation are Kd (MeIm-GMP) 23 mM, Kdh (GMP) 27 mM, kcov = 0.020 h−1 and khydr = 0.013 h−1; as well as Kd (OAt-GMP) 10 mM, kcov = 0.095 h−1 and khydr = 0.147 h−1. We note that neither of the two monomers is expected to yield full conversion, even at unrealistically high monomer concentration. Please also note that these are not kinetics, but concentration versus yield curves.
CONCLUSIONS
In conclusion, dissociation constants for activated nucleotides binding to their complementary template base are reported for the first time. Together with the rate constants for hydrolysis as well as that of the chemical step of primer extension, a good quantitative agreement between theory and experiments is achieved. This confirms that a set of four parameters is sufficient to describe enzyme-free primer extension reaction, namely the binding constants for activated and unactivated nucleotides and the rate constants for hydrolysis and the covalent step of primer extension. For successful primer extension, a monomer should bind tightly with leaving group, but less tightly as hydrolyzed, free nucleotide to avoid inhibition, and should show a large kcov and a small khydr value. Our study shows how this necessary set of data can be obtained and that for the current cases, these criteria are best met by OAt esters reacting with aminoterminal primers. While our quantitative description of enzyme-free copying steps explains why some reactions are successful and others stall after incomplete conversion, a more systematic search for higher-yielding extension of RNA primers is desirable, including studies on multiple extensions with their more complex kinetics. The current values are for one set of experimental conditions and no more than typical sequence contexts. Other monomer-primer-template combinations exist, and the reaction conditions, including temperature, pH and salt content of the buffer affect the outcome of enzyme-free extension reactions. Independent of what chemistry will ultimate succeed in inducing replication, future work should include quantitative data on the binding equilibria and reactivity of nucleotides with organic leaving groups, as well as data on fidelity (32,43). Overall, it is tempting to conclude that enzyme-free primer extension with the type of leaving groups and primers chosen is now understood, and that an experimental approach for quantitatively evaluating novel leaving groups has been established.
Supplementary Material
Acknowledgments
The authors thank Prof. Ulrich Steiner for discussions and Dr Birgit Claasen for help with the acquisition of NMR spectra.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft (DFG) [RI 1063/8-2 to CR]. Funding for open access charge: U. Stuttgart.
Conflict of interest statement. None declared.
REFERENCES
- 1.Kornberg A., Baker T.A. DNA Replication. 2nd edn. Mill Valley: University Science Books; 2005. [Google Scholar]
- 2.Inoue T., Orgel L.E. A nonenzymatic RNA polymerase model. Science. 1983;219:859–862. doi: 10.1126/science.6186026. [DOI] [PubMed] [Google Scholar]
- 3.Rojas Stütz J.A., Kervio E., Deck C., Richert C. Chemical primer extension - individual steps of spontaneous replication. Chem. Biodiv. 2007;4:784–802. doi: 10.1002/cbdv.200790064. [DOI] [PubMed] [Google Scholar]
- 4.Orgel L.E. Prebiotic chemistry and the origin of the RNA world. Crit. Rev. Biochem. Mol. Biol. 2004;39:99–123. doi: 10.1080/10409230490460765. [DOI] [PubMed] [Google Scholar]
- 5.Sulston J., Lohrmann R., Orgel L.E., Miles H.T. Nonenzymatic synthesis of oligoadenylates on a polyuridylic acid template. Prod. Natl. Acad. Sci. U.S.A. 1968;59:726–733. doi: 10.1073/pnas.59.3.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanavarioti A., Bernasconi C.F., Baird E.E. Effects of monomer and template concentration on the kinetics of nonenzymatic template-directed oligoguanylate synthesis. J. Am. Chem. Soc. 1998;120:8575–858. [Google Scholar]
- 7.Matteucci M.D., Caruthers M.H. Synthesis of deoxyoligonucleotides on a polymer support. J. Am. Chem. Soc. 1981;103:3185–3191. [PubMed] [Google Scholar]
- 8.Wu T., Orgel L.E. Nonenzymatic template-directed synthesis on hairpin oligonucleotides. Incorporation of adenosine and uridine residues. J. Am. Chem. Soc. 1992;114:7963–7969. doi: 10.1021/ja00047a001. [DOI] [PubMed] [Google Scholar]
- 9.Kozlov I.A., Orgel L.E. Nonenzymatic template-directed synthesis of RNA from monomers. Mol. Biol. 2000;34:781–789. [PubMed] [Google Scholar]
- 10.Hill A.R., Orgel L.E., Wu T.F. The Limits of Template-Directed Synthesis with Nucleoside-5′-Phosphoro(2-methyl)imidazolides. Orig. Life Evol. Biosphere. 1993;23:285–290. doi: 10.1007/BF01582078. [DOI] [PubMed] [Google Scholar]
- 11.Zielinski M., Kozlov I.A., Orgel L.E. A comparison of RNA with DNA in template-directed synthesis. Helv. Chim. Acta. 2000;83:1678–1684. doi: 10.1002/1522-2675(20000809)83:8<1678::AID-HLCA1678>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 12.Kervio E., Hochgesand A., Steiner U., Richert C. Templating efficiency of naked DNA. Proc. Natl. Acad. Sci. U.S.A. 2010;107:12074–12079. doi: 10.1073/pnas.0914872107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adelfinskaya O., Terrazas M., Froeyen M., Marlière P., Nauwelaerts K., Herdewijn P. Polymerase-catalyzed synthesis of DNA from phosphoramidate conjugates of deoxynucleotides and amino acids. Nucleic Acids Res. 2007;35:5060–5072. doi: 10.1093/nar/gkm498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herdewijn P., Marlière P. Redesigning the leaving group in nucleic acid polymerization. FEBS Lett. 2012;586:2049–2056. doi: 10.1016/j.febslet.2012.02.033. [DOI] [PubMed] [Google Scholar]
- 15.Lohrmann R., Orgel L.E. Preferential formation of (2′–5′)-linked internucleotide bonds in non-enzymatic reactions. Tetrahedron. 1978;34:853–855. [Google Scholar]
- 16.Joyce G.F., Inoue T., Orgel L.E. Non-enzymatic template-directed synthesis on RNA random copolymers: Poly(C,U) templates. J. Mol. Biol. 1984;176:279–286. doi: 10.1016/0022-2836(84)90425-x. [DOI] [PubMed] [Google Scholar]
- 17.Hagenbuch P., Kervio E., Hochgesand A., Plutowski U., Richert C. Chemical Primer Extension: Efficiently Determining Single Nucleotides in DNA. Angew. Chem. Int. Ed. 2005;44:6588–6592. doi: 10.1002/anie.200501794. [DOI] [PubMed] [Google Scholar]
- 18.Prabahar K.J., Ferris J.P. Adenine derivatives as phosphate-activating groups for the regioselective formation of 3′,5′-linked oligoadenylates on Montmorillonite: possible phosphate activating groups for the prebiotic synthesis of RNA. J. Am. Chem. Soc. 1997;119:4330–4337. doi: 10.1021/ja9700764. [DOI] [PubMed] [Google Scholar]
- 19.Röthlingshöfer M., Richert C. Chemical primer extension at submillimolar concentration of deoxynucleotides (featured article) J. Org. Chem. 2010;75:3945–3952. doi: 10.1021/jo1002467. [DOI] [PubMed] [Google Scholar]
- 20.Jauker M., Griesser H., Richert C. Copying of RNA sequences without pre-activation. Angew. Chem. Int. Ed. 2015;54:14559–14563. doi: 10.1002/anie.201506592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vogel H., Gerlach C., Richert C. Reactions of Buffers in Cyanogen Bromide-Induced Ligations. Nucleosides Nucleotides Nucl. Acids. 2013;32:17–27. doi: 10.1080/15257770.2012.744036. [DOI] [PubMed] [Google Scholar]
- 22.Deck C., Jauker M., Richert C. Efficient enzyme-free copying of all four nucleobases templated by immobilized RNA. Nature Chem. 2011;3:603–608. doi: 10.1038/nchem.1086. [DOI] [PubMed] [Google Scholar]
- 23.Lohrmann R., Orgel L.E. Template-directed synthesis of high molecular weight polynucleotide analogues. Nature. 1976;261:342–344. doi: 10.1038/261342a0. [DOI] [PubMed] [Google Scholar]
- 24.Röthlingshöfer M., Kervio E., Lommel T., Plutowski U., Hochgesand A., Richert C. Chemical primer extension in seconds. Angew. Chem. Int. Ed. 2008;47:6065–606. doi: 10.1002/anie.200801260. [DOI] [PubMed] [Google Scholar]
- 25.Rojas Stütz J.A., Richert C. A steroid cap adjusts the selectivity and accelerates the rates of non-enzymatic single nucleotide extensions of an oligonucleotide. J. Am. Chem. Soc. 2001;123:12718–12719. doi: 10.1021/ja011448i. [DOI] [PubMed] [Google Scholar]
- 26.Schrum J.P., Ricardo A., Krishnamurthy M., Blain J.C., Szostak J.W. Efficient and rapid template-directed nucleic acid copying using 2′-amino-2′,3′-dideoxyribonucleoside−5′-phosphorimidazolide monomers. J. Am. Chem. Soc. 2009;131:14560–14570. doi: 10.1021/ja906557v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang S., Zhang N., Blain J.C., Szostak J.W. Synthesis of N3′-P5′-linked phosphoramidate DNA by nonenzymatic template-directed primer extension. J. Am. Chem. Soc. 2013;135:924–932. doi: 10.1021/ja311164j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szostak J.W. The eightfold path to non-enzymatic RNA replication. J. Syst. Chem. 2012;3:2. [Google Scholar]
- 29.Griesang N., Giessler K., Lommel T., Richert C. Four color, enzyme-free interrogation of DNA sequences with chemically activated, 3′-fluorophore-labeled nucleotides. Angew. Chem. Int. Ed. 2006;45:6144–6148. doi: 10.1002/anie.200600804. [DOI] [PubMed] [Google Scholar]
- 30.Kervio E., Claasen B., Steiner U.E., Richert C. The strength of the template effect attracting nucleotides to naked DNA. Nucleic Acids Res. 2014;42:7409–7420. doi: 10.1093/nar/gku314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Izgu E.C., Fahrenbach A.C., Zhang N., Li L., Zhang W., Larsen A.T., Blain J.C., Szostak J.W. Uncovering the thermodynamics of monomer binding for rna replication. J. Am. Chem. Soc. 2015;137:6373–6382. doi: 10.1021/jacs.5b02707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leu K., Kervio E., Obermayer B., Turk-MacLeod R.M., Yuan C., Luevano J.-M., Jr, Chen E., Gerland U., Richert C., Chen I.A. Cascade of reduced speed and accuracy after errors in enzyme-free copying of nucleic acid sequences. J. Am. Chem. Soc. 2013;135:354–366. doi: 10.1021/ja3095558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogel S.R., Deck C., Richert C. Accelerating chemical replication steps of RNA involving activated ribonucleotides and downstream-binding elements. Chem. Comm. 2005:4922–4924. doi: 10.1039/b510775j. [DOI] [PubMed] [Google Scholar]
- 34.Eisenhuth R., Richert C. Convenient syntheses of 3′-amino-2′,3′-dideoxynucleosides, their 5′-monophosphates, and 3′-aminoterminal oligodeoxynucleotide primers. J. Org. Chem. 2009;74:26–37. doi: 10.1021/jo8018889. [DOI] [PubMed] [Google Scholar]
- 35.Sarracino D., Richert C. Quantitative MALDI-TOF MS of oligonucleotides and a nuclease assay. Bioorg. Med. Chem. Lett. 1996;6:2543–2548. [Google Scholar]
- 36.Rojas Stütz J.A., Richert C. Tuning the reaction site for enzyme-free primer extension reactions through small molecule substituents. Chem. Eur. J. 2006;12:2472–2481. doi: 10.1002/chem.200501008. [DOI] [PubMed] [Google Scholar]
- 37.Kanavarioti A., Chang S., Alberas D.J. Limiting concentrations of activated mononucleotides necessary for poly(C)-directed elongation of oligoguanylates. J. Mol. Evol. 1990;31:462–469. doi: 10.1007/BF02102072. [DOI] [PubMed] [Google Scholar]
- 38.Haug R., Kramer M., Richert C. Three-pronged probes: high-affinity DNA binding with cap, β-alanines and oligopyrrolamides. Chem. Eur. J. 2013;19:15822–15826. doi: 10.1002/chem.201302972. [DOI] [PubMed] [Google Scholar]
- 39.Inoue T., Orgel L.E. Oligomerization of (guanosin 5′-phosphor)-2-methylimidazolide on poly(C), an RNA polymerase model. J. Mol. Biol. 1982;162:201–217. doi: 10.1016/0022-2836(82)90169-3. [DOI] [PubMed] [Google Scholar]
- 40.Zhang S., Zhang N., Blain J.C., Szostak J.W. Synthesis of N3′-P5′-linked phosphoramidate DNA by nonenzymatic template-directed primer extension. J. Am. Chem. Soc. 2013;135:924–932. doi: 10.1021/ja311164j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carpino L.A. 1-Hydroxy-7-azabenzotriazole. An efficient peptide coupling additive. J. Am. Chem. Soc. 1993;115:4397–4398. [Google Scholar]
- 42.Vogel S.R., Richert C. Adenosine residues in the template do not block spontaneous replication steps of RNA. Chem. Commun. 2007:1896–1898. doi: 10.1039/b702768k. [DOI] [PubMed] [Google Scholar]
- 43.Rajamani S., Ichida J.K., Antal T., Treco D.A., Leu K., Nowak M.A., Szostak J.W., Chen I.A. Effect of stalling after mismatches on the error catastrophe in nonenzymatic nucleic acid replication. J. Am. Chem. Soc. 2010;132:5880–5885. doi: 10.1021/ja100780p. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.