


Abstract
The motility of MCF-7 cells increases following expression of a human PMR1 transgene and the current study sought to identify the molecular basis for this phenotypic change. Ensemble and single cell analyses show increased motility is dependent on the endonuclease activity of hPMR1, and cells expressing active but not inactive hPMR1 invade extracellular matrix. Nanostring profiling identified 14 microRNAs that are downregulated by hPMR1, including all five members of the miR-200 family and others that also regulate invasive growth. miR-200 levels increase following hPMR1 knockdown, and changes in miR-200 family microRNAs were matched by corresponding changes in miR-200 targets and reporter expression. PMR1 preferentially cleaves between UG dinucleotides within a consensus YUGR element when present in the unpaired loop of a stem–loop structure. This motif is present in the apical loop of precursors to most of the downregulated microRNAs, and hPMR1 targeting of pre-miRs was confirmed by their loss following induced expression and increase following hPMR1 knockdown. Introduction of miR-200c into hPMR1-expressing cells reduced motility and miR-200 target gene expression, confirming hPMR1 acts upstream of Dicer processing. These findings identify a new role for hPMR1 in the post-transcriptional regulation of microRNAs in breast cancer cells.
INTRODUCTION
PMR1 is an endoribonuclease that was originally identified by its role in catalyzing the destabilization of serum protein mRNAs in Xenopus (1). The subsequent purification (2) and cloning of PMR1 identified this RNA degradative enzyme as a product of the peroxidase gene family (3). PMR1 differs from the peroxidases in several important aspects, the most notable of which is the absence of covalently-bound heme. In PMR1 the histidine residues that would otherwise coordinate protoporphyrin-bound iron instead function as general acid and general base for RNA strand scission. Changing either or both histidines to alanine generates a catalytically inactive form of PMR1 (4).
Human PMR1 (hPMR1) is a 57 kDa protein that is expressed from an alternatively spliced form of peroxidasin homolog (Drosophila)-like protein (PXDNL) mRNA (5). PXDNL, also known as cardiac peroxidase, is a 164 kDa membrane-bound protein that is found predominately in heart and aorta. The 57 kDa hPMR1 protein is cytoplasmic, and it is the only form of PXDNL detectable in a number of cancer cell lines, including U2OS, K562, MCF-7 and MDA-MB-231. We previously showed that the motility of U2OS cells was increased following expression of Xenopus PMR1 from a tetracycline-inducible promoter (6), and similar results were seen for hPMR1 in MCF-7 breast cancer cells (5). MCF-7 cells are not particularly motile or invasive, but become both motile and invasive following suppression of miR-200 family microRNAs (7). The miR-200 family regulates a network of genes that control invasive growth of breast cancer cells (8,9), and we wondered if this had any relationship to hPMR1 simulation of motility. Up until now there have been no reports describing post-transcriptional regulation of miR-200. We show that the increased motility of hPMR1-expressing MCF-7 cells is associated with development of an invasive phenotype, that this is a function of hPMR1 catalytic activity, and that hPMR1 selectively reduces the levels of 14 microRNAs, notably those of the miR-200 family. hPMR1 acts upstream of Dicer processing by cleaving within a consensus sequence in the apical loop of the corresponding pre-miRs, and we show the impact of hPMR1 on cell motility is reversed by introduction of mature miR-200c. These findings provide the first evidence for hPMR1 regulating microRNAs and for post-transcriptional regulation of the miR-200 family of microRNAs.
MATERIALS AND METHODS
Cell culture
The creation of tetracycline-inducible lines of MCF-7 cells and cells knocked down for hPMR1 were described in (5). These were maintained in RPMI-1640 supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 1.0 mM sodium pyruvate, and 10 mM Hepes and 4.5 g/l glucose until 3 days before the start of each experiment. At that time they were shifted into estrogen-free medium to minimize the possible impact of this hormone. This consisted of phenol red-free RPMI-1640 containing the same supplements plus 1% ITS-G (insulin, transferrin, selenium, Invitrogen), and charcoal-stripped FBS. hPMR1 induction was achieved by adding 100 or 400 ng/ml doxycycline to the medium at the indicated times. siRNA knockdowns were performed as described previously (5).
Preparation of cytoplasmic extracts for protein and RNA analysis
Cytoplasmic extracts were prepared as described previously (5). Briefly, cells were harvested with trypsin, washed with ice-cold PBS and transferred into sterile 12 × 75mm polystyrene tube. Cells were collected by centrifugation for 1 min at 50 × g and resuspended in 100 μl CER I from the NE-PER kit (Pierce). Cytoplasmic extracts were prepared followed the manufacturer's protocol except that gentle mixing was used in place of vortexing. RNA used for Nanostring microRNA profiling and Taqman assays was recovered from these extracts using the Norgen microRNA purification kit (small fraction) and RNA for RT-qPCR of mRNA targets was recovered using the large fraction of the Norgen microRNA purification kit or the Norgen total RNA purification kit.
Analysis of hPMR1-mediated changes in microRNAs, pre-miRs and target mRNAs
TaqMan probes for RNU48, miR-200a, miR-200c, miR-26a and miR-26b were purchased from Applied Biosystems. cDNA was synthesized using the TaqMan microRNA RT kit (Applied Biosystems), and 1 μl aliquots of each product were added to 10 μl reactions containing TaqMan Universal Master mix with uracil N-glycosidase (UNG). PCR reactions were incubated at 50°C for 2 min, then for 45 cycles at 95°C for 15 s, 60°C for 1 min). RT-qPCR for pre-miR-26a, 200a and 200c was performed using the miScript precursor assay (Qiagen) consisting of primers specific to each RNA. cDNA was prepared from cytoplasmic RNA using the miScript II RT Kit (Qiagen) and two-step RT-qPCR was carried out with QuantiTect SYBR Green PCR Kit (Qiagen). PCR reactions were incubated for 45 cycles at 94°C for 15 s, 60°C for 30 s and 72°C for 30 s. Results were normalized as above to Taqman assay for RNU48.
ZEB1, ZEB2, FHOD1 and PPM1F and hPMR1 were assayed by RT-qPCR and results were normalized to RPLP0 using primer sets in Supplementary Table S1. cDNA was synthesized with SuperScript III reverse transcriptase (Invitrogen) following the manufacturer's protocol. One microliter of each product was used as template in 10 μl qPCR reactions performed with SensiFAST SYBR® No-Rox mix (Bioline). All reactions were performed with biological triplicates. qPCR was performed using Eco Real-Time PCR System (Illumina). Reactions were incubated at 95°C for 3 min, then for 40 cycles at two step-cycling (95°C for 10 s, 60°C for 20 s), and melting curves were used to confirm the specificity of each amplification product. To monitor functional changes in miR-200 the 222 bp FHOD1 3′-UTR, which has a single miR-200b/c binding site, was cloned into the Xba1 site of pGL3-control firefly luciferase vector. This was cotransfected with plasmid expressing Renilla luciferase (pRL-TK), doxycycline was added to half of the cultures and activity was measured 24 h later using the Dual Luciferase assay (Promega).
NanoString microRNA profiling
Nanostring profiling was performed with the nCounter human v2 microRNA expression assay using total RNA from duplicate cultures of MCF-7 cells in which hPMR1 transgene was uninduced or induced for 24 h. Raw read data from the nCounter microRNA assay were analyzed using the methods described in the nCounter Expression Data Analysis Guide (NanoString Technologies). First, the geometric mean for positive control counts across all lanes was divided by the geometric mean for positive control counts in each lane to obtain a lane specific technical scaling factor (TSF). Counts for each microRNA in a lane were multiplied by the TSF to obtain technically normalized counts that are corrected for variation in counting efficiency in each lane. Next, the counts of all endogenous microRNAs across all lanes were averaged and divided by the counts of all endogenous microRNAs for each specific lane to obtain a global normalization factor (GNF). Technically normalized counts for each microRNA were multiplied by the GNF to obtain globally normalized counts that are corrected for variations in purity or amount of input sample. After normalization, the average counts for negative control probes in each lane were averaged and subtracted from counts of each microRNA to correct for background. Those microRNAs with average counts across all four samples less than 3-fold above background were excluded from further analysis in order to minimize changes due to noise. For each microRNA, average counts for duplicate samples from doxycycline-treated cells were divided by average counts for duplicate untreated samples to identify which microRNAs are up- or down-regulated upon hPMR1 expression.
Antisera and Western blotting
Western blotting was performed as described in (10). A rabbit polyclonal antiserum to hPMR1 was prepared by New England Peptide using peptide Ac-CRESQALRDPSVP-amide (hPMR1 104–115). Prior to use this was affinity purified against immobilized peptide and qualified by western blotting against endogenous and recombinant protein. Mouse monoclonal anti-FLAG antibody was obtained from Sigma. Secondary antibodies consisted of AlexaFluor®680-coupled goat anti-mouse IgG and AlexaFluor®790-coupled goat anti-rabbit IgG (Jackson Immunoresearch).
Ensemble motility assay
Ensemble motility experiments were performed as described in (5). Briefly, 3 × 105 cells were inoculated into each well of a six-well dish and cultured as described above. Cells received mitomycin C (0.4 μg/ml) to block replication and the semi-confluent monolayer was scored with a sterile 1000 μl blue pipette tip. The plates were washed three times with phosphate-buffered saline (PBS) followed by medium ± doxycycline. Motility was determined by photographing three fields in each of triplicate cultures over time using a Nikon Eclipse Ti inverted microscope equipped with an XY encoded stage. The distances across each gap were determined using T-scratch motility image analysis software (11), and the percent of the gap closed over time was calculated using the formula: 100 × (1 – wt-x/wt-0). The results were plotted as mean ± standard deviation. The same approach was used to determine the impact of miR-200c complementation on cell motility except that cells were transfected 24 h prior to scoring with 50 nM miR-200c mimic or control miR (miR-C2, miRIDIAN microRNA Mimic Negative Control #2, Thermo) using Lipofectamine RNAimax transfection reagent. Efficient transfection was confirmed by monitoring nuclear fluorescence of a parallel set of cultures that were transfected with the same miRs plus 5 nmol siGLO green transfection indicator (Thermo).
Single cell motility assay
Biomimetic micropatterned polydimethylsiloxane (PDMS) surfaces designed to resemble structural extracellular matrix were fabricated through a replica-molding process from a photolithographically-patterned Si master (Supplementary Figure S1) (12). Briefly, an array of 2 × 1.5 μm (width × height) lines (spaced by 2 μm) was patterned in S1813 photoresist on a Si wafer using standard contact photolithography. Then, a 10:1 mixture of PDMS:curing agent (Sylgard 184, Dow Corning) was poured over the patterned Si surface, degassed, and allowed to cure at room temperature for 48–72 h. The patterned PDMS substrate was subsequently peeled off of the Si surface and cut into 12 mm diameter circles for the single cell motility assays. These substrates were sterilized in 70% ethanol, and to insure optimal attachment were pre-incubated for 2 days in medium containing 25% FBS. Cells were allowed to attach for 2 days, after which the medium was changed with half of the treatment groups receiving 400 ng/ml doxycycline in the medium. Statistical analysis was performed using the Kruskal-Wallis and Fisher LSD Methods.
Matrix invasion assay
Invasive growth was determined using 24-well ThinCert™ cell culture inserts with translucent PET membranes and 8 μm pores (Gibco) that were coated with Biomatrix® extracellular matrix from Engelbreth–Holm–Swarm mouse tumors (Sigma). Six hundred microliters of estrogen-free medium was added to the bottom chamber of a 24-well plate, and half of the wells also had 400 ng/ml doxycycline to induce hPMR1 expression. Two hundred microliters of a suspension of 106 cells/ml was added to each of the upper chamber of matrix-coated inserts, which were then placed into the 24-well plate. To control for matrix-dependent differences in invasion the experiment was performed in parallel with inserts that had not been coated with extracellular matrix. After 30 h, the medium in the bottom compartment was replaced with 450 μl of medium containing 8 μM Calcein AM (Life Technologies) and the plates were returned to the incubator for an additional 45 min. Medium in the upper chamber was removed, the inserts were transferred into 24-well plates containing 500 μl trypsin–EDTA per well, and the plates were again returned to the cell culture incubator for 10 min with occasional agitation. 200 μl of the trypsin–EDTA solution was then transferred to individual wells of a flat-bottom black 96-well plate and fluorescence was determined using a fluorescence plate reader (Tecan) with excitation at 485 nm and emission at 520 nm. Invasion index was calculated according to the formula: (RFU1-RFU0)/(RFU2-RFU0), where RFU1 represents the relative fluorescence units obtained from cells that migrated through an ECM coated membrane towards lower chamber with doxycycline, RFU2 denotes the relative fluorescence units obtained from cells that migrated through an uncoated membrane towards lower chamber in the presence of doxycycline, and RFU0 signifies the relative fluorescence units obtained from cells that passed through an uncoated membrane in the absence of doxycycline.
RESULTS
hPMR1 stimulation of cell motility is a function of its catalytic activity
Previous work described the creation of lines of MCF-7 cells stably expressing tetracycline-inducible forms of hPMR1 with an N-terminal FLAG tag (5). MCF-7 cells natively express hPMR1, and to minimize the potential for artifacts resulting from overexpressing an active endonuclease the concentration of doxycycline used to induce the hPMR1 transgene was adjusted to yield a level of FLAG-hPMR1 that was 1.5–2-fold that of the endogenous protein. An ensemble assay was used to confirm that this level of induction was sufficient to increase motility (Figure 1A). In this experiment a semiconfluent monolayer was scored, doxycycline was added on one-half of the cultures, and motility was determined by photographing the same fields at intervals over the next 24 h. As in our previous work cell motility increased ∼2-fold following induction of the hPMR1 transgene.
Figure 1.

Increased motility of MCF-7 cells following induced expression of catalytically active hPMR1. (A) Semi-confluent monolayers of cells were treated with mitomycin C to block DNA replication and scored prior to addition of doxycycline to one-half of the cultures. After washing to remove non-adherent cells multiple fields of three individual wells were photographed using an inverted microscope equipped with an XY encoded stage. Results are plotted as the percent of the gap closed over time and represent the mean ± standard deviation (n = 9). B) Cells carrying catalytically active hPMR1 transgene were plated onto biomimetic micropatterned polydimethylsiloxane (PDMS) surfaces designed to resemble structural extracellular matrix. Two days later the medium was changed, half of the matrices were treated with doxycycline and the movement of 20 individual cells was tracked under phase contrast with a 10× objective lens for approximately 16 h using an inverted microscope fitted with a 37°C cell culture chamber. Images were collected every 10 min and analyzed using the manual tracker plugin in Fiji. (C) The ensemble motility experiment in (A) was repeated using a parallel line of MCF-7 cells that was stably transfected with a catalytically-inactive hPMR1 transgene. (D) Single cell analysis of cells carrying a catalytically-inactive hPMR1 transgene was performed as in (B).
We next turned to single cell analysis to obtain a more quantitative picture of the impact of hPMR1 on cell motility. Cells carrying the hPMR1 transgene were cultured on matrices consisting of an array of lithographically-generated 2 μm wide tracks (12). These tracks mimic the remodeled extracellular matrix structure associated with invasive cancers (12–16). Half of the cultures received doxycycline to induce hPMR1 and 20 cells on each matrix were then continuously monitored by phase contrast microscopy for 16 h (Figure 1B, Supplementary Movie S1.mp4). Our results indicate that compared to control cells, hPMR1 induction led to an overall increase in directional single-clone motility, with velocities of ≥40 μm/h versus ≥30 μm/h for the uninduced cells.
To determine if the increase in motility upon induction of hPMR1 is due to its activity as an endoribonuclease or some other feature of the protein we examined a parallel line of MCF-7 cells that carries a tetracycline-inducible hPMR1 transgene in which the two active site histidines were changed to alanines to generate a protein that lacks endonuclease activity (17). Although the basal motility of this particular cell line is higher than of the cell line carrying active hPMR1 expression of catalytically-inactive hPMR1 had no impact on collective motility (Figure 1C), or on directional velocity as determined by movement of single cells on the microtextured PDMS substrates (Figure 1D). We conclude that the hPMR1-mediated increase in cell motility is a consequence of its activity as an endoribonuclease.
MCF-7 cells become invasive upon induced expression of hPMR1
Because MCF-7 cells have limited ability to invade extracellular matrix in vitro or into the tissue surrounding mouse xenografts (18) we asked whether the observed increase in motility might also indicate the acquisition of an invasive phenotype. In the experiment in Figure 2 cells with active and inactive hPMR1 transgenes were placed in the upper portion of an invasion chamber. In half of the cultures doxycycline was added to the lower chamber and invasion was determined by Calcein AM staining of cells that moved through extracellular matrix and accumulated on the underside of the membrane. The invasion index for cells expressing catalytically active hPMR1 was three times greater than that of uninduced cells or cells expressing catalytically-inactive hPMR1. Together with the results in Figure 1 these findings indicate that MCF-7 cells acquire a more motile and invasive phenotype in response to an increase in hPMR1.
Figure 2.
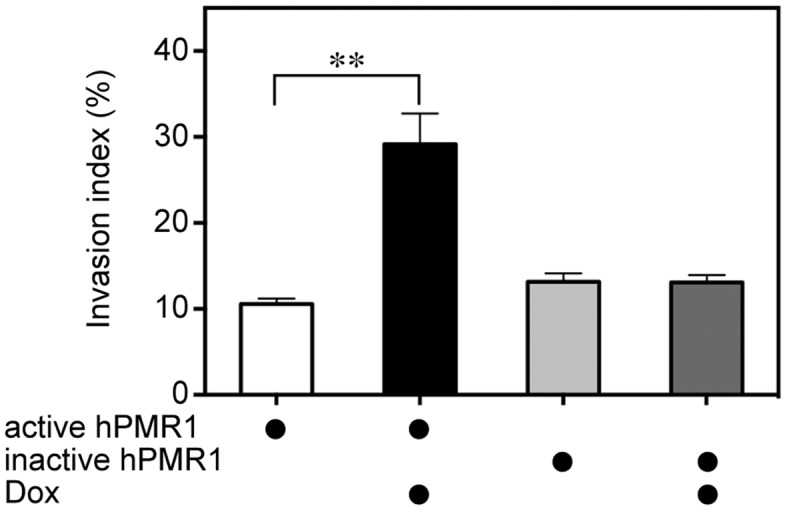
Elevated expression of hPMR1 converts MCF-7 cells from non-invasive to invasive growth in extracellular matrix. MCF-7 cells carrying active and inactive hPMR1 transgenes were grown on cell culture inserts containing membranes coated with extracellular matrix. These were placed into 6-well dishes containing medium ± doxycycline and cultured for 30 hr, after which cells that migrated through the membrane were stained with Calcein-AM. An invasion index was determined by comparing the fluorescence intensity after trypsin/EDTA treatment of cells that migrated through coated membranes versus uncoated membranes. Shown is the mean ± standard deviation (n = 6). The double asterisk (∗∗) indicates P < 0.001 by Student's t-test. There was no statistically significant difference between treatment groups for cells expressing inactive hPMR1.
hPMR1 suppresses microRNAs that regulate a network controlling motility and invasiveness
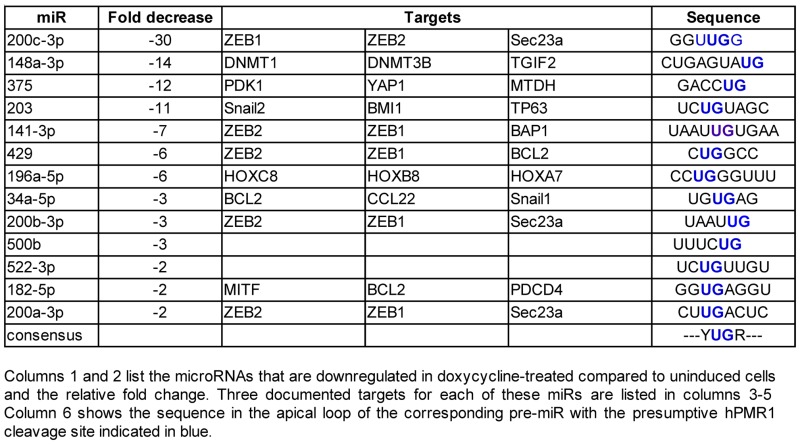
The preceding results are consistent with changes in breast cancer cells seen with loss of the miR-200 family of microRNAs (8,9,19). Nanostring profiling of cytoplasmic RNA from hPMR1-expressing cells identified 14 miRs that were downregulated at least 2-fold compared to uninduced controls (Table 1), including all five members of the miR-200 family. This was confirmed for miR-200a and miR-200c by Taqman assay (Figure 3A). In addition, hPMR1 had little impact on miR-26a and miR-26b, neither of which were shown by Nanostring to be downregulated by hPMR1. Targets of the miR-200 family constitute a network controlling cell motility and invasion (8,9) that includes the ZEB1 and 2 transcriptional repressors (19–21) and the cytoskeleton regulators FHOD1 and PPM1F (22). Results in Figure 3B show that induction of active hPMR1 transgene results in increased steady-state levels of ZEB1, ZEB2, FHOD1 and PPM1F mRNA. The increase in steady-state levels of these mRNAs changes with the concentration of doxycycline used to induce hPMR1 (Supplementary Figure S2) and hence the level of hPMR1. Functional evidence for hPMR1 regulation of miR-200 was obtained using a firefly luciferase reporter assay. FHOD1 mRNA has a single 3′-UTR miR-200b/c binding site, and in Figure 3C cells carrying the active hPMR1 transgene were transfected with a plasmids expressing firefly luciferase fused to the FHOD1 3′-UTR and Renilla luciferase. Reporter gene expression was 7 times higher in hPMR1 expressing cells than in the uninduced control, thus confirming the functional impact of hPMR1 expression on miR-200.
Table 1. Nanostring identification of downregulated microRNAs.
|
Figure 3.

Complementary changes in miR-200 and miR-200 target mRNAs following hPMR1 induction or knockdown. (A) Cytoplasmic RNA from uninduced and hPMR1-expressing cells was analyzed by Taqman assay for miRs-200a and 200c and two unaffected miRs (miRs-26a and 26b). Results are the mean ± standard deviation (n = 6). (B) RNA from triplicate cultures of uninduced and hPMR1-expressing cells was analyzed by RT-qPCR for ZEB1, ZEB2, PPM1F and FHOD1 mRNA. Results represent the mean ± standard deviation (n = 3). The double asterisks (∗∗) indicate P < 0.001 by Student's t-test. (C) Cells carrying the inducible hPMR1 transgene were transfected with plasmids expressing firefly luciferase fused to the FHOD1 3′-UTR and Renilla luciferase. The next day half of the cultures were induced with doxycycline and luciferase activity was determined 24 h later. Results represent the mean ± standard deviation (n = 7). (D) Cytoplasmic extracts from non-transduced MCF-7 cells and cells transduced with lentivirus expressing control or hPMR1 shRNA were analyzed by Western blotting with anti-hPMR1 antibody (upper panel) or antibody to GAPDH (lower panel). Changes in hPMR1 were normalized to GAPDH by LiCor. (E) Cytoplasmic RNA from cells expressing control or hPMR1 shRNAs was analyzed by Taqman assay as in (A) for miRs-200a, 200c, 26a and 26b. (F) Parental MCF-7 cells were transfected with control or hPMR1 siRNAs and analyzed as in (A) for changes in miR-26a and miR-200c. Results represent the mean ± standard deviation (n = 3). The relative extent of knockdown was determined by Western blotting for hPMR1 and GAPDH (lower panels) and normalized results were determined by LiCor.
RNA interference was used to determine if miR-200 microRNAs are natively regulated by endogenous hPMR1. The parental line of MCF-7 cells was transduced with lentiviral vectors expressing a control shRNA or an shRNA targeting hPMR1. Western blotting with anti-hPMR1 antibody indicated this reduced the level of hPMR1 to 30% of control (Figure 3D). Taqman assays performed as in Figure 3A showed hPMR1 knockdown is accompanied by an 8-fold increase in miR-200a and a 15-fold increase in miR-200c (Figure 3E) but had little impact on miR-26a or miR-26b. In keeping with the observed changes in miR-200 family microRNAs the levels of ZEB1 and ZEB2 mRNA were lower in cells knocked down for hPMR1 (Supplementary Figure S3B). These findings were confirmed by transient knockdown with hPMR1 siRNAs (5) (Figure 3F), which resulted in a 5-fold increase in miR-200c. To determine the generality of the preceding findings to other members of the miR-200 family we examined the impact of hPMR1 induction and knockdown on miR-141 (Supplementary Figure S3A). Although there is substantially less of this microRNA in MCF-7 cells (23) it too declined (5-fold) following induction of active hPMR1 and increased 5-fold following hPMR1 knockdown. Taken together the preceding results indicate that miR-200 family microRNAs are natively regulated by hPMR1.
hPMR1 targets the precursors to miR-200 microRNAs
Given their small size and protection within the RISC complex it seemed unlikely that mature microRNAs were the proximal targets of hPMR1. We previously showed that PMR1 preferentially cleaves between UG dinucleotides when these are present in a consensus YUGR motif in the apical loop of a stem–loop structure (24). This motif is present in the apical loop of the pre-miRs for 12 of the microRNAs in Table 1 and is shown for pre-miR-200c in Supplementary Figure S4A. There is precedent in (25,26) for endonuclease-mediated downregulation of select microRNAs by cleavage within the apical loop of their corresponding pre-miRs. To determine if hPMR1 targets precursors to the miR-200 family we looked first at the impact of inducing active and inactive forms of the enzyme on levels of pre-miR-200a and 200c. Expression of active hPMR1 resulted in a 2-fold decrease in pre-miR-200a and an almost 6-fold decrease in pre-miR-200c (Figure 4A). Each of these pre-miRs was unaffected by induction of catalytically inactive hPMR1 (Figure 4B), thus indicating that hPMR1-mediated downregulation of pre-miRs-200a and 200c is a function of its endonuclease activity. These findings were complemented by results with cells knocked down for hPMR1, where loss of hPMR1 resulted in an 80% increase in both pre-miR-200a and pre-miR-200c (Figure 4C). Support for hPMR1 targeting the apical loop is shown in Supplementary Figure S4, where hPMR1 recovered from MCF-7 cells generated a product of the size expected for cleavage at that location when incubated in vitro with pre-miR-200c. Previous work showed that PMR1 does not cleave RNA when U or G in the YUGR element were changed to A or C, respectively (24). Pre-miR-200c with each of these changes (U34A, G35C) was refractory to in vitro cleavage, thus confirming that hPMR1 cleaves within the apical loop of pre-miR-200c.
Figure 4.

hPMR1 downregulates pre-miR-200a and 200c. (A) Cytoplasmic RNA from untreated and doxycycline-treated cells carrying the active hPMR1 transgene was analyzed by RT-qPCR for pre-miR-26a, pre-miR-200a and pre-miR-200c. (B) The experiment in A was repeated using cytoplasmic RNA from cells carrying the catalytically-inactive hPMR1 transgene. (C) RT-qPCR for pre-miR-26a, pre-miR-200a and pre-miR-200c was performed using cytoplasmic RNA from cells that were transduced with lentivirus expressing control (Ctrl) shRNA or shRNA targeting hPMR1. In each case the results represent the mean ± standard deviation (n = 3), and P values by Student's t-test for each determination having statistically significant differences are shown above the respective datasets.
Downregulation of miR-200 is responsible for the PMR1-mediated increase in cell motility and target gene expression
The preceding results point to hPMR1 acting upstream of Dicer processing. If that is correct, the introduction of mature miR-200 into hPMR1-expressing cells should reverse the impact of hPMR1 on cell motility and target gene expression. In the experiment in Figure 5 cells carrying the active hPMR1 transgene were transfected with miR-200c or a control miR (miR-C2) prior to adding doxycycline. Cells that received control miR (miR-C2) showed the expected hPMR1 stimulation of motility whereas no increase in motility was observed for cells that received miR-200c (Figure 5A).
Figure 5.

miR-200c reverses hPMR1 stimulation of motility and increases in miR-200 target gene expression. MCF-7 cells carrying the active hPMR1 transgene were transfected with miR-200c or a control miR (miR-C2) 24 hr prior to addition of doxycycline to half of the cultures. (A) The matrix was scored at the time of doxycycline treatment and motility was monitored as in Figure 1B. (B) Cytoplasmic RNA recovered from a parallel set of cultures was analyzed as in Figure 3B by RT-qPCR for ZEB1, ZEB2, PPM1F and FHOD1 mRNA. The results represent the mean ± standard deviation (n = 3). The double asterisks (∗∗) indicate P < 0.001 by Student's t-test.
These results were complemented by changes in ZEB1, ZEB2, FHOD1 and PPM1F mRNA (Figure 5B), where the hPMR1-mediated increase of each mRNA was reversed by the introduction of mature miR-200c into hPMR1-expressing cells. Levels of ZEB1, FHOD1 and PPM1F were reduced below that of uninduced controls, and although ZEB2 mRNA induction was reversed, it was unexpectedly higher in uninduced cells that received miR-200c compared to uninduced cells that received control microRNA. The reason for this observation is not known. Taken together, these data show hPMR1 acts upstream of Dicer to reduce levels of miR-200 family microRNAs by cleaving within the apical loop of their corresponding pre-miRs, and that loss of miR-200 microRNAs in these cells is responsible for hPMR1 stimulation of motility and invasiveness.
DISCUSSION
A number of post-transcriptional mechanisms for down-regulating miRs have been identified (27,28), and while some of these directly affect the decay of mature miRs (29,30), others target their precursors (31–35). To date there are two published reports describing endonuclease cleavage within the apical loop of pre-miRs as a mechanism for downregulating steady-state levels of the corresponding microRNAs. MCPIP1 (also called Zc3h12a or regnase-1, 36) downregulates a number of microRNAs in lung cancer (25), and IRE1 downregulates a different group of microRNAs during ER stress (26). In each of these examples, cleavage within the apical loop reduced availability of pre-miR for Dicer processing into the mature microRNA. Results presented here identify hPMR1 as the third example of an endonuclease regulating microRNA levels by cleaving within the apical loop of the corresponding pre-miR, and the first such enzyme shown to do so in breast cancer cells. In vivo complementation experiments in which introduction of mature miR-200c reversed the hPMR1-mediated increase in motility and target gene expression provided additional evidence for hPMR1 acting upstream of Dicer.
Human PMR1 is the product of an alternatively spliced form of peroxidasin-like protein (PXDNL, 5). Data in the Oncomine™ database show PXDNL transcripts are elevated in a limited number of cancers, most notably invasive breast cancers, where the level is ∼75% higher than that of normal tissue. This was intriguing because breast cancer cells become more motile and invasive when miR-200 family microRNAs are suppressed (7,20–22,37), and they acquire these properties when hPMR1 was expressed from an inducible transgene. Thus, our identification of miR-200 family of microRNAs as targets of hPMR1 regulation is consistent with this well-established paradigm. The fact that these changes were only seen with catalytically active enzyme also meant that these RNAs or their precursors were the most likely proximal targets of hPMR1. Induced expression of active (but not inactive) hPMR1 also resulted in increased expression the well characterized miR-200 targets ZEB1, ZEB2, FHOD1 and PPM1F. Of equal importance were results from knockdown experiments that showed the miR-200 family microRNAs are natively regulated by hPMR1. Because MCF-7 cells are not particularly motile it was not surprising that loss of hPMR1 had no discernable impact on motility.
Increased motility is a characteristic feature of invasive cancers (38) and recent modeling studies highlighted the importance of even small increases in motility in the formation, growth and evolution of tumors (39). While it is premature to speculate whether the results presented here extend beyond cells in culture, these findings identify a new avenue for investigating the involvement of post-transcriptional processes in cancer.
Supplementary Material
Acknowledgments
We wish to thank Kay Huebner for her helpful comments. Nanostring microRNA profiling was performed in the Genomics Shared Resource of the Ohio State University Comprehensive Cancer Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of The Ohio State University or the National Institutes of Health.
Contributions: S.G.: Performed all of the experiments except for single cell motility experiments, the initial identification of miR-200 as a PMR1 target, and provided RNA for Nanostring analysis. D.G.P.: Fabricated microtextured surfaces, performed single-cell analysis experiments and analyzed single-cell motility data. S.M.: Performed analysis of Nanostring data to identify miRs that are downregulated by hPMR1. J.S.: Performed single-cell analysis experiments and analyzed single-cell motility data. J.H.: Made the initial observation that miR-200c is downregulated by hPMR1, performed Taqman and luciferase activity assays, generated luciferase reporter constructs and analyzed hPMR1-mediated changes in FHOD1 via miR-200c. L.J.L.: Provided guidance (e.g. experimental design, data interpretation) and support for the single-cell motility experiments. D.R.S.: Wrote the manuscript and generated all of the figures except for single cell motility experiments.
Footnotes
Present addresses:
Sean P. McClory, Department of Biochemistry and Biophysics, University of Pennsylvania, USA.
Joonhee Han, MeiraGTx Limited, 450 East 29th Street, New York, NY 10016, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [GM038277 and GM084177 to D.R.S.]; National Science Foundation [NSEC EEC-0914790 to L.J.L.]; Ohio State University College of Medicine. Funding for Shared Resources comes from the OSUCCC and the National Cancer Institute [CA016058] to the Ohio State University Comprehensive Cancer. Funding for open access charge: NIH [GM084177].
Conflict of interest statement. None declared.
REFERENCES
- 1.Pastori R.L., Moskaitis J.E., Schoenberg D.R. Estrogen-induced ribonuclease activity in Xenopus liver. Biochemistry. 1991;30:10490–10498. doi: 10.1021/bi00107a018. [DOI] [PubMed] [Google Scholar]
- 2.Dompenciel R.E., Garnepudi V.R., Schoenberg D.R. Purification and characterization of an estrogen-regulated Xenopus liver polysomal nuclease involved in the selective destabilization of albumin mRNA. J. Biol. Chem. 1995;270:6108–6118. doi: 10.1074/jbc.270.11.6108. [DOI] [PubMed] [Google Scholar]
- 3.Chernokalskaya E., DuBell A.N., Cunningham K.S., Hanson M.N., Dompenciel R.E., Schoenberg D.R. A polysomal ribonuclease involved in the destabilization of albumin mRNA is a novel member of the peroxidase gene family. RNA. 1998;4:1537–1548. doi: 10.1017/s1355838298980451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cunningham K.S., Hanson M.N., Schoenberg D.R. Polysomal ribonuclease 1 exists in a latent form on polysomes prior to estrogen activation of mRNA decay. Nucleic Acids Res. 2001;29:1156–1162. doi: 10.1093/nar/29.5.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gu S.Q., Bakthavachalu B., Han J., Patil D.P., Otsuka Y., Guda C., Schoenberg D.R. Identification of the human PMR1 mRNA endonuclease as an alternatively processed product of the gene for peroxidasin-like protein. RNA. 2012;18:1186–1196. doi: 10.1261/rna.031369.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng Y., Murray E.L., Sarkar M., Liu X., Schoenberg D.R. The cytoskeleton-associated Ena/VASP proteins are unanticipated partners of the PMR1 mRNA endonuclease. RNA. 2009;15:576–587. doi: 10.1261/rna.1206209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song S.J., Poliseno L., Song M.S., Ala U., Webster K., Ng C., Beringer G., Brikbak N.J., Yuan X., Cantley L.C., Richardson A.L., Pandolfi P. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell. 2013;154:311–324. doi: 10.1016/j.cell.2013.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bracken C.P., Li X., Wright J.A., Lawrence D.M., Pillman K.A., Salmanidis M., Anderson M.A., Dredge B.K., Gregory P.A., Tsykin A., et al. Genome-wide identification of miR-200 targets reveals a regulatory network controlling cell invasion. EMBO J. 2014;33:2040–2056. doi: 10.15252/embj.201488641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perdigão-Henriques R., Petrocca F., Altschuler G., Thomas M.P., Le M.T., Tan S.M., Hide W., Lieberman J. miR-200 promotes the mesenchymal to epithelial transition by suppressing multiple members of the Zeb2 and Snail1 transcriptional repressor complexes. Oncogene. 2016;35:158–172. doi: 10.1038/onc.2015.69. [DOI] [PubMed] [Google Scholar]
- 10.Mukherjee C., Bakthavachalu B., Schoenberg D.R. The cytoplasmic capping complex assembles on adapter protein NCK1 bound to the proline-rich C-terminus of mammalian capping enzyme. PLoS Biol. 2014;12:e1001933. doi: 10.1371/journal.pbio.1001933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geback T., Schulz M.M., Koumoutsakos P., Detmar M. TScratch: a novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques. 2009;46:265–274. doi: 10.2144/000113083. [DOI] [PubMed] [Google Scholar]
- 12.Gallego-Perez D., Higuita-Castro N., Denning L., DeJesus J., Dahl K., Sarkar A., Hansford D.J. Microfabricated mimics of in vivo structural cues for the study of guided tumor cell migration. Lab Chip. 2012;12:4424–4432. doi: 10.1039/c2lc40726d. [DOI] [PubMed] [Google Scholar]
- 13.Doyle A.D., Wang F.W., Matsumoto K., Yamada K.M. One-dimensional topography underlies three-dimensional fibrillar cell migration. J. Cell Biol. 2009;184:481–490. doi: 10.1083/jcb.200810041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petrie R.J., Doyle A.D., Yamada K.M. Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell. Biol. 2009;10:538–549. doi: 10.1038/nrm2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson J., Nowicki M.O., Lee C.H., Chiocca E.A., Viapiano M.S., Lawler S.E., Lannutti J.J. Quantitative analysis of complex glioma cell migration on electrospun polycaprolactone using time-lapse microscopy. Tissue Eng. Part C Methods. 2009;15:531–540. doi: 10.1089/ten.tec.2008.0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fei Z., Wu Y., Sharma S., Gallego-Perez D., Higuita-Castro N., Hansford D., Lannutti J.J., Lee L.J. Gene delivery to cultured embryonic stem cells using nanofiber-based sandwich electroporation. Anal. Chem. 2013;85:1401–1407. doi: 10.1021/ac302140p. [DOI] [PubMed] [Google Scholar]
- 17.Yang F., Schoenberg D.R. Endonuclease-mediated mRNA decay involves the selective targeting of PMR1 to polyribosome-bound substrate mRNA. Mol. Cell. 2004;14:435–445. doi: 10.1016/j.molcel.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 18.Holliday D.L., Speirs V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011;13:215. doi: 10.1186/bcr2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korpal M., Lee E.S., Hu G., Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 2008;283:14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park S.M., Gaur A.B., Lengyel E., Peter M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gregory P.A., Bert A.G., Paterson E.L., Barry S.C., Tsykin A., Farshid G., Vadas M.A., Khew-Goodall Y., Goodall G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 22.Jurmeister S., Baumann M., Balwierz A., Keklikoglou I., Ward A., Uhlmann S., Zhang J.D., Wiemann S., Sahin O. MicroRNA-200c represses migration and invasion of breast cancer cells by targeting actin-regulatory proteins FHOD1 and PPM1F. Mol. Cell. Biol. 2012;32:633–651. doi: 10.1128/MCB.06212-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fix L.N., Shah M., Efforts T., Farrell M.A., Zhang B. MicroRNA expression profile of MCF-7 human breast cancer cells and the effect of green tea polyphenon-60. Cancer Genomics Proteomics. 2010;7:261–276. [PubMed] [Google Scholar]
- 24.Chernokalskaya E., Dompenciel R.E., Schoenberg D.R. Cleavage properties of a polysomal ribonuclease involved in the estrogen-regulated destabilization of albumin mRNA. Nucleic Acids Res. 1997;25:735–742. doi: 10.1093/nar/25.4.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki H.I., Arase M., Matsuyama H., Choi Y.L., Ueno T., Mano H., Sugimoto K., Miyazono K. MCPIP1 ribonuclease antagonizes Dicer and terminates microRNA biogenesis through precursor microRNA degradation. Mol. Cell. 2011;44:424–436. doi: 10.1016/j.molcel.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 26.Upton J.P., Wang L., Han D., Wang E.S., Huskey N.E., Lim L., Truitt M., McManus M.T., Ruggero D., Goga A., et al. IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic caspase-2. Science. 2012;338:818–822. doi: 10.1126/science.1226191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruegger S., Grosshans H. MicroRNA turnover: when, how, and why. Trends Biochem. Sci. 2012;37:436–446. doi: 10.1016/j.tibs.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Z., Qin Y.W., Brewer G., Jing Q. MicroRNA degradation and turnover: regulating the regulators. Wiley Interdiscip. Rev. RNA. 2012;3:593–600. doi: 10.1002/wrna.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo Y., Liu J., Elfenbein S.J., Ma Y., Zhong M., Qiu C., Ding Y., Lu J. Characterization of the mammalian miRNA turnover landscape. Nucleic Acids Res. 2015;43:2326–2341. doi: 10.1093/nar/gkv057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katoh T., Hojo H., Suzuki T. Destabilization of microRNAs in human cells by 3′ deadenylation mediated by PARN and CUGBP1. Nucleic Acids Res. 2015;43:7521–7534. doi: 10.1093/nar/gkv669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bajan S., Hutvagner G.H. Another “loophole” in miRNA processing. Mol. Cell. 2011;44:345–347. doi: 10.1016/j.molcel.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 32.Medina P.P., Slack F.J. microRNAs and cancer: an overview. Cell Cycle. 2008;7:2485–2492. doi: 10.4161/cc.7.16.6453. [DOI] [PubMed] [Google Scholar]
- 33.Viswanathan S.R., Daley G.Q., Gregory R.I. Selective blockade of microRNA processing by Lin28. Science. 2008;320:97–100. doi: 10.1126/science.1154040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heo I., Joo C., Kim Y.K., Ha M., Yoon M.J., Cho J., Yeom K.H., Han J., Kim V.N. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell. 2009;138:696–708. doi: 10.1016/j.cell.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 35.Chang H.M., Triboulet R., Thornton J.E., Gregory R.I. A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28-let-7 pathway. Nature. 2013;497:244–248. doi: 10.1038/nature12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uehata T., Akira S. mRNA degradation by the endoribonuclease Regnase-1/ZC3H12a/MCPIP-1. Biochim. Biophys. Acta. 2013;1829:708–713. doi: 10.1016/j.bbagrm.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 37.Howe E.N., Cochrane D.R., Richer J.K. Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res. 2011;13:R45. doi: 10.1186/bcr2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 39.Waclaw B., Bozic I., Pittman M.E., Hruban R.H., Vogelstein B., Nowak M.A. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature. 2015;525:261–264. doi: 10.1038/nature14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.