

Abstract
The genetic code is the cellular translation table for the conversion of nucleotide sequences into amino acid sequences. Changes to the meaning of sense codons would introduce errors into almost every translated message and are expected to be highly detrimental. However, reassignment of single or multiple codons in mitochondria and nuclear genomes, although extremely rare, demonstrates that the code can evolve. Several models for the mechanism of alteration of nuclear genetic codes have been proposed (including “codon capture,” “genome streamlining,” and “ambiguous intermediate” theories), but with little resolution. Here, we report a novel sense codon reassignment in Pachysolen tannophilus, a yeast related to the Pichiaceae. By generating proteomics data and using tRNA sequence comparisons, we show that Pachysolen translates CUG codons as alanine and not as the more usual leucine. The Pachysolen tRNACAG is an anticodon-mutated tRNAAla containing all major alanine tRNA recognition sites. The polyphyly of the CUG-decoding tRNAs in yeasts is best explained by a tRNA loss driven codon reassignment mechanism. Loss of the CUG-tRNA in the ancient yeast is followed by gradual decrease of respective codons and subsequent codon capture by tRNAs whose anticodon is not part of the aminoacyl-tRNA synthetase recognition region. Our hypothesis applies to all nuclear genetic code alterations and provides several testable predictions. We anticipate more codon reassignments to be uncovered in existing and upcoming genome projects.
The genetic code determines the translation of nucleotide sequences into amino acid sequences. It is commonly assumed that, as any change in the code altering the meaning of a codon would introduce errors into almost every translated message, such codon reassignments would be highly detrimental or lethal (Knight et al. 2001a). Therefore, regardless of whether it is optimal or not (Freeland and Hurst 1998; Freeland et al. 2000), the canonical genetic code was long thought to be immutable and was termed a “frozen accident” of history (Crick 1968). However, reassignment of single or multiple codons in mitochondria (Knight et al. 2001b) and nuclear genomes (Lozupone et al. 2001; Miranda et al. 2006) demonstrates that the code can evolve (Knight et al. 2001a; Koonin and Novozhilov 2009; Moura et al. 2010). Several codons have been reassigned in independent lineages. Most nuclear code alterations reported so far are stop codon and CUG-codon reassignments.
Three theories have been proposed to explain reassignments in the genetic code. The codon capture hypothesis states that first a codon and, subsequently, its then meaningless cognate tRNA must disappear from the coding genome before a tRNA with a mutated anticodon appears, changing the meaning of the codon (Osawa and Jukes 1989; Osawa et al. 1992). Genome GC or AT pressure (for reasons often unclear) is thought to cause codon disappearance. In contrast, the ambiguous intermediate hypothesis postulates that either mutant tRNAs, which are charged by more than one aminoacyl-tRNA synthetase, or misreading tRNAs drive genetic code changes (Schultz and Yarus 1994, 1996). The ambiguous codon decoding leads to a gradual codon identity change that is completed upon loss of the wild-type cognate tRNA. The alternative CUG encoding as serine instead of leucine in Candida and Debaryomyces species (the so-called alternative yeast code [AYCU]) has been strongly promoted as an example for the ambiguous intermediate theory. It is supposed that CUG-codon decoding is ambiguous in many extant Candida species (Tuite and Santos 1996; Suzuki et al. 1997), that the CUG-codon decoding can—at least in part—be converted (Santos et al. 1996; Bezerra et al. 2013), and that the origin of the has been estimated to precede the separation of the Candida and Saccharomyces genera by ∼100 Myr (Massey et al. 2003). However, the ambiguous decoding of the CUG triplet in extant “CTG clade” species is caused by slightly inaccurate charging of the and not by competing tRNAs.
The genome streamlining hypothesis notes that codon changes are driven by selection to minimize the translation machinery (Andersson and Kurland 1995). This best explains the many codon reassignments and losses in mitochondria. In Saccharomycetaceae mitochondria, for example, 10 to 25 sense codons are unused, and the CUG codons are usually translated as threonine by a tRNAThr, which has evolved from a tRNAHis ancestor (Su et al. 2011). In the Eremothecium subbranch, the CUG codons are decoded by alanine, but this modified code did not originate by capture of the CUG codons by an anticodon-mutated tRNAAla but by switching the acceptor stem identity determinants of the Saccharomycetaceae tRNAThr from threonine to alanine (Ling et al. 2014).
From the analysis of the conservation of amino acid types and CUG-codon positions in motor and cytoskeletal proteins, we recently showed that these proteins allow us to unambiguously assign the code employed by any given species of yeast (Mühlhausen and Kollmar 2014). Plotting the assigned code onto the yeast phylogeny demonstrates that the AYCU appears to be polyphyletic in origin, with the “CTG clade” species and Pachysolen tannophilus grouping in different branches. Pachysolen is especially noteworthy with regards to its possible genetic code. Sequence conservation–based analysis indicates that Pachysolen does not encode leucine by CUG and that its CUG-encoded residues are also not present at conserved serine positions. In addition, Pachysolen shares only a few CUG-codon positions with yeasts using the standard genetic code and no CUG-codon positions with “CTG clade” species. This prompted us to determine the identity of the Pachysolen CUG encoding by molecular phylogenetic and proteome analyses.
Results
A new nuclear genetic code in the yeast P. tannophilus
We determined the tRNACAGs in 60 sequenced yeast species (Supplemental Table S1) and aligned them against known and . While and could clearly be classified, the identified Pachysolen tRNACAG sequence was dissimilar to both (Fig. 1A). Comparison to all Candida albicans cytoplasmic tRNAs suggested a close relationship to alanine tRNAs. The alanine identity of the Pachysolen tRNACAG was verified by molecular phylogenetic analyses based on extensive sequence and taxonomic sampling (Fig. 1B; Supplemental Figs. S1–S4; Supplemental Table S1). The sequence identity between the Pachysolen tRNACAG and all identified yeast GCN-decoding tRNAs (752 Saccharomycetales clade sequences) is on average 69.3%, ranging from 62.1%–77.0%. This is slightly below the average sequence identity within the GCN-decoding tRNAs (83.6%; minimum sequence identity is 50.6%), reflecting some sequence divergence beyond the different anticodons (Supplemental Fig. S1). The major alanine tRNA identity determinants, the discriminator base “A73” and the invariant “G3:U70” wobble base pair as part of the conserved 5′ sequence G1GGC4 (Musier-Forsyth et al. 1991; Saks et al. 1994; Giegé et al. 1998; Giegé and Eriani 2015), are also present in the Pachysolen tRNACAG (Fig. 1A). In contrast, serine tRNAs are characterized by a conserved variable loop sequence (Fig. 1A); leucine tRNANAGs, by highly conserved A35 and m1G37 nucleotides and extended variable loops (Fig. 1A; Saks et al. 1994; Giegé et al. 1998; Giegé and Eriani 2015). Although both contain extended variable loop sequences, not the anticodon sequences but different tertiary structures seem to be important for discriminating serine and leucine tRNAs (Asahara et al. 1993, 1994).
Figure 1.

The Pachysolen CUG-tRNA is an Ala-tRNA. (A) Secondary structures of the Pachysolen tannophilus , the Candida albicans , and the Yarrowia lipolytica . The consensus Saccharomycetales (based on 752 sequences), (748 sequences), and (300 sequences) are shown for comparison. The discriminator base “N73” is denoted by the corresponding nucleotide number of the respective tRNA gene. It is obvious that the Pachysolen and the Candida share more consensus elements with the alanine and serine tRNAs, respectively, than with the leucine tRNAs. tRNA identity determinants are indicated by red boxes and dashed lines. (B) Unrooted phylogenetic network of 172 tRNA sequences generated using the neighbor-net method as implemented in SplitsTree v4.1.3.1. Methionine, isoleucine, arginine, and threonine tRNAs were included as outgroup. , , and the Pachysolen are highlighted in dark green, blue, and red, respectively.
Although identity elements are highly conserved within the respective codon family box tRNAs, the same elements might be present in other tRNAs where they are located in variable regions. For example, the “G3:U70” wobble base pair, which is usually only found in alanine tRNAs, is also present in Phaffomycetaceae and some Saccharomycetaceae , although these tRNAs clearly belong to the CTN codon box family tRNAs (Fig. 1B; Supplemental Figs. S1–S4), and CUG codons in these species are translated as leucine (Mühlhausen and Kollmar 2014). The presence of m1G37 in most Candida was shown to be necessary for CUG decoding accuracy and efficiency but also has been shown to cause partial mischarging by leucine-tRNA synthetases (Suzuki et al. 1997). However, the Candida cylindracea contains an A37 but shows no decoding ambiguity (Kawaguchi et al. 1989; Tuite and Santos 1996), indicating that multiple and overlapping discriminators determine CUG decoding accuracy and tRNACAG acetylation efficiency. The anticodon and the neighboring G37 nucleotide of the Pachysolen tRNACAG are identical to leucine tRNAs, while the remainder of the Pachysolen sequence is similar to alanine tRNAs (Fig. 1A). The absence of an extended variable loop sequence should prevent the Pachysolen becoming misacetylated.
CUG codons can unambiguously translate as alanine
To verify the translation of the CUG codons to alanine, we analyzed a cytoplasmic extract of laboratory-grown Pachysolen by high-resolution tandem mass spectrometry (LC-MS/MS), generating approximately 460,000 high-quality mass spectra (Supplemental Fig. S5). Spectra processing resulted in 27,126 nonredundant peptide matches with a median mass measurement error of about 240 parts per billion (Fig. 2A; Supplemental Fig. S6). We identified 53% (2817) of the 5288 predicted proteins with median protein sequence coverage of ∼20%. The median numbers of peptides and corresponding peptide spectrum matches (PSMs) identified per protein are six and nine, respectively (Supplemental Fig. S6).
Figure 2.

Pachysolen translates CUG codons as alanine and not as leucine. (A) Distribution of the mass errors of randomly selected 10% of all peptides (gray dots), 10% of peptides with CUG translated as alanine (red), and 10% of peptides with CUG translated as other amino acids (blue). (B) Proteins identified in the soluble cell extract of Pachysolen sorted by sequence coverage. (C) Number of PSMs covering CUG-codon positions per protein. (D) Percentage of the CUG-codon positions per protein covered by the proteomics data. (E) Number and distribution of observed CUG translations in all peptide spectrum matches from MS/MS analysis. (F) Representative MS/MS spectrum of a peptide containing a CUG translated as alanine. The peptide sequence is shown below the spectrum, with the annotation of the identified matched N-terminal fragment ions (b-type ions) in blue and the C-terminal fragment ions (y-type ions) in red. Only major peaks are labeled for clarity (full annotation of the spectrum is shown in Supplemental Fig. S7).
CUG-codon translation affects 4210 (80%) of Pachysolen protein coding genes (Supplemental Fig. S5). Of the 16,824 CUG-codon positions in Pachysolen protein coding genes, 1433 (8.5%) are covered by nonredundant PSMs (3835 PSMs in total, 2.9-fold average coverage) (Fig. 2B–E). Of these unique CUG positions, 907 are covered by PSMs containing sequences with CUG codons fully supported by b- and/or y-type fragment ions. Almost all of these (97.2%) contained the CUG codons translated as alanine (Fig. 2E,F; Supplemental Fig. S7).
The remaining rare incidences can be classified into two groups. For eight of the fully supported CUG-codon positions, PSMs with ambiguously translated CUGs (alanine plus another amino acid) were found (Fig. 2E). For an additional 18 CUG-codon positions, PSMs were found that translate CUG as an amino acid other than alanine (Fig. 2E). Both of the above minority cases might be due to differences between our and the sequenced Pachysolen strain (Liu et al. 2012), might be due to transcription and translation errors, or might represent spurious mischarging of the Pachysolen . For comparison, we analyzed the unambiguously decoded AUG codon and found similar numbers of differences (Supplemental Fig. S8). Accordingly, the CUG codon is as unambiguous as the unambiguous, related codon AUG. Substantial mischarging of Candida by leucines has been shown in vitro and in vivo (Suzuki et al. 1997), but other potential mischargings have never been analyzed.
History of the CUG-decoding tRNA
To reconstruct the history and origin of all yeast tRNACAGs, we performed in-depth phylogenetic analyses of all UCN-decoding tRNAs (serine), GCN-decoding tRNAs (alanine), and CUN-decoding tRNAs (leucine) (Supplemental Figs. S9–S11). These analyses support our previous assumption (Mühlhausen and Kollmar 2014) that all “CTG clade” species’ tRNACAGs are serine-tRNAs (Supplemental Fig. S9) and that all Saccharomycetaceae, Phaffomycetaceae, and “Pichiaceae” species’ tRNACAGs are leucine-tRNAs (Fig. 3; Supplemental Fig. S10). Monophyly of the indicates a common origin in the ancestor of the “CTG clade.”
Figure 3.

The history of CUG-codon reassignment in yeasts. Decoding and origin of the tRNACAGs were plotted onto a time-calibrated yeast phylogeny adapted from Mühlhausen and Kollmar (2014). Colored lines denote the different tRNACAG types, with green representing , blue denoting the “CTG clade” , red marking the Pachysolen , and black denoting the absence of a tRNACAG in the respective branch. The schemes with the four circles represent the reconstructed CUN tRNA family at each branch, with filled and empty circles denoting the presence and absence, respectively, of the CUN family box tRNAs. Arrows indicate tRNA gene duplications followed by anticodon mutations, except for the duplication in the ancestor of the SP–PC branches that resulted in orthologous subtypes. Branches with identical decoding schemes have been collapsed. The percentage of CUG codons per species/taxon were derived from Mühlhausen and Kollmar (2014). “Pichiaceae,” as shown here, is not a commonly agreed taxon. Because all species of the respective branch in the presented tree have at least one synonymous name starting with “Pichia,” we termed the entire branch “Pichiaceae” for simplicity. The scheme at the bottom shows how the four CUN codons can be decoded by several combinations of isoacceptor tRNAs using standard and wobble base-pairing.
The UCN-decoding tRNAs split into two major subbranches: a group of UCG-decoding tRNAs to which the belong, and a group of UCU-decoding tRNAs. This supports the prior notion (Massey et al. 2003) that the ancient originated from an UCG-tRNA by insertion of an A into the anticodon rather than from an UCU-tRNA by insertion of a C directly before the anticodon.
In contrast to the monophyletic , the are polyphyletic, and many yeasts contain multiple derived from gene duplication of cognate and isoacceptor tRNAs (Fig. 3). For example, Yarrowia lipolytica contains 13 , of which two were most probably derived from either an ancestral or and 11 were derived by gene duplication and mutation from a . The Phaffomycetaceae, Kluyveromyces, Lachancea kluyveri, and Eremothecium were derived from an ancestral ; the Lachancea thermotolerans and Lachancea waltii were derived by a recent gene duplication and anticodon mutation from a ; and the “Pichiaceae” species have derived their by species-specific events (Fig. 3). The Pachysolen most probably originated by duplication of a tRNAUGC (Supplemental Fig. S11), followed by mutation to the isoacceptor tRNACGC and, finally, insertion of an A into the anticodon similar to the origin of the ancient .
History of the CUN family box tRNAs
The CUN family box tRNAs are split into two major groups: a group of and a group of . The tRNACAGs are only present in Saccharomycetaceae and have been derived from an ancestral (Fig. 3). The form two subgroups, which most probably originated after the split of Y. lipolytica. One of the subgroups is restricted to Yarrowia, Pachysolen, and the “Pichiaceae”; the other is common to all yeasts and contains the Saccharomyces . The latter is unique because it is the only Saccharomyces cerevisiae tRNA with an unmodified uridine in the wobble position of the anticodon triplet (Randerath et al. 1979; Johansson and Byström 2005). Unmodified U34s have otherwise only been found in mitochondria, chloroplasts, and Mycoplasma species. The Saccharomyces is also unique as it is able to translate all six leucine codons (Weissenbach et al. 1977).
All sequenced yeasts have distinct NNA-decoding and NNG-decoding tRNAs for all respective two-codon families and most four-codon families. Modifications at U34 in NNA-decoding tRNAs enable these to also read G-ending codons, and accordingly, many NNA-decoding tRNAs are competing with NNG-decoding tRNAs when reading NNG codons (Johansson et al. 2008). However, it is also known that some U34-modified tRNAs, such as 5′-methoxycarbonylmethyl-2-thiouridine (mcm5s2)–modified Gln- and Glu-decoding tRNAs in most if not all prokaryotic and eukaryotic species, are not able to read G-ending codons in vivo (Johansson et al. 2008; Rezgui et al. 2013). While all Saccharomycetaceae and Phaffomycetaceae contain the unique U34-unmodified , in Pachysolen and the “Pichiaceae,” the tRNA of this subbranch is mutated to or (Fig. 3). The reason for the unmodified U34 in the Saccharomyces is unknown, but given that all other U-wobble tRNAs contain modified U34 nucleotides, it is tempting to assume that the ancient yeast also contained a modified U34. Given the unambiguous decoding of the CUG codons by the in Pachysolen, the Pachysolen may be presumed to contain a U34 modification, such as mcm5s2 in Glu-decoding tRNAs or pseudouridine in Ile-decoding tRNAUAUs, to prevent competitive decoding of the CUG codons.
Discussion
How did such diversity in tRNA origin and CUG-codon decoding (leucine vs. serine vs. alanine) evolve? While several testable predictions of each of the codon reassignment hypotheses have been summarized (Knight et al. 2001a), these predictions, however, did not include decoding of CUG codons by an Ala-tRNACAG. Does the presence of the Pachysolen still fit into the existing models?
The yeast CUG-codon reassignments do not accord with the codon capture theory
According to the codon capture theory, CUG codons need to have disappeared before their reassignment at the split of the “CTG clade” (Fig. 4; Supplemental Fig. S12). The time frame for codon disappearance is defined by the split of the Saccharomycetaceae/Phaffomycetaceae and Pachysolen/“CTG clade” branches (hereafter called SP and PC branches, respectively). Codon disappearance must have happened either in the very short time frame between the split of the SP and PC branches and the divergence of the “CTG clade” (Supplemental Fig. S12) or before the SP–PC split (Fig. 4), which would then necessarily include reappearance of the and CUG codons at original positions in the SP branch. Subsequently, the Ser-, Ala-, and Leu-tRNACAGs could have captured the still unassigned CUG codon in the “CTG clade,” Pachysolen, and the “Pichiaceae” branches independently from each other. However, disappearance of an entire codon from a genome by neutral mutations is extremely unlikely to happen in such a short time. It is similarly unlikely that AT/GC bias, the main force driving codon reassignment according to the codon capture theory, caused only one codon to disappear.
Figure 4.

The mechanism of CUG-codon reassignment. The scheme contrasts the presence of tRNACAGs and evolution of CUG codons according to the tRNA loss driven codon reassignment hypothesis with assumptions based on the codon capture and ambiguous intermediate theories. Only the most probable sequence of events is shown for each hypothesis. Alternative but less likely scenarios are presented in Supplemental Figure S12. Mixed models, for example, ambiguous decoding of the CUG codons in the ancestor of the SP and CP clades followed by loss of the in the ancient Pachysolen and capture by the , seem extremely unlikely and are not shown. The codon capture theory distinguishes from the tRNA loss driven codon reassignment hypothesis mainly by the order of events (tRNA loss after or before the reduction of CUG codons, respectively), the degree of CUG-codon loss (loss of all codons vs. a reduction of CUG-codon usage, respectively), and the cause of reduction of CUG codons (AT pressure vs. tRNA loss and decreased decoding fidelity, respectively). In contrast to the ambiguous intermediate hypothesis, the tRNA loss driven codon reassignment theory does not assume any ambiguous decoding of the CUG codons. In addition, capture of the CUG codon by different tRNAs is an elemental characteristic of the tRNA loss driven codon reassignment theory and does not need to be split into independent events.
The yeast CUG-codon reassignments do not accord with the ambiguous intermediate theory
The ambiguous intermediate theory assumes the simultaneous assignment of a codon to two tRNAs. In case of the yeasts, it was proposed that both the cognate and the new were present before the split of the SP and PC branches (Massey et al. 2003). In order to fit the into this scenario, one has to assume either the presence of the at the same time or two successive ambiguous intermediate states. The presence of three tRNACAGs at the same time would give rise to an even more ambiguous decoding and seems to be highly unlikely. More likely seems the scenario including two successive ambiguous intermediates (Fig. 4; Supplemental Fig. S12). In this scenario, a time span with another ambiguous CUG-codon decoding ( competing with ) would have followed the split of the “CTG clade” or would have started in the Pachysolen branch. If the ambiguous intermediate theory were true, and in extant species should have been derived from the same ancestral Ser- and Leu-tRNAs. While this is true for the of the “CTG clade,” the are polyphyletic and clearly have different origins (Fig. 3; Supplemental Figs. S9, S10). Accordingly, the ambiguous intermediate theory would at least require—in case of the more likely separate ambiguous intermediate events—the independent loss of the in the SP and the Pachysolen/“Pichiaceae” branches (PP branches) and the independent loss of the in multiple branches, including the branches with altered decoding. The polyphyly of the is not explained in either of the scenarios.
The scenario of two successive CUG-codon reassignments is further weakened by the frequent nature of the CUG codon. Although phylogenetic mapping of variant codes has shown that the same codons have independently been reassigned both in nuclear genomes and in mitochondrial genomes, those reassignments only affected rare codons (Knight et al. 2001a). However, the CUG codon is not a rare codon, and it seems extremely unlikely that the same frequently used sense codon became ambiguous in two subbranches of the same taxon within a very short time in independent events, as it would be required for the two CUG-codon reassignments in yeasts according to the ambiguous intermediate theory. A preference for further ambiguous intermediate events because of CUG-codon usage reduction is similarly unlikely. If this preference would exist, many more CUG-codon reassignment events would be expected in all branches of the Saccharomycetes.
In the other scenario assuming the simultaneous ambiguous CUG decoding by three tRNACAGs, one would expect to observe CUG-codon positions conserved between species decoding CUG as leucine, serine, and alanine. A comparison of cytoskeletal and motor protein sequences from 60 yeast species, however, showed that CUG codons from “CTG-clade” species were not found even at moderately (≥50%) conserved leucine positions with CUG codons and that CUG codons from yeasts using standard codon usage were not found at moderately conserved serine positions with CUG codons (Mühlhausen and Kollmar 2014). Pachysolen did not share any CUG-codon positions with the “CTG-clade” species but did share some CUG-codon positions with CUG codons from yeasts using standard codon usage (Mühlhausen and Kollmar 2014). However, these shared positions were at alignment positions with low sequence conservation. The few shared CUG codons at nonconserved sequence positions probably do not represent original CUG codons but more likely resulted from random reassignments/mutations. Thus, Pachysolen and “CTG-clade” species have independently reassigned the CUG codons: Pachysolen mainly at conserved alanine positions and “CTG-clade” species mainly at conserved serine positions. These findings contradict a scenario of simultaneous ambiguous CUG decoding by three tRNACAGs.
The tRNA loss driven codon reassignment mechanism presents a parsimonious explanation
The observed polyphyly of the tRNACAGs is best described by a tRNA loss driven codon reassignment process as follows (Fig. 4): The ancestor of the SP and PC clades lost its by gene loss or mutation. This loss was accompanied or followed by the appearance of the Saccharomyces type by gene duplication of a “normal” . This new evolved the characteristic unmodified U34 after its appearance or later in the SP branch. The loss of the might not have caused considerable viability issues because CUG codons could still be decoded as leucine by, although probably inefficiently, wobble base-pairing (Crick 1966) involving the ancestral U34-modified . This process was presumably supported by the doubling in tRNA copy number.
Subsequent to the loss of the original tRNA, reduction in translational fidelity (Gromadski et al. 2006) might have caused the number of CUG codons to gradually decrease. Compared with the most ancient yeast species Yarrowia, all analyzed yeasts have considerably decreased numbers of CUG codons (Fig. 3; Mühlhausen and Kollmar 2014). Even the highly GC-rich genomes of Ogataea parapolymorpha (Ravin et al. 2013) and Kuraishia capsulata (Morales et al. 2013) have fewer CUG codons, suggesting a general strong reduction of CUG-codon usage after the split of Yarrowia.
Many subbranches of the Saccharomycetaceae independently lost their , which was not accompanied by further CUG-codon losses (Fig. 3). These subbranches demonstrate that the CUG codons can efficiently be translated by noncognate tRNAs, most probably the U34-unmodified . The tRNACAG-independent CUG decoding might have developed into full functionality during the time from the split of Yarrowia to the divergence of the SP branch, and until achievement of full functionality, CUG-codon usage considerably decreased. CUG-codon reduction most probably happened by transitions and transversions to other leucine codons before the divergence of the SP branch. Within protein coding regions, codon changes within the CTN family box, and also between CTG and TTG, are extremely frequent. Indeed, even closely related Saccharomyces species have few conserved leucine codons (Mühlhausen and Kollmar 2014).
Subsequent to the loss of the ancient and the reduction of CUG-codon usage, the unassigned CUG codon became free to be captured by other tRNAs with mutated anticodons. Capturing by isoacceptor tRNAs is most straightforward and happened in the ancestor of the SP branch and within the “Pichiaceae” species independently from each other as shown by the polyphyly of the (Fig. 3; Supplemental Fig. S8). In addition to isoacceptor tRNAs, only tRNAs whose anticodon is not part of the identity determinants of their respective aminoacyl-tRNA synthetases (aaRSs) could also capture the free CUG codon. In the “CTG clade,” a mutated Ser-tRNA captured the CUG codon. This event was triggered or supported by loss of the , which significantly reduced the possibility of further ambiguous CUG decoding. In an independent event, Pachysolen acquired the by duplication and subsequent mutation of a GCU-decoding tRNA.
Characteristics of the capturing tRNAs
In principle, it is highly unlikely that sense codons are captured by mutant tRNAs charged with noncognate amino acids because of the high recognition accuracy of the respective tRNAs by the aaRSs (Crick 1968; Saks et al. 1994). Aminoacyl-tRNA synthetases usually recognize their cytoplasmic cognate tRNAs in at least two different regions: the most prominent being the discriminator nucleotide “N73” and the acceptor stem, and the other consisting of the anticodon and neighboring nucleotides (Saks et al. 1994; Giegé et al. 1998; Giegé and Eriani 2015). There are four principal scenarios to account for how codons could be captured by noncognate tRNAs and aaRS still be maintained. All four scenarios have been observed in nature (Fig. 5): (1) tRNAs could be mutated in the anticodon retaining recognition by the original aaRSs; (2) tRNAs could be mutated at other discriminator bases disrupting tRNA recognition by the cognate aaRSs and enabling acylation by other aaRSs; (3) mutations in the respective aaRSs might relax anticodon discrimination, and this might happen either without or in combination with tRNA anticodon mutations; and (4) new orthogonal tRNA/aaRS pairs might evolve.
Figure 5.
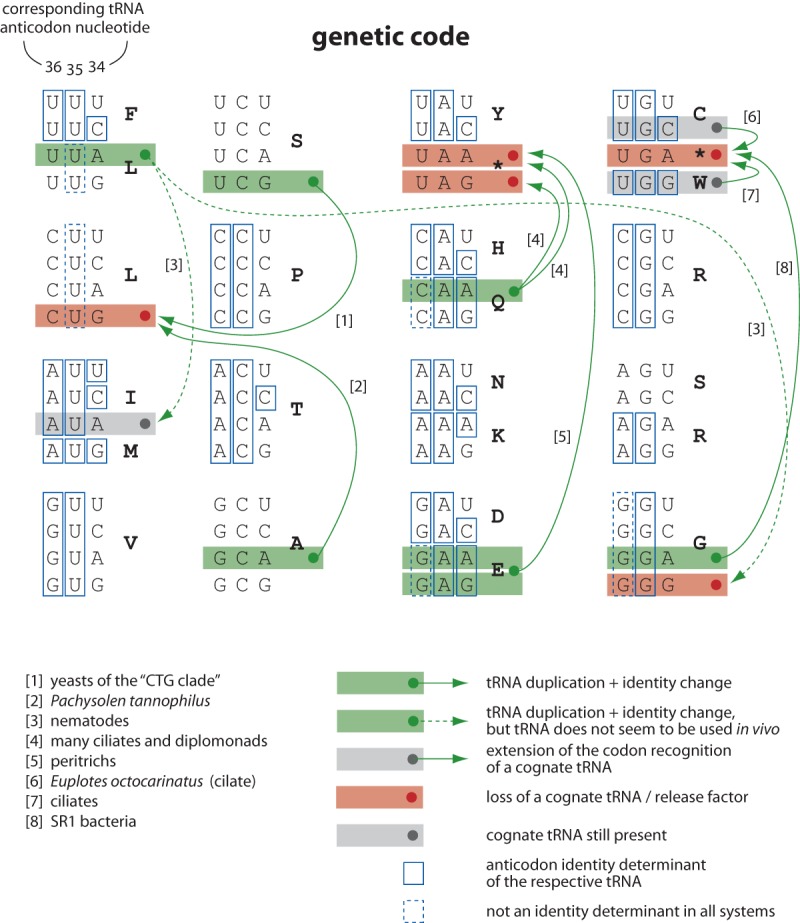
Compilation of nuclear codon reassignments. The scheme shows tRNA anticodon identity determinants plotted onto the genetic code. If mutated, Ala- and Ser-tRNAs (and in some systems also the Leu-tRNAs) should, in principle, be able to capture any other codon. Plotting all known cases of nuclear genetic code reassignments onto the codon table shows that most cases resulted from extending the decoding capabilities of near-cognate tRNAs. Reassignments reported for Mycoplasma capricolum, which was thought to lack a dedicated tRNA for decoding CGG although still containing six CGG codons in its genome (Oba et al. 1991), and Micrococcus luteus, which was thought to lack AUA and AGA codons (Kano et al. 1993), are not supported by whole-genome sequencing data (Young et al. 2010; Chu et al. 2011). The available data show that the Caenorhabditis elegans GGG codons are translated as glycine in vivo and not as leucine (Hamashima et al. 2015).
The first scenario includes the capture of the CUG codons in “CTG clade” yeasts and Pachysolen. The recognition sites of seryl- and alanyl-RSs do not include the respective anticodons, providing an explanation as to why it is that only these tRNAs (in addition to other leucine tRNAs) could capture the free CUG codon (Fig. 5). This also means that tRNASers and tRNAAlas could potentially capture any other free codon. The anticodon nucleotide A35 in leucine tRNAs is a system-dependent identity determinant (e.g., it is a determinant in yeasts but not in human). This might explain the presence of many leucine tRNAs with noncognate anticodons in nematodes (Fig. 5; Hamashima et al. 2012). Because the anticodon identity determinants are not entirely conserved but could vary from species to species or taxon to taxon, it would be necessary to determine the orthogonal tRNA/aaRS pairs for each species to generate species-specific sets of potentially capturing tRNAs.
The second scenario is represented by the mitochondrial in Eremothecium, which originated from a by acquiring the acceptor stem identity determinant “G3:U70” (Supplemental Fig. S13; Ling et al. 2014). This is charged by the AlaRS, which is nondiscriminative against the anticodon. The itself originated from a according to the fourth scenario by mutating the anticodon and the acceptor stem and by acquiring a dedicated ThrRS for correct charging (Pape et al. 1985; Su et al. 2011).
The third scenario, extending the aminoacylation potential of aaRS by removing tRNA recognition sites, is found, for example, in the reassignment of the UAA and UAG stop codons in the nuclear codes of ciliates and diplomonads (Fig. 5; Knight et al. 2001a; Lozupone et al. 2001). Here, the stop codons were captured by single-base mutated Gln- or Glu-tRNAs, and both the cognate tRNAs and the new tRNAs decoding stop codons are correctly charged (Hanyu et al. 1986; Sánchez-Silva et al. 2003). Although the details of the molecular mechanism are unknown, it is tempting to assume that the respective aaRSs do not discriminate the third position of the anticodon, similar to the bacterial and archaeal glutamyl-tRNA synthetases (Nureki et al. 2010). Reassignment of the stop codons might have happened according to the tRNA loss driven codon reassignment hypothesis by mutation of the single eukaryotic release factor eRF1 freeing respective stop codons. In contrast to bacteria that often have polycistronic mRNAs, eukaryotic mRNAs are usually monocistronic. Thus, stop codon read-through of eukaryotic mRNAs does not impose any consequences other than elongation of protein tails by usually a few residues.
The identity determinants for mitochondrial tRNAs are largely unknown (Salinas-Giegé et al. 2015), and the origins of many of the tRNAs with altered anticodons have never been determined (Supplemental Fig. S13). Nevertheless, the current data suggest a close connection between the tRNAs capturing a free codon and the respective aaRSs being able to correctly charge the cognate and the newly assigned tRNAs.
Predictions based on the tRNA loss driven codon reassignment theory
Our tRNA loss driven codon reassignment hypothesis presents several testable predictions that are mutually exclusive with the codon capture and ambiguous intermediate theories. We predict identification of (1) additional yeast species with branching before the SP and PC split or within the group of “Pichiaceae” and Pachysolen species, (2) species with evolved from serine AGN-decoding tRNAs, and (3) species with derived from tRNAYAAs. Furthermore, we anticipate finding additional yeasts with , and finding species without within the PP group. According to the ambiguous intermediate theory, such findings would be considered as additional independent ambiguous intermediate events. While this might be theoretically possible, it becomes exponentially unlikely that it is always the same codon that is affected. Based on our assumption that tRNAs can only capture CUG codons if the respective aminoacyl-tRNA synthetases are nondiscriminative against the mutated anticodon, we do not expect the CUG codon to be captured by tRNAs other than leucine-, serine-, and alanine-encoding ones. The predictions of the tRNA loss driven codon reassignment model might best be tested by sequencing and analyzing further yeast species.
Methods
Growth and lysis of P. tannophilus NRRL Y-2460
P. tannophilus NRRL Y-2460 was obtained from ATCC (LGC Standards). Cells were grown in YFPD medium at 30°C, harvested by centrifugation (20 min at 5000g), and washed and resuspended in lysis buffer (50 mM HEPES at pH 6.8, 100 mM KCl). The cells were disrupted by three passages through a French press (20,000 lb/in2) at 4°C, and intact cells and the cell debris were removed by centrifugation (10 min at 15,000g). The supernatant was subjected to SDS-PAGE gel electrophoresis.
Genome annotation
The Pachysolen genome assembly (Liu et al. 2012) has been obtained from NCBI (GenBank accessions CAHV01000001–CAHV01000267). Gene prediction was done with AUGUSTUS (Stanke and Waack 2003) using the parameter “genemodel=complete,” the gene feature set of C. albicans, and the standard codon translation table. The gene prediction resulted in 5288 predicted proteins, out of which 4210 contain at least one CUG codon. For the mass spectrometry database search, the database was multiplied so that each new database contains the CUG codons translated by another amino acid.
Mass spectrometry analysis
SDS-PAGE–separated protein samples were processed as described previously (Shevchenko et al. 1996). The resuspended peptides in sample loading buffer (2% acetonitrile and 0.1% trifluoroacetic acid) were fractionated and analyzed by an online UltiMate 3000 RSLCnano HPLC system (Thermo Fisher Scientific) coupled online to the Q Exactive HF mass spectrometer (Thermo Fisher Scientific). Firstly, the peptides were desalted on a reverse-phase C18 precolumn (3 cm long, 100 μm inner diameter, 360 µm outer diameter) for 3 min. After 3 min the precolumn was switched online with the analytical column (30 cm long, 75 μm inner diameter) prepared in-house using ReproSil-Pur C18 AQ 1.9 μm reverse-phase resin (Dr. Maisch). The peptides were separated with a linear gradient of 5%–35% buffer (80% acetonitrile and 0.1% formic acid) at a flow rate of 300 nL/min (with back pressure 500 bars) over a 90-min gradient time. The precolumn and the column temperature were set to 50°C during the chromatography. The MS data were acquired by scanning the precursors in mass range from 350–1600 m/z at a resolution of 70,000 at m/z 200. Top 30 precursor ions were chosen for MS2 by using data-dependent acquisition (DDA) mode at a resolution of 15,000 at m/z 200 with maximum IT of 50 msec. Data analysis and search were performed using MaxQuant v.1.5.2.8 as search engine with 1% FDR against the Pachysolen genome annotation database as annotated above. The search parameters for searching the precursor and fragment ion masses against the database were as previously described (Oellerich et al. 2011) except that all peptides shorter than seven amino acids were excluded. To increase confidence in the amino acids translated from CUG codons, we determined the observed fragment ions around each CUG-encoded residue. Only amino acids with fragment ions at both sides of the amino acid, which allows the determination of the mass of the respective amino acid, were regarded as supported by the data. If fragment ions at both sides of the CUG-encoded residue are missing, the respective CUG translation can be misinterpreted because the potential post-translational modifications and chemical reactions (as result from the data generation process) at neighboring residues are not included in the database search.
tRNA phylogeny
tRNA genes in 60 sequenced yeast species and four Schizosaccharomyces species, which were used as outgroup (Supplemental Table S1; Mühlhausen and Kollmar 2014), were identified with tRNAscan (Lowe and Eddy 1997) using standard parameters. All genomes in which tRNACAGs were not found by tRNAscan, were searched with BLAST and respective tRNAs reconstructed manually. This especially accounts for the many tRNACAGs having long introns (up to 287 bp). The intron-free tRNACAGs were aligned against all C. albicans cytoplasmic tRNAs to identify the closest related tRNA types for in-depth analysis. While the and were easily identified, the Pachysolen tRNACAG grouped within the Candida alanine and valine tRNAs. To finally resolve tRNA codon type relationships and reconstruct tRNACAG evolution, we increased sequence and taxonomic sampling. Therefore, we randomly selected three to 10 homologs from all leucine, serine, and alanine isoacceptor tRNAs from all 60 yeast species, as well as similar numbers of tRNAs from a selection of valine, phenylalanine, methionine, arginine, isoleucine, and threonine codon types. Identical tRNACAG sequences from gene duplications were removed, resulting in an alignment of 172 tRNA sequences (Supplemental Fig. S2). To refine the resolution of tRNA relationships within codon family boxes, we manually removed mitochondrial Leu-, Ser-, and Ala-tRNAs from the data sets and performed separate phylogenetic analyses of all NAG-tRNAs (320 leucine isoacceptor tRNAs), NGA-tRNAs (776 serine isoacceptor tRNAs), and NGC-tRNAs (824 alanine isoacceptor tRNAs) (Supplemental Figs. S7–S9). Sequence redundancy was removed using the CD-HIT suite (Li and Godzik 2006), generating reduced alignments of representative sequences of <95% identity (80 Leu-tRNAs, 76 Ser-tRNAs, and 70 Ala-tRNAs).
Phylogenetic trees were inferred using neighbor joining–based, Bayesian-based, and maximum likelihood–based methods as implemented in ClustalW v.2.1 (Chenna et al. 2003), Phase v. 2.0 (Jow et al. 2002; Hudelot et al. 2003) and FastTree v. 2.1.7 (Price et al. 2010), respectively. The most appropriate model of nucleotide substitution was determined with JModelTest v. 2.1.5 (Darriba et al. 2012). Accordingly, FastTree was run with the GTR model for estimating the proportion of invariable sites and the GAMMA model to account for rate heterogeneity. Bootstrapping in ClustalW and FastTree was performed with 1000 replicates. Phase was used with a mixed model, the REV-Γ model for the loops. and the RNA7D-Γ model for the stem regions, which were given by a manually generated consensus tRNA secondary structure. Phase was run with 750,000 burn-in and 3 million sampling iterations, as well as a sampling period of 150 cycles. The phylogenetic network was generated with SplitsTree v.4.1.3.1 (Huson and Bryant 2006) using the neighbor-net method to identify alternative splits.
Data access
The mass spectrometry data from this study have been submitted to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE (Vizcaíno et al. 2016) partner repository with the data set identifier PXD003898.
Acknowledgments
We thank Prof. Dr. Christian Griesinger for his continuous generous support, Dr. Hans-Dieter Schmitt for his help in growing Pachysolen, Dr. Christof Lenz and M.Sc. Samir Karaca for help in analyzing the mass spectrometry data, and Prof. Dr. Laurence Hurst for helpful comments. This project was funded by the Deutsche Forschungsgemeinschaft DFG Grant KO 2251/13-1 to M.K. and a Synaptic Systems fellowship to S.M.
Author contributions: M.K. initiated the study. S.M. performed MS/MS data and phylogenetic analyses. P.F. prepared experimental samples. U.P. performed MS/MS experiments. H.U. was involved in MS/MS data interpretation. M.K. assembled, aligned, and analyzed tRNA sequences. S.M. and M.K. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.200931.115.
References
- Andersson SG, Kurland CG. 1995. Genomic evolution drives the evolution of the translation system. Biochem Cell Biol Biochim Biol Cell 73: 775–787. [DOI] [PubMed] [Google Scholar]
- Asahara H, Himeno H, Tamura K, Nameki N, Hasegawa T, Shimizu M. 1993. Discrimination among E. coli tRNAs with a long variable arm. Nucleic Acids Symp Ser 1993: 207–208. [PubMed] [Google Scholar]
- Asahara H, Himeno H, Tamura K, Nameki N, Hasegawa T, Shimizu M. 1994. Escherichia coli seryl-tRNA synthetase recognizes tRNASer by its characteristic tertiary structure. J Mol Biol 236: 738–748. [DOI] [PubMed] [Google Scholar]
- Bezerra AR, Simões J, Lee W, Rung J, Weil T, Gut IG, Gut M, Bayés M, Rizzetto L, Cavalieri D, et al. 2013. Reversion of a fungal genetic code alteration links proteome instability with genomic and phenotypic diversification. Proc Natl Acad Sci 110: 11079–11084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. 2003. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 31: 3497–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Gao P, Zhao P, He Y, Liao N, Jackman S, Zhao Y, Birol I, Duan X, Lu Z. 2011. Genome sequence of Mycoplasma capricolum subsp. capripneumoniae strain M1601. J Bacteriol 193: 6098–6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick FH. 1966. Codon–anticodon pairing: the wobble hypothesis. J Mol Biol 19: 548–555. [DOI] [PubMed] [Google Scholar]
- Crick FH. 1968. The origin of the genetic code. J Mol Biol 38: 367–379. [DOI] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9: 772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeland SJ, Hurst LD. 1998. The genetic code is one in a million. J Mol Evol 47: 238–248. [DOI] [PubMed] [Google Scholar]
- Freeland SJ, Knight RD, Landweber LF, Hurst LD. 2000. Early fixation of an optimal genetic code. Mol Biol Evol 17: 511–518. [DOI] [PubMed] [Google Scholar]
- Giegé R, Eriani G. 2015. Transfer RNA recognition and aminoacylation by synthetases. In eLS, Molecular Biology (ed. Candi E). John Wiley & Sons, Hoboken, NJ: http://onlinelibrary.wiley.com/doi/10.1002/9780470015902.a0000531.pub3/abstract. [Google Scholar]
- Giegé R, Sissler M, Florentz C. 1998. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res 26: 5017–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromadski KB, Daviter T, Rodnina MV. 2006. A uniform response to mismatches in codon-anticodon complexes ensures ribosomal fidelity. Mol Cell 21: 369–377. [DOI] [PubMed] [Google Scholar]
- Hamashima K, Fujishima K, Masuda T, Sugahara J, Tomita M, Kanai A. 2012. Nematode-specific tRNAs that decode an alternative genetic code for leucine. Nucleic Acids Res 40: 3653–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamashima K, Mori M, Andachi Y, Tomita M, Kohara Y, Kanai A. 2015. Analysis of genetic code ambiguity arising from nematode-specific misacylated tRNAs. PLoS One 10: e0116981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanyu N, Kuchino Y, Nishimura S, Beier H. 1986. Dramatic events in ciliate evolution: alteration of UAA and UAG termination codons to glutamine codons due to anticodon mutations in two Tetrahymena tRNAsGln. EMBO J 5: 1307–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudelot C, Gowri-Shankar V, Jow H, Rattray M, Higgs PG. 2003. RNA-based phylogenetic methods: application to mammalian mitochondrial RNA sequences. Mol Phylogenet Evol 28: 241–252. [DOI] [PubMed] [Google Scholar]
- Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23: 254–267. [DOI] [PubMed] [Google Scholar]
- Johansson MJO, Byström AS. 2005. Transfer RNA modifications and modifying enzymes in Saccharomyces cerevisiae. In Topics in current genetics: fine-tuning of RNA functions by modification and editing (ed. Grosjean H), pp. 87–120. Springer, Berlin, Heidelberg. [Google Scholar]
- Johansson MJO, Esberg A, Huang B, Björk GR, Byström AS. 2008. Eukaryotic wobble uridine modifications promote a functionally redundant decoding system. Mol Cell Biol 28: 3301–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jow H, Hudelot C, Rattray M, Higgs PG. 2002. Bayesian phylogenetics using an RNA substitution model applied to early mammalian evolution. Mol Biol Evol 19: 1591–1601. [DOI] [PubMed] [Google Scholar]
- Kano A, Ohama T, Abe R, Osawa S. 1993. Unassigned or nonsense codons in Micrococcus luteus. J Mol Biol 230: 51–56. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Honda H, Taniguchi-Morimura J, Iwasaki S. 1989. The codon CUG is read as serine in an asporogenic yeast Candida cylindracea. Nature 341: 164–166. [DOI] [PubMed] [Google Scholar]
- Knight RD, Freeland SJ, Landweber LF. 2001a. Rewiring the keyboard: evolvability of the genetic code. Nat Rev Genet 2: 49–58. [DOI] [PubMed] [Google Scholar]
- Knight RD, Landweber LF, Yarus M. 2001b. How mitochondria redefine the code. J Mol Evol 53: 299–313. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Novozhilov AS. 2009. Origin and evolution of the genetic code: the universal enigma. IUBMB Life 61: 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Godzik A. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22: 1658–1659. [DOI] [PubMed] [Google Scholar]
- Ling J, Daoud R, Lajoie MJ, Church GM, Söll D, Lang BF. 2014. Natural reassignment of CUU and CUA sense codons to alanine in Ashbya mitochondria. Nucleic Acids Res 42: 499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Kaas RS, Jensen PR, Workman M. 2012. Draft genome sequence of the yeast Pachysolen tannophilus CBS 4044/NRRL Y-2460. Eukaryot Cell 11: 827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25: 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Knight RD, Landweber LF. 2001. The molecular basis of nuclear genetic code change in ciliates. Curr Biol 11: 65–74. [DOI] [PubMed] [Google Scholar]
- Massey SE, Moura G, Beltrão P, Almeida R, Garey JR, Tuite MF, Santos MAS. 2003. Comparative evolutionary genomics unveils the molecular mechanism of reassignment of the CTG codon in Candida spp. Genome Res 13: 544–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda I, Silva R, Santos MAS. 2006. Evolution of the genetic code in yeasts. Yeast 23: 203–213. [DOI] [PubMed] [Google Scholar]
- Morales L, Noel B, Porcel B, Marcet-Houben M, Hullo M-F, Sacerdot C, Tekaia F, Leh-Louis V, Despons L, Khanna V, et al. 2013. Complete DNA sequence of Kuraishia capsulata illustrates novel genomic features among budding yeasts (Saccharomycotina). Genome Biol Evol 5: 2524–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura GR, Paredes JA, Santos MAS. 2010. Development of the genetic code: insights from a fungal codon reassignment. FEBS Lett 584: 334–341. [DOI] [PubMed] [Google Scholar]
- Mühlhausen S, Kollmar M. 2014. Molecular phylogeny of sequenced Saccharomycetes reveals polyphyly of the alternative yeast codon usage. Genome Biol Evol 6: 3222–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musier-Forsyth K, Usman N, Scaringe S, Doudna J, Green R, Schimmel P. 1991. Specificity for aminoacylation of an RNA helix: an unpaired, exocyclic amino group in the minor groove. Science 253: 784–786. [DOI] [PubMed] [Google Scholar]
- Nureki O, O'Donoghue P, Watanabe N, Ohmori A, Oshikane H, Araiso Y, Sheppard K, Söll D, Ishitani R. 2010. Structure of an archaeal non-discriminating glutamyl-tRNA synthetase: a missing link in the evolution of Gln-tRNAGln formation. Nucleic Acids Res 38: 7286–7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oba T, Andachi Y, Muto A, Osawa S. 1991. CGG: an unassigned or nonsense codon in Mycoplasma capricolum. Proc Natl Acad Sci 88: 921–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oellerich T, Bremes V, Neumann K, Bohnenberger H, Dittmann K, Hsiao H-H, Engelke M, Schnyder T, Batista FD, Urlaub H, et al. 2011. The B-cell antigen receptor signals through a preformed transducer module of SLP65 and CIN85. EMBO J 30: 3620–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa S, Jukes TH. 1989. Codon reassignment (codon capture) in evolution. J Mol Evol 28: 271–278. [DOI] [PubMed] [Google Scholar]
- Osawa S, Jukes TH, Watanabe K, Muto A. 1992. Recent evidence for evolution of the genetic code. Microbiol Rev 56: 229–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape LK, Koerner TJ, Tzagoloff A. 1985. Characterization of a yeast nuclear gene (MST1) coding for the mitochondrial threonyl-tRNA1 synthetase. J Biol Chem 260: 15362–15370. [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP. 2010. FastTree 2: approximately maximum-likelihood trees for large alignments. PLoS One 5: e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randerath E, Gupta RC, Chia LL, Chang SH, Randerath K. 1979. Yeast tRNALeuUAG. Purification, properties and determination of the nucleotide sequence by radioactive derivative methods. Eur J Biochem 93: 79–94. [DOI] [PubMed] [Google Scholar]
- Ravin NV, Eldarov MA, Kadnikov VV, Beletsky AV, Schneider J, Mardanova ES, Smekalova EM, Zvereva MI, Dontsova OA, Mardanov AV, et al. 2013. Genome sequence and analysis of methylotrophic yeast Hansenula polymorpha DL1. BMC Genomics 14: 837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezgui VAN, Tyagi K, Ranjan N, Konevega AL, Mittelstaet J, Rodnina MV, Peter M, Pedrioli PGA. 2013. tRNA tKUUU, tQUUG, and tEUUC wobble position modifications fine-tune protein translation by promoting ribosome A-site binding. Proc Natl Acad Sci 110: 12289–12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saks ME, Sampson JR, Abelson JN. 1994. The transfer RNA identity problem: a search for rules. Science 263: 191–197. [DOI] [PubMed] [Google Scholar]
- Salinas-Giegé T, Giegé R, Giegé P. 2015. tRNA biology in mitochondria. Int J Mol Sci 16: 4518–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Silva R, Villalobo E, Morin L, Torres A. 2003. A new noncanonical nuclear genetic code: translation of UAA into glutamate. Curr Biol 13: 442–447. [DOI] [PubMed] [Google Scholar]
- Santos MA, Perreau VM, Tuite MF. 1996. Transfer RNA structural change is a key element in the reassignment of the CUG codon in Candida albicans. EMBO J 15: 5060–5068. [PMC free article] [PubMed] [Google Scholar]
- Schultz DW, Yarus M. 1994. Transfer RNA mutation and the malleability of the genetic code. J Mol Biol 235: 1377–1380. [DOI] [PubMed] [Google Scholar]
- Schultz DW, Yarus M. 1996. On malleability in the genetic code. J Mol Evol 42: 597–601. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Jensen ON, Podtelejnikov AV, Neubauer G, Shevchenko A, Mortensen P, Mann M. 1996. A strategy for identifying gel-separated proteins in sequence databases by MS alone. Biochem Soc Trans 24: 893–896. [DOI] [PubMed] [Google Scholar]
- Stanke M, Waack S. 2003. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19: ii215–ii225. [DOI] [PubMed] [Google Scholar]
- Su D, Lieberman A, Lang BF, Simonovic M, Söll D, Ling J. 2011. An unusual tRNAThr derived from tRNAHis reassigns in yeast mitochondria the CUN codons to threonine. Nucleic Acids Res 39: 4866–4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Ueda T, Watanabe K. 1997. The “polysemous” codon: a codon with multiple amino acid assignment caused by dual specificity of tRNA identity. EMBO J 16: 1122–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuite MF, Santos MA. 1996. Codon reassignment in Candida species: an evolutionary conundrum. Biochimie 78: 993–999. [DOI] [PubMed] [Google Scholar]
- Vizcaíno JA, Csordas A, Del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, et al. 2016. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44: D447–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenbach J, Dirheimer G, Falcoff R, Sanceau J, Falcoff E. 1977. Yeast tRNALeu (anticodon U–A–G) translates all six leucine codons in extracts from interferon treated cells. FEBS Lett 82: 71–76. [DOI] [PubMed] [Google Scholar]
- Young M, Artsatbanov V, Beller HR, Chandra G, Chater KF, Dover LG, Goh E-B, Kahan T, Kaprelyants AS, Kyrpides N, et al. 2010. Genome sequence of the Fleming strain of Micrococcus luteus, a simple free-living actinobacterium. J Bacteriol 192: 841–860. [DOI] [PMC free article] [PubMed] [Google Scholar]