

Abstract
The gene PFKFB3 encodes for inducible 6-phosphofructo-2-kinase, a glycolysis-regulatory enzyme that protects against diet-induced intestine inflammation. However, it is unclear how nutrient overload regulates PFKFB3 expression and inflammatory responses in intestinal epithelial cells (IECs). In the present study, primary IECs were isolated from small intestine of C57BL/6J mice fed a low-fat diet (LFD) or high-fat diet (HFD) for 12 weeks. Additionally, CMT-93 cells, a cell line for IECs, were cultured in low glucose (LG, 5.5 mmol/L) or high glucose (HG, 27.5 mmol/L) medium and treated with palmitate (50 μmol/L) or bovine serum albumin (BSA) for 24 hr. These cells were analyzed for PFKFB3 and inflammatory markers. Compared with LFD, HFD feeding decreased IEC PFKFB3 expression and increased IEC proinflammatory responses. In CMT-93 cells, HG significantly increased PFKFB3 expression and proinflammatory responses compared with LG. Interestingly, palmitate decreased PFKFB3 expression and increased proinflammatory responses compared with BSA, regardless of glucose concentrations. Furthermore, HG significantly increased PFKFB3 promoter transcription activity compared with LG. Upon PFKFB3 overexpression, proinflammatory responses in CMT-93 cells were decreased. Taken together, these results indicate that in IECs glucose stimulates PFKFB3 expression and palmitate contributes to increased proinflammatory responses. Therefore, PFKFB3 regulates IEC inflammatory status in response to macronutrients.
It is well established that inactivity and overnutrition are major determinants in the development of obesity and contribute to obesity-related metabolic diseases such as type 2 diabetes, fatty liver disease, and inflammatory atherosclerosis1,2,3,4,5. High saturated fat intake is an especially causative factor as it is known to directly contribute to the growth of individual adipocytes6,7 which results in impaired lipid storage abilities and the generation of inflammation locally8 and systemically in chronic conditions9. It is now accepted that the obesity-associated chronic, low-grade systemic inflammation is a major underlying factor for the development of many metabolic diseases10,11,12,13,14. As such, much research has investigated the mechanisms of diet-induced inflammation, particularly in adipose tissue.
The intestine has recently been implicated as another key organ that critically contributes to the development of obesity-associated chronic inflammation and systemic insulin resistance, and metabolic dysregulation15,16,17,18. While investigating how nutrient overload interacts with the intestine to cause systemic inflammation, a number of studies have shown that feeding a high-fat diet (HFD) to mice alters the composition of the gut microbiota and leads to increased intestinal permeability19,20. This in turn increases the levels of endotoxin in the intestinal lumen and circulation, thereby accelerating obesity and its related metabolic dysregulation. A recent study even indicated a role for HFD-induced intestinal eosinophil depletion, not inflammation, in contributing to defective barrier integrity and the onset of metabolic disease21. Considering that the intestine is responsible for digestion, absorption, and assimilation of nutrients and that the nutrients absorbed by intestine have also undergone metabolism whose dysregulation accounts for increased proinflammatory responses, intestinal cells, in particular intestinal epithelial cells (IECs), may respond to nutrient overload to regulate its own inflammatory status prior to regulating inflammatory responses in distal organs. Indeed, in a mouse model of diet-induced obesity (DIO), feeding an HFD activated nuclear factor kappa B (NFκB) activity in intestine cells including epithelial cells, immune cells, and endothelial cells of the small intestine18. While showing a similar finding in small intestine of HFD-fed mice, the study by Guo et al.22 further indicated that the anti-inflammatory responses in the intestine accounted for, at least in part, the insulin-sensitizing effect of peroxisome proliferator-activated receptor gamma (PPARγ) activation. Given this, there is a need to address the responses of IECs to nutrient overload in order to better understand the mechanisms of obesity-associated inflammation.
In the intestine, the gene 6-phosphofructo-2-kinase/fructose-2, 6-bisphophatase 3 (PFKFB3) is abundantly expressed22. As the product of PFKFB3, inducible 6-phosphofructo-2-kinase (iPFK2) generates fructose-2,6-bisphophate. The latter, as the most powerful activator of 6-phosphofructokinase-1, stimulates glycolysis. Recent studies by Huo et al.13,14,23 and Guo et al.13,14,23 have further demonstrated that PFKFB3/iPFK2 critically determines the balance of metabolic fluxes through glycolysis and fatty acid oxidation, thereby suppressing the generation of reactive oxygen species and proinflammatory responses in adipocytes13,14,23. Unlike its role in adipose tissue/adipocytes, the role for PFKFB3/iPFK2 in the small intestine is less known. A previous study by Guo et al.22 showed increased PFKFB3 expressions in the small intestine in response to HFD feeding, as well as increased inflammation. However, the regulation of PFKFB3 within IECs in relation to IEC inflammatory responses has not been investigated. Therefore, this study sought to first determine how macronutrients influence PFKFB3/iPFK2 expression in IECs and secondly, how this relates to the IEC inflammatory status.
Results
Induction of obesity-related insulin resistance and glucose intolerance
To investigate nutritional regulation of IEC PFKFB3/iPFK2 expression and inflammatory responses in the context of obesity and insulin resistance, we fed C57BL/6J mice a HFD for 12 weeks. Compared with low-fat diet (LFD)-fed control mice, HFD-fed mice gained much more body weight (Fig. 1A; P < 0.01) after only 5 weeks on the respective diet. During the feeding period, the mice consumed comparable amount of foods, but displayed a significant increase in energy efficiency, which was calculated as milli-gram BW gained/Kcal consumed (Fig. 1B). Along with obesity, HFD-fed mice displayed overt insulin resistance as indicated by the results from insulin tolerance tests (Fig. 1C), in which HFD-fed mice received twice the amount of insulin injection compared with LFD-fed mice. Consistently, HFD-fed mice also showed impairment of glucose tolerance (Fig. 1D). As such, obesity-related insulin resistance and glucose dysregulation were successfully induced in these mice.
Figure 1. HFD induction of obesity-related insulin resistance and glucose intolerance.

Male C57BL/6J mice, at 5–6 weeks of age, were fed an HFD or LFD for 12 weeks, n = 9–10. (A) Body weight. (B) Energy efficiency. (C) Insulin tolerance tests. (D) Glucose tolerance tests. For (C,D), areas under curves (AUC) were calculated based on the corresponding tolerance tests. For (A–D), data are means ± SEM. *P < 0.05; **P < 0.01; and ***P < 0.001 HFD vs. LFD (AUC in C,D) for the same time (A,C,D). HFD, high-fat diet; LFD, low-fat diet.
Reduction of PFKFB3/iPFK2 in primary IECs
In the present study, we confirmed the previous findings22 that HFD feeding increased iPFK2 amount and inflammatory responses in intestine extracts (Fig. 2A). Next, we examined PFKFB3/iPFK2 expression in primary IECs isolated from DIO- and/or control mice. Compared with those in IECs from LFD-fed mice, the mRNA levels of PFKFB3 in IECs from HFD-fed mice were significantly lower (Fig. 2B; P < 0.05). Consistently, the amount of iPFK2 was reduced in primary IECs isolated from HFD-fed mice compared with those from LFD-fed mice (Fig. 2C,D). Thus, HFD feeding lowered PFKFB3/iPFK2 expression in IECs, which is opposite to the effect of HFD feeding on increasing PFKFB3/iPFK2 in intestine extracts22. The latter includes various types of cells.
Figure 2. Dietary effects on intestinal PFKFB3/iPFK2 expression.

Small intestine and primary IECs were isolated following the feeding period. (A) Intestine extracts were examined for iPFK2 amount and JNK signaling using Western blot analysis. (B) IEC expression of PFKFB3 mRNAs was determined using real-time PCR. (C) Western blot analysis of IEC iPFK2 amount. (D) Quantification of IEC iPFK2 amount. For bar graphs, data are means ± SEM, n = 4–6. HFD, high-fat diet; LFD, low-fat diet; JNK, c-Jun n-terminal kinase.
Stimulation of proinflammatory responses in primary IECs
We examined if reduced IEC PFKFB3/iPFK2 expression was associated with greater proinflammatory responses. Relative to that of primary IECs of control mice, the proinflammatory signaling through c-Jun N-terminal kinase 1 (JNK1) was much higher in primary IECs from DIO mice (Fig. 3A,B). In addition, the mRNA levels of proinflammatory cytokines, e.g., interleukin-6 (IL-6) and tumor necrosis factor alpha (TNFα), in IECs of DIO mice were significantly higher than in IECs of control mice (Fig. 3C). Similar changes were also observed in the mRNA levels of Toll-like receptor 4 (TLR4) (Fig. 3C), whose activation promotes proinflammatory responses. Together, these results suggest that HFD feeding increased IEC proinflammatory responses, which were associated with a reduction in PFKFB3/iPFK2 expression in IECs.
Figure 3. Dietary effects on IEC proinflammatory responses.

Primary IECs were isolated following the feeding period. (A) Western blot analysis of IEC JNK signaling. (B) Quantification of IEC Pp46/p46. (C) IEC expression of IL-6, TNFα, and TLR4 mRNAs. For (C) data are means ± SEM, n = 4–6. *P < 0.05; **P < 0.01 HFD vs. LFD for the same gene. HFD, high-fat diet; IL-6, interleukin-6; iPFK2, inducible 6-phosphofructo-2-kinase; LFD, low-fat diet; Pp46, phosphorylated c-Jun n-terminal kinase 1 (JNK1); p46, total JNK1; TLR4, Toll-like receptor 4; TNFα, tumor necrosis factor alpha.
Direct effects of glucose and palmitate on IEC PFKFB3/iPFK2 expression and proinflammatory responses
To gain nutritional insight into PFKFB3 regulation, we examined the direct effects of glucose and palmitate, two major macronutrients associated with overnutrition, on IEC responses. When the effects of glucose were examined, treatment of CMT-93 cells with 27.5 mmol/L glucose led to greater iPFK2 amount relative to treatment of CMT-93 cells with 5.5 mmol/L glucose, indicating a stimulatory effect of glucose on PFKFB3 expression (Fig. 4A,B). In contrast, palmitate appeared to mainly account for higher proinflammatory responses in cultured CMT-93 cells. Indeed, two-way ANOVA results indicated an interaction between glucose concentration and palmitate treatment (P = 0.010). Specifically, in the presence of low levels of glucose, palmitate did not alter JNK1 signaling. However, in the presence of high levels of glucose, palmitate treatment caused a significant increase in JNK1 signaling (Fig. 4A,B). When the expression of proinflammatory cytokines was examined, palmitate treatment led to significantly higher mRNA levels of TNFα and TLR4 regardless of glucose concentration (Fig. 4C). This finding was confirmed by two-way ANOVAs where glucose by treatment interactions were non-significant (TNFα P = 0.852; TLR4 P = 0.847). There was a slight interaction of glucose concentration and palmitate treatment regarding IL-6 mRNA levels (P = 0.048), where mRNA levels were slightly higher in the presence of high concentrations of glucose. Taken together, these results indicate that palmitate, more so than glucose, is responsible for inducing inflammatory responses.
Figure 4. Effects of glucose and palmitate on IEC iPFK2 and proinflammatory responses.

CMT-93 cells were treated as described in methods. (A) Western blot analyses of IEC iPFK2 amount and JNK signaling. (B) Quantification of IEC iPFK2 and Pp46/p46. (C) IEC expression of IL-6, TNFα, and TLR4 mRNAs. For (B,C), data are means ± SEM, n = 4. *P < 0.05 and **P < 0.01 High glucose vs. Low glucose for the same treatment (BSA or Pal); †P < 0.05 and ††P < 0.01 Pal vs. BSA for the same condition. BSA, bovine serum albumin; HFD, high-fat diet; IL-6, interleukin-6; iPFK2, inducible 6-phosphofructo-2-kinase; LFD, low-fat diet; Pal, palmitate; Pp46, phosphorylated c-Jun n-terminal kinase 1 (JNK1); p46, total JNK1; TLR4, Toll-like receptor 4; TNFα, tumor necrosis factor alpha.
Stimulatory effect of glucose on PFKFB3 transcription
We further explored the effect of glucose on stimulating IEC PFKFB3 expression. In a time-course study, treatment of CMT-93 cells with low levels of glucose for 24 hr did not alter the mRNA levels of PFKFB3 compared with treatment of CMT-93 cells with low levels of glucose for 4 hr (Fig. 5A). However, in the presence of high levels of glucose, treatment of CMT-93 cells for 24 hr significantly increased the mRNA levels of PFKFB3 relative to treatment of CMT-93 cells for 4 hr (Fig. 5A). Next, we examined the effects of palmitate on PFKFB3 expression in the presence of low or high levels of glucose. Two-way ANOVA analyses indicated a significant interaction between glucose concentration and palmitate treatment (P = 0.002). Specifically, high levels of glucose remained a dominant effect in stimulating PFKFB3 expression even in the presence of palmitate, which appeared to decrease the mRNA levels of PFKFB3 (Fig. 5B). To gain transcription insights, a reporter assay was performed and showed that high levels of glucose stimulated the transcription activity of the PFKFB3 promoter (Fig. 5C). Unlike glucose, palmitate treatment did not alter the transcription activity of the PFKFB3 promoter. No interaction between glucose concentration and palmitate treatment was found (P = 0.558).
Figure 5. Effects of glucose and palmitate on PFKFB3 gene transcription.

CMT-93 cells were treated as described in the methods. (A) Time course and dose responses of glucose regulation of PFKFB3 expression. (B) Effects of glucose and palmitate on PFKFB3 expression. (C) Nutrient regulation of PFKFB3 promoter activity. For (A–C), data are means ± SEM, n = 4–6. *P < 0.05 and **P < 0.01 High glucose vs. Low glucose for the same time/treatment (BSA or Pal); ††P < 0.01 24 hr vs 4 hr (A) or Pal vs. BSA (B) for the same glucose condition. BSA, bovine serum albumin; Pal, palmitate.
Suppression of proinflammatory responses by PFKFB3/iPFK2 overexpression
We investigated the effect of PFKFB3/iPFK2 overexpression on the proinflammatory responses in IECs. Overexpression of PFKFB3/iPFK2 was associated with a decrease in JNK1 signaling (Fig. 6A,B). Also, real-time PCR analyses showed similar results (Fig. 6C). Specifically, LPS-stimulated mRNA levels of all three inflammatory markers in PFKFB3-overexpressing cells were significantly lower than those in control cells. PFKFB3 overexpression also resulted in decreased superoxide production (Fig. 6D), indicated by a decrease in NBT production. Therefore, PFKFB3 overexpression appeared to decrease the severity of the inflammatory response induced by stress stimuli. Also, it should be pointed out that the relatively low transfection efficiency may lead to underestimation of the effect of iPFK2 overexpression on suppressing IEC proinflammatory responses.
Figure 6. Suppression of proinflammatory responses by PFKFB3/iPFK2 overexpression.

CMT-93 cells were treated as described in methods. (A,B) Western blot analyses for PFKFB3/iPFK2 overexpression and quantification of IEC iPFK2 and Pp46/p46. (C) Changes in mRNA levels. (D) Changes in NBT production. For bar graphs, data are means ± SEM, n = 4. *P < 0.05 and **P < 0.01 iPFK2 vs GFP (B,D) under the same condition (C, PBS or LPS); ††P < 0.01 and †††P < 0.001 LPS vs PBS for the same treatment (C, GFP or iPFK2). For (A–D), iPFK2, inducible 6-phosphofructo-2-kinase; Pp46, phosphorylated c-Jun n-terminal kinase 1 (JNK1); p46, total JNK1; IL-6, interleukin-6; TLR4, Toll-like receptor 4; TNFα, tumor necrosis factor alpha.
Discussion
Recent studies have established that PFKFB3/iPFK2 links nutrient metabolism and inflammatory responses in several tissues and cell types, e.g., adipocytes and endothelial cells13,14,23,24. In the intestine, PFKFB3/iPFK2 has also been implicated as a regulator that critically controls the development of intestinal inflammation during obesity22. Significantly, PFKFB3/iPFK2 is involved in the effect of rosiglitazone, one of the only two currently prescribed medicines as insulin-sensitizers for the treatment of type 2 diabetes, on suppressing intestinal inflammation. The current study presented here builds upon this finding by investigating PFKFB3/iPFK2 specifically within IECs, a topic which has not been previously studied.
In DIO mice, the mRNA levels of PFKFB3 and the amount of iPFK2 were significantly decreased in IECs compared with their respective levels in IECs from LFD-fed mice. Surprisingly, these changes were opposite to the previous finding that the iPFK2 amount was increased in intestine extracts of DIO mice22. Considering that the intestine includes various types of cells, it is possible that HFD feeding increased PFKFB3/iPFK2 in cells other than IECs, and those cells had higher abundance of PFKFB3/iPFK2 than IECs. Additionally, it is possible that during and after the digestion, absorption, and metabolism of nutrients, the composition of nutrients and the metabolites of nutrients were different across IECs and cells other than IECs. As a result, IECs and cells other than IECs likely interacted, respectively, with different nutrients and/or metabolites, thereby displaying differential consequences on PFKFB3/iPFK2. While these possibilities need to be further examined, it is important to consider that LFD provides a significantly high amount of carbohydrates (i.e. corn starch). Subsequently, IECs from this diet group would display increased expressions of PFKFB3/iPFK2 and thus, exhibit reduced levels in diets with less carbohydrate stimuli (e. g., HFD). This outcome was evident in both PFKFB3 mRNA levels and iPFK2 amount in primary IECs. As additional evidence, glucose and palmitate showed differential effects on PFKFB3/iPFK2 expression in cultured cells (see below). To be noted, the markers of proinflammatory responses, however, were significantly higher with HFD. This effect appeared to be due to the high amount of saturated fat, e.g., palmitate, in the diet, which is known to elevate proinflammatory responses in many tissues/cells25,26,27. In fact, the findings from cultured IEC cell line studies further confirmed this postulation as evidenced by increases in JNK1 signaling and the mRNA levels of several proinflammatory markers in response to palmitate treatment. Therefore, palmitate/saturated fats appeared to serve as the primary underlying factor as to why IECs displayed an increase in inflammatory responses with HFD feeding. Of importance, the status of proinflammatory responses in IECs was reversely correlated with PFKFB3/iPFK2 expression, suggesting that PFKFB3/iPFK2 also has an anti-inflammatory role in IECs.
To better understand the underlying mechanisms of dietary effects, we investigated the effects of individual major macronutrients on PFKFB3/iPFK2 expression and showed that dietary components exerted differential effects on PFKFB3/iPFK2. Notably, glucose, at high levels, significantly increased the mRNA levels of PFKFB3 and the amount of iPFK2. This stimulatory effect of glucose was expected as PFKFB3 is highly involved in the stimulation of glycolysis when carbohydrates, in particular glucose, are in excess. In fact, the role of PFKFB3/iPFK2 in this manner has been demonstrated in numerous cell types13,24,28. Mechanistically, glucose stimulation of PFKFB3/iPFK2 expression was attributable to the effect of glucose on increasing the transcription activity of the PFKFB3 promoter. In support of this, high levels of glucose significantly increased the activity of luciferase whose expression was driven by a 6.1 kb fragment of the PFKFB3 promoter. A previous study by Sans et al.29 had shown that the PFKFB3 promoter region contains the CACGTG-containing regulatory elements that interact with MondoA:Mlx complex. The latter mediates the effect of glucose on stimulating the expression of a number of genes that participate in glycolysis and lipogenesis when nutrients are in excess. Therefore, within IECs glucose acts to directly stimulate PFKFB3/iPFK2 expression. This data further validated the high PFKFB3/iPFK2 levels seen in LFD-fed mice.
As a critical component of HFD, palmitate has been previously shown to serve as a ligand to TLR4 in IECs and therefore directly stimulates the expression of proinflammatory cytokines via the TGFβ and NFκB pathways30. Consistently, we showed that palmitate treatment significantly increased TLR4 and proinflammatory cytokine expression in cultured IECs. Of interest, palmitate treatment also decreased the mRNA levels of PFKFB3 and the amount of iPFK2. However, this effect of palmitate was not sufficient to counter against the effect of glucose on increasing PFKFB3/iPFK2. Nonetheless, this finding further confirmed that the HFD-induced decrease in PFKFB3/iPFK2 expression in primary IECs was due to low levels of carbohydrates (glucose) relative to LFD. In addition, consistent with the results from primary IECs of HFD-fed mice, the decrease in PFKFB3/iPFK2 was also correlated with increased proinflammatory responses in cultured IECs. Based on these findings, it is likely that one mechanism for palmitate to increase IEC proinflammatory responses was to decrease PFKFB3/iPFK2 expression, thereby leading to a decrease in the anti-inflammatory effect. As a side note, although high glucose (HG) treatment was associated with increased PFKFB3/iPFK2, inflammatory markers also remained relatively high at this condition. This was expected as the supra-physiological glucose concentration used (27.5 mmol/L) models overnutrition and was necessary for this study to best demonstrate glucose regulation of PFKFB3. However, the control of inflammatory responses induced by such conditions likely requires more than one anti-inflammatory mechanism and thus, the anti-inflammatory ability of PFKFB3/iPFK2 alone may not be sufficient to fully suppress an inflammatory response of this nature.
It should be pointed out that the HFD-induced decrease in PFKFB3/iPFK2 expression in the primary IECs was reversely correlated with systemic insulin resistance. Based on this relationship, it is likely that PFKFB3/iPFK2 in IECs participates in the regulation of obesity-associated insulin resistance and dysregulation of glucose homeostasis. Furthermore, the role played by PFKFB3/iPFK2 appears to be attributable to its anti-inflammatory properties as indicated by the results of the present study in both primary IECs and cultured CMT-93 cells, and by the findings of previous studies in adipocytes13,23. The present study further confirmed this anti-inflammatory role within IECs given that overexpression of PFKFB3/iPFK2 decreased proinflammatory markers. Although future studies are needed to validate a specific role for PFKFB3/iPFK2 in IECs in the control of obesity-associated insulin resistance and metabolic dysregulation, targeting PFKFB3/iPFK2 in IECs through nutritional intervention could offer novel approaches for prevention and/or treatment of inflammatory responses, which contribute to the development of obesity-associated metabolic diseases.
In summary, this study provides evidence for the first time that major macronutrients, e.g., glucose and palmitate, differentially influence the expression of PFKFB3/iPFK2 within IECs. As outlined in the proposed scheme (Fig. 7), glucose directly stimulates PFKFB3/iPFK2 expression whereas palmitate more so contributes to the generation of inflammation. The present study also provides the insight into the potential role for PFKFB3/iPFK2 in protecting against the proinflammatory responses within IECs. Overall, diet has a significant impact on PFKFB3/iPFK2 expression within IECs in the context of obesity-associated inflammation. Because of this, activating PFKFB3/iPFK2 in IECs through nutritional approaches could be beneficial for obesity-associated metabolic diseases.
Figure 7. PFKFB3/iPFK2 mediates nutritional control of IEC inflammatory responses.
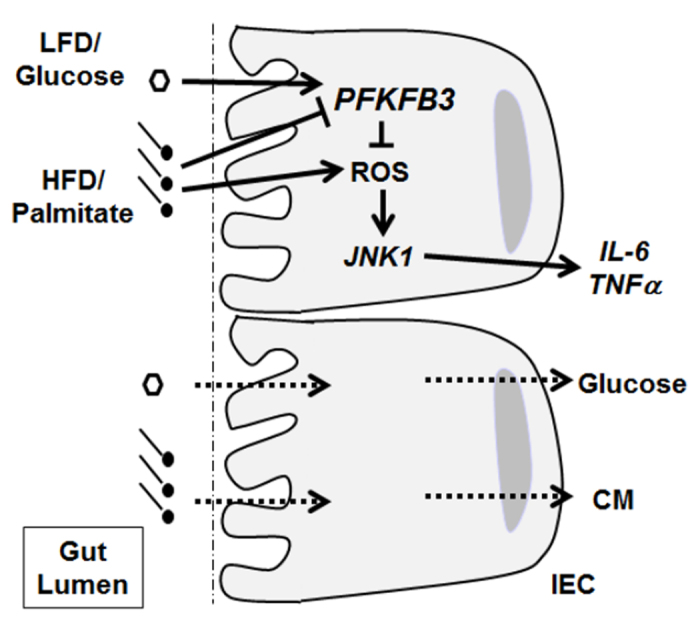
The proposed scheme summarizes the differential effects of major macronutrients, e.g., glucose and palmitate, on PFKFB3/iPFK2 expression and the inflammatory responses within IECs. Upon LFD feeding, glucose, as a major macronutrient at high concentration, stimulates PFKFB3/iPFK2. This in turn inhibits the proinflammatory responses in IECs, likely through suppressing the generation of ROS. Upon HFD feeding, the stimulatory effect on PFKFB3/iPFK2 is not present due to low concentrations of glucose. This in turn de-inhibits the proinflammatory responses in IECs. In addition, palmitate, as a major macronutrient of HFD, has a direct proinflammatory effect on IECs. The combined effects exacerbate IEC proinflammatory responses, which may contribute to HFD-induced systemic inflammation. IEC, intestinal epithelial cells; LFD, low-fat diet; HFD, high-fat diet; ROS, reactive oxygen species; JNK1, c-Jun N-terminal kinase 1; IL-6, interleukin-6; TNFα, tumor necrosis factor alpha; TLR4, Toll-like receptor 4; CM, chylomicrons.
Materials and Methods
Animal experiments
C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and housed under a 12 hr light/dark cycle with free access to water and fed ad libitum. At 5–6 weeks of age, male mice were fed either an LFD or HFD for 12 weeks. LFD (Research Diets, New Brunswick, NJ product #D12450B) and HFD (Research Diets, #D12492) consisted of 10% and 60% calories from fat, respectively. The complete macro- and micronutrient composition of the diets is provided in Supplemental Table 1. During the feeding period, body weight and food intake were monitored weekly. After the feeding period, the mice were fasted for 4 hr before sacrifice for collection of blood and tissue samples31. Some mice were fasted similarly and used for insulin and glucose tolerance tests and/or IEC isolation as described below. All animals received human care and all study protocols were approved by the Institutional Animal Care and Use Committee of Texas A&M University. In addition, all experiments were performed in accordance with relevant guidelines and regulations.
Insulin and glucose tolerance tests
Insulin and glucose tolerance tests were performed as previously described27,31. For insulin tolerance, the mice received an intraperitoneal injection of insulin (0.5 U/kg body weight for LFD-fed mice and 1 U/kg body weight for HFD-fed mice). LFD-fed mice were injected with half the insulin amount as HFD-fed mice since LFD animals are at risk for hypoglycemia following a 1 U/kg dose.
Isolation of primary IEC
During tissue harvest, the small intestine was removed and cleaned of fecal debris. A small portion of the ileum was prepared for mRNA and protein analyses. The remaining intestine was first flushed with warm DMEM (Sigma-D5523; containing 4 mmol/L L-glutamine, 4.5 g/L glucose, 1.5 g/L sodium bicarbonate, 10% FBS and 1% penicillin/streptomycin), and then placed in warm medium and transferred to a biosafety cabinet. The intestine was cut into 3–4, ~4 cm sections and inverted over bamboo splints. The splints were incubated at 37 °C in DMEM + 2 mmol/L EDTA for 10 min, with gentle shaking every 3 min. After incubation, the splints were discarded, and the medium was filtered through a 70-μm filter and centrifuged at 478 × g for 5 min at 4 °C. After removal of the medium, the IEC pellets were re-suspended in 3 mL warm DMEM and divided into 2, 1.5 mL tubes. After an additional centrifugation, the cell pellets were re-suspended in lysis buffer or STAT-60 for protein and mRNA analyses, respectively.
Cell culture and treatment
The mouse-derived IEC line, CMT-93 (passage 10–30), was purchased from the American Type Culture Collection (ATCC, Catalog # CRL 223) and grown to confluence in DMEM (Sigma-D5523; containing 4 mmol/L L-glutamine, 4.5 g/L glucose, 1.5 g/L sodium bicarbonate, 10% FBS and 1% penicillin/streptomycin) in 100-mm cell culture dishes in a humidified 5% CO2 atmosphere at 37 °C. Confluent cells were transferred to 60-mm cell culture dishes and conditioned in low glucose (LG; 5.5 mmol/L) medium for 24 hr prior to treatment. Thereafter, cells were incubated in LG or high glucose (HG; 27.5 mmol/L) DMEM and treated with palmitate (50 μmol/L) or bovine serum album (BSA, control) for an additional 24 hr. Palmitate was chosen for in vitro experiments to mimic the major fat source provided by HFDs. After the treatment period, the cells were harvested for protein and mRNA and stored at −80 °C for further analyses.
To confirm the role of PFKFB3/iPFK2 in regulating inflammatory responses, we conducted a gain-of-function in vitro experiment. Briefly, cells at 80% confluence were transfected with a plasmid containing the cDNA of iPFK2 with Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA) following the manufacturer’s protocol. Cells were also transfected with a vector expressing green fluorescent protein (GFP) as a control. The transfected cells were then treated with LPS (100 ng/mL) or PBS (control) and harvested and saved in −80 °C for protein and mRNA analyses of proinflammatory markers.
RNA isolation, reverse transcription, and real-time PCR
Total RNA was isolated from IECs and cultured CMT-93 cells. Reverse transcription was performed using the GoScript™ Reverse Transcription System (Promega) and real-time PCR analysis was performed using SYBR Green (LightCycler® 480 system; Roche Life Science, Indianapolis, IN). The mRNA levels were analyzed for PFKFB3, IL-6, TNFα, and TLR4. A total of 0.1 μg RNA was used for the determination. Results were normalized to 18s ribosomal RNA and plotted as relative expression to the mean expression in LG-treated cells or cells of LFD-fed mice, which were set as 1.
Western blot analysis
Lysates were prepared from frozen IEC samples and cultured cells. Western blot analyses were performed as previously described32. Protein amount of iPFK2 (Proteintech Group, Catalog # 13763-1-ap), JNK1 (p46; Santa Cruz, Catalog # sc-571), and phosphorylated-JNK1(pJNK1, Pp46; Santa Cruz, Catalog # sc-6254) was examined. The maximum intensity of each band was quantified using ImageJ software. Ratios of Pp46/p46 were normalized to GAPDH (Santa Cruz, Catalog # sc-25778) and adjusted relative to the mean of control LFD-fed IEC or LG-treated control cells, which were arbitrarily set as 1 (AU).
Gene transcription reporter assay
A luciferase reporter assay was performed as previously described27. Briefly, a reporter construct in which luciferase expression is driven by an empty promoter (pGL3-luc) or PFKFB3 promoter (pPFKFB3-luc) was transfected into CMT-93 cells. After transfection for 24 h, the cells were incubated with LG or HG medium and treated with palmitate (50 μmol/L) or BSA for an additional 24 hr. Cell lysates were prepared and used to measure luciferase activity using a kit from Promega (Madison, WI). The luciferase activity was normalized to protein concentrations and adjusted relative to the mean of LG- and BSA-treated pGL3 controls, which were arbitrarily set as 1 (AU).
Nitroblue tetrazolium assay
A nitroblue tetrazolium (NBT) assay was conducted to investigate the role of PFKFB3/iPFK2 in regulating superoxide generation. The present study followed a modification of methods previously described33.
Statistical analysis
Numerical data are presented as means ± SEM (standard error). Two-tailed ANOVA or Student’s t tests were used to evaluate differences between diet or glucose concentration. To test whether glucose concentration interacted with treatment effects, two-way ANOVAs, and Tukey’s post hoc tests when necessary, were performed. In case of interactions, treatments at individual glucose concentrations were compared. Differences were considered significant at P < 0.05.
Additional Information
How to cite this article: Botchlett, R. et al. Glucose and Palmitate Differentially Regulate PFKFB3/iPFK2 and Inflammatory Responses in Mouse Intestinal Epithelial Cells. Sci. Rep. 6, 28963; doi: 10.1038/srep28963 (2016).
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (HL108922 and HL095556 to Y.H., and R01DK095828 and R01DK095862 to C.W.). Also, J. Zheng is supported by National Natural Science Foundation of China (81100562/H0711) and C.W. is supported by the Hatch Program of the National Institutes of Food and Agriculture (NIFA).
Footnotes
Author Contributions C.W. and Y.H. designed the research; R.B. conducted the research; H.L., X.G., T.Q., J.Zhao, J.Zheng, S.-L.W., Y.P., M.L., X.H., G.C. and T.G. assisted with animal care and cell culture methods; R.B. and C.W. analyzed the data; R.B. wrote the paper; S.Y., Q.L. and X.X. were critically involved in scientific discussion; C.W. and Y.H. had primary responsibility for the final content. All authors read and approved the final manuscript.
References
- Berg A. H. & Scherer P. E. Adipose tissue, inflammation, and cardiovascular disease. Circ Res 96, 939–949 (2005). [DOI] [PubMed] [Google Scholar]
- Ota T. et al. Insulin resistance accelerates a dietary rat model of nonalcoholic steatohepatitis. Gastroenterology 132, 282–293 (2007). [DOI] [PubMed] [Google Scholar]
- Ohman M. K. et al. Visceral adipose tissue inflammation accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation 117, 798–805 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen M. K. et al. Obesity, behavioral lifestyle factors, and risk of acute coronary events. Circulation 117, 3062–3069 (2008). [DOI] [PubMed] [Google Scholar]
- Fantuzzi G. & Mazzone T. Adipose tissue and atherosclerosis: exploring the connection. Arterioscler Thromb Vasc Biol 27, 996–1003 (2007). [DOI] [PubMed] [Google Scholar]
- Sansbury B. E., Bhatnagar A. & Hill B. G. Impact of nutrient excess and endothelial nitric oxide synthase on the plasma metabolite profile in mice. Front Physiol 5, 453 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T. et al. Growth hormone receptor antagonist transgenic mice are protected from hyperinsulinemia and glucose intolerance despite obesity when placed on a HF diet. Endocrinology 156, 555–564 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen K. E. & Hotamisligil G. S. Obesity-induced inflammatory changes in adipose tissue. J. Clin. Invest. 112, 1785–1788 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidhu M. P., Anthony N. M., Patel P., Hawke T. J. & Ceddia R. B. Dysregulation of lipolysis and lipid metabolism in visceral and subcutaneous adipocytes by high-fat diet: role of ATGL, HSL, and AMPK. Am J Physiol Cell Physiol 298, C961–C971 (2010). [DOI] [PubMed] [Google Scholar]
- Dandona P., Aljada A. & Bandyopadhyay A. Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol 25, 4–7 (2004). [DOI] [PubMed] [Google Scholar]
- Greenberg A. S. & Obin M. S. Obesity and the role of adipose tissue in inflammation and metabolism. Am J Clin Nutr 83, 461S–465 (2006). [DOI] [PubMed] [Google Scholar]
- Kanda H. et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Invest. 116, 1494–1505 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo Y. et al. Disruption of inducible 6-phosphofructo-2-kinase ameliorates diet-induced adiposity but exacerbates systemic insulin resistance and adipose tissue inflammatory response. J. Biol. Chem. 285, 3713–3721 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X. et al. Involvement of inducible 6-phosphofructo-2-kinase in the anti-diabetic effect of PPARg activation in mice. J. Biol. Chem. 285, 23711–23720 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley R. E. et al. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA 102, 11070–11075 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Membrez M. et al. Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. FASEB J. 22, 2416–2426 (2008). [DOI] [PubMed] [Google Scholar]
- Turnbaugh P. J. et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1131 (2006). [DOI] [PubMed] [Google Scholar]
- Ding S. et al. High-fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS ONE 5, e12191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani P. D. et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57, 1470–1481 (2008). [DOI] [PubMed] [Google Scholar]
- Kim K.-A., Gu W., Lee I.-A., Joh E.-H. & Kim D.-H. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE 7, e47713 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A. M. F. et al. High fat diet causes depletion of intestinal eosinophils associated with intestinal permeability. PLoS ONE 10, e0122195 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X. et al. Disruption of inducible 6-phosphofructo-2-kinase impairs the suppressive effect of PPARg activation on diet-induced intestine inflammatory response. J Nutr Biochem 24, 770–775 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo Y. et al. Targeted overexpression of inducible 6-phosphofructo-2-kinase in adipose tissue increases fat deposition but protects against diet-induced insulin resistance and inflammatory responses. J. Biol. Chem. 287, 21492–21500 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y. et al. Endothelial PFKFB3 plays a critical role in angiogenesis. Arterioscler Thromb Vasc Biol 34, 1231–1239 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajuwon K. M. & Spurlock M. E. Palmitate activates the NF-kB transcription factor and induces IL-6 and TNFa expression in 3T3-L1 adipocytes. J. Nutr. 135, 1841–1846 (2005). [DOI] [PubMed] [Google Scholar]
- Nakamura S. et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J. Biol. Chem. 284, 14809–14818 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X. et al. Palmitoleate induces hepatic steatosis but suppresses liver inflammatory response in mice. PLoS ONE 7, e39286 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. A role for inducible 6-phosphofructo-2-kinase in the control of neuronal glycolysis. J Nutr Biochem 24, 1153–1158 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans C. L., Satterwhite D. J., Stoltzman C. A., Breen K. T. & Ayer D. E. MondoA-Mlx heterodimers are candidate sensors of cellular energy status: mitochondrial localization and direct regulation of glycolysis. Mol. Cell. Biol. 26, 4863–4871 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N. et al. Expression and activity of the TLR4/NF-κB signaling pathway in mouse intestine following administration of a short-term high-fat diet. Exp Ther Med 6, 635–640 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H. et al. Myeloid cell-specific disruption of Period1 and Period2 exacerbates diet-induced inflammation and insulin resistance. J. Biol. Chem. 289, 16374–16388 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo S.-L. et al. Metformin ameliorates hepatic steatosis and inflammation without altering adipose phenotype in diet-induced obesity. PLoS ONE 9, e91111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G. et al. Berberine differentially modulates the activities of ERK, p38 MAPK, and JNK to suppress Th17 and Th1 T cell differentiation in type 1 diabetic mice. J. Biol. Chem. 284, 28420–28429 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.