

Abstract
Aberrant modifications of proteins occur during disease development and elicit disease-specific antibody responses. We have developed a protein array platform that enables the modification of many proteins in parallel and assesses their immunogenicity without the need to express, purify, and modify proteins individually. We used anticitrullinated protein antibodies (ACPAs) in rheumatoid arthritis (RA) as a model modification and profiled antibody responses to ∼190 citrullinated proteins in 20 RA patients. We observed unique antibody reactivity patterns in both clinical anticyclic citrullinated peptide assay positive (CCP+) and CCP- RA patients. At individual antigen levels, we detected antibodies against known citrullinated autoantigens and discovered and validated five novel antibodies against specific citrullinated antigens (osteopontin (SPP1), flap endonuclease (FEN1), insulin like growth factor binding protein 6 (IGFBP6), insulin like growth factor I (IGF1) and stanniocalcin-2 (STC2)) in RA patients. We also demonstrated the utility of our innovative array platform in the identification of immune-dominant epitope(s) for citrullinated antigens. We believe our platform will promote the study of post-translationally modified antigens at a breadth that has not been achieved before, by both identifying novel autoantigens and investigating their roles in disease development. The developed platforms can potentially be used to study many autoimmune disease-relevant modifications and their immunogenicity.
Post-translational modification (PTM)1 is a common biological process that alters specific residues of a protein after its synthesis. Disease often leads to increased activity of enzymes that perform PTM, which in turn can create neoantigens that elicit disease-specific host humoral immune responses including the release of circulating antibodies (1, 2). Autoantibodies against PTM proteins are well established in many diseases, such as methylated SmD1 and SmD3 in systemic lupus erythematosus, deamidated transglutaminase in celiac disease, and glycosylated MUC1 in various cancers (3–6). Their discoveries often relied on Western blots using either serum samples or cell lysates followed by separation by either one dimensional (1D) or 2D gel electrophoresis followed by the identification/characterization of reactive protein bands/spots using mass spectrometry (7–12). Alternatively, modified antigens were immunoprecipitated instead of using Western blots (11). To further confirm the presence of antibodies against a citrullinated protein or a citrullinated epitope on a protein, immunoassays were performed against in vitro citrullinated recombinant proteins using PAD or synthetic peptides with arginine substituted by citrulline (11, 12). This approach has proven to be effective, and has the advantage that it examines proteins from natural cell lysates. However, highly abundant proteins in such lysates can obscure the detection of low abundance proteins, identifying the relevant protein in a reactive spot is cumbersome because multiple proteins are often present in the same location, and interpretation of gels can be complicated because multiple isoforms of the same protein may migrate differently (13, 14). Although all these technical challenges can be overcome, the level of effort needed to thoroughly analyze even a single sample prevents this approach from use in large clinical sample sets (13, 15). Therefore, the depth and breadth of our knowledge of antimodified protein antibodies (AMPA) in autoimmune diseases is still limited.
The lack of high-throughput, unbiased methods to test different candidate antigens has hindered the identification of AMPA (16). To address the need for high throughput screening methods, protein microarrays were developed and the first real breakthrough on protein arrays came in 2000 when MacBeath and Schreiber developed a protein microarray for high throughput screening (17). Conventional human proteome arrays require the spotting of purified proteins (18). Protein arrays have been successfully used to study antimodified antigens (19–22); however, these were limited to refined studies on a set of known modified antigens using modified purified proteins or synthetic modified peptides (23). In addition to the challenges associated with proteome level purification of full-length human proteins, it is often impractical to modify thousands of individual recombinant proteins to print conventional protein arrays (24). Printed proteins are usually immobilized on array substrates through nonspecific hydrophobic/electrostatic interactions, which do not survive modification protocols that often include extended incubation at high temperatures (11, 22). In addition, the exposed amine groups of the lysine residues are prone to reactivity and may result in steric hindrance that mask reactive epitopes.
In this study, we have developed a novel protein microarray platform that enables the examination of AMPAs in high-throughput without the need to modify individually purified recombinant proteins. We used ACPA in RA as a model modification for our development because citrullination exemplifies the challenges of modification with a harsh protocol (55°C for 3 h) as well as the potential benefits to better understand RA by identifying novel ACPAs. Citrullination is a PTM that converts arginine to citrulline catalyzed by peptidyl arginine deiminase (PAD) (25). ACPA have been specifically detected in RA patients and show utility in RA risk assessment and diagnosis (26). ACPA levels parallel with RA disease activity, prognosticate erosive diseases and serve as a surrogate marker for treatment efficacy (27, 28). Clinically, ACPA are assayed using cyclic citrullinated peptides (29, 30), which measures a generalized reactivity with citrulline containing peptides but does not provide information about reactivity to disease-specific RA antigens. Given that CCP+ patients have antibodies against specific citrullinated peptides, it is speculated that specific endogenous citrullinated antigens may drive immune responses and subsequent RA development (9, 31, 32). The identification of these specific antigens in individual RA patients may help elucidate disease pathogenesis (33–35). For example, it was reported that cigarette smoking is primarily associated with the anti-cit-enolase positive subset of CCP+ RA (36). Furthermore, reported diagnostic sensitivities for anti-CCP assay in RA patients range from 68–79% at 86–95% specificity (37, 38). The identification of additional citrullinated antigens and elucidation of their immunodominant epitopes will help develop more sensitive diagnostic assays, and by comparing citrullinated antigens in different diseases, the assays will achieve higher disease specificity (39, 40). We have also developed a complementary enzyme-linked immunosorbent assay (ELISA) validation platform that allows the assessment of antibody reactivity of a large number of patients to full-length citrullinated antigens of interest. Additionally, there is a wide spectrum of disease-related PTMs. We wanted to test the question whether the platform we have developed can be generalized to study antibodies against other PTMs. We also showed proof-of-concept application of our platform for the identification of antiglycosylated antigens. We believe our platform will greatly promote the study of post-translationally modified antigens at a breadth that has not been achieved before to help understand their roles in disease development.
EXPERIMENTAL PROCEDURES
Serum Samples
All serum samples were collected after receipt of informed patient consent at Benaroya Research Institute under protocols approved by the Institutional Review Board at Arizona State University and Benaroya Research Institute. 8.5–10mls of serum were collected in a red or tiger-top vacutainer tube via venipuncture and the blood collection tubes contained no anticoagulants, preservatives, or protease inhibitors. Collected samples were processed within 2 h of receipt and frozen immediately at −80 °C. If collected samples were not able to be processed immediately in the above mentioned timeline, the samples were stored at 4 °C overnight and processed immediately the following morning. A total of 30 serum samples from three different groups (CCP+ RA (10), CCP- RA (10), and healthy controls (10)) were used for the study on protein array and initial testing on ELISA. Two CCP- RA subjects (S341 and S451) were found to have ACPA reactivity on protein array, and upon re-review of their charts they were found to be CCP+ on clinical assays performed after initial sample collection. A total of 150 samples (CCP+ (50), CCP- (50) and healthy controls (50)) were used for further validation of novel ACPAs and studying anti-cit-MBP using ELISA. Sample information for serums used in validation set is summarized in Table I and detailed information of all samples are available in supplemental Table S1.
Table I. Sample information for the validation set.
| Characteristics | CCP+ | CCP- | HC | p value |
|---|---|---|---|---|
| No. subjects | 50 | 50 | 50 | |
| Mean age | 52.22 (18–82) | 53.64 (19–89) | 52.54 (19–81) | 0.87 (one-way ANOVA) |
| Gender | ||||
| Male | 7 | 7 | 7 | 1 (Chi-square test) |
| Female | 43 | 43 | 43 | |
| No data | 0 | 0 | 0 | |
| Race | ||||
| White | 49 | 48 | 50 | 0.77 (Fisher's exact test) |
| Asian | 0 | 0 | 0 | |
| Indian | 0 | 1 | 0 | |
| other | 1 | 0 | 0 | |
| No data | 0 | 1 | 0 |
DNA Preparation and MBP Deletion Mutants and Tiling Fragments Construction
All genes of interest were cloned in the nucleic acid-programmable protein array (NAPPA) compatible expression vectors, pJFT7_nHALO or pJFT7_cHALO. Plasmid DNA was prepared and mixed with NAPPA printing buffer prior to printing as previously described (41). These expression vectors allow the in vitro expression of proteins of interest with a terminal HaloTag.
C-terminal deletion mutants for both myelin basic protein (MBP) isoforms and fibrinogen alpha chain (FGA) were designed based on the following rule: if two arginines were separated by fewer than 9 amino acids, they were treated as a unit; if two arginines were separated by more than 9 amino acids, the amino acids in the middle was used to divide the two arginines into two mutants (supplemental Table S2). These rules were designed so that enough numbers of native amino acids surrounding arginines was maintained in our deletion mutants to facilitate their recognition after citrullination by serum antibodies. This also limited the total number of deletion mutants to be assayed for this preliminary study. Sequences for all mutants are shown in supplemental Table S2. Based on these rules, primers for each mutant were designed (Integrated DNA Technologies, Coralville, Iowa) and mutant genes were PCR amplified and cloned into pJFT7_nHALO expression vector using Gateway recombinational cloning as previously described (42). In total, eight deletion mutants were constructed for each MBP isoforms.
Contra Capture Protein Array Production
The conceptual design of contra capture protein arrays is illustrated in Fig. 1 and described in protein capture and citrullination on arrays in the results section. Briefly, plasmid DNA encoding proteins with a HaloTag are printed in polydimethylsiloxane (PDMS) wells followed by in vitro protein expression in the wells using a cell free expression system. All the PDMS wells during expression are sealed using a hydrogel glass slide coated with HaloTag ligand. Proteins expressed in the wells with HaloTag are covalently bound to the HaloTag ligand on the cover glass slide. After the capture of expressed proteins, the cover slide is used for subsequent PTM and screening studies.
Fig. 1.
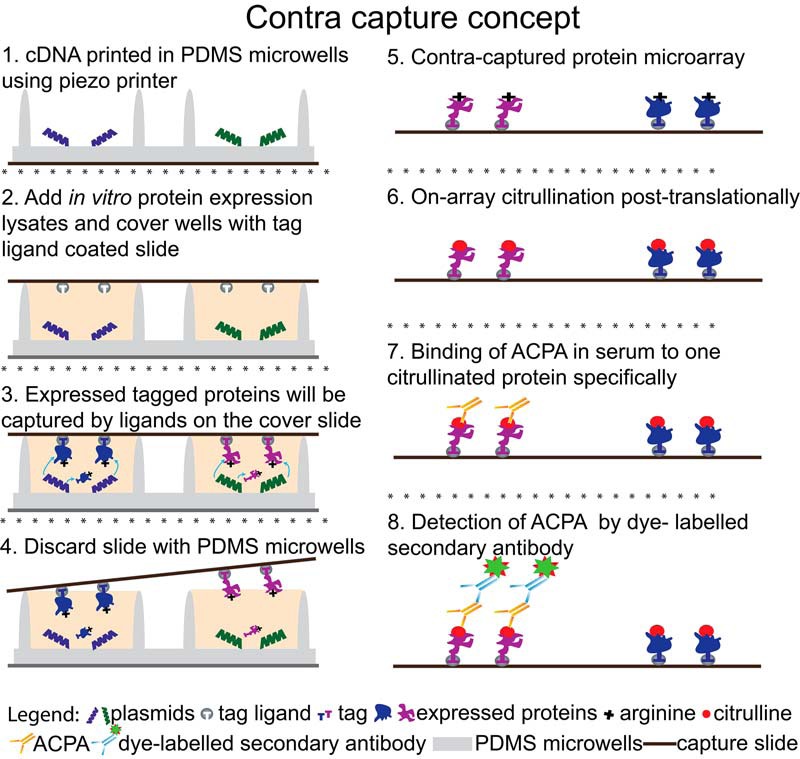
Principle of the contra capture protein array platform.
PDMS Microwell Substrate Production
Fused silica glass slides (25mm × 75 mm) with etched pillars from Trianja Technologies were used as the negative pattern to create PDMS microwells. PDMS substrates were prepared using Sylgard 184 silicone elastomer kit (Dow Corning, Auburn, Michigan) with a mixing ratio of 10:1 (elastomer to curing agent, w/w) and poured onto the fused silica glass slides with etched pillars followed by the application of a regular microscopic glass slide on the top. The microscopic glass slide served as the solid support for the plastic PDMS substrate and was pretreated with 1200 OS primer (Dow Corning) to enable it to bind with the PDMS during curing. We designed an aluminum fabrication fixture to hold the fused silica glass slide with pillars and the microscope glass slide in position with exactly a 2 mm thick PDMS in between during curing. In this way, we were able to produce PDMS substrates with microwells with micron precision fused onto a glass slide for easy manipulation and minimal distortion during down streaming process. After curing at 60 °C for 3 h, PDMS microwell substrates were cut out of the aluminum mold and cleaned with 70% ethanol to remove any residual organic debris on the surface. The overall size of one PDMS well is 650 μm, where the inner well is 450 μm and the walls which confined every well to itself is 100 μm. The well has a depth of 85 μm.
For functionalization with amine group, PDMS microwell substrates were immersed in 2% solution of amino-propyl-triethoxy-silane (APTES) (Thermo Scientific) in 95% ethanol for 30 min followed by a thorough rinse with 95% ethanol and DI water. APTES treated-PDMS microwell substrates were dried using nitrogen and stored in a nitrogen chamber until use.
High Speed Piezo Printing in Microwells
Piezo printing was done using an au302 piezo dispense system (www.engineeringarts.com) with an integrated alignment system for microwells. The alignment system consists of a micrometer angular alignment fixture, look down camera, transfer arm and vacuum tray. PDMS microwells slides were aligned one at a time on the alignment fixture using the look down camera and then transferred using the transfer arm to the vacuum tray where it was held in place. Plasmid DNA was then dispensed, in the aligned PDMS microwells on the vacuum tray, “on-the-fly” using 6 dispense head. Each microwell is filled with 4800 picoliters of printing solution (Plasmid DNA+ printing buffer). Following the piezo printing, NAPPA PDMS slides were stored in nitrogen containers until use.
Protein Expression and Capture
PDMS microwell substrates were blocked with superblock (Thermo Scientific) for 15 min at room temperature with gentle rocking and rinsed thoroughly with deionized water. After the application of in vitro protein expression lysates (Human in vitro transcription and translation kit- Thermo Scientific) into the PDMS wells, the wells were covered with a HaloTag ligand (Promega, Madison, WI) coated hydrogel slides (SCHOTT). The hydrogel slide and the PDMS microwell substrate were held together by placing them between the plates of the hybridization chamber DT-1001 (Die-Tech, York Haven, Pennsylvania) clamped together using screws from DT-2002 (Die-Tech) (43). The substitution of screws helped us to accommodate the PDMS microwell substrate and capture slide setup (∼3.5 mm) better between the hybridization plates. This setup was then placed at 30 °C for 3 h and proteins expressed in the wells were covalently immobilized on the HaloTag ligand coated hydrogel slides through the HaloTag on each protein. After disassembly, contra captured protein arrays, i.e. the hydrogel slide immobilized with individually addressable HaloTagged proteins, were rinsed with an appropriate buffer and ready for the down-stream process.
Citrullination of FGA or H3 in Solution and Detection by Commercial Antibodies
FGA or histone 3 (H3) were expressed using in vitro protein expression kit from Thermo Fisher, Waltham, Massachusetts. Expression mixture was incubated with Halolink beads (Promega) to pull down HaloTagged FGA or H3. After washing, HaloTagged FGA or H3 bound on the beads were citrullinated by Peptidyl Arginine Deiminase 2 from rabbit muscle (rmPAD2, P1584 Sigma, St. Louis, Missouri) at 40U/mg in the citrullination buffer (100 mm TRIS, pH 7.6, 10 mm CaCl2, 5 mm DTT (Sigma) at 55 °C for 3 h (11). Citrullinated FGA or H3 were released from the beads by incubation with TEV protease (Promega) at 4 °C overnight. Citrullinated FGA or H3 in the supernatant was collected and separated by SDS-PAGE and probed with appropriate antibodies: anti-FGA (CA-1023 Cambio, Cambridge, United Kingdom), anticitrullinated fibrinogen antibody (MQ13.102 Modiquest, Moleneind, The Netherlands), antihistone H3 XP (4499 Abcam, Cambridge, MA) and anti-cit-Histone H3 (ab5103 Abcam).
Specific Native and Citrullinated Protein Detection On Arrays
Specific proteins such as FGA, fibrinogen beta chain (FGB), fibrinogen gamma chain (FGG) and vimentin (VIM) on the arrays were detected using antibodies: anti-FGA (CA-1023 cambio), anti-FGB (HPA001900 Sigma), anti-FGG (CA-1027-RA cambio) and anti-VIM (ab8978 abcam). Contra capture protein arrays were citrullinated by rmPAD2 (40U/mg) in the citrullination buffer at 55 °C for 3 h (11). After citrullination, arrays were either probed with antibodies against anti-citrullinated fibrinogen antibody or anti-cit-Histone H3 to confirm successful on-array citrullination. To assay sero-reactivity to citrullinated proteins, citrullinated arrays were assayed with 30 serum samples (CCP+, CCP- and healthy control) with dilution of 1:300 overnight at 4 °C with gentle rocking. The presence of antibodies at each spot was detected by Alexa Fluor 647 labeled Goat α Human IgG (A21445 Invitrogen, Carlsbad, CA).
On Array Glycosylation and Detection
Genes encoding protein fragments were spotted to display positive and negative controls for Tn-glycosylation on contra capture arrays. These included: (1) a 60-mer MUC1-TR comprising 3 repeats of the 20 AA tandem repeat (PDTRPAPGSTAPPAHGVTSA) of MUC1; (2) an NRP2 fragment (SKPTVETLGPTVKSEETTTP); (3) a POMC fragment (PGNGDEQPLTENPRKYVMGHFR); (4) a CD55 (SRTTKHFHETTPNKGSGTTS) fragment, (5) a wild type APOE fragment (VEQGRVRAATVGSLAGQP), and (6) a mutant APOE fragment (VEQGRVRAAAVGALAGQ) with the glycosylation site Thr and Ser mutated to Ala. Tn glycosylation was performed using GalNac-T2, T3 or T11 (kindly provided by Dr. Henrik Clausen) with Uridine 5′-diphospho-N-acetylgalactosamine disodium salt (U5252 Sigma) (2 mm) and MnCl2 (Sigma) (10 mm) in glycosylation buffer (25 mm TRIS (MP Biomedicals, Santa Ana, CA), pH7.5, 10 mm MnCl2, 0.25% Triton X-100 (Sigma) at 37 °C for 18 h (22, 44).
Successful on-array Tn glycosylation of in vitro expressed protein was confirmed by biotinylated lectin Vicia Villosa Lectin (VVA, B-1235, Vector laboratories, Burlingame, CA) followed by Alexa Fluor 555-conjugated streptavidin.
Data Analysis
Arrays were scanned using Tecan PowerScanner and intensity data were quantified using the ArrayPro image analysis software (MediaCybernetics). Local background subtracted median intensity for replicate spots were used for the analysis. Spots with obvious defects were excluded and corresponding spots from replicate arrays were used for analysis instead. GraphPad Prism software was used for generating jitter plots and un-paired t test analysis. Heat maps of sample reactivity to proteins were generated using MeV software and a hierarchical cluster analysis using Pearson correlation was performed on the log transformed data.
ELISA
We adapted rapid ELISA (45) to allow citrullination of target antigen before assessing its sero-reactivity. HaloTag ligand coated 96 well plates (Promega) were preblocked with superblock (Thermo Scientific) overnight. Antigen of interest was produced in vitro as described above. Protein expression mixture was added into the plates and incubated for 1 h at room temperature with shaking at 500 RPM to allow the antigen to bind to the HaloTag ligand in each well. Plates were washed and covalently bound antigens were citrullinated by rmPAD2 at 55 °C for 3 h. Citrullinated antigens were incubated with diluted serum samples (1:1000) overnight at 4 °C followed by the addition of HRP conjugated Goat α Human IgG (Jackson Immunoresearch) with shaking at 500RPM. The plates were developed with the TMB substrate (Thermo Scientific) for 20 min. Absorbance at 450 nm was read on Perkin Elmer EnVision Multilabel Reader.
RESULTS
Concept of Contra Capture Protein Array
Our contra capture protein array was built on NAPPA platform that enables the display of thousands of full-length human proteins without the need for labor-intensive protein expression and purification (41). Proteins are synthesized from printed plasmids bearing genes encoding proteins of interest using the in vitro protein expression system. We initially attempted to perform citrullination of proteins displayed on standard NAPPA followed by probing with serum samples for responses. However, we encountered high background (data not shown). We believed this was at least partially because of the reagents in the NAPPA printing mixture required for DNA immobilization and in vitro protein synthesis as well as impurities in the plasmid DNA preparation.
To address this issue, we reconfigured the design of our array production to separate the printed DNA template from the captured protein (Fig. 1). We printed cDNAs encoding proteins of interest fused to HaloTag into microwells. HaloTag is a modified halo alkane dehalogenase designed to covalently bind to synthetic ligands (HaloTag ligands). After the application of cell free protein expression lysates, these microwells were sealed with a cover slide coated with HaloTag ligands that bind to the HaloTag on each expressed protein. Proteins expressed in each microwell were captured onto the cover slide and then separated from the background-inducing NAPPA printing mixture. Proteins displayed on the cover slide created a protein microarray that could be citrullinated and assessed for antibody binding in RA patients' serum samples.
We implemented this concept in micro PDMS wells. PDMS is a well-established plastic bio-compatible material. PDMS microwells were produced by the use of a fused silica master mold etched with thousands of pillars in the center surrounded by a moat shaped structure. Different DNAs were printed into each well precisely and accurately using a noncontact piezoelectric arrayer (46).
Protein Capture and Citrullination on Arrays
To demonstrate the specific confined capture of proteins expressed in PDMS microwells and the citrullination on contra capture arrays, we produced an array with 24 proteins (supplemental Fig. S1) including the best known citrullinated antigens as positive controls: histones, MBP, FGA, FGB, FGG, VIM, enolase-1 (ENO1), and clusterin (CLU). Epstein-Barr Nuclear Antigen (EBNA) was included as an immune-response positive control because most adults have antibodies against EBNA. Proteins were specifically captured and citrullinated using rmPAD2 on the cover slides as demonstrated by antibodies that recognize either native or citrullinated proteins (supplemental Fig. S2A-S2H). Cross-reactivity of anti-FGA with CLU and anti-FGG with FGB and cyclic AMP-responsive element-binding protein 3-like protein 1 (CREB3L1) was also observed (supplemental Fig. S2A and S2C).
Serum Reactivity to Contra Captured Protein Arrays
To demonstrate the utility of our novel platform in assessing antibody reactivity against citrullinated antigens in RA patients, we tested 30 serum samples against the above described 24-protein arrays in their native and citrullinated forms (Fig. 2). Three different categories of serum samples (supplemental Table. S1) were used: healthy controls, CCP+ RA patients, and CCP- RA patients. Representative array images are shown in Fig. 3.
Fig. 2.

Heat map of quantitative analysis of sero-reactivity to 11 known citrullinated auto-antigens and Epstein-Barr Nuclear Antigen (EBNA) on contra captured protein arrays. Thirty serum samples (12 CCP+ RA patients, 8 CCP- RA patients, 10 healthy controls) were used for this study. The left half of the heat map shows sero-reactivity to native protein arrays and the right half shows sero-reactivity to citrullinated protein arrays. EBNA was used as a positive sero-profiling control because most adults show sero-positivity to this antigen. Blue, low reactivity; red, high reactivity.
Fig. 3.
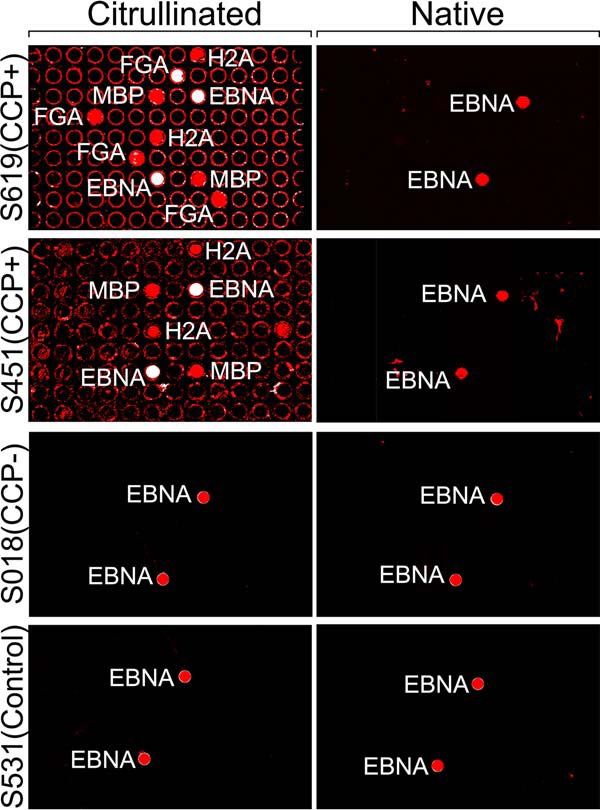
Representative images of native and citrullinated arrays probed with serum samples. All samples showed reactivity to both citrullinated and native EBNA.
Most CCP+ sera showed strong reactivity to citrullinated MBP and FGA. Interestingly, no serum showed strong reactivity to fibrinogen alpha chain shorter version (FGAs) as seen for FGA. In addition, some samples showed responses to other citrullinated proteins, including moderate reactivity to Histone H2A type 1 (H2A) and ENO1. The fractions of samples reactive to other citrullinated autoantigens such as VIM and Histone H2B (H2B) were lower than those reported in the literature (47). Sampling bias due to our small sample size and over-representation of rheumatoid factor negative (RF-) RA subtypes in our samples (50% versus ∼25% in the typical RA population (48)) may have contributed to the difference. CCP+ serum did not react strongly to any native proteins except EBNA (Fig. 3). All healthy controls and a majority of the CCP- serum samples showed minimal or no reactivity to either native or citrullinated form of proteins except EBNA. Unexpectedly, two serum samples (S451, S341), which was clinically classified as CCP-, showed citrullination-specific reactivity, the former reacting with citrullinated MBP and H2A and the later with cit-MBP (Fig. 2, 3). These findings prompted a review of the classification of these subjects; S451 and S341 were subjects who on chart review were found to be CCP+ through clinical testing, in the case of S451 these findings occurred after the research sample was obtained. This change of classification caused a redistribution of our sample set to 12 CCP+ and 8 CCP- samples instead of the equal number originally designed, however it also brings to light the ability of this assay format to observe individualized immune responses to different citrullinated proteins and the potential to improve assay sensitivity over the clinically employed anticyclic citrullinated peptide assay.
ELISA Validation of Contra Capture Array Data
Protein arrays efficiently assay many proteins in parallel. Conversely, to test many samples against a few targeted antigens to generate clinical assay-quality data, we turned to enzyme-linked immunosorbent assay (ELISA) (19). Conventional ELISA requires purified recombinant proteins and is not compatible with the harsh citrullination assay conditions. Thus, we developed a HaloTag version of RAPID-ELISA (Methods) and confirmed the array data for MBP for all 30 samples by a HaloTag version of RAPID-ELISA (Fig. 4).
Fig. 4.
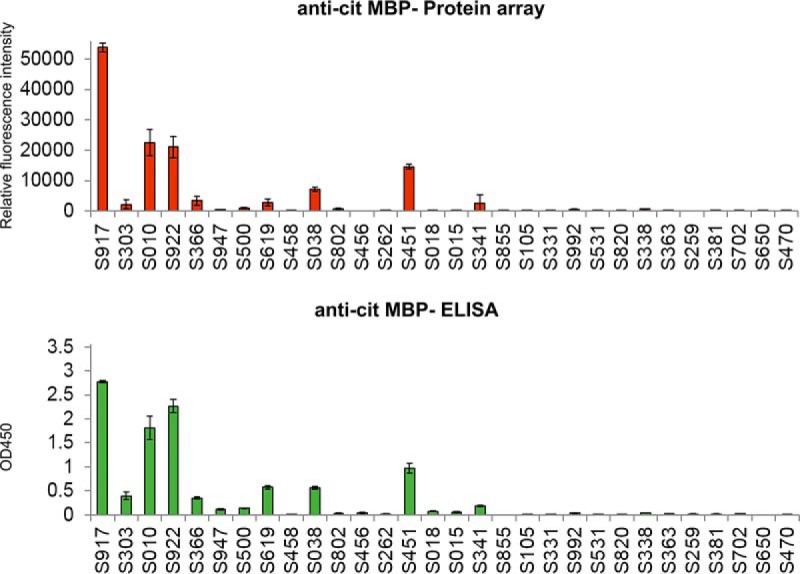
Quantitative comparison of sero-reactivity of MBP. Results obtained from the contra capture platform versus the ELISA platform against anti-citrullinated MBP for 30 serum samples. Please refer to Methods for assay details.
Profiling Anticitrullinated Antigen Antibodies in RA Patients
We applied our novel array platform to profile ACPAs with RA patient sera. For this purpose, we printed a total of 190 genes (supplemental Table. S3) into PDMS microwells (supplemental Fig. S3). We selected 96 of the 190 genes based on their reported high or differential expression in synovial fluid from RA patients (49–60). The rest were randomly selected from our human gene collection (http://dnasu.org). Citrullinated protein arrays were probed with all CCP+ and CCP- serum samples from RA patients (Fig. 5A). Representative images for two CCP+ serum samples (S366 and S451) are shown Fig. 5B.
Fig. 5.

Sero-profiling of ACPAs in RA patients on contra capture protein arrays against ∼190 antigens and validation of novel ACPAs identified on contra capture arrays by ELISA. A, Heat map depicting overall reactivity of 20 CCP+ and CCP- serum samples to 190 genes printed on array. CCP- negative samples were assay as pools of two samples as annotated on the top of the figure. Blue, low reactivity; red, high reactivity. B, Example array images probed with CCP+ RA patient serum samples (S366 and S451). C, Blinded validation of selected novel ACPAs by ELISA using an independent set of 150 sera comprising 50 CCP+, 50 CCP- and 50 control samples. HC, healthy control. * indicates t test p < 0.05, **, p < 0.01 and ****, p < 0.0001. D, Heat map depicting sero-reactivity against citrullinated antigens assayed by ELISA in different groups of the independent sample set.
CCP+ serum samples generally displayed antibody reactivity to more citrullinated proteins than CCP- serum samples. Eleven (11) out of 12 CCP+ samples are clustered on the left side of the dendrogram and the CCP- samples are clustered toward the right of the dendrogram; however, the distinction between the 2 clusters is subtle (Fig. 5A). Among the CCP+ samples, a subset of CCP+ samples (S619, S922, and S917) had markedly higher reactivity than the others (S341, S947, and S303). Some CCP- samples (S105, S331) showed higher reactivity compared with other CCP- samples. It is worth noting that most samples, including CCP-, had their unique reactivity patterns.
We provided the first glimpse of reactivity patterns of 20 RA patients against ∼190 individual citrullinated antigens. We observed two clusters with subtle distinctions of samples clinically classified as CCP+ and CCP-; additionally, each sample seemed to have its own unique reactivity pattern (Fig. 5A)
Among the 190 citrullinated antigens, the majority were not recognized by RA serum. However, we were able to observe antibodies to novel citrullinated antigens from this pool of antigens. These included antibodies against citrullinated FEN1, SPP1 and IGFBP6 that were present in several RA samples. There were also antibodies against other antigens that were found only in a single sample. These included Bcl-2 homologous antagonist/killer (BAK1) that sample S917 had high reactivity to and trefoil factor 1 (TFF1) for which samples S619 and S500 showed high reactivity. We also observed reactivity to novel citrullinated proteins for CCP- samples. For example, both pooled CCP- sample of S015/S855 and S802/S456 reacted with RAD51 while only one CCP+ (S010) reacted less strongly with RAD51.
Based on the array data, we selected 11 antigens (SPP1, FEN1, IGFBP6, IGF1, STC2, cyclin-A1 (CCNA1), calumenin (CALU), F-actin-capping protein subunit alpha-1 (CAPZA1), protein S100-A11 (S100A11), peroxiredoxin-2 (PRDX2), and glutamate decarboxylase 2 (GAD65)) that showed high antibody reactivity against their citrullinated form in several RA patients and assessed the antibody reactivity of their citrullinated form in all thirty clinical samples by ELISA (supplemental Fig. S4). Antibodies against citrullinated SPP1, FEN1, IGFBP6, IGF1 and STC2 showed the best differential reactivity between RA patients and healthy controls suggesting the identification of 5 novel autoantibodies against citrullinated antigens (supplemental Fig. S4). Of these, SPP1 is a protein selected based on its high expression in synovial fluid in patients with erosive RA. To our knowledge it is not known whether SPP1 is citrullinated in RA. Antibodies against citrullinated SPP1 showed the best differential reactivity between RA patients and healthy controls.
To validate these novel ACPA, we performed a blinded study of responses in ELISA to SPP1, FEN1, IGFBP6, IGF1, and STC2 in an independent set of 150 serum samples (supplemental table 1) from groups of subjects with three different clinical characteristics: CCP+ RA patients (50); CCP- RA patients (50); and healthy controls (50). We observed significant differences for the antigens' reactivity between the CCP+ RA and healthy control groups (Fig. 5C and 5D). At 95% specificity, the sensitivity of predicting RA with the new ACPAs was 50%, 42%, 56%, 52%, and 66% in CCP+ RA patients for SPP1, FEN1, IGFBP6, IGF1 and STC2, respectively. These results compare favorably with historical citrullinated antigens such as ENO1 (46% sensitivity), citrullinated vimentin (anti-Sa) (22–43% sensitivity), and fibrinogen (56–75%) (9, 61–63) Surprisingly, response levels to these five ACPAs were also higher in a number of CCP- RA patients, who were clinically categorized as not responsive to standard citrullinated antigens. The sensitivities in the CCP- RA patients were 12%, 10%, 18%, 12%, and 12%, respectively (Table II). We also assayed anti-cit-MBP antibodies in these samples by ELISA. At 95% specificity, the sensitivity for cit-MBP was 82% for CCP+ serum and 22% for CCP- serum samples.
Table II. Sensitivities of novel antigens.
| Antigen | Sensitivity at 95% specificity |
|
|---|---|---|
| CCP+ | CCP- | |
| MBP | 82% | 22% |
| SPP1 | 50% | 12% |
| FEN1 | 42% | 10% |
| IGFBP6 | 56% | 18% |
| IGF1 | 52% | 12% |
| STC2 | 66% | 12% |
Among these six ACPAs, there was a significantly higher chance for subjects who had a positive antibody response to one antigen (greater than 95 percentile of healthy controls) to have responses to the others. Quantitatively, anti-cit-IGFBP6, anti-cit-STC2 and anti-cit-IGF1 had higher correlations but the correlations between other antigens were relatively low.
Epitope Mapping
It is often useful to determine which epitopes are recognized by antibodies in an immune response. For epitope mapping, ELISA is usually performed on overlapping peptides. This requires the synthesis of multiple native and modified peptides and is a costly process. Recognizing that our platform can manipulate epitopes at the DNA level, followed by display and citrullination of the peptides expressed in vitro, we asked if we could map immune-dominant citrullinated epitopes for antigens of interest. We used MBP as an example. We were particularly interested in mapping the epitope(s) for MBP, because it represented a poorly studied citrullinated antigen for RA and there was only one report of anti-cit-MBP in RA patients (64). Two isoforms of MBP were used for epitope mapping. Isoform 1 (Uniprot: P02686–2) represents the N-terminal 197 AAs (N1- C197) and isoform 2 (Uniprot: P02686–6) the C-terminal 160 AAs (N134-C293) of the full length MBP, with a partial overlap between them (MBP 134–197; Fig. 6). C-terminal serial deletion mutants of these two MBP isoforms were constructed, expressed, citrullinated, and probed with MBP reactive serum samples on arrays.
Fig. 6.
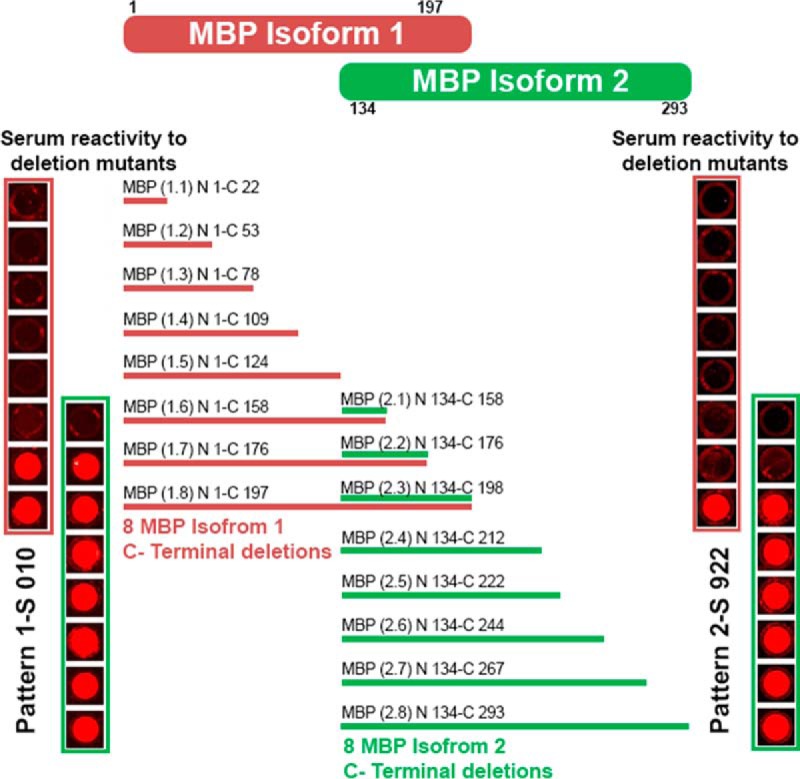
Epitope mapping for MBP. Eight C-terminal deletion fragments were constructed for each isoform of MBP. Two response patterns were observed. In pattern 1, reactivity to deletion mutants MBP 1.7, MBP 1.8, MBP 2.2 and MBP 2.3 were observed. In pattern 2, reactivity to deletion mutants MBP 1.8 and MBP 2.3 were observed, but not to MBP 1.7 and MBP 2.2. MBP 1.7 and MBP 2.2 shared the same sequence and MBP 1.8 and MBP 2.3 shared same sequence.
We observed two representative immune response patterns to these MBP deletion mutants. Within each pattern, the reactivity to isoform 1 and isoform 2 deletion mutants was consistent in the overlapping regions (Fig. 6). In pattern 1, a response was always detected for polypeptides including MBP N144-C176 with the corresponding sequence HGSKYLATASTMDHARHGFLPRHRDTGILDSIG and in pattern 2, response was observed for polypeptides MBP N168-C198, which includes the sequence DTGILDSIGRFFGGDRGAPKRGSGKVSSEE. To pinpoint the reactive epitope in the region N131-C205, we constructed tiling fragments of MBP T (2.1) N134-C158, T (2.2) N144-C176, T (2.3) N168-C198, and assayed antibody reactivity using all serum samples. We found several serum samples reacting to region MBP N168-C198 and the rest to region MBP N131-C205 (supplemental Fig. S5). No reactivity was seen on other deletion mutants of either isoform lacking this sequence. Thus we were able to use this platform to rapidly identify two distinct B cell epitopes in citrullinated MBP.
Contra-capture Array to Assess AAbs Targeting Glycosylated Proteins
Protein glycosylation is the most common and complex PTM (65). Incomplete synthesis of O-glycans leads to the expression of truncated O-glycans Tn (GalNAc), STn (NeuAc (α2,6) GalNAc) and T (Gal (β3) GalNAc) that become enriched in cancer (66). There are more than 15 different GalNac-transferases (GalNac-Ts) that catalyze the addition GalNac to a Thr or Ser residue with different yet overlapping peptide substrate sequence specificity. To demonstrate on-array O-glycosylation, we produced arrays with 24 different proteins (or protein fragments) including MUC1-TR encoding the 20 aa repeats of MUC1, which is a universal O-glycosylation substrate (supplemental Fig. S6). General Tn-glycosylation was detected by the lectin VVA. There was no signification Tn-glycosylation on freshly produced proteins (Fig. 7A). We tested three different GalNac-Ts (T2, T3 and T11) on our arrays. MUC1-TR was Tn-glycosyalted by all three GalNac-Ts (column 1, row 2 (Fig. 7A, green boxes)). On these arrays, we have spotted genes encoding specific substrates for the three different GALNTs based on studies on synthetic peptides, an NRP2 fragment for GalNac-T2, a POMC fragment for GalNac-T11, an APOE and a CD55 fragment for GalNac-T3 (44, 67, 68). Besides wild type APOE, we have also spotted a gene encoding an APOE mutant with the glycosylation sites Thr and Ser by GALNT3 mutated to Ala that cannot be glycosylated by GalNac-T3. All but the mutant were specifically glycosylated by T2, T3 and T11 on the contra capture arrays as shown in Fig. 7A. GalNac-T3 could specifically glycosylate CD55 and wild type APOE but not the APOE mutant. GalNac-T11 could specifically glycosylate POMC. The expressed NRP2 fragment displayed on the array was also glycosylated by GalNac-T2 and T3 although a corresponding synthetic peptide was glycosylated only by GalNac-T2 in earlier studies (68). This may be due to a better presentation of the NRP2 fragment on the array instead of as an isolated synthetic peptide (68).
Fig. 7.

Detection of antibodies against Tn-glycosylated proteins on contra capture arrays. A, Tn glycosylation by GalNac-T2, T3 and T11. After protein expression, arrays were untreated (native) or treated with one of three GalNac-Ts, labeled as GalNac-T2, GalNac-T3 or GalNac-T11, respectively. Features in boxes represents positive and negative control proteins. Green boxes on GalNac-T2, -T3 and -T11 represent MUC1-TR. The yellow box on GalNac-T2 represents NRP2 and that on GalNac-T11 represents POMC. The two yellow boxes on GalNac-T3 represent APOE (right) and CD55 (left), respectively. The APOE mutant (unmodifiable) is shown in a purple box. B, VVA and anti-HaloTag staining of arrays displaying 87 different proteins Tn-glycosylated by GalNac-T3. C, Antibody profiling against the 87 proteins shown in (B) with (left column) and without (right column) Tn-glycosylation by GALNT3. S1571-IgA and S1571-IgG were the same array detecting AAb of IgA and IgG classes in sample S1571, simultaneously. Same for S1660-IgG and S1660-IgA.
After demonstration of successful protein glycosylation, we assessed reactivity to a set of 87 proteins (supplemental Table S4), with and without Tn-glycosylation, for two ovarian cancer patients S1571 and S1660 (Fig. 7B and 7C). Protein display and successful Tn-glycosylation by GALNT3 was confirmed by anti-HaloTag and VVA staining, respectively (Fig. 7B). Both IgG and IgA classes of autoantibodies were assayed simultaneously (Fig. 7C). Patient S1571showed higher antibody reactivity of IgA class to Tn-glycosylated MUC7 and FER, and patient S1660 showed higher antibody reactivity of IgG class to glycosylated MUC7 and RPA2. It is interesting to note that patient S1660 had high IgG reactivity to Tn-glycosylated MUC1-TR but patient S1571 had higher IgG reactivity to native MUC1-TR. Taken together, these data demonstrate successful glycosylation of proteins on contra capture protein arrays, as well as glycosylation-specific antibody responses in both directions.
DISCUSSION
Post-translational modifications of protein antigens profoundly affect the development of autoimmunity, particularly in the setting of RA where the ACPA has become a marker of disease risk as well as a diagnostic tool. However, neither the breadth of citrullinated antigens nor the differences of response signatures between patients have been comprehensively explored. A comprehensive examination of the PTM autoantigenome would facilitate our understanding of the role of this form of autoimmunity in the pathogenesis of disease, the development of more sensitive and specific diagnostic assays, the stratification of patients for more appropriate treatment and the design of novel prevention and clinical management strategies.
Using ACPA in RA patients as a model, we applied serum samples to arrays displaying hundreds of citrullinated proteins in parallel. We were able to detect specific antibody reactivity to known citrullinated proteins in RA patients. We provided the first glimpse of reactivity patterns of 20 RA patients against ∼190 individual citrullinated antigens. We observed two clusters of samples clinically classified as CCP+ and CCP-; additionally, each sample seemed to have its own unique reactivity pattern (Fig. 5A). We believe future studies to profile longitudinal samples against an expanded set of thousands of antigens and track the changes of anticitrullinated antibody-ome during RA development will provide new biomarkers that have prognostic value to predict disease severity or treatment efficacy when these profiles are analyzed in the context of other clinical parameters and promote our understanding the autoimmunity in RA development beyond that can be provided by a single CCP test. We are encouraged to identify subjects who were CCP- and yet still responded to modified proteins on the array suggesting the opportunity to improve sensitivity of detection of ACPA in RA patients by expanding the test antigen set (Fig. 5).
Until now, there are no reliable methods to study antibodies that target specific PTMed antigens at a proteome level. In this study, we have developed a platform that produces fresh proteins with associated modifications using human ribosomes and chaperone proteins. Once displayed, these proteins can be probed with patient serum or other molecules to identify interactions of interest. Compared with classical proteomics approaches, such as 2D gels and Western blotting, this approach supports the rapid screening of hundreds of samples, and requires only a few microliters of serum. Protein identification is instantaneous based on the location on the array and this method is not subject to protein abundance bias, because each protein is displayed separately at similar abundance levels (41).
Our contra capture protein array platform builds upon NAPPA and enables convenient production of customized protein arrays with any known DNA sequence. Our method avoids the time consuming, labor intensive and technologically challenging need for full-length protein purification and modification in individual sample tubes. HaloTagged proteins were simultaneously but individually expressed in solution in PDMS microwells, covalently captured by HaloTag ligand on a separate glass slide, and then subjected to PTM in a process that takes only several hours. Currently one protein array is produced using a PDMS microwell array. Although we have not yet tested this approach, it might be even possible to produce multiple contra capture protein arrays using the same PDMS microwell array, in a manner similar to that reported by Stoevesandt et al. (69). This will likely require some development, including retaining the transcription efficacy of spotted plasmid DNA, filling fresh lysates into PDMS wells effectively, and re-establishing a seal between the new capture surface and the PDMS array and this could allow multiple arrays to be produced from the same set of printed DNAs. However, in standard NAPPA, proteins are displayed among reagents required for capture and plasmids prepared from E. coli at each feature. Components of the printing mixture usually do not interfere with protein interaction and antibody assays. However, they may interfere with enzymatic modification and subsequent detection that may contribute to unusually high background and low signals. Thus, an important improvement is the capture of naked protein in folded conformation in a manner that isolates it from the printed mixture and covalently attaches it to the solid surface. This removes any interference from the printing mixture and allows complex chemical modifications of captured proteins under harsh conditions.
Another important advance was the addition of the HaloTag capture step for the proteins. On conventional protein arrays, the use of non-covalent capture of proteins to the array matrix, such as by hydrophobic interactions or electrostatic interactions, creates an attachment that cannot withstand stringent conditions, like elevated temperatures or high salt conditions. Whereas, nonspecific covalent attachments, such as between lysines and amine reactive surfaces, will bring proteins too close to the matrix surface and tend to sterically block enzymatic access to the proteins. Our strategy employed a covalent attachment through a single specific site on the fusion tag that is distant from the important epitopes on our target protein. We tested this strategy using citrullination, which requires both enzymatic modification and a reaction temperature above the physiological temperature, as an example that demonstrates the versatility of our platform.
Whereas DNA array to protein array (DAPA) also immobilizes expressed proteins away from the production site (70), the lack of covalent protein capture would limit the ability to use high temperature modification protocols and the free diffusion of proteins prior to capture may limit the potential feature density due do to cross contamination (46). The citrullinated peptide/protein arrays developed by Robinson and Utz, which display tens of in vitro purified antigens after citrullination in solution as well as hundreds of citrullinated synthetic peptides has proved useful for the refined analysis of known ACPAs in autoimmune diseases (23, 71). However, its reliance on known citrullinated antigens limits its utility in the discovery of novel antigens. Nonetheless, their modified peptide/protein arrays would provide a complementary and useful platform for follow up studies after the discovery of novel antigens and their epitopes on our contra capture arrays.
Anti-cit-MBP has the highest prevalence among the ACPAs that we assayed in this study. Citrullinated MBP has been reported in other diseases such as multiple sclerosis (MS) and other demyelinating diseases and is believed to be involved in the pathogenesis of CNS autoimmune diseases. Previous studies using arrays spotted with cit-MBP peptides detected anti-cit-MBP-peptide antibodies in the experimental autoimmune encephalomyelitis (EAE) mouse model for MS but not in collagen-induced arthritis (CIA) mouse model (71). Here, we detected anti-cit-MBP antibodies in a large fraction of RA patients (∼80% of CCP+ samples), which agrees with a previous report on the expression of MBP in the synovial lining and the presence of anti-cit-MBP in RA, although the prevalence of response has not been previously determined (64).
We acknowledge that more experiments will be required to confirm that cit-MBP was the triggering antigen for the production of anti-cit-MBP antibodies. However, in conjunction with the fact that the prevalence to anti-cit-MBP (82%) was much higher than the other known or novel citrullinated antigens (40–60%) in our sample set, it seems likely that these two citrullinated epitopes on MBP may represent novel immunodominant epitopes in RA, whose biological implications warrant further investigations.
We discovered and validated five novel ACPAs in RA patients against antigens SPP1, FEN1, IGFBP6, IGF1 and STC2. Antibodies against citrullinated SPP1 are particularly interesting because SPP1, also known as osteopontin, is a candidate marker elevated in synovial fluid of patients with erosive RA (50), though it has never been shown to be a citrullinated antigen. We observed immune reactivity specific to citrullinated SPP1 in 50% of CCP+ patients and 12% of CCP- patients at a specificity of 95% in our blinded validation study. SPP1 is recognized as a potential proinflammatory cytokine associated with inflammatory processes (50, 54, 58). SPP1 is an extracellular protein containing an Arg-Gly-Asp (RGD) sequence which is up-regulated and secreted by activated macrophages, leukocytes and activated T lymphocytes and is present in extracellular fluid and at sites of inflammation (72). SPP1 has been suggested as a potential mediator of the promotion of joint destruction in RA patients through its interaction with the αVβ3 and αVβ5 integrins (72). Its discovery as a specific antibody target in RA, from among almost 200 candidates, suggests the possibility of a role that it or the immune response to it might play in the disease. It would be interesting to assay for the presence of citrullinated SPP1 in the synovial joints of RA patients.
The physiological relevance of these novel ACPAs warrants future investigation. More importantly we believe that the global profiling of ACPAs against antigens in the human proteome and the mapping of their immunodominant epitopes will provide a more comprehensive picture of ACPA responses in RA. The clinical characteristics of RA are heterogeneous, which may be in part due to the specificity of the immune response directed against post translationally modified antigens. With our contra-capture array platform, we can easily assess reactivity to these immune-dominant antigens/epitopes in longitudinal samples collected from high-risk subjects or in patients with different clinical parameters (such as environmental exposures). This will promote a better understanding of RA pathogenesis and stratification of RA patients into subtypes based on their response signatures, which based on this initial study may impact the CCP- RA population as it is now defined.
Protein arrays display many proteins, which make them suitable to screen thousands of proteins against multiple samples for discovery studies. A key expectation is that they will discover new candidate targets which will need to be validated in many more samples. To address the need to test a small number of candidate antigens against many samples (hundreds to thousands), we adapted this method to produce an ELISA assay that supports protein modification and is compatible with biomarker validation/verification studies. This method dovetails with arrays by exploiting the same plasmid clones, without the need to transfer the gene, and it avoids the need to optimize and execute the expression and purification of recombinant proteins as needed by conventional ELISA. Instead, it uses the same human ribosomal machinery to produce the proteins just-in-time for assay and captures the protein covalently to the surface, allowing harsh treatment if needed. Because the broad availability of ELISA, this method may prove useful to more applications and can be employed to assess target proteins generated from hypothesis-driven approaches or data-driven high-throughput screening as described in this study.
The dysregulation of glycosylation enzymes result in aberrant glycosylation in diseases like cancer. Antibodies against well studied glycosylated proteins, such as MUC1, have been reported; however, their utility in early detection was challenged (73). The lack of an appropriate platform has limited anti-glycosylated protein antibody studies to synthetic peptides. Our successful demonstration of specific glycosylation with GalNac-Ts of different substrate specificities and the detection of glycosylation specific antibodies in cancer patients paves the way for future studies of this important type of autoantibodies.
In summary, we have developed a platform that can assay sero-reactivity to many post-translationally modified antigens in parallel. In this initial proof of concept we screened 190 proteins, and utilized rmPAD2 to determine reactivity in RA sera to known and novel citrullinated protein epitopes. As different PADs have different specificities, we have the opportunity to identify different citrullinated autoantigens if we employ different PADs. The high-throughput nature of protein arrays makes it convenient to assess immune reactivity to citrullinated protein/epitope substrates of different PAD enzymes at the proteome level. This platforms has the capacity to expand to the assessment of ∼2000 different proteins on one slide and with the potential of ∼10,000 by reducing well dimension from the current 600 μm to 300 μm. In addition this platform allows for the modification of proteins to examine other types of posttranslational modification or other diseases that PTMs play important roles. We have successfully performed there PTMs such as glycosylation and acetylation on this novel platform (data not shown). The ability to examine the antibody-ome across the human and microbial proteome promises to deepen and enhance our understanding of the immune response in disease (74, 75).
Supplementary Material
Acknowledgments
We thank Justin Saul and Jie Wang for their expert support on vectors used for protein expression and their technical assistance and Ian Shoemaker for the initial design of the molds used to produce the PDMS substrates. We thank Dr. Henrik Clausen for generously providing enzymes required for glycosylation and guidance provided for on array glycosylation. We thank Eddie James for reviewing the manuscript.
Footnotes
Author Contributions: J.Q. and J.L. conceived, designed, oversaw all of the studies and contributed to analyzing data and writing the manuscript; K.K. designed and performed experiments, analyzed data, and wrote the manuscript; K.B. cloned and prepared all the DNA used in the experiments and performed all ELISA assays; Y.T. performed ELISA for new targets validation; P.K, P.W., A.B. helped in developing a system to produce PDMS substrates, provided input on the engineering aspects of this project and printed DNA in the PDMS wells using the piezo-electric dispenser; B.T. conceptualized the contra capture idea; V.K. was involved in the development of in vitro citrullination; J.B. and J.N. provided samples for this study, helped with the biological aspects of the project and edited of the manuscript.
* This work was funded by the National Institutes of Health (NIH) grant R21 (ZDS0078), partially by National Institutes of Health (NIH) grant R42GM106704 awarded to Engineering Arts LLC. and Virginia G. Piper Center for Personalized Diagnostics at the Biodesign Institute (Arizona State University). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental material.
This article contains supplemental material.
The authors declare no competing financial interests.
1 The abbreviations used are:
- PTM
- post-translational Modification
- ACPA
- anticitrullinated protein antibodies
- RA
- rheumatoid arthritis
- CCP+
- anti-cyclic citrullinated peptide assay positive
- PAD
- peptidyl arginine deiminase.
REFERENCES
- 1. Zaenker P., and Ziman M. R. (2013) Serologic autoantibodies as diagnostic cancer biomarkers–a review. Cancer Epidemiol. Biomarkers Prevention 22, 2161–2181 [DOI] [PubMed] [Google Scholar]
- 2. Anderton S. M. (2004) Post-translational modifications of self antigens: implications for autoimmunity. Curr. Opinion Immunol. 16, 753–758 [DOI] [PubMed] [Google Scholar]
- 3. Cantaert T., De Rycke L., Bongartz T., Matteson E. L., Tak P. P., Nicholas A. P., and Baeten D. (2006) Citrullinated proteins in rheumatoid arthritis: crucial.but not sufficient! Arthritis Rheumatism 54, 3381–3389 [DOI] [PubMed] [Google Scholar]
- 4. Brahms H., Raymackers J., Union A., de Keyser F., Meheus L., and Luhrmann R. (2000) The C-terminal RG dipeptide repeats of the spliceosomal Sm proteins D1 and D3 contain symmetrical dimethylarginines, which form a major B-cell epitope for anti-Sm autoantibodies. J. Biol. Chem. 275, 17122–17129 [DOI] [PubMed] [Google Scholar]
- 5. Dieterich W., Ehnis T., Bauer M., Donner P., Volta U., Riecken E. O., and Schuppan D. (1997) Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat. Med. 3, 797–801 [DOI] [PubMed] [Google Scholar]
- 6. Taylor-Papadimitriou J., Burchell J., Miles D. W., and Dalziel M. (1999) MUC1 and cancer. Biochim. Biophys. Acta 1455, 301–313 [DOI] [PubMed] [Google Scholar]
- 7. Pratesi F., Tommasi C., Anzilotti C., Chimenti D., and Migliorini P. (2006) Deiminated Epstein-Barr virus nuclear antigen 1 is a target of anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheumatism 54, 733–741 [DOI] [PubMed] [Google Scholar]
- 8. Hagiwara T., Nakashima K., Hirano H., Senshu T., and Yamada M. (2002) Deimination of arginine residues in nucleophosmin/B23 and histones in HL-60 granulocytes. Biochem. Biophys. Res. Commun. 290, 979–983 [DOI] [PubMed] [Google Scholar]
- 9. Kinloch A., Tatzer V., Wait R., Peston D., Lundberg K., Donatien P., Moyes D., Taylor P. C., and Venables P. J. (2005) Identification of citrullinated alpha-enolase as a candidate autoantigen in rheumatoid arthritis. Arthritis Res. Therapy 7, R1421–R1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Menard H. A., Lapointe E., Rochdi M. D., and Zhou Z. J. (2000) Insights into rheumatoid arthritis derived from the Sa immune system. Arthritis Res. 2, 429–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vossenaar E. R., Despres N., Lapointe E., van der Heijden A., Lora M., Senshu T., van Venrooij W. J., and Menard H. A. (2004) Rheumatoid arthritis specific anti-Sa antibodies target citrullinated vimentin. Arthritis Res. Therapy 6, R142–R150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Masson-Bessiere C., Sebbag M., Girbal-Neuhauser E., Nogueira L., Vincent C., Senshu T., and Serre G. (2001) The major synovial targets of the rheumatoid arthritis-specific antifilaggrin autoantibodies are deiminated forms of the alpha- and beta-chains of fibrin. J. Immunol. 166, 4177–4184 [DOI] [PubMed] [Google Scholar]
- 13. Jenkins R. E., and Pennington S. R. (2001) Arrays for protein expression profiling: towards a viable alternative to two-dimensional gel electrophoresis? Proteomics 1, 13–29 [DOI] [PubMed] [Google Scholar]
- 14. Bunai K., and Yamane K. (2005) Effectiveness and limitation of two-dimensional gel electrophoresis in bacterial membrane protein proteomics and perspectives. J. Chromatogr. B Analyt Technol Biomed Life Sci 815, 227–236 [DOI] [PubMed] [Google Scholar]
- 15. Gonzalez-Gonzalez M., Jara-Acevedo R., Matarraz S., Jara-Acevedo M., Paradinas S., Sayagues J. M., Orfao A., and Fuentes M. (2012) Nanotechniques in proteomics: protein microarrays and novel detection platforms. Eur. J. Pharm. Sci. 45, 499–506 [DOI] [PubMed] [Google Scholar]
- 16. Pruijn G. J. (2015) Citrullination and carbamylation in the pathophysiology of rheumatoid arthritis. Front. Immunol. 6, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. MacBeath G., and Schreiber S. L. (2000) Printing proteins as microarrays for high-throughput function determination. Science 289, 1760–1763 [DOI] [PubMed] [Google Scholar]
- 18. Zhu H., Bilgin M., Bangham R., Hall D., Casamayor A., Bertone P., Lan N., Jansen R., Bidlingmaier S., Houfek T., Mitchell T., Miller P., Dean R. A., Gerstein M., and Snyder M. (2001) Global analysis of protein activities using proteome chips. Science 293, 2101–2105 [DOI] [PubMed] [Google Scholar]
- 19. Robinson W. H., DiGennaro C., Hueber W., Haab B. B., Kamachi M., Dean E. J., Fournel S., Fong D., Genovese M. C., de Vegvar H. E., Skriner K., Hirschberg D. L., Morris R. I., Muller S., Pruijn G. J., van Venrooij W. J., Smolen J. S., Brown P. O., Steinman L., and Utz P. J. (2002) Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat. Med. 8, 295–301 [DOI] [PubMed] [Google Scholar]
- 20. Uchida T., Fukawa A., Uchida M., Fujita K., and Saito K. (2002) Application of a novel protein biochip technology for detection and identification of rheumatoid arthritis biomarkers in synovial fluid. J. Proteome Res. 1, 495–499 [DOI] [PubMed] [Google Scholar]
- 21. Pedersen J. W., Blixt O., Bennett E. P., Tarp M. A., Dar I., Mandel U., Poulsen S. S., Pedersen A. E., Rasmussen S., Jess P., Clausen H., and Wandall H. H. (2011) Seromic profiling of colorectal cancer patients with novel glycopeptide microarray. Int. J. Cancer 128, 1860–1871 [DOI] [PubMed] [Google Scholar]
- 22. Blixt O., Clo E., Nudelman A. S., Sorensen K. K., Clausen T., Wandall H. H., Livingston P. O., Clausen H., and Jensen K. J. (2010) A high-throughput O-glycopeptide discovery platform for seromic profiling. J. Proteome Res. 9, 5250–5261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hueber W., Kidd B. A., Tomooka B. H., Lee B. J., Bruce B., Fries J. F., Sonderstrup G., Monach P., Drijfhout J. W., van Venrooij W. J., Utz P. J., Genovese M. C., and Robinson W. H. (2005) Antigen microarray profiling of autoantibodies in rheumatoid arthritis. Arthritis Rheumatism 52, 2645–2655 [DOI] [PubMed] [Google Scholar]
- 24. Nand A., Gautam A., Perez J. B., Merino A., and Zhu J. (2012) Emerging technology of in situ cell free expression protein microarrays. Protein Cell 3, 84–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goldman K., Gertel S., and Amital H. (2013) Anti-citrullinated peptide antibodies is more than an accurate tool for diagnosis of rheumatoid arthritis. Isr. Med. Assoc. J. 15, 516–519 [PubMed] [Google Scholar]
- 26. Kokkonen H., Mullazehi M., Berglin E., Hallmans G., Wadell G., Ronnelid J., and Rantapaa-Dahlqvist S. (2011) Antibodies of IgG, IgA and IgM isotypes against cyclic citrullinated peptide precede the development of rheumatoid arthritis. Arthritis Res. Therapy 13, R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alessandri C., Bombardieri M., Papa N., Cinquini M., Magrini L., Tincani A., and Valesini G. (2004) Decrease of anti-cyclic citrullinated peptide antibodies and rheumatoid factor following anti-TNFalpha therapy (infliximab) in rheumatoid arthritis is associated with clinical improvement. Anna. Rheumatic Dis. 63, 1218–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aotsuka S., Okawa-Takatsuji M., Nagatani K., Nagashio C., Kano T., Nakajima K., Ito K., and Mimori A. (2005) A retrospective study of the fluctuation in serum levels of anti-cyclic citrullinated peptide antibody in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 23, 475–481 [PubMed] [Google Scholar]
- 29. Wiik A. S., van Venrooij W. J., and Pruijn G. J. (2010) All you wanted to know about anti-CCP but were afraid to ask. Autoimmunity Rev. 10, 90–93 [DOI] [PubMed] [Google Scholar]
- 30. Forslind K., Ahlmen M., Eberhardt K., Hafstrom I., Svensson B., and Group B. S. (2004) Prediction of radiological outcome in early rheumatoid arthritis in clinical practice: role of antibodies to citrullinated peptides (anti-CCP). Anna. Rheumatic Dis. 63, 1090–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kuhn K. A., Kulik L., Tomooka B., Braschler K. J., Arend W. P., Robinson W. H., and Holers V. M. (2006) Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J. Clin. Investig. 116, 961–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fernandes-Cerqueira C., Ossipova E., Gunasekera S., Hansson M., Mathsson L., Catrina A. I., Sommarin Y., Klareskog L., Lundberg K., Ronnelid J., Goransson U., and Jakobsson P. J. (2015) Targeting of anti-citrullinated protein/peptide antibodies in rheumatoid arthritis using peptides mimicking endogenously citrullinated fibrinogen antigens. Arthritis Res. Therapy 17, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schwenzer A., Jiang X., Mikuls T. R., Payne J. B., Sayles H. R., Quirke A. M., Kessler B. M., Fischer R., Venables P. J., Lundberg K., and Midwood K. S. (2015) Identification of an immunodominant peptide from citrullinated tenascin-C as a major target for autoantibodies in rheumatoid arthritis. Anna. Rheumatic Dis. 0, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Demoruelle M. K., and Deane K. (2011) Antibodies to citrullinated protein antigens (ACPAs): clinical and pathophysiologic significance. Curr. Rheumatol. Reports 13, 421–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Klareskog L., Ronnelid J., Lundberg K., Padyukov L., and Alfredsson L. (2008) Immunity to citrullinated proteins in rheumatoid arthritis. Annu. Revi. Immunol. 26, 651–675 [DOI] [PubMed] [Google Scholar]
- 36. Lundberg K., Bengtsson C., Kharlamova N., Reed E., Jiang X., Kallberg H., Pollak-Dorocic I., Israelsson L., Kessel C., Padyukov L., Holmdahl R., Alfredsson L., and Klareskog L. (2013) Genetic and environmental determinants for disease risk in subsets of rheumatoid arthritis defined by the anticitrullinated protein/peptide antibody fine specificity profile. Anna. Rheumatic Dis. 72, 652–658 [DOI] [PubMed] [Google Scholar]
- 37. Lutteri L., Malaise M., and Chapelle J. P. (2007) Comparison of second- and third-generation anti-cyclic citrullinated peptide antibodies assays for detecting rheumatoid arthritis. Clin. Chim. Acta 386, 76–81 [DOI] [PubMed] [Google Scholar]
- 38. Santiago M., Baron M., Miyachi K., Fritzler M. J., Abu-Hakima M., Leclercq S., Bell M., Hudson M., Mathieu J. P., Taillefer S., Jones N., Docherty P., Khraishi M., Markland J., Pope J., Robinson D., Smith D., and Sutton E. (2008) A comparison of the frequency of antibodies to cyclic citrullinated peptides using a third generation anti-CCP assay (CCP3) in systemic sclerosis, primary biliary cirrhosis and rheumatoid arthritis. Clin. Rheumatol. 27, 77–83 [DOI] [PubMed] [Google Scholar]
- 39. Szekanecz Z., Soos L., Szabo Z., Fekete A., Kapitany A., Vegvari A., Sipka S., Szucs G., Szanto S., and Lakos G. (2008) Anti-citrullinated protein antibodies in rheumatoid arthritis: as good as it gets? Clin. Rev. Allergy Immunol. 34, 26–31 [DOI] [PubMed] [Google Scholar]
- 40. van Venrooij W. J., van Beers J. J., and Pruijn G. J. (2008) Anti-CCP antibody, a marker for the early detection of rheumatoid arthritis. Ann. N.Y. Acad. Sci. 1143, 268–285 [DOI] [PubMed] [Google Scholar]
- 41. Ramachandran N., Raphael J. V., Hainsworth E., Demirkan G., Fuentes M. G., Rolfs A., Hu Y., and LaBaer J. (2008) Next-generation high-density self-assembling functional protein arrays. Nat. Methods 5, 535–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Park J., and LaBaer J. (2001) Recombinational cloning. Curr. Protoc. Mol. Biol., John Wiley & Sons, Inc. [DOI] [PubMed] [Google Scholar]
- 43. Ogunniyi A. O., Story C. M., Papa E., Guillen E., and Love J. C. (2009) Screening individual hybridomas by microengraving to discover monoclonal antibodies. Nat. Protocols 4, 767–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tarp M. A., Sorensen A. L., Mandel U., Paulsen H., Burchell J., Taylor-Papadimitriou J., and Clausen H. (2007) Identification of a novel cancer-specific immunodominant glycopeptide epitope in the MUC1 tandem repeat. Glycobiology 17, 197–209 [DOI] [PubMed] [Google Scholar]
- 45. Ramachandran N., Anderson K. S., Raphael J. V., Hainsworth E., Sibani S., Montor W. R., Pacek M., Wong J., Eljanne M., Sanda M. G., Hu Y., Logvinenko T., and Labaer J. (2008) Tracking humoral responses using self assembling protein microarrays. Proteomics Clin. Appl. 2, 1518–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Takulapalli B. R., Qiu J., Magee D. M., Kahn P., Brunner A., Barker K., Means S., Miersch S., Bian X., Mendoza A., Festa F., Syal K., Park J. G., LaBaer J., and Wiktor P. (2012) High density diffusion-free nanowell arrays. J. Proteome Res. 11, 4382–4391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sokolove J., Bromberg R., Deane K. D., Lahey L. J., Derber L. A., Chandra P. E., Edison J. D., Gilliland W. R., Tibshirani R. J., Norris J. M., Holers V. M., and Robinson W. H. (2012) Autoantibody epitope spreading in the pre-clinical phase predicts progression to rheumatoid arthritis. PloS One 7, e35296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van Boekel M. A., Vossenaar E. R., van den Hoogen F. H., and van Venrooij W. J. (2002) Autoantibody systems in rheumatoid arthritis: specificity, sensitivity and diagnostic value. Arthritis Res. 4, 87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sinz A., Bantscheff M., Mikkat S., Ringel B., Drynda S., Kekow J., Thiesen H. J., and Glocker M. O. (2002) Mass spectrometric proteome analyses of synovial fluids and plasmas from patients suffering from rheumatoid arthritis and comparison to reactive arthritis or osteoarthritis. Electrophoresis 23, 3445–3456 [DOI] [PubMed] [Google Scholar]
- 50. Liao H., Wu J., Kuhn E., Chin W., Chang B., Jones M. D., O'Neil S., Clauser K. R., Karl J., Hasler F., Roubenoff R., Zolg W., and Guild B. C. (2004) Use of mass spectrometry to identify protein biomarkers of disease severity in the synovial fluid and serum of patients with rheumatoid arthritis. Arthritis Rheumatism 50, 3792–3803 [DOI] [PubMed] [Google Scholar]
- 51. Matsuo K., Xiang Y., Nakamura H., Masuko K., Yudoh K., Noyori K., Nishioka K., Saito T., and Kato T. (2006) Identification of novel citrullinated autoantigens of synovium in rheumatoid arthritis using a proteomic approach. Arthritis Res. Therapy 8, R175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gobezie R., Kho A., Krastins B., Sarracino D. A., Thornhill T. S., Chase M., Millett P. J., and Lee D. M. (2007) High abundance synovial fluid proteome: distinct profiles in health and osteoarthritis. Arthritis Res. Therapy 9, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Weiler T., Du Q., Krokhin O., Ens W., Standing K., El-Gabalawy H., and Wilkins J. A. (2007) The identification and characterization of a novel protein, c19orf10, in the synovium. Arthritis Res. Therapy 9, R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cano L., and Arkfeld D. (2009) Targeted Synovial Fluid Proteomics for Biomarker Discovery in Rheumatoid Arthritis. Clin. Proteom. 5, 75–102 [Google Scholar]
- 55. Chang X., Cui Y., Zong M., Zhao Y., Yan X., Chen Y., and Han J. (2009) Identification of proteins with increased expression in rheumatoid arthritis synovial tissues. J. Rheumatol. 36, 872–880 [DOI] [PubMed] [Google Scholar]
- 56. Low J. M., Chauhan A. K., Gibson D. S., Zhu M., Chen S., Rooney M. E., Ombrello M. J., and Moore T. L. (2009) Proteomic analysis of circulating immune complexes in juvenile idiopathic arthritis reveals disease-associated proteins. Proteomics Clin. Appl. 3, 829–840 [DOI] [PubMed] [Google Scholar]
- 57. Rosenkranz M. E., Wilson D. C., Marinov A. D., Decewicz A., Grof-Tisza P., Kirchner D., Giles B., Reynolds P. R., Liebman M. N., Kolli V. S., Thompson S. D., and Hirsch R. (2010) Synovial fluid proteins differentiate between the subtypes of juvenile idiopathic arthritis. Arthritis Rheumatism 62, 1813–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ademowo O. S., Staunton L., FitzGerald O., and Pennington S. R. (2013) Biomarkers Inflammatory Arthritis Proteomics [Google Scholar]
- 59. Liao W., Li Z., Wang H., Wang J., Fu Y., and Bai X. (2013) Proteomic analysis of synovial fluid: insight into the pathogenesis of knee osteoarthritis. Int. Orthopaed. 37, 1045–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ritter S. Y., Subbaiah R., Bebek G., Crish J., Scanzello C. R., Krastins B., Sarracino D., Lopez M. F., Crow M. K., Aigner T., Goldring M. B., Goldring S. R., Lee D. M., Gobezie R., and Aliprantis A. O. (2013) Proteomic analysis of synovial fluid from the osteoarthritic knee: comparison with transcriptome analyses of joint tissues. Arthritis Rheumatism 65, 981–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Van Steendam K., Tilleman K., and Deforce D. (2011) The relevance of citrullinated vimentin in the production of antibodies against citrullinated proteins and the pathogenesis of rheumatoid arthritis. Rheumatology 50, 830–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nielen M. M., van der Horst A. R., van Schaardenburg D., van der Horst-Bruinsma I. E., van de Stadt R. J., Aarden L., Dijkmans B. A., and Hamann D. (2005) Antibodies to citrullinated human fibrinogen (ACF) have diagnostic and prognostic value in early arthritis. Anna. Rheumatic Dis. 64, 1199–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hill J. A., Al-Bishri J., Gladman D. D., Cairns E., and Bell D. A. (2006) Serum autoantibodies that bind citrullinated fibrinogen are frequently found in patients with rheumatoid arthritis. J. Rheumatol. 33, 2115–2119 [PubMed] [Google Scholar]
- 64. Terao C., Ohmura K., Katayama M., Takahashi M., Kokubo M., Diop G., Toda Y., Yamamoto N., Human Disease Genomics Working, G., Rheumatoid Arthritis, C., Genetic Study, C., Shinkura R., Shimizu M., Gut I., Heath S., Melchers I., Manabe T., Lathrop M., Mimori T., Yamada R., and Matsuda F. (2011) Myelin basic protein as a novel genetic risk factor in rheumatoid arthritis–a genome-wide study combined with immunological analyses. PloS One 6, e20457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Apweiler R., Hermjakob H., and Sharon N. (1999) On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta 1473, 4–8 [DOI] [PubMed] [Google Scholar]
- 66. Tarp M. A., and Clausen H. (2008) Mucin-type O-glycosylation and its potential use in drug and vaccine development. Biochim. Biophys. Acta 1780, 546–563 [DOI] [PubMed] [Google Scholar]
- 67. Bennett E. P., Mandel U., Clausen H., Gerken T. A., Fritz T. A., and Tabak L. A. (2012) Control of mucin-type O-glycosylation: a classification of the polypeptide GalNAc-transferase gene family. Glycobiology 22, 736–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kong Y., Joshi H. J., Schjoldager K. T., Madsen T. D., Gerken T. A., Vester-Christensen M. B., Wandall H. H., Bennett E. P., Levery S. B., Vakhrushev S. Y., and Clausen H. (2015) Probing polypeptide GalNAc-transferase isoform substrate specificities by in vitro analysis. Glycobiology 25, 55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stoevesandt O., Vetter M., Kastelic D., Palmer E. A., He M., and Taussig M. J. (2011) Cell free expression put on the spot: advances in repeatable protein arraying from DNA (DAPA). Nat. Biotechnol. 28, 282–290 [DOI] [PubMed] [Google Scholar]
- 70. He M., Stoevesandt O., Palmer E. A., Khan F., Ericsson O., and Taussig M. J. (2008) Printing protein arrays from DNA arrays. Nat. Methods 5, 175–177 [DOI] [PubMed] [Google Scholar]
- 71. Kidd B. A., Ho P. P., Sharpe O., Zhao X., Tomooka B. H., Kanter J. L., Steinman L., and Robinson W. H. (2008) Epitope spreading to citrullinated antigens in mouse models of autoimmune arthritis and demyelination. Arthritis Res. Therapy 10, R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gravallese E. M. (2003) Osteopontin: a bridge between bone and the immune system. J. Clin. Invest. 112, 147–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Burford B., Gentry-Maharaj A., Graham R., Allen D., Pedersen J. W., Nudelman A. S., Blixt O., Fourkala E. O., Bueti D., Dawnay A., Ford J., Desai R., David L., Trinder P., Acres B., Schwientek T., Gammerman A., Reis C. A., Silva L., Osorio H., Hallett R., Wandall H. H., Mandel U., Hollingsworth M. A., Jacobs I., Fentiman I., Clausen H., Taylor-Papadimitriou J., Menon U., and Burchell J. M. (2013) Autoantibodies to MUC1 glycopeptides cannot be used as a screening assay for early detection of breast, ovarian, lung or pancreatic cancer. Br. J. Cancer 108, 2045–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bian X., Wiktor P., Kahn P., Brunner A., Khela A., Karthikeyan K., Barker K., Yu X., Magee M., Wasserfall C. H., Gibson D., Rooney M. E., Qiu J., and LaBaer J. (2015) Antiviral antibody profiling by high-density protein arrays. Proteomics 15, 2136–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bian X., Wallstrom G., Davis A., Wang J., Park J., Throop A., Steel J., Yu X., Wasserfall C., Schatz D., Atkinson M., Qiu J., and LaBaer J. (2015) Immunoproteomic profiling of anti-viral antibodies in new-onset type 1 diabetes using protein arrays. Diabetes 65(1), 285–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.