

Abstract
Exposure to inorganic arsenic, a ubiquitous environmental toxic metalloid, leads to carcinogenesis. However, the mechanism is unknown. Several studies have shown that inorganic arsenic exposure alters specific gene expression patterns, possibly through alterations in chromatin structure. While most studies on understanding the mechanism of chromatin-mediated gene regulation have focused on histone post-translational modifications, the role of histone variants remains largely unknown. Incorporation of histone variants alters the functional properties of chromatin. To understand the global dynamics of chromatin structure and function in arsenic-mediated carcinogenesis, analysis of the histone variants incorporated into the nucleosome and their covalent modifications is required. Here we report the first global mass spectrometric analysis of histone H2B variants as cells undergo arsenic-mediated epithelial to mesenchymal transition. We used electron capture dissociation-based top-down tandem mass spectrometry analysis validated with quantitative reverse transcription real-time polymerase chain reaction to identify changes in the expression levels of H2B variants in inorganic arsenic-mediated epithelial-mesenchymal transition. We identified changes in the expression levels of specific histone H2B variants in two cell types, which are dependent on dose and length of exposure of inorganic arsenic. In particular, we found increases in H2B variants H2B1H/1K/1C/1J/1O and H2B2E/2F, and significant decreases in H2B1N/1D/1B as cells undergo inorganic arsenic-mediated epithelial-mesenchymal transition. The analysis of these histone variants provides a first step toward an understanding of the functional significance of the diversity of histone structures, especially in inorganic arsenic-mediated gene expression and carcinogenesis.
Millions of people worldwide are chronically exposed to inorganic arsenic (iAs)1, a ubiquitous environmental carcinogen, through drinking water and food, with consequences ranging from acute toxicities to malignant transformations (1, 2). Despite well-known deleterious health effects, the molecular mechanisms in iAs-mediated toxicity and disease are not fully understood. Several possible mechanisms have been proposed, including oxidative stress and genotoxic DNA damage (2–5). Recent evidence suggests that an even less understood—but equally important—mechanism is toxicity produced via changes in epigenetic-regulated gene expression. Proper gene expression requires regulatory proteins to bind to their target sites on DNA, found in eukaryotic cells as chromatin, many of which are directed by epigenetic marks.
In eukaryotes, genomic DNA is organized into nucleosomes, the fundamental repeating unit of chromatin (6). Each nucleosome consists of 147 base pairs of DNA wrapping 1.7 turns around a histone octamer comprised of two copies each of H2A, H2B, H3, and H4 (7–9). The packaging of eukaryotic genomes with histones to form chromatin is essential for the necessary condensation and protection of DNA. Changes in the structure of chromatin are essential for the proper regulation of cellular processes, including gene activation and silencing, DNA repair, replication and recombination. Alterations of the chromatin template during these processes can occur through at least 3 interrelated mechanisms: post-translational modifications of histones, ATP-dependent chromatin remodeling, and the incorporation of specialized histone variants into chromatin. Of these mechanisms, the exchange of variants into and out of chromatin is the least understood or studied. It is now known that the exchange of conventional histones for variant histones has distinct and profound consequences within the cell (10, 11).
The intricate mechanisms of regulation that involve chromatin are largely based on the enormous complexity built into its structure. A number of studies have demonstrated that iAs exposure induces global and gene-specific post-translational histone modifications such as reduction of acetylation in histone H3 and H4: loss of H4Lys16ac, increases in H3Lys14ac, H3Lys4me3, changes in methylation: increases in H3Lys4me2, H3Lys4me3, loss of H3Lys27me3, loss or gain of H3Lys9me2, increases in H3Ser10 and H2AX phosphorylation, and decreases in H2B ubiquitination (12–26).
Although histone post-translational modifications are clearly an important source of complexity, each of the canonical histones has multiple variants that can be incorporated into various chromatin loci at different times of the cell cycle (27). These histone variants can be divided into two groups based on whether their expression is regulated during replication—replication-dependent and replication-independent. Histone variants are found in distinct gene clusters within the human genome (11, 28). Interestingly, although they differ in their primary sequences, some of these encode similar proteins, whereas others do not (28, 29). For example, 22 genes code for replication-dependent histone H2B variants and they produce 16 distinct polypeptides.
The function and physiological roles of some histone variants have been well studied. For example, the H2A replication-independent histone variant H2A.X is phosphorylated near the sites of DNA double-strand breaks, facilitating the assembly of DNA repair factors (30). Another replication-independent variant, H2A.Z, is known to compact chromatin and poise genes for activation (reviewed in (31)). Two well-studied H3 replication-independent variants are H3.3, best known for its association with active transcription, and CenH3, found at the centromere and involved in kinetocore assembly (32). Expression of these replication-independent variants is not limited to any specific time during the cell cycle. In contrast, replication-dependent variants are expressed solely during S-phase of the cell cycle (33–35). All five classes of histones (H1, H2A, H2B, H3, and H4) have replication-dependent variants. The expression, integration and functional roles of most histone variants, especially the replication-dependent ones, have not been identified (33–35). Of the 16 unique H2B variants, 13 are somatic and 3 are testis-specific (H2BFWT, H2B1A, and H2BFM). These testis-specific variants have been characterized and have very different protein sequences (36–38). We hypothesize that alterations in the expression and incorporation of these variants depends on developmental and environmental cues.
How do Histone Variants Get Incorporated?
Due to the weaker contacts between H2A/H2B dimers and DNA, the DNA at the entry/exit sites of the nucleosome is more prone to transiently unwrap (39). This allows for higher turnover rates for histones H2A and H2B than for H3 and H4 (40, 41). In this study, we focused on the variants of histone H2B, in particular, as their expression changes during iAs-mediated epithelial-to-mesenchymal transition (EMT) (42). To date, no functional role for the H2B variants has been elucidated (35, 43). However, the clinical importance of histone variants is highlighted by recent reports that identified alterations in specific histone H2B variants in specific cancer cell types, suggesting cancer or tissue-specific H2B variant regulation (38). In fact, there are 22 genes encoding the different H2B variants within the human genome spread across the three major histone gene clusters. Fifteen of the H2B variants are found within the HIST1 gene cluster, encoding 11 unique variants, five are located on the HIST2 cluster and the last two are located in the HIST3 cluster (28, 29).
Histone H2B variants are highly sequence-conserved, differing only by a few amino acids (supplemental Fig. S1A). This high degree of sequence identity makes the H2B variants challenging to study with standard protein techniques that would require the use of variant-specific antibodies. However, MS can readily resolve them because of the changes in mass from the differences in amino acids found in the variants (35, 38, 43). Thus, in this study, we used top-down ultrahigh resolution Fourier transform ion cyclotron resonance (FT-ICR) tandem mass spectrometry (MS/MS) to quantify the relative abundance of histone H2B variants in cells undergoing iAs-mediated EMT. By comparing the histone H2B variant profiles between normal cells and iAs-transformed (iAs-T) cells, we identified significant changes in the levels of expression of specific histone H2B variants in iAs-T cells. Our studies highlight the enormous heterogeneity of histone variants present in human cells and indicate the importance of quantifying the histone proteome as cells go through differentiation to understand the impact of chromatin structure on carcinogenesis.
EXPERIMENTAL PROCEDURES
BEAS-2B and HeLa Growth Conditions and Cell Transformation Through Sodium Arsenite Exposure
BEAS-2B and HeLa cells were grown in Dulbecco's modified Eagle medium (Sigma-Aldrich, St. Louis, MO), supplemented with 10% fetal bovine serum (Sigma-Aldrich), 1% MEM nonessential amino acids (Sigma-Aldrich) and 1% penicillin-streptomycin (Sigma-Aldrich). Cells were grown to ∼80% confluency in a humidified chamber at 37 °C, 5% CO2. Cells were transformed (iAs-T) with sodium arsenite (Sigma-Aldrich) as previously described (42). Thus, cells were continuously treated with a low dose of iAs, either 0.5 μm or 1 μm, until transformation (about 36 days). For reversed cells (iAs-Rev), at ∼36 days of exposure to iAs, exposure was stopped and cells continued to grow under non-iAs conditions. Time matched control cells were treated with water (nontreated or NT cells).
Preparation of Nuclei and Extraction of Histones
Cells were released from plates by trypsinization, and pelleted by centrifugation at 1000 × g. Cell pellets were washed twice with phosphate buffered saline (PBS), and then resuspended in standard nuclei isolation buffer (15 mm Tris-HCl, pH 7.5, 60 mm KCl, 15 mm NaCl, 5 mm MgCl2, 1 mm CaCl2, 250 mm sucrose, and 0.3% Nonidet P-40. 1 mm dithiothreitol, 10 nm microcystin, 0.5 mm 4-(2-aminoethyl) benzenesulfonyl fluoride, and 10 mm sodium butyrate were added as inhibitors). After incubation on ice for 5 min at 4 °C, pellets were centrifuged at 600 × g for 5 min to obtain nuclei. The isolated nuclei were resuspended in 0.2 m H2SO4 at a ratio of 10:1 for ∼2 h while rotating at 4 °C. After centrifugation at 3400 × g, supernatant was precipitated with 25% (final concentration) of trichloroacetic acid on ice for 45 min. After centrifugation, the recovered histones were washed with 0.1% hydrochloric acid in acetone, with subsequent acetone washes. Thereafter, histone pellets were air-dried overnight at room temperature for high-performance liquid chromatography (HPLC) purification.
HPLC Separation of Histones
Extracted histones were further purified by reversed-phase (RP) HPLC on a Dionex UltiMate 3000 × 2 Dual Analytical system (Thermo Scientific, Waltham, MA) with a 4.6 × 250 mm, 5 μm, 300 Å pore size C18 column (218TP52, GRACE/Vydac, Columbia, Maryland). Individual histones were eluted from the column by applying a linear gradient of acetonitrile (30–60% B with a 0.3% B increase over 105 min; solvent A: 0.2% trifluoroacetic acid in 5% acetonitrile; solvent B: 0.188% trifluoroacetic acid in 95% acetonitrile. The flow rate was set at 0.8 ml/min. All histone fractions were collected with a fraction collector and dried under vacuum centrifugation (model SPD121P; Thermo Scientific).
9.4 T FT-ICR MS
Prepared histone samples were dissolved in 50% acetonitrile (H2O/CH3CN) (50/50) with 1.0% formic acid to reach a final concentration of 1.0 μm and introduced into a custom-built 9.4 Tesla FT-ICR mass spectrometer via positive electrospray ionization (44). Both precursor and product ion spectra were acquired by direct infusion at the flow rate of 0.35 μl/min. Precursor ions of interest were first selected by a quadrupole mass filter and further isolated by Stored Waveform Inverse Fourier Transform (SWIFT) ejection in the ICR cell (45). Electron capture dissociation (ECD) experiments were performed at a potential of 5 V dc on an accelerating grid and −2.5 V on the cathode during electron ejection. Prior to ECD analysis, a 50 ms delay was added to compensate for ion magnetron motion in the ICR cell (46). 500–1000 time-domain transients were averaged to improve signal to noise ratio for each product ion spectrum. The spectrometer was controlled by a modular ICR data acquisition station (Predator) (47). Each time-domain ICR signal is Hanning apodized, zero-filled once, fast Fourier transformed to magnitude mode, and converted to a mass-to-charge ratio spectrum by a two-term calibration equation (48).
qRT-PCR
RNA was isolated from HeLa and BEAS-2B cell lines, 1 × 107 cells, by use of the RNeasy MiniKit (Qiagen, Hilden, Germany). One microgram of RNA was used in a reverse transcriptase reaction with iScript Reverse Transcriptase (Bio-Rad, Hercules, CA #1708891) to prepare cDNA. 25 ng of the cDNA was used in qRT-PCR reactions. The protocol used for H2B variants was as follows: (1) 94 °C 5 min, (2) 94 °C 30 s, (3) 52 °C 30 s, (4) 72 °C 45 s, 2–4 repeated 40 cycles, 72 °C 10 min. HIST2H2BE and HIST2H2BF used the following protocol: (1) 94 °C 5 min, (2) 94 °C 30 s, (3) 60 °C 30 s, (4) 72 °C 45 s, 2–4 repeated 40 cycles, 72 °C 10 min. H2B variant primers were used as described in a previous study (49). EMT markers were amplified according to the following protocol: (1) 94 °C 5 min, (2) 94 °C 30 s, (3) 56 °C 45 s, (4) 72 °C 45 s, 2–4 repeated 40 cycles, 72 °C 10 min. Primers for the housekeeping gene (GAPDH) and primers used for EMT markers were made for this study by use of PrimerBank (50–52) (http://pga.mgh.harvard.edu/primerbank) and are available in supplemental Table S1. qRT-PCR data was analyzed by the 2−ΔΔCt method, and Student's t-tests were performed for significance.
Western Blot Analysis
Total protein was extracted from 107 HeLa or BEAS-2B cells. Cells were sonicated for 30 s followed by a 30 s off period for a total of 12 cycles (Diagenode Biodisruptor 300), and centrifuged at 12,800 × g at 4 °C for 15 min to pellet cellular debris. Total protein concentration was measured by use of a BCA kit (ThermoFisher #23221) and 25 ng of total protein were loaded into each lane of a 10% SDS-PAGE gel. Gels were run at 120 V until the loading dye reached the end of the gel and then transferred to polyvinylidene fluoride membranes (PVDF) at 65 V for 90 min on ice. Membranes were blocked with 5% Milk + phosphate buffered saline with Tween 20 (PBST) and incubated with primary antibodies (Cell Signaling Technologies, Danvers, MA #9782, EMT Antibody Sampler Kit) (in 0.5% Milk + PBST) overnight at 4 °C. A secondary antibody (α-Rabbit or α-Mouse) was applied the next day and developed by use of ECF (GE-Typhoon FLA9500).
RESULTS
Transformation of Cells by Low-dose Chronic Exposure to iAs
Chronic low-dose exposure to iAs is known to cause cells to undergo EMT (42, 53, 54). As cells go through EMT, changes in cell morphology and expression patterns of EMT markers occur. In this study, we used a similar procedure as previously shown to induce EMT, by exposing cells to low-dose iAs for 5 weeks (42). We used two types of cells, HeLa and BEAS-2B cells, and monitored changes in growth and morphology. After 5 weeks of exposure to these low doses of iAs, BEAS-2B cells became elongated and spindle-shaped, indicating EMT transformation (Fig. 1A). HeLa cells showed similar changes (data not shown). We confirmed EMT transformation in two ways: (1) at the protein level, using Western blot analyses, to measure the protein levels of known EMT markers (Fig. 1B); and (2) at the transcript level, quantitative reverse transcription real-time PCR (qRT-PCR) was used to measure the transcript levels of these known EMT markers (Fig. 1C). Previously, we have shown that iAs-mediated EMT correlated with alterations in chromatin structure and epigenomic changes. These studies showed broad chromatin structural changes (42). However, in the current study, we attempted to better define the details of these changes in chromatin structure. Thus, after confirming transformation, we asked whether there are differences in histone H2B variants in iAs-T cells compared with normal cells.
Fig. 1.

Chronic low-dose exposure to iAs causes cells to undergo EMT. A, Morphology changes seen in iAs-transformed BEAS-2B cells. BEAS-2B cells become elongated and spindle-shaped with 0.5 μm iAs treatment. B, Western blot analyses of EMT markers showing changes in the EMT markers. For instance, β-Catenin levels decrease and Slug levels increase in iAs transformed cells. C, qRT-PCR confirmation of iAs-induced EMT markers in HeLa (top) and BEAS-2B (bottom). qRT-PCR confirms the changes in expression of EMT markers in iAs-transformed cells. Error bars represent standard deviation (S.D.) from three biological replicates, each containing three technical replicates. Asterisks denote p values < 0.05 as determined by Student's t test.
Top-down MS/MS Analysis of Histone H2B
We purified histone H2B from acid-extracted histones by C18 reversed-phase chromatography (Fig. 2A). The purified H2B was injected directly into the mass spectrometer (see Experimental Procedures) and the collected spectra were used to determine the masses of the H2B variants. Five major peaks were observed in the broadband spectrum of histone H2B from NT and iAs-T cells as shown in Fig. 2B. The molecular mass of each peak was determined by deconvolution of multiply charged ion species and then compared with the calculated masses of all histone H2B sequence variants (Table I). All raw ECD product ion mass spectra were phase-corrected and analyzed by our in-house software in a variant-specific manner to determine each proteoform (55, 56). Briefly, a library of all known H2B sequence variants from Uniprot was imported into our software for identification and characterization. The software determines the isotopic distribution of all potential fragment ions of all variants by combining possible post-translational modifications and then checking for the presence of each possible fragment ion of all variants. All possible proteoforms are assigned and ranked by an objective function, which is generated based on the sum of peak heights of all assigned peaks within a given mass error tolerance of 10 ppm for every possible proteoform. Usually, a higher score indicates a better match, but some of the top assignments are not valid based on the intact protein mass obtained from the broadband spectrum. We manually filter out these unreasonable assignments. For example, H2B1C sometimes has a higher score than H2B1H for the first peak in Fig. 2 because of the highly similar sequences of H2B1C and H2B1H/1K (Table I). However, the intact experimental mass for the first peak does not agree with the calculated H2B1C mass; thus, producing a false positive result, which we eliminate. ProSight Lite was conducted at the same time to validate our identifications (44).
Fig. 2.
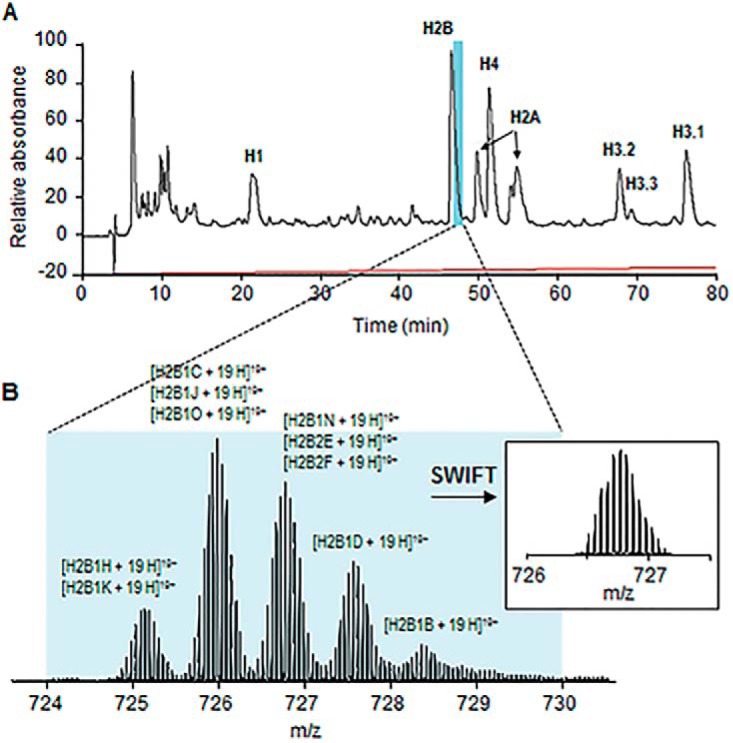
Electron Capture Dissociation top-down MS/MS of H2B isoforms. A, Off-line reversed-phase liquid chromatogram for histones. B, Representative mass spectrum of histone H2B sequence variant isotopic distributions for charge state, Z = 19. Precursor m/z 726.75 ions were isolated by SWIFT and fragmented with ECD.
Table I. Comparison of H2B variants. Experimental and calculated neutral masses of H2B variants that showed changes in expression in iAs-induced EMT transformed cells. Also shown are the amino acids, accession number of the variants, and their positions that differ between the various variants.
| Sequence variant | Accession number | Neutral mass (Da) |
Amino acid positions |
|||||
|---|---|---|---|---|---|---|---|---|
| Experimental | Calculated | 2 4 | 18 | 21 | 39 | 124 | ||
| H2B1H | Q93079 | 13758.72 | 13760.92 | DPA | V | A | V | S |
| H2B1K | O60814 | 13758.72 | 13758.95 | EPA | V | A | V | A |
| H2B1C | P62807 | 13774.87 | 13774.95 | EPA | V | A | V | S |
| H2B1J | P06899 | 13774.87 | 13772.97 | EPA | V | A | I | A |
| H2B1O | P23527 | 13774.87 | 13774.95 | DPA | V | A | I | S |
| H2B1N | Q99877 | 13789.12 | 13790.95 | EPS | V | A | V | S |
| H2B2E | Q16778 | 13789.12 | 13788.97 | EPA | V | A | I | S |
| H2B2F | Q5QNW6 | 13789.12 | 13788.97 | DPA | V | V | V | S |
| H2B1D | P58876 | 13804.89 | 13804.97 | EPT | I | A | V | S |
| H2B1B | P33778 | 13819.52 | 13819.00 | EPS | I | A | I | S |
Because H2B variants have highly similar sequences (Table I and supplemental Fig. S1), some of them have the same or very similar molecular masses (∼2 Da difference), which are unresolved by quadrupole MS precursor ion isolation. However, we were able to perform ECD experiments to differentiate them based on their specific c and z fragment ions. For example, H2B2E and H2B2F are structural isomers (i.e. they share the same molecular mass) differing by only 3 amino acids at sites 2, 21 and 39, whereas the nominal mass of H2B1N is 2 Da greater than H2B2E with variations in 2 amino acids at sites 4 and 39 (Table I). These similar molecular weights make them difficult to resolve by quadrupole MS (Fig. 2A, 2B). Thus, the isotopic distribution of the overlapping isotope distributions was isolated by SWIFT, as shown in Fig. 2B, and then subjected to ECD fragmentation. Because H2B1N, H2B2E, and H2B2F differ by amino acids 2, 4, 21, and 39, and the rest of the sequences are exactly the same we can distinguish them by their critical fragment ions, c2-c38 and z105-z123. From the ECD product ion mass spectra, we identified 19 critical c ions for H2B1N (Fig. 3; supplemental Fig. S2), 34 for H2B2E (Fig. 3; supplemental Fig. S3) and 26 for H2B2F (Fig. 3, supplemental Fig. S4), which confirmed the co-existence of these three sequence variants within the initial peak. For quantification, we calculate their fragment ion relative ratios based on the common ions from those critical c ions of all three variants. The same principle applies for H2B1H/1K and H2B1C/1J/1O (supplemental Fig. S5). Of all known variants, we identified 10 with differential expression in one top-down experiment of which eight are located in the HIST1 gene cluster HIST1H2BH (H2B1H), HIST1H2BK (H2B1K), HIST1H2BC (H2B1C), HIST1H2BJ (H2B1J), HIST1H2BO (H2B1O), HIST1H2BN (H2B1N), HIST1H2BD (H2B1D), and HIST1H2BB (H2B1B) and two are located in the HIST2 gene cluster HIST2H2BE (H2B2E) and HIST2H2BF (H2B2F). Further, these 10 variants were quantified according to their normalized abundance in the same broadband mass spectrum and their fragment ion relative abundance ratios (35, 57). No post-translational modifications were observed for any of the sequence variants in either HeLa or BEAS-2B cell lines.
Fig. 3.

Upper left. ECD product ion spectrum for precursor (H2B1N/2E/2F) m/z 726. 75 ions. Upper right and bottom. ECD fragmentation maps generated by our custom software. When peptides are dissociated, c ions contain the charge at the N terminus, whereas z ions contain the charge at the C terminus.
Mass Spectrometry Profiling H2B Variants in iAs-mediated EMT Cells
Previous studies have demonstrated variation in the expression of the mRNA of different H2B variants across cancer cell lines (49, 58). This suggests that there may be different abundances of H2B variant proteins between cell lines and possibly cell-type-specific functions for the different H2B variants. We therefore asked whether there are variations in expression of these variants as cells go through iAs-mediated EMT. Cells were collected at days 4, 8, and 16 during the process of iAs-mediated EMT to examine variant expression. Histones were extracted and subjected to MS/MS. Of the H2B variants that were identified through mass spectrometry, ten were found to have expression level changes during the iAs-mediated EMT compared with NT HeLa cells. Seven of the H2B variants were up-regulated (H2B1H, H2B1K, H2B1C, H2B1J, H2B1O, H2B2E, H2B2F) and three were downregulated (H2B1N, H2B1D, H2B1B) (Fig. 4A). We also transformed these cells with two different concentrations of iAs (0.5 μm and 1 μm). For the majority of the histone variants, there was no difference in expression between the iAs doses. We did observe changes for three variants (H2B1C, H2B2F, and H2B1B) with the 0.5 μm treatment having an enhanced effect compared with the 1 μm treatment of iAs (Fig. 4A). However, this might not be biologically significant because in both doses there is a significant change in expression level compared with the nontreated cells. Whether these slight differences in the 0.5 μm compared with the 1 μm is biologically significant remains to be explored.
Fig. 4.
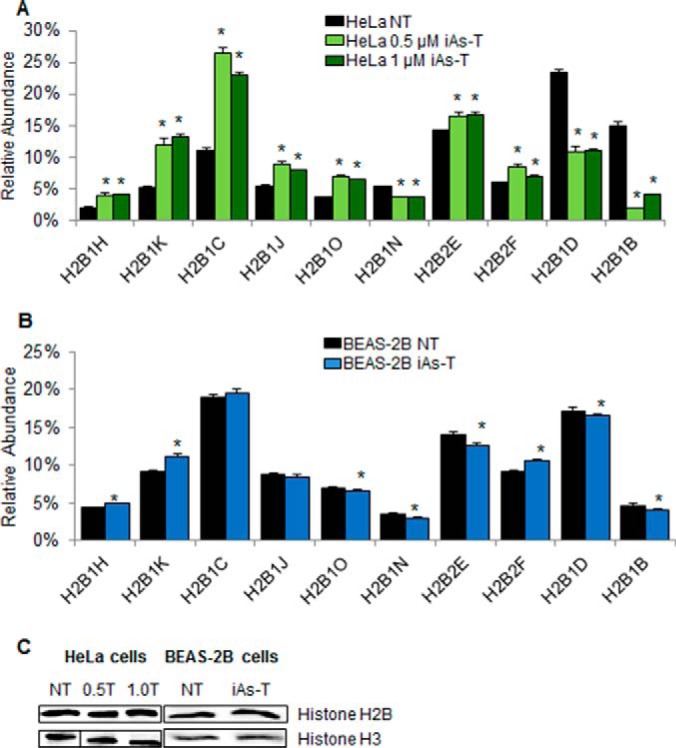
Mass spectrometry profiling of H2B variants in NT and iAs-T cells. A, Profiles of H2B variants in HeLa cells show differential abundance of 10 histone H2B variants in iAs-transformed cells compared with time-matched NT cells, after 16 days of treatment. Additionally, there were no major changes in the expression levels in cells transformed with 0.5 μm and 1 μm iAs. B, Changes in the abundance of these variants were confirmed in BEAS-2B cells. Although most of the variants show patterns similar to iAs-T HeLa cells, H2B1J, H2B1O, and H2B2E, show different patterns of expression in the iAs-T BEAS-2B cells. C, Western blots of total histones H2AB and H3 show equal expression in treated and nontreated cells. Equal amounts of nuclear protein (25 ng) were loaded onto a 15% SDS-PAGE gel and blotted for H2B or H3. Error bars represent standard deviation from three technical replicates. Biological replicates in HeLa and BEAS-2B cells show a similar trend (supplemental Fig. 7). Asterisks denote p values < 0.05 as determined by Student's t test.
Next we asked if the observed changes in the levels of these variants were cell-type specific. To answer this question, we extracted histones from iAs-T BEAS-2B cells and the abundance of the H2B variants was investigated through mass spectrometry (Fig. 2). The expression levels of the H2B variants in BEAS-2B cells followed the same patterns of up- or down-regulation seen previously in the iAs-T HeLa cells (Fig. 4A, supplemental Fig. S6). On the other hand some of the changes seen were not as drastic as in the iAs-T HeLa cells (Fig. 4B). To determine if overall levels of histones were changing in cells exposed to iAs, Western blot analysis for total histone H2B and total histone H3 were performed. As shown (Fig. 4C), there is no difference in the expression of H2B or H3 proteins in nontreated and iAs transformed cells. In addition, three of the histone variants in the BEAS-2B iAs-T cells showed an expression pattern that differed from the HeLa iAs-T cell expression pattern. H2B1J, H2B1O, and H2B2E in HeLa cells showed an overall up-regulation compared with NT cells. However, in the BEAS-2B transformed cells, there is a decrease in the abundance of these variants compared with the NT cells (Fig. 4, supplemental Fig. S6). These differences could be because of a couple of possibilities: (1) The process of EMT involves a number of stages (59, 60). Although HeLa cells are carcinogenic, they can still, under specific conditions, undergo further EMT (Fig. 1) (61, 62). On the other hand, BEAS-2B cells are considered normal and thus the process of transformation will involve many more steps than HeLa cells. (2) The differences we see could be cell-type specific. In summary, the differences in the H2B variant levels in the iAs-mediated EMT suggest that these variants are being expressed and regulated for specific purposes in the cells, especially those that have undergone iAs-mediated EMT.
Time-dependent Mass Spectrometry Profiling in H2B Variants as Cells Go Through iAs-mediated EMT
We next asked if there were changes in the abundance of the histone variants as cells go through iAs-mediated transformation. H2B variants levels were analyzed at days 4, 8, and 16 of iAs-treatment. Six variants exhibited significant differences in their levels throughout the time course as cells went through iAs-mediated EMT (supplemental Fig. S7). Compared with time-matched controls (NT), the abundance of H2B1C increased at each day of exposure. Likewise, H2B1K levels increased with the days of iAs exposure (Fig. 5). On the other hand, H2B1D abundance overall decreased across all three time points of exposure, but interestingly on day 8 the levels in iAs-treated cells increased to near those of NT cells, and by day 16 they decreased to well below the levels in NT cells (Fig. 5). H2B1B abundance in iAs-treated cells decreased similarly throughout the exposure times (Fig. 5). In summary, the changes in protein levels seen in H2B1K and H2B1C suggest that they are actively being added into chromatin as the cells undergo iAs-mediated EMT. The changes in abundance seen in H2B1D and H2B1B suggest that as cells undergo the changes in iAs-mediated EMT, they are actively excluded from chromatin. As there was little difference seen between the cells exposed to the two concentrations, we used only the 1.0 μm concentration for HeLa cells from this point forward (iAs-T). The directionality in the changes in the abundance of these histone variants are consistent with cells treated for 5 weeks (iAs-T cells - Fig. 4) and could indicate that these changes in the abundance of histone variants occur early and drive the gene expression patterns in iAs-EMT.
Fig. 5.
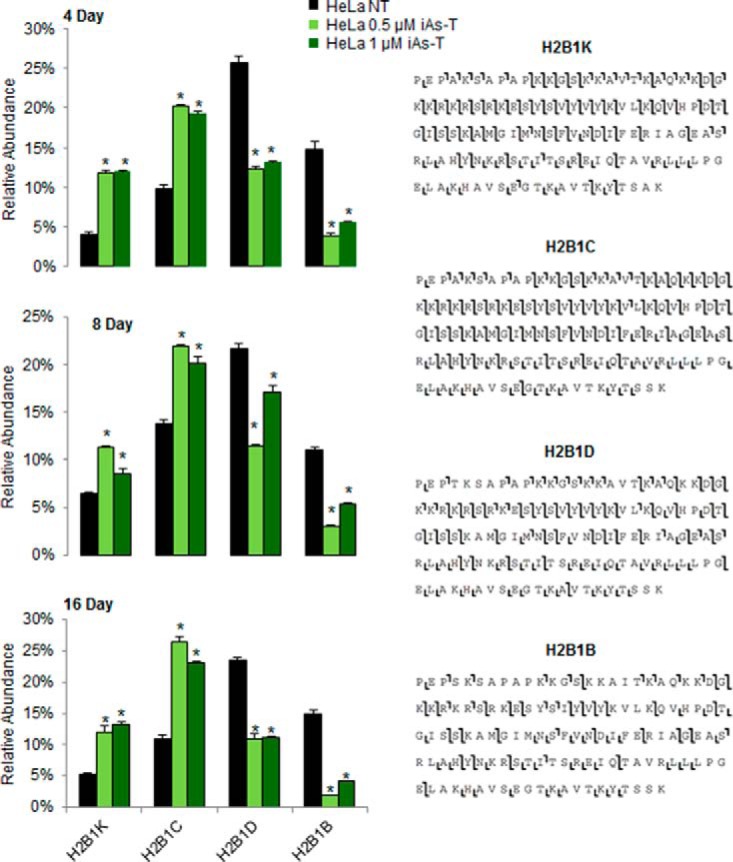
Time-dependent changes in H2B variants as cells undergo iAs-mediated EMT. The abundance of histone H2B variants was monitored at different times during the iAs-transformation process. Changes by day (Top-4 days, Middle-8 days, Bottom-16 days) of the H2B variants showed that the most overall up-regulated histone H2B variants were H2B1K and H2B1C, whereas the most downregulated variants were H2B1D and H2B1B. Shown next to the graphs are the obtained ECD spectra for these four variants. The full extent of the changes in the relative abundance of the 10 observed H2B variants changes and a consequence of exposure time (days of exposure) are shown in supplemental Fig. 6 and ECD fragmentation maps in supplemental Fig. 5. Untreated cells are from the same days as the treated groups. Error bars represent S.D. from three technical replicate experiments. *, p value < 0.05 as determined by Student's t test.
mRNA Profiling of the H2B Variants by qRT-PCR
As there were changes in protein levels of the H2B variants seen in the MS data, we were interested to see if these changes were correlated with changes in mRNA expression. To investigate the gene expression levels of the H2B variants, we used qRT-PCR on technical as well as biological replicates. RNA was extracted from the same cell population used in the mass spectrometry analyses, reverse transcribed and used for qRT-PCR. In both cell types, the overall gene expression patterns were similar to those of the mass spectrometry data (protein levels). In HeLa cells (Fig. 6A), the expression levels of 7 out of the 10 H2B variants (70%) were confirmed with qRT-PCR. The expression levels of all ten of the H2B variants observed in mass spectrometry data were confirmed for the BEAS-2B cells (Fig. 6B).
Fig. 6.
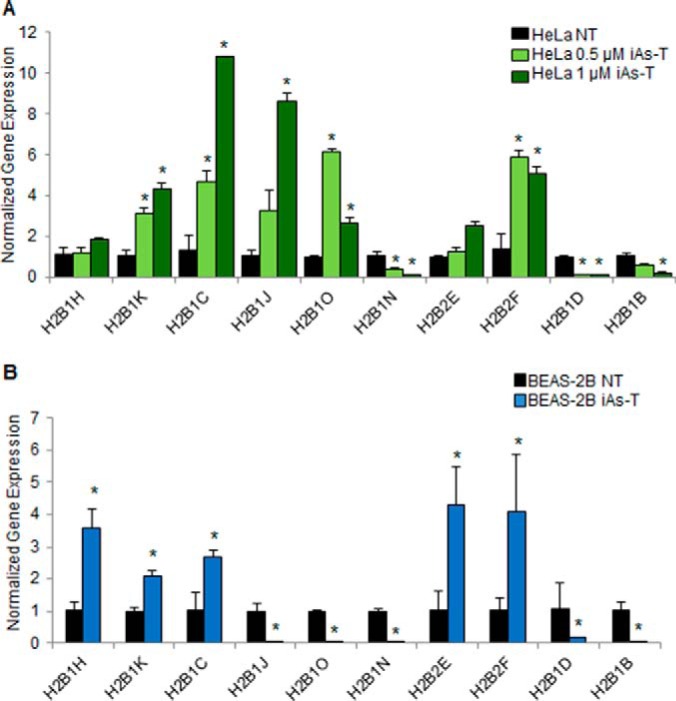
Validation of H2B changes with qRT-PCR. A, qRT-PCR in HeLa cells validated the expression patterns of 7 of the 10 H2B variants investigated through MS. B, qRT-PCR in BEAS-2B cells validates the expression patterns of all 10 H2B variants. Error bars represent S.D. from three biological replicates, each containing three technical replicates. *, p value < 0.05, determined by Student's t test.
Effects of Removal of iAs in Transformed Cells
Once cells were transformed and changes to morphology and EMT were measured, we asked what would happen to EMT and histone expression when the iAs exposure is removed (iAs-Rev). We have previously shown that cell morphology and expression of certain genes reverted to normal levels after removal of iAs from iAs-T cells (42). We thus carried out similar studies and confirmed in these iAs-Rev cells that the morphology of the cells reverted toward NT cells, though not completely (not shown). Furthermore, we also had previously reported that some gene expression patterns reverted completely whereas others did not. We proposed that the gene expression patterns that did not revert completely were responsible for the sustenance of the oncogenic potential of these cells (42). In this study, we also observed that in iAs-Rev cells, the expression levels of some EMT markers did not completely revert to nontreated levels, substantiating our initial observation. In Western blot analysis for EMT markers, we show that in HeLa-iAs-Rev cells, the expression levels of some EMT markers showed intermediate protein levels, most notably in Claudin-3 and Slug (Fig. 7A). When qRT-PCR was performed to investigate gene expression of these EMT markers, we saw mixed levels in the HeLa RT. For instance, the data for β-Catenin, E-cadherin, and ZO-1 show that these genes are expressed at similar levels as the iAs-T HeLa cells. N-cadherin shows an increase in expression in the transformed cells; however in the iAs-Rev cells, its expression was higher. Claudin-1, Slug, and Vimentin, as well as Snail and ZEB1, all show the opposite gene expression patterns as the iAs-T cells (Fig. 7B). This opposite expression pattern would suggest the cells are reverting to a normal HeLa phenotype, albeit incompletely. We then asked whether in iAs-Rev cells, the alterations in the levels of histone variants were also observable. We carried out qRT-PCR to measure the expression of the above 10 histone variants whose expression was altered in iAs-T cells. Here too, we observed some of these variants reverting to NT levels, whereas others stayed altered. We observed that the expression levels for H2B1N, H2B1B, and H2B1D were decreased similarly in iAs-Rev cells as in iAs-T cells (Fig. 7C). On the other hand, the expression levels for both H2B1H and H2B1O in iAs-Rev cells were at levels that were increased above and beyond the iAs-T cells. H2B1K, H2B1C, H2B1J, H2B2F, and H2B2E all show a decrease in iAs-Rev expression of the variant compared with levels in the iAs-T cells (Fig. 7C). Thus, these five variants seem to be reverting to the levels in HeLa NT cells.
Fig. 7.
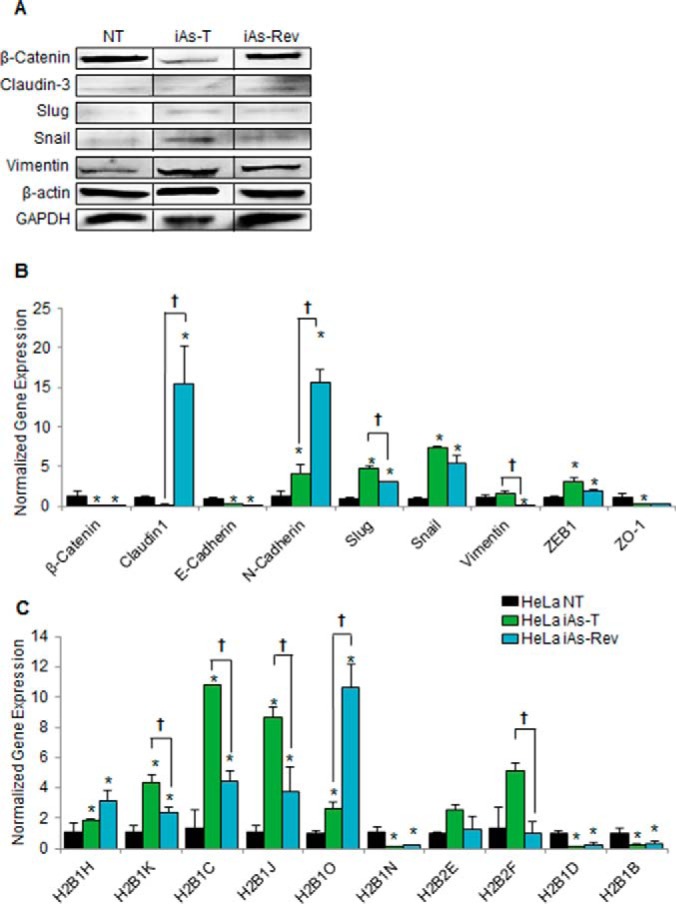
Effects with the removal of iAs-treatment after transformation (iAs-rev cells). A, Western blot analyses of EMT markers. Claudin-3 and Slug in iAs-rev cells show an intermediate level of expression, whereas others such as β-Catenin and Vimentin revert to near NT levels. B, qRT-PCR for EMT markers. qRT-PCR for EMT markers in iAs-Rev cells mostly show changes in expression opposite of the iAs-T cells. C, qRT-PCR of the H2B variants. qRT-PCR for H2B variants in iAs-Rev cells mostly show changes in expression reverting to near NT cells. Error bars represent S.D. from three biological replicates, each containing three technical replicates. *, p < 0.05 compared with NT; †, p < 0.05 between iAs-T and iAs-Rev.
Although these cells had the iAs removed, some of the key EMT genes remained altered, possibly allowing the cells to remain tumorigenic (42). It is possible that some histone variants, just like with those genes that did not revert completely, could be driving the metastatic potential of these cells, even when they are no longer exposed to iAs. One possibility is that the incorporation of these histone variants allows for an epigenetic program that drives specific gene expression patterns with carcinogenic potential. More studies will be needed to elucidate this hypothesis.
DISCUSSION
Epigenetic modifications in response to environmental disruptors include changes in histone modifications, DNA methylation, ATP-dependent chromatin remodeling, and incorporation of histone variants (63). While so many studies have focused on other epigenetic factors, the impact of histone variants in this process has remained largely understudied. In this study, we present the first analyses of its kind, determining the changes in the levels of histone H2B variants in iAs-mediated cancer pathogenesis. Using top-down MS/MS, we identified 10 histone H2B variants that showed differential expression in iAs-T HeLa cells (Fig. 4A). Most striking were the large differences in the levels of four H2B variants: down-regulation of H2B1B and H2B1D and up-regulation for H2B1C and H2B1K (Figs. 5 and 6). We confirmed alterations in H2B variants in a second iAs-T cell line, the lung epithelial BEAS-2B cells, suggesting that turnover of these variants is important in iAs-dependent EMT pathology and not cell-type dependent (Fig. 4B). Lastly, we validated the changes in gene expression of these variants using qRT-PCR (Fig. 6). These studies indicate that incorporation of variants into chromatin might be responsible for the specific epigenetic reprogramming in iAs-mediated EMT pathology.
Several recent reviews have highlighted the epigenetic role of histone variants (64–67) and have shown the importance of these variants in the progression of cancer to different stages (68–73). Underscoring that importance, the expression of H2B histone variants is cell-type specific, differentiation stage-dependent as well as cancer-stage and type dependent (38). However, the study of these variants has remained difficult largely because they differ by very few amino acids (Table I; supplemental Fig. S1), making variant-specific antibodies virtually impossible to develop. However, the use of top-down MS/MS has proven to be an invaluable tool to study such closely related histone variants (38). This type of mass spectrometry characterizes the histone proteome in an unbiased approach, providing a sequence that does not rely on the existence of modification-specific antibodies. The top-down method, which allows for whole protein sequencing, is a great tool for distinguishing histone variants (74).
Importance of Histone Variants in Gene Regulation
H2B variants are involved in the indexing of the genome, in this way establishing different regulated regions that can be activated or repressed for transcription. As such, histone variants may confer a new level of gene expression regulation, important for cell-type or tissue-specificity and adaptation to specific cellular events. The understanding of the various histone variants and their incorporation in each toxin-associated disease may be a useful tool for identifying diseases and may unveil their pathogenesis because incorporation of histone variants can change the functionality of these proteins.
Why Does the Turnover of Histone H2B Matter?
The heterodimer of histone H2A-H2B forms a core component of the nucleosome. Structurally, the dissociation of this H2A-H2B dimer from the nucleosome core results in the exposure of a region around the DNA pseudo-dyad axis of symmetry in the nucleosome (39–41). This exposure, with consequences in factor accessibility to the DNA, has functional consequences in gene regulation. This important observation was proposed to explain the subnucleosomal structures observed upon transcriptional activation of the heat-shock inducible HSP82 gene in yeast (75). Interestingly, a recent genome-wide analysis in budding yeast has provided further support to the idea of a widespread presence of these (and other) subnucleosomal particles (76). Thus, the association of the histone H2A-H2B dimer with the nucleosome, including its many variants, plays a critical role in nucleosome dynamics (77) and functional adaptability (78). However, the structural role of the different H2B variants contributing to this dynamic has resulted in a new epigenetic paradigm.
In humans, there are 16 reported H2B variants (13 somatic and 3 testis-specific), adding to the complexity of this paradigm. It will be of interest to understand when and where these variants are located genome-wide and the functional consequences of this incorporation. In these studies, we did not observe any post-translational modifications on the H2B variants and it was not surprising, as relatively very few post-translational modifications have been reported for histone H2B compared with other core histones.
H2B Variants During Differentiation and Dedifferentiation
In organisms, little is known about the distribution of H2B variants during processes that involve alterations in chromatin function, such as differentiation and dedifferentiation. In this study, we evaluated the ratio of each histone H2B variant as cells go through dedifferentiation after arsenic removal (iAs-Rev), in order to define possible alterations either during iAs-EMT (differentiation) or iAs-Rev (dedifferentiation). We propose that the alterations in these histone variant ratios are correlated with iAs-mediated EMT (differentiation) and the physiological status of the cell. Indeed, we show that when iAs is removed from iAs-T cells, some of the induced epigenetic events, gene expression and chromatin structural changes reverted to pre-iAs exposure levels, albeit incompletely. We also show that some of the histone variants revert to pre-iAs exposure, whereas others stay altered (Fig. 7). It is possible that the continuous incorporation of a subset of histone variants sustain the pathogenic state in iAs-Rev cells. However, further studies will be needed to test that hypothesis.
Lastly, although the functional analysis of these distinct replication-dependent H2B variants is only beginning, the observation that their abundances are altered in iAs-mediated EMT process supports the intriguing possibility that they may perpetuate specific expression patterns. Additionally, identification of the molecular make-up of these histone variants and their target genes will be needed to determine whether the various forms of histone H2B play a functional role in disease progression. However, the high amount of sequence identity among the histone variants provides challenges for studies aimed at understanding the physiological roles of the H2B variants in iAs-mediated transformation.
The changes in expression that are induced by these histone variants appear to be important to the changes that are seen in the iAs-mediated EMT. These H2B variants, even though located within gene clusters, have individual promoter regions (37). Our results indicate that the patterns of histones H2B variants found in iAs-mediated EMT models are highly dynamic. For example, the pattern of H2B variants observed in HeLa iAs-T cells was very similar to BEAS-2B lung epithelial cells, suggesting perhaps that the same mechanism is used by iAs-signaling resulting in the deposition of these specific histone variants. Moreover, the possibility exists that there may be iAs-mediated EMT specific variations in the patterns of H2B species. These changes in the expression patterns of the H2B variants may be responsible for the chromatin rearrangements that occur for iAs-mediated pathogenesis. Recent studies have highlighted the role of iAs on the expression of histones. Arsenic exposure induced the polyadenylation of the canonical histone mRNAs, which otherwise do not have this tail (79). Implicated as a possible mechanism is the depletion of the stem-loop binding protein by iAs. These histone variants, in our studies, all have the polyadenylated tails, thus protected from degradation. In fact expressing of canonical histone proteins with polyadenyalted tails, recapitulated the iAs-transformation state. These studies and ours suggest that the dynamic control of histone proteins is important and could play a role in arsenic-induced transformation. Further studies will be needed to test this idea.
In conclusion, we identified 10 H2B variants that showed differential expression patterns in iAs-transformed versus nontransformed cells. The use of top-down mass spectrometry allowed us to achieve this high level of resolution on the H2B variants, using another type of analysis would not have given results conducive to these studies. A combination of mass spectrometry and qRT-PCR was used to investigate and validate changes in H2B levels that are induced through the iAs-mediated transformation. The changes seen in H2B variants with iAs-mediated transformation suggest that H2B levels are well regulated and their incorporation into chromatin during this process might be important in the carcinogenic potential of iAs.
Supplementary Material
Acknowledgments
We thank Nicolas L. Young and Xibei Dang (Florida State University) for their help with software, and the Markey Cancer Center's Research Communications Office for manuscript editing. Raw mass spectrometry data was deposited into ProteomeXchange via the PRIDE database and the accession number is PXD003503.
Footnotes
Author contributions: Conceived and designed the experiments: YNF-M. Performed the experiments: MR, TJ, RE, ME, YFN-M. Analyzed the data: MR, TJ, AGM, YNF-M. Contributed reagents/materials: AGM, YNF-M. Wrote the paper: MR, TJ, RE, ME, AGM, YNF-M.
* This work was supported by NSF grant MCB 1517986 to YFN-M, and NIEHS grant R01-ES024478 to YNF-M. A portion of this work was performed at the National High Magnetic Field Laboratory, which is supported by National Science Foundation Cooperative Agreement # DMR-1157490 and the State of Florida.
 This article contains supplemental material.
This article contains supplemental material.
MR: matthew.rea@uky.edu; TJ: tingjiang@magnet.fsu.edu; RE: rebekah.eleazer@uky.edu; ME: meredith.eckstein@uky.edu; AGM: marshall@magnet.fsu.edu; YNF-M: y.fondufe-mittendorf@uky.edu.
1 The abbreviations used are:
- iAs
- inorganic arsenic
- ECD
- electron capture dissociation
- EMT
- epithelial-to-mesenchymal transition
- FT-ICR
- Fourier transform ion cyclotron resonance
- HPLC
- high-performance liquid chromatography
- iAs-Rev
- inorganic arsenic reverse cells
- iAs-T
- inorganic arsenic transformed cells
- MS/MS
- tandem mass spectrometry
- NP-40
- nonyl phenoxypolyethoxylethanol
- PBS
- phosphate buffered saline
- PBST
- phosphate buffered saline with Tween 20
- qRT-PCR
- quantitative reverse transcription real-time polymerase chain reaction
- SWIFT
- Stored Waveform Inverse Fourier Transform.
REFERENCES
- 1. Salnikow K., and Zhitkovich A. (2008) Genetic and Epigenetic Mechanisms in Metal Carcinogenesis and Cocarcinogenesis: Nickel, Arsenic, and Chromium. Chem. Res. Tox 21, 28–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cheng T. F., Choudhuri S., and Muldoon-Jacobs K. (2012) Epigenetic targets of some toxicologically relevant metals: a review of the literature. JAT 32, 643–653 [DOI] [PubMed] [Google Scholar]
- 3. Huang C., Ke Q., Costa M., and Shi X. (2004) Molecular mechanisms of arsenic carcinogenesis. Mol. Cell. Biochem. 255, 57–66 [DOI] [PubMed] [Google Scholar]
- 4. Lynn S., Lai H. T., Gurr J. R., and Jan K. Y. (1997) Arsenite retards DNA break rejoining by inhibiting DNA ligation. Mutagenesis 12, 353–358 [DOI] [PubMed] [Google Scholar]
- 5. Hei T. K., Liu S. X., and Waldren C. (1998) Mutagenicity of arsenic in mammalian cells: Role of reactive oxygen species. Proc. Natl. Acad. Sci. U.S.A. 95, 8103–8107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kornberg R. D. (1974) Chromatin Structure: A Repeating Unit of Histones and DNA. Science 184, 868–871 [DOI] [PubMed] [Google Scholar]
- 7. Li E. (2002) Chromatin modification and epigenetic reprogramming in mammalian development. Nat. Rev. Gen. 3, 662–673 [DOI] [PubMed] [Google Scholar]
- 8. Strahl B. D., and Allis C. D. (2000) The language of covalent histone modifications. Nature 403, 41–45 [DOI] [PubMed] [Google Scholar]
- 9. Talbert P. B., and Henikoff S. (2014) Environmental responses mediated by histone variants. Trends Cell Biol. 24, 642–650 [DOI] [PubMed] [Google Scholar]
- 10. Loyola A., and Almouzni G. (2004) Histone chaperones, a supporting role in the limelight. Biochim. Biophys. Acta 1677, 3–11 [DOI] [PubMed] [Google Scholar]
- 11. Henikoff S., and Ahmad K. (2005) Assembly of Variant Histones into Chromatin. Ann. Rev. Cell Devl. Biol. 21, 133–153 [DOI] [PubMed] [Google Scholar]
- 12. Arita A., and Costa M. (2009) Epigenetics in metal carcinogenesis: nickel, arsenic, chromium and cadmium. Metallomics 1, 222–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arita A., Niu J., Qu Q., Zhao N., Ruan Y., Nadas A., Chervona Y., Wu F., Sun H., Hayes R. B., and Costa M. (2012) Global levels of histone modifications in peripheral blood mononuclear cells of subjects with exposure to nickel. Env. Health Perspect. 120, 198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou X., Li Q., Arita A., Sun H., and Costa M. (2009) Effects of nickel, chromate, and arsenite on histone 3 lysine methylation. Toxicol. Appl. Pharmacol. 236, 78–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jo W., Ren X., Chu F., Aleshin M., Wintz H., Burlingame A., Smith M., Vulpe C., and Zhang L. (2009) Acetylated H4K16 by MYST1 protects UROtsa cells from arsenic toxicity and is decreased following chronic arsenic exposure. Toxicol. Appl. Pharmacol. 241, 294–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cantone L., Nordio F., Hou L., Apostoli P., Bonzini M., Tarantini L., Angelici L., Bollati V., Zanobetti A., Schwartz J., Bertazzi P. A., and Baccarelli A. (2011) Inhalable metal-rich air particles and histone H3K4 dimethylation and H3K9 acetylation in a cross-sectional study of steel workers. Env. Health Perspect. 119, 964–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramirez T., Brocher J., Stopper H., and Hock R. (2007) Sodium arsenite modulates histone acetylation, histone deacetylase activity and HMGN protein dynamics in human cells. Chromosoma 117, 147–157 [DOI] [PubMed] [Google Scholar]
- 18. Jensen T. J., Wozniak R. J., Eblin K. E., Wnek S. M., Gandolfi A. J., and Futscher B. W. (2009) Epigenetic mediated transcriptional activation of WNT5A participates in arsenical-associated malignant transformation. Toxicol. Appl. Pharmacol. 235, 39–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Arrigo A.-P. (1983) Acetylation and methylation patterns of core histones are modified after heat or arsenite treatment of Drosophila tissue culture cells. Nucleic Acids Res. 11, 1389–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jensen T. J., Novak P., Eblin K. E., Gandolfi A. J., and Futscher B. W. (2008) Epigenetic remodeling during arsenical-induced malignant transformation. Carcinogenesis 29, 1500–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li J., Chen P., Sinogeeva N., Gorospe M., Wersto R. P., Chrest F. J., Barnes J., and Liu Y. (2002) Arsenic trioxide promotes histone H3 phosphoacetylation at the chromatin of CASPASE-10 in acute promyelocytic leukemia cells. J. Biol. Chem. 277, 49504–49510 [DOI] [PubMed] [Google Scholar]
- 22. Li J., Gorospe M., Barnes J., and Liu Y. (2003) Tumor Promoter Arsenite Stimulates Histone H3 Phosphoacetylation of Proto-oncogenes c-fos and c-jun Chromatin in Human Diploid Fibroblasts. J. Biol. Chem. 278, 13183–13191 [DOI] [PubMed] [Google Scholar]
- 23. Ray P. D., Huang B. W., and Tsuji Y. (2015) Coordinated regulation of Nrf2 and histone H3 serine 10 phosphorylation in arsenite-activated transcription of the human heme oxygenase-1 gene. Biochim. Biophys. Acta 1849, 1277–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reichard J. F., and Puga A. (2010) Effects of arsenic exposure on DNA methylation and epigenetic gene regulation. Epigenomics 2, 87–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ren X., McHale C. M., Skibola C. F., Smith A. H., Smith M. T., and Zhang L. (2011) An emerging role for epigenetic dysregulation in arsenic toxicity and carcinogenesis. Env. Health Perspect. 119, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang F., Paramasivam M., Cai Q., Dai X., Wang P., Lin K., Song J., Seidman M. M., and Wang Y. (2014) Arsenite binds to the RING finger domains of RNF20-RNF40 histone E3 ubiquitin ligase and inhibits DNA double-strand break repair. J. Am. Chem. Soc. 136, 12884–12887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alvelo-Ceron D., Niu L., and Collart D. (2000) Growth regulation of human variant histone genes and acetylation of the encoded proteins. Mol. Biol. Rep. 27, 61–71 [DOI] [PubMed] [Google Scholar]
- 28. Albig W., Trappe R., Kardalinou E., Eick S., and Doenecke D. (1999) The human H2A and H2B histone gene complement. J. Biol. Chem. 380, 7. [DOI] [PubMed] [Google Scholar]
- 29. Marzluff W. F., Gongidi P., Woods K. R., Jin J., and Maltais L. J. (2002) The human and mouse replication-dependent histone genes. Genomics 80, 487–498 [PubMed] [Google Scholar]
- 30. van Attikum H., and Gasser S. M. (2009) Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 19, 207–217 [DOI] [PubMed] [Google Scholar]
- 31. Law C., and Cheung P. (2013) Histone variants and transcription regulation. In: Kundu T. K., ed. Epigenetics: Development and Disease, pp. 319–341, Springer; Netherlands: [DOI] [PubMed] [Google Scholar]
- 32. Allshire R. C., and Karpen G. H. (2008) Epigenetic regulation of centromeric chromatin: old dogs, new tricks? Nat. Rev. Genet. 9, 923–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Black B. E., Foltz D. R., Chakravarthy S., Luger K., Woods V. L., and Cleveland D. W. (2004) Structural determinants for generating centromeric chromatin. Nature 430, 578–582 [DOI] [PubMed] [Google Scholar]
- 34. Ramakrishnan V., Finch J. T., Graziano V., Lee P. L., and Sweet R. M. (1993) Crystal structure of globular domain of histone H5 and its implications for nucleosome binding. Nature 362, 219–223 [DOI] [PubMed] [Google Scholar]
- 35. Siuti N., Roth M. J., Mizzen C. A., Kelleher N. L., and Pesavento J. J. (2006) Gene-specific characterization of human histone H2B by electron capture dissociation. J. Proteome Res. 5, 233–239 [DOI] [PubMed] [Google Scholar]
- 36. Zalensky A. O., Siino J. S., Gineitis A. A., Zalenskaya I. A., Tomilin N. V., Yau P., and Bradbury E. M. (2002) Human testis/sperm-specific histone H2B (hTSH2B): Molecular Cloning Characterization. J. Biol. Chem. 277, 43474–43480 [DOI] [PubMed] [Google Scholar]
- 37. Boulard M., Gautier T., Mbele G. O., Gerson V., Hamiche A., Angelov D., Bouvet P., and Dimitrov S. (2006) The NH2 tail of the novel histone variant H2BFWT exhibits properties distinct from conventional H2B with respect to the assembly of mitotic chromosomes. Mol. Cell. Biol. 26, 1518–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Molden R. C., Bhanu N. V., LeRoy G., Arnaudo A. M., and Garcia B. A. (2015) Multi-faceted quantitative proteomics analysis of histone H2B isoforms and their modifications. Epigenetics Chromatin 8, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li G., Levitus M., Bustamante C., and Widom J. (2005) Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 12, 46–53 [DOI] [PubMed] [Google Scholar]
- 40. Kimura H., and Cook P. R. (2001) Kinetics of core histones in living human cells: Little exchange of H3 and H4 and some rapid exchange of H2b. J. Cell Biol. 153, 1341–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jamai A., Imoberdorf R. M., and Strubin M. (2007) Continuous histone H2B and transcription-dependent histone H3 exchange in yeast cells outside of replication. Mol. Cell 25, 345–355 [DOI] [PubMed] [Google Scholar]
- 42. Riedmann C., Ma Y., Melikishvili M., Godfrey S., Zhang Z., Chen K., Rouchka E. C., and Fondufe-Mittendorf Y. N. (2015) Inorganic Arsenic-induced cellular transformation is coupled with genome wide changes in chromatin structure, transcriptome and splicing patterns. BMC Genomics 16, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bonenfant D., Coulot M., Towbin H., Schindler P., and van Oostrum J. (2006) Characterization of histone H2A and H2B variants and their post-translational modifications by mass spectrometry. Mol. Cell. Proteomics 5, 541–552 [DOI] [PubMed] [Google Scholar]
- 44. Kaiser N., Quinn J., Blakney G., Hendrickson C., and Marshall A. (2011) A Novel 9.4 Tesla FTICR mass spectrometer with improved sensitivity, mass resolution, and mass range. J. Am. Soc. Mass Spectrom. 22, 1343–1351 [DOI] [PubMed] [Google Scholar]
- 45. Guan S., and Marshall A. G. (1996) Stored waveform inverse Fourier transform (SWIFT) ion excitation in trapped-ion mass spectometry: Theory and applications. Int. J. Mass Spectrometry Ion Processes 158, 5–37 [Google Scholar]
- 46. Tsybin Y. O., Hendrickson C. L., Beu S. C., and Marshall A. G. (2006) Impact of ion magnetron motion on electron capture dissociation Fourier transform ion cyclotron resonance mass spectrometry. Int. J. Mass Spectrometry 256, 144–149 [Google Scholar]
- 47. Blakney G. T., Hendrickson C. L., and Marshall A. G. (2011) Predator data station: A fast data acquisition system for advanced FT-ICR MS experiments. Int. J. Mass Spectrometry 306, 246–252 [Google Scholar]
- 48. Ledford E. B., Rempel D. L., and Gross M. L. (1984) Space charge effects in Fourier transform mass spectrometry. II. Mass calibration. Anal. Chem. 56, 2744–2748 [DOI] [PubMed] [Google Scholar]
- 49. Kari V., Karpiuk O., Tieg B., Kriegs M., Dikomey E., Krebber H., Begus-Nahrmann Y., and Johnsen S. A. (2013) A subset of histone H2B genes produces polyadenylated mRNAs under a variety of cellular conditions. PLoS ONE 8, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Spandidos A., Wang X., Wang H., and Seed B. (2010) PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res. 38, D792–D799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang X., and Seed B. (2003) A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res. 31, e154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Spandidos A., Wang X., Wang H., Dragnev S., Thurber T., and Seed B. (2008) A comprehensive collection of experimentally validated primers for polymerase chain reaction quantitation of murine transcript abundance. BMC Genomics 9, 633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stueckle T. A., Lu Y., Davis M. E., Wang L., Jiang B. H., Holaskova I., Schafer R., Barnett J. B., and Rojanasakul Y. (2012) Chronic occupational exposure to arsenic induces carcinogenic gene signaling networks and neoplastic transformation in human lung epithelial cells. Toxicol. Appl. Pharmacol. 261, 204–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tokar E. J., Diwan B. A., and Waalkes M. P. (2010) Arsenic exposure transforms human epithelial stem/progenitor cells into a cancer stem-like phenotype. Env. Health Perspectives 118, 108–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. DiMaggio P. A., Young N. L., Baliban R. C., Garcia B. A., and Floudas C. A. (2009) A mixed integer linear optimization framework for the identification and quantification of targeted post-translational modifications of highly modified proteins using multiplexed electron transfer dissociation tandem mass spectrometry. Mol. Cell. Proteomics 8, 2527–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xian F., Hendrickson C. L., Blakney G. T., Beu S. C., and Marshall A. G. (2010) Automated broadband phase correction of fourier transform ion cyclotron resonance mass spectra. Anal. Chem. 82, 8807–8812 [DOI] [PubMed] [Google Scholar]
- 57. Pesavento J. J., Mizzen C. A., and Kelleher N. L. (2006) Quantitative analysis of modified proteins and their positional isomers by tandem mass spectrometry: human histone H4. Anal. Chem. 78, 4271–4280 [DOI] [PubMed] [Google Scholar]
- 58. Doenecke D., Albig W., Bode C., Drabent B., Franke K., Gavenis K., and Witt O. (1997) Histones: genetic diversity and tissue-specific gene expression. Histochemistry 107, 1–10 [DOI] [PubMed] [Google Scholar]
- 59. Guo F., Kerrigan B. C., Yang D., Hu L., Shmulevich I., Sood A. K., Xue F., and Zhang W. (2014) Post-transcriptional regulatory network of epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions. J. Hematol. Oncol. 7, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wu C. Y., Tsai Y. P., Wu M. Z., Teng S. C., and Wu K. J. (2012) Epigenetic reprogramming and post-transcriptional regulation during the epithelial–mesenchymal transition. Trends Genetics 28, 454–463 [DOI] [PubMed] [Google Scholar]
- 61. Lee M. Y., and Shen M. R. (2012) Epithelial-mesenchymal transition in cervical carcinoma. Am. J. Transl. Res. 4, 1–13 [PMC free article] [PubMed] [Google Scholar]
- 62. Xiao W., Zhou S., Xu H., Li H., He G., Liu Y., Qi Y (2013) Nogo-B promotes the epithelial-mesenchymal transition in HeLa cervical cancer cells via Fibulin-5. Oncol. Reports 29, 109–116 [DOI] [PubMed] [Google Scholar]
- 63. Turner B. M. Decoding the nucleosome. (1993) Cell 75, 5–8 [PubMed] [Google Scholar]
- 64. Maze I., Noh K. M., Soshnev A. A., and Allis C. D. (2014) Every amino acid matters: essential contributions of histone variants to mammalian development and disease. Nat. Rev. Genet. 15, 259–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Henikoff S., and Smith M. M. (2015) Histone variants and epigenetics. Cold Spring Harbor Perspect. Biol. 7 a019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cheung P., and Lau P. (2005) Epigenetic regulation by histone methylation and histone variants. Mol. Endocrinol. 19, 563–573 [DOI] [PubMed] [Google Scholar]
- 67. Henikoff S., Furuyama T., and Ahmad K. (2004) Histone variants, nucleosome assembly and epigenetic inheritance. Trends Genetics 20, 320–326 [DOI] [PubMed] [Google Scholar]
- 68. Vardabasso C., Hasson D., Ratnakumar K., Chung C. Y., Duarte L., and Bernstein E. (2014) Histone variants: emerging players in cancer biology. Cell. Mol. Life Sci. 71, 379–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Monteiro F., Baptista T., Amado F., Vitorino R., Jerónimo C., and Helguero L. A. (2014) Expression and functionality of histone H2A variants in cancer. Oncotarget 5, 3428–3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Dryhurst D., and Ausió J. (2014) Histone H2A.Z deregulation in prostate cancer. Cause or effect? Cancer Metastasis Rev. 33, 429–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Skene P. J., and Henikoff S. (2013) Histone variants in pluripotency and disease. Development 140, 2513–2524 [DOI] [PubMed] [Google Scholar]
- 72. Cantariño N., Douet J., and Buschbeck M. (2013) MacroH2A – An epigenetic regulator of cancer. Cancer Lett. 336, 247–252 [DOI] [PubMed] [Google Scholar]
- 73. Svotelis A., Gévry N., and Gaudreau L. (2009) Regulation of gene expression and cellular proliferation by histone H2A.Z. Biochem. Cell Biol. 87, 179–188 [DOI] [PubMed] [Google Scholar]
- 74. Yuan Z.-F., Arnaudo A. M., and Garcia B. A. (2014) Mass spectrometric analysis of histone proteoforms. Ann. Rev. Anal.l Chem. 7, 113–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lee M. S., and Garrard W. T. (1991) Transcription-induced nucleosome ‘splitting’: an underlying structure for DNase I sensitive chromatin. EMBO J. 10, 607–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rhee H. S., Bataille A. R., Zhang L., and Pugh B. F. (2014) Subnucleosomal structures and nucleosome asymmetry across a genome. Cell 159, 1377–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ausió J. (2006) Histone variants—the structure behind the function. Briefings Funct. Genomics Proteomics 5, 228–243 [DOI] [PubMed] [Google Scholar]
- 78. Shaytan A. K., Landsman D., and Panchenko A. R. (2015) Nucleosome adaptability conferred by sequence and structural variations in histone H2A–H2B dimers. Current Opinion Structural Biol. 32, 48–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Brocato J., Fang L., Chervona Y., Chen D., Kiok K., Sun H., Tseng H.-C., Xu D., Shamy M., Jin C., and Costa M. (2014) Arsenic induces polyadenylation of canonical histone mRNA by down-regulating stem-loop-binding protein gene expression. J. Biol. Chem. 289, 31751–31764 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.