

Abstract
Polycomb repressive complex 2 (PRC2) modifies chromatin to maintain genes in a repressed state during development. PRC2 is primarily associated with CpG islands at repressed genes and also possesses RNA binding activity. However, the RNAs that bind PRC2 in cells, the subunits that mediate these interactions, and the role of RNA in PRC2 recruitment to chromatin all remain unclear. By performing iCLIP for PRC2 in comparison with other RNA binding proteins, we show here that PRC2 binds nascent RNA at essentially all active genes. Although interacting with RNA promiscuously, PRC2 binding is enriched at specific locations within RNAs, primarily exon–intron boundaries and the 3′ UTR. Deletion of other PRC2 subunits reveals that SUZ12 is sufficient to establish this RNA binding profile. Contrary to prevailing models, we also demonstrate that the interaction of PRC2 with RNA or chromatin is mutually antagonistic in cells and in vitro. RNA degradation in cells triggers PRC2 recruitment to CpG islands at active genes. Correspondingly, the release of PRC2 from chromatin in cells increases RNA binding. Consistent with this, RNA and nucleosomes compete for PRC2 binding in vitro. We propose that RNA prevents PRC2 recruitment to chromatin at active genes and that mutual antagonism between RNA and chromatin underlies the pattern of PRC2 chromatin association across the genome.
Polycomb group (PcG) proteins modify chromatin to maintain developmental regulator genes in a repressed state and are required for embryogenesis and embryonic stem cell (ESC) differentiation (for review, see Margueron and Reinberg 2011; Di Croce and Helin 2013). There are two main PcG complexes, Polycomb repressive complex (PRC) 1 and PRC2. PRC2 is composed of the histone methyltransferase EZH2, EED, SUZ12, and the histone binding proteins RBBP4 and RBBP7. JARID2, AEBP2, PHF1, MTF2, and PHF19 are also present in PRC2 in substoichiometric amounts. PRC2 trimethylates lysine 27 of histone H3 (H3K27me3), creating a binding site for CBX-containing PRC1, which ubiquitinates H2A at K119 (H2AK119ub) and induces formation of a repressive chromatin structure.
Much research has been focused on identifying the determinants responsible for the pattern of PRC2 association with chromatin in mammals. In ESC, PRC2 and PRC1 are primarily associated with CpG islands at repressed developmental regulator genes (Tanay et al. 2007; Ku et al. 2008). Insertion of CpG islands or GC-rich sequences into the genome reveals them to be sufficient for PRC2 recruitment (Mendenhall et al. 2010; Lynch et al. 2011; Jermann et al. 2014). PRC2 binding to CpG islands is favored by a lack of DNA methylation (Bartke et al. 2010; Lynch et al. 2011; Jermann et al. 2014), perhaps due to recognition of H2AK119ub deposited by a KDM2B-containing form of PRC1 (Blackledge et al. 2014; Cooper et al. 2014; Kalb et al. 2014). EED also binds to H3K27me3, generating a positive-feedback loop and suggesting a form of epigenetic memory (Margueron et al. 2009). Potentially destabilizing association with active genes, the interaction of PRC2 with histone H3 in vitro is inhibited by H3K4me3 (Schmitges et al. 2011), and PRC2 methyltransferase activity is repressed by H3K36me2/3 (Schmitges et al. 2011; Yuan et al. 2011). Consistent with this, the association of PRC2 with CpG islands at active genes is increased upon RNA polymerase (Pol) II inhibition (Riising et al. 2014) and correspondingly reduced at CpG islands positioned next to an active promoter (Lynch et al. 2011; Jermann et al. 2014).
The association of PRC2 with chromatin is also thought to be regulated by interaction with RNA. Recombinant EZH2, SUZ12, and JARID2 all exhibit RNA binding activity (Zhao et al. 2008, 2010; Kanhere et al. 2010; Cifuentes-Rojas et al. 2014; Kaneko et al. 2014a). By use of native RNA immunoprecipitation (RIP) and in vitro methods, PRC2 was first found to interact with the long noncoding RNAs (lncRNAs) HOTAIR, which modulates H3K27me3 in trans at the HOXD locus (Rinn et al. 2007), and Xist RepA, potentially recruiting PRC2 to the X Chromosome during X-inactivation (Zhao et al. 2008). PRC2 has since been found to interact with many other lncRNAs in a variety of systems (for review, see Wang and Chang 2011; Lee 2012). Knockdown or overexpression of a number of these transcripts leads to changes in PRC2 chromatin association, and these data have led to models in which specific RNAs recruit PRC2 to chromatin in cis or in trans.
Such models suggest that PRC2 preferentially binds certain RNAs, but whether this is the case has been unclear. In vitro, PRC2 exhibits higher affinity for Xist RepA compared with nonphysiological RNAs, but this depends on buffer composition, potentially limiting the relevance for PRC2 RNA binding specificity in cells (Davidovich et al. 2013, 2015; Cifuentes-Rojas et al. 2014). Unbiased genome-wide screens for RNAs bound by PRC2 in cells using RIP have identified thousands of lncRNA and mRNA transcripts (Khalil et al. 2009; Zhao et al. 2010). This has led to the “junk-mail” model in which promiscuous binding to nascent RNA allows PRC2 recruitment to the subset of Polycomb target genes that have escaped repression (Davidovich et al. 2013; Davidovich and Cech 2015). However, this model has yet to be tested experimentally, and RIP is compromised by the low-stringency conditions required, the inability to discriminate direct from indirect interactions, and the potential formation of nonphysiological protein–RNA interactions after cell lysis (Brockdorff 2013).
The limitations of RIP can be addressed by UV crosslinking followed by IP (CLIP), which identifies direct protein–RNA interactions as they occur in living cells (Hafner et al. 2010; Konig et al. 2010). Studies using photoactivatable-ribonucleoside–enhanced CLIP (PAR-CLIP) have reported that EZH2 and JARID2 directly interact with RNA in cells (Kaneko et al. 2013, 2014a,b). It was concluded that EZH2 interacted with a specific set of nascent RNAs (ezRNAs) and that this inhibits PRC2 methyltransferase activity without affecting its association with chromatin, leaving PRC2 in a poised state (Kaneko et al. 2013, 2014b). However, because total RNA abundance and gene length were not controlled for, whether the high PRC2 crosslinking frequency at ezRNAs reflects genuine PRC2 binding specificity or the detection of only highly abundant, long RNAs is not clear. Thus, the RNAs bound by PRC2 in cells, the subunits that mediate this interaction, and the role of Polycomb–RNA interactions in the cell remain under debate (Wang and Chang 2011; Lee 2012; Brockdorff 2013; Davidovich and Cech 2015).
Results
Identification of PRC2 RNA crosslink sites using iCLIP
We sought to address these issues by using individual-nucleotide resolution CLIP (iCLIP) to identify the RNAs directly bound by endogenous PRC2 in cells at unprecedented depth. iCLIP has a number of advantages over other CLIP-based methods, including the ability to precisely identify the crosslink site and the incorporation of a random barcode that permits quantitative comparisons between libraries (Konig et al. 2010). Because SUZ12 and EZH2 are coprecipitated under the stringent conditions used for CLIP (Supplemental Fig. S1A–C) and because they have the same apparent molecular weight by SDS-PAGE, CLIP measures direct RNA binding by both factors and thus by core PRC2. To confirm that PRC2 directly interacted with RNA, we performed CLIP and PAR-CLIP with an antibody to SUZ12 in wild-type (WT) and Suz12−/− mouse ESCs, which also lack EZH2 due to protein degradation (Pasini et al. 2007). This revealed the presence of a SUZ12-dependent RNP at the molecular weight shared by SUZ12 and EZH2, with a smear of trimmed RNA extending above, which diminished as the RNase concentration was increased (Fig. 1A; Supplemental Fig. S1D–E). No radiolabeled bands were observed in the absence of UV crosslinking, demonstrating that the interaction with RNA was direct. These results confirm that PRC2 directly interacts with RNA in ESCs.
Figure 1.

PRC2 has a characteristic RNA binding profile with enrichment at exon–intron boundaries and the 3′ UTR. (A) SDS-PAGE for RNPs enriched by CLIP for endogenous SUZ12 in the wild type (WT) and Suz12−/− ESCs. The autoradiogram is shown at the top; the corresponding SUZ12 and EZH2 immunoblots, below. CLIP was performed with and without UV crosslinking and T4-polynucleotide kinase (T4-PNK) and with increasing concentrations of RNase I (1, 2, 4, 10, and 20 U/mL). (B) Significant PRC2 RNA crosslink sites (FDR < 0.05) at Hnrnpa2b1. Significant crosslinks are also marked for FUS, HNRNPC, GFP, and IgG controls and for PRC2 in Suz12−/− cells. Counts of Watson and Crick strand crosslinks per base are shown by positive and negative integers, respectively. Nuclear and total RNA-seq read densities (reads per million [RPM]) are shown below. Scale is denoted by the bar above. (C) As B, except for Lamc1. (D) Percentage of significant crosslink sites or RNA-seq reads in different gene segments. The percentage of bases within each segment is shown for comparison. (E) Composite crosslink density profiles (crosslinks per million) across an average gene, divided into exons and introns and 1 kb of flanking sequence, with the first and last exons additionally divided into UTR and CDS. Total and nuclear RNA-seq read densities are shown for comparison. Segment length is arbitrary. (F, top) Cartoon showing the identification of reads which span exon–intron boundaries or exon–exon boundaries. (Bottom) Relative proportions of iCLIP or RNA-seq reads that span exon–intron (unspliced; blue) or exon–exon boundaries (spliced; red), as a percentage of all reads spanning a junction.
To identify the RNAs directly bound by PRC2, we performed six biological replicate iCLIP experiments (Supplemental Fig. S2; Supplemental Tables S1,S2). Since crosslink frequency is related to RNA abundance, we also gathered total and nuclear RNA-seq data. Furthermore, in order to relate PRC2 RNA binding activity to other RNA binding proteins and to control for potential biases in the methodology, we performed iCLIP for the splicing regulators FUS and HNRNPC (Konig et al. 2010; Rogelj et al. 2012). Lastly, to control for nonspecific crosslink events, we performed iCLIP for SUZ12 in Suz12−/− cells, for GFP in transfected cells (Supplemental Fig. S1G), and with a nonspecific IgG antibody control. Reads mapping to unique nonrepeat sites were identified and PCR duplicates (with identical barcodes) removed, leaving 16,269,035 individual nucleotides (unique cDNA molecules) crosslinked to PRC2 (Supplemental Table S1). The control iCLIP in Suz12−/− cells identified only 0.25% (10,297) of the crosslink sites of the WT cell iCLIP performed in parallel (Supplemental Fig. S3A). Similarly, only 1157 and 334 unique cDNAs were retained for IgG and GFP iCLIP, respectively. Only RNAs directly bound by PRC2 in cells could be detected; in the absence of UV crosslinking, no RNA could be visualized by autoradiography, and no library could be generated (Supplemental Fig. S2C–E). The PRC2 iCLIP replicates were combined, and comparison to a randomized distribution of sites across the gene (Konig et al. 2010) identified 3,856,538 significant PRC2 crosslink sites. This is over 330 times greater than the 11,503 RNA contact sites previously identified for EZH2 (Kaneko et al. 2013), allowing us to measure PRC2 binding at a greater proportion of the transcriptome.
PRC2 has a specific RNA crosslinking profile characterized by binding to exon–intron boundaries and the 3′ UTR
We first sought to determine how PRC2 RNA crosslinking events were distributed across RNAs. Plotting crosslink sites at individual genes revealed PRC2 binding across introns (Fig. 1B,C; Supplemental Fig. S3B), consistent with previous reports that the complex binds to nascent RNA (Kanhere et al. 2010; Guil et al. 2012; Kaneko et al. 2013). We observed, rather than an even distribution of crosslinks across genes, specific peaks of PRC2 binding, some which overlapped peaks of FUS or HNRNPC but others that were unique to PRC2. Calculating the distribution of significant crosslink sites across all genes confirmed the majority of sites were within introns but revealed a higher crosslinking frequency in exons than for FUS or HNRNPC (Fig. 1D). To gain a high-resolution view, we plotted the average crosslink density across gene segments. This revealed that PRC2 possessed a characteristic RNA binding profile distinct from that of FUS and HNRNPC and what would be expected if RNA were being enriched nonspecifically (Fig. 1E). The highest density of PRC2 crosslinks was within the 3′ UTR, with a sharp reduction in binding at the CDS. Similar to FUS, PRC2 binding also peaked at exon–intron boundaries, indicative of binding to nascent RNA soon after its transcription (Rogelj et al. 2012). However, compared with FUS, PRC2 exhibited a higher crosslink density in exons. In contrast, HNRNPC crosslink density peaked in introns downstream from the exon–intron boundary, as shown previously (Konig et al. 2010).
The enrichment of PRC2 crosslinking at exons could either indicate binding to spliced mRNA or binding to exons within nascent unspliced transcripts. To distinguish between these, we calculated the number of iCLIP reads that spanned exon–exon versus exon–intron junctions. This revealed that only 4.7% of PRC2 iCLIP reads that crossed a 3′ exon boundary were spliced to a downstream exon, similar to the proportion for FUS (4.8%) and lower than that for HNRNPC (15.1%) (Fig. 1F). By taking these data together, we conclude that PRC2 has a distinctive RNA crosslinking signature, binding to unspliced nascent RNA but with enrichment at exon–intron boundaries and the 3′ UTR.
PRC2 interacts with nascent transcripts at essentially all active genes
We next considered which RNAs were bound by PRC2. Mapping significant crosslink sites for PRC2 identified 11,827 genes (Supplemental Table S2). PRC2 interacted with 94% and 95% of the RNAs bound by the general RNA binding proteins FUS and HNRNPC, respectively, suggesting that, like these factors, PRC2 makes contact with nascent RNA at essentially all active genes (Fig. 2A). Genes at which PRC2 crosslinks were not detected were either transcriptionally silent or very lowly expressed (Fig. 2B) and lacked RNA Pol II and H3K4me3 occupancy (Supplemental Fig. S4A). This suggested that PRC2 bound nascent RNA promiscuously. To test this further, we examined PRC2 crosslinking to two nonmammalian RNAs. Ezh2fl/fl cells express the CreERT2 transgene, and iCLIP revealed PRC2, FUS, and HNRNPC crosslinking to this RNA (Supplemental Fig. S4B). Similarly, UV RIP for GFP RNA in GFP-expressing WT and Suz12−/− ESC lines revealed UV- and SUZ12-dependent enrichment for GFP RNA, supporting promiscuous PRC2 binding to RNA in cells (Supplemental Fig. S4C). In contrast, in vitro transcribed GFP RNA spiked-in after cell lysis was not precipitated with PRC2, confirming that only PRC2 interactions occurring in cells were detected (Supplemental Fig. S4D).
Figure 2.

PRC2 interacts with nascent RNA at essentially all active genes. (A) The overlap between genes with significant PRC2, FUS, and HNRNPC RNA crosslinks. (B) Scatter plot showing gene length (kb) and nuclear RNA abundance (reads) for all genes (black), genes with significant PRC2 crosslinks identified by iCLIP (red), and genes previously reported to produce PRC2 binding ezRNAs (blue) (Kaneko et al. 2013). (C) Cumulative frequency distribution of PRC2 (red), FUS (blue), and HNRNPC (green) crosslinks (RPKM) and total and nuclear RNA-seq RPKM for lincRNA genes (top) and protein-coding genes (bottom). (D) Scatter plot of nuclear RNA abundance (RPKM) against PRC2 crosslink density (RPKM). Genes marked in red are significantly enriched for PRC2 crosslinks compared with nuclear RNA abundance (FDR < 0.05). (E) Scatter plots showing frequency of significant crosslinks (RPKM) for FUS (left) and HNRNPC (right) versus PRC2. Genes significantly enriched for PRC2 crosslinks compared with RNA (from D) are marked in red and are not enriched compared with FUS and HNRNPC. (F) Ratio of PRC2 significant crosslink density to FUS (left) or HNRNPC significant crosslink density (right) at genes ranked by their relative proportion of exonic sequence (percentage). The trends (Loess) are marked by red lines.
The detection of PRC2 binding at essentially all active genes was seemingly in contrast to the results of PAR-CLIP for HA-tagged EZH2 that identified 774 “ezRNAs” bound by PRC2 (Kaneko et al. 2013). We identified PRC2 binding to 766 (99%) of these RNAs. Comparison between the data sets revealed that the increased number of RNAs identified here was due to increased sensitivity; while our iCLIP data allowed detection of PRC2 crosslinks to essentially all nascent RNAs, ezRNAs are limited to abundant, long transcripts, for which crosslinks are generally easier to detect by CLIP (Fig. 2B; Supplemental Fig. S4E). Consistent with this, the characteristic PRC2 RNA binding profile identified in our data (Fig. 1E) was also discernable in the EZH2 PAR-CLIP data (Supplemental Fig. S4F). Similarly, iCLIP for SUZ12 and EZH2 immunoprecipitated by MYC-tagged EED gave an identical RNA binding profile (Supplemental Fig. S4G,H). The recapitulation of the PRC2 RNA binding signature in these multiple data sets using different methods and antibodies to different subunits provides confidence that it reflects genuine PRC2 RNA binding in cells.
We then turned our attention to long intergenic RNAs (lincRNAs), another RNA class for which PRC2 has been suggested to have particular specificity. Generating cumulative frequency plots of PRC2, FUS, and HNRNPC crosslink density and RNA-seq read density, we found that PRC2 crosslinking was not enriched at lincRNAs, instead binding at similar levels to FUS and HNRNPC (Fig. 2C). This reveals that PRC2 does not possess any particular specificity for lincRNAs as a class.
We next examined our data to determine whether PRC2 preferentially bound any RNAs. Although comparison of PRC2 crosslink frequency with nuclear RNA-seq read density suggested that there were a set of RNAs at which PRC2 crosslinking was significantly enriched, these RNAs were also enriched for FUS and HNRNPC (Fig. 2D; Supplemental Fig. S4I), revealing this to be a general property of CLIP versus RNA-seq analysis. Direct comparison between PRC2, FUS, and HNRNPC demonstrated that the vast majority of RNAs showed comparable levels of crosslinking for each factor, with RNA from only 170 genes exhibiting significantly higher crosslink frequency (FDR < 0.05) for PRC2 compared with both FUS and HNRNPC (Fig. 2E). Rather than being indicative of preferential binding of PRC2, however, RNAs with high levels of PRC2 binding compared with FUS or HNRNPC exhibited a greater proportion of exonic sequence (Fig. 2F), the RNA feature at which PRC2 crosslinks are enriched (Fig. 1E). These results confirm that PRC2 promiscuously interacts with nascent RNA, but with specific enrichment at exon–intron boundaries and the 3′ UTR.
SUZ12 directly interacts with RNA in cells and can do so in the absence of other PRC2 subunits
Which PRC2 subunits directly interact with RNA in cells is unknown. Because of the coprecipitation of SUZ12 and EZH2 and because they have similar molecular weights, CLIP for either protein does not distinguish which subunit binds RNA (Fig. 1A; Supplemental Fig. S1A–C). To address this, we performed PAR-CLIP in Ezh2fl/fl ESCs before and after addition of tamoxifen, which induces deletion of DNA encoding the EZH2 SET domain and degradation of the truncated protein (Ezh2Δ/Δ cells) (Supplemental Fig. S5A; Kanhere et al. 2010). IP for SUZ12 in Ezh2fl/fl cells precipitated both RNA and EZH2 (Fig. 3A; Supplemental Fig. S5B). Upon treatment with tamoxifen, neither full-length nor truncated EZH2 was precipitated by SUZ12, but the crosslinked RNA remained, revealing that SUZ12 directly interacted with RNA and that it could do so in the absence of EZH2. Co-IP experiments demonstrated that SUZ12 does not bind the alternative PRC2 subunit EZH1 in Ezh2Δ/Δ cells (Supplemental Fig. S5C) and thus revealed that SUZ12 RNA binding is independent of both H3K27 methyltransferases.
Figure 3.
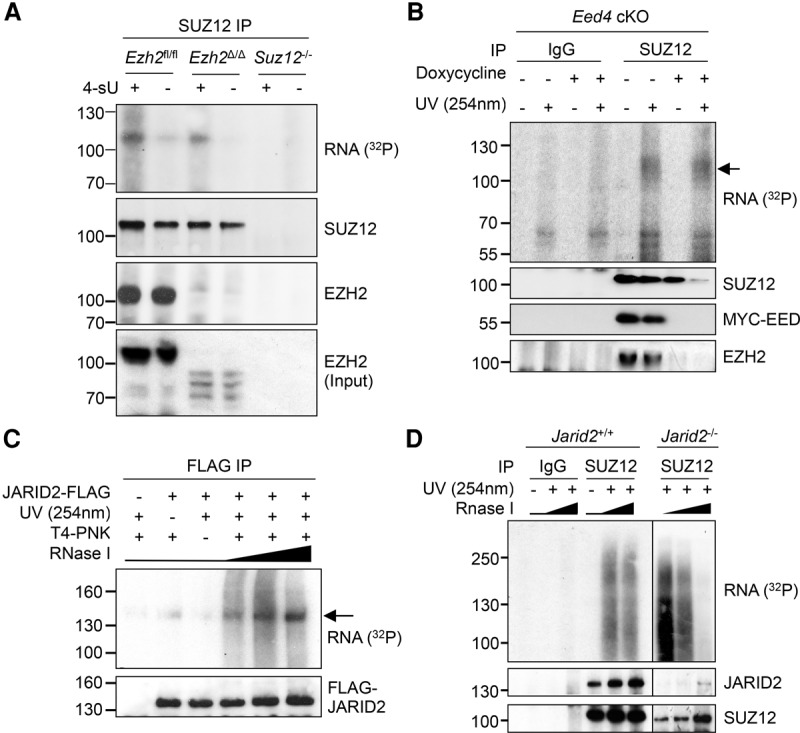
SUZ12 directly binds RNA independently of other PRC2 subunits. (A) PAR-CLIP for SUZ12 in Ezh2fl/fl (WT), Ezh2Δ/Δ, and Suz12−/− cells, with and without 4-sU. (B) CLIP for SUZ12 in Eed4 cKO cells before or after the addition of doxycycline to repress expression of MYC-EED4. An arrow marks the SUZ12 RNP. Immunoblots for SUZ12, EZH2, and MYC-EED are shown below. (C) CLIP for JARID2 with anti-Flag antibody in WT (JM8) and JARID2-Flag ESCs. RNase I was titrated at 1, 4, and 20 U/mL. An arrow marks an RNP of the expected molecular weight. (D) CLIP for SUZ12 and IgG in WT (JM8) or Jarid2−/− ESCs. JARID2 and SUZ12 immunoblots are shown below.
To confirm that SUZ12 could bind RNA in the absence of other core PRC2 subunits, we performed CLIP for SUZ12 in the presence and absence of EED by using an Eed conditional-null ESC line in which the EED4 variant is expressed from a Dox-repressible promoter on an Eed−/− background (Ura et al. 2008). Upon down-regulation of EED, IP for SUZ12 also no longer coimmunoprecipitated EZH2, but again, SUZ12 RNA crosslinking remained (Fig. 3B). Thus, SUZ12 is able to bind RNA in the absence of two core PRC2 subunits. We also did not observe any radiolabeled RNA bands that were dependent on EED, consistent with in vitro experiments suggesting that this subunit does not directly interact with RNA (Cifuentes-Rojas et al. 2014).
We next asked whether core PRC2 could bind RNA in the absence of the nonstoichiometric subunit JARID2, which has also been reported to bind RNA (Cifuentes-Rojas et al. 2014; Kaneko et al. 2014a). CLIP for Flag-JARID2 in a stable ESC line (Landeira et al. 2010) identified an RNP of around ≥130 kDa (Fig. 3C), confirming that JARID2 directly interacts with RNA. CLIP for SUZ12 in Jarid2+/+ and Jarid2−/− ESCs revealed that SUZ12/EZH2 directly interacted with RNA in both lines (Fig. 3D; Supplemental Fig. S5D), demonstrating that, even though JARID2 binds RNA, the interaction of core PRC2 with RNA is independent of this factor.
PRC2 RNA binding specificity is unchanged in the absence of EZH2 or JARID2
Given that SUZ12 interacted with RNA in cells in the absence of other PRC2 subunits, we next considered the role of SUZ12 in defining the characteristic PRC2 RNA binding profile. To do this, we performed iCLIP for SUZ12 in Ezh2Δ/Δ cells and Jarid2−/− cells and compared the data with those from WT (Ezh2fl/fl and Jarid2+/+) cells. We found that the distribution of PRC2 RNA crosslinks was unchanged in Ezh2Δ/Δ and Jarid2−/− cells, with enrichment at exon–intron boundaries and in the 3′ UTR as before (Fig. 4A,B; Supplemental Fig. S6A,B). Clustering confirmed that PRC2 maintained a common pattern of binding across RNAs in the absence of EZH2 or JARID2 (Fig. 4C). We then asked whether there was any change in the set of RNAs bound by PRC2 when EZH2 or JARID2 was deleted. Overall, the number of crosslinks per gene was highly correlated between WT and Ezh2- or Jarid2-null cells (Fig. 4D; Supplemental Fig. S6C). Some genes exhibited differences in crosslinking efficiency, potentially indicating changes in RNA binding specificity. However, comparison to the changes in gene expression that occur upon Ezh2 deletion showed that the changes in PRC2 crosslinking were reflective of changes in RNA abundance (Fig. 4D) and therefore do not represent altered RNA binding preferences due to changes in PRC2 composition.
Figure 4.

PRC2 RNA binding specificity is unchanged in the absence of EZH2 or JARID2. (A) Composite crosslink density profiles for PRC2 in Ezh2fl/fl, Ezh2Δ/Δ, Jarid2+/+, and Jarid2−/− cells. Details as for Figure 1E. (B) Significant PRC2 crosslinks at the gene Hnrnpa2b1 in Ezh2fl/fl, Ezh2Δ/Δ, Jarid2+/+, and Jarid2−/− cells. Total RNA reads in Ezh2fl/fl and Ezh2Δ/Δ cells are shown below. (C) Dendrogram showing the correlation between composite crosslink density profiles between different iCLIP experiments. (D) Scatter plot comparing the number of significant PRC2 crosslinks per gene between Ezh2fl/fl and Ezh2Δ/Δ cells. Overlaid is log2 fold-change in RNA abundance in Ezh2Δ/Δ cells compared with Ezh2fl/fl cells, with shades of red indicating an increase and yellow a decrease, as shown by the scale on the right. (E) Composite crosslink site density profile for SUZ12 in Ezh2Δ/Δ cells at the set of genes up-regulated in Ezh2Δ/Δ cells compared with Ezh2fl/fl cells (Supplemental Fig. S6D, red).
We next sought to determine whether the PRC2 RNA binding profile could be established by SUZ12 de novo in the absence of EZH2. To do this, we identified the genes at which transcription was induced upon Ezh2 deletion, and examined SUZ12 binding at the newly transcribed RNAs (Fig. 4E; Supplemental Fig. S6D). At these genes, SUZ12 formed the same characteristic RNA crosslinking pattern as PRC2 in WT cells, indicating that SUZ12 can establish PRC2 RNA binding in the absence of EZH2.
PRC2 chromatin association antagonizes RNA binding
The binding of PRC2 to nascent RNA at essentially all active genes is difficult to reconcile with models in which RNA binding plays a positive role in the recruitment of PRC2 to chromatin. Indeed, at genes with CpG islands, PRC2 RNA binding is anti-correlated with chromatin association (Fig. 5A). Furthermore, although SUZ12 remained in the nucleus in Ezh2Δ/Δ cells (Supplemental Fig. S7A), it no longer interacted with chromatin (Fig. 5B; Supplemental Fig. S7B). Therefore, the invariance of the PRC2 RNA binding profile between WT and Ezh2Δ/Δ cells (Fig. 4) reveals that the normal pattern of PRC2 RNA binding does not require PRC2 association with chromatin. We therefore considered that PRC2 RNA binding and chromatin association might instead be mutually antagonistic, with RNA and chromatin competing for PRC2 interaction.
Figure 5.

Release of PRC2 from chromatin increases binding to RNA. (A) Heat map showing SUZ12 chromatin binding (input subtracted reads/million, according to the scale on the right) 2 kb to either side of the TSS of CpG island–associated genes with significant PRC2 RNA crosslinks. Genes are ordered by PRC2 RNA crosslink density. (B) Average chromatin binding profiles (reads/million) of EZH2 and SUZ12 across gene bodies in Ezh2fl/fl and Ezh2Δ/Δ cells. (C, left) CLIP for IgG, FUS, and SUZ12 in Ezh2fl/fl and Ezh2Δ/Δ cells. The fold-change in the levels of each protein and crosslinked RNA are indicated below. (Right) Fold-change in RNA crosslinked to FUS and SUZ12 in biological replicate experiments (normalized to protein; mean and SD, n = 3). (D) CLIP for IgG, FUS, and SUZ12/EZH2 in Jarid2+/+ and Jarid2−/− cells. Details as for C.
We reasoned that if RNA binding and chromatin association are mutually antagonistic, then reducing one should increase the other. We first tested whether release of PRC2 from chromatin leads to increased RNA binding by performing CLIP for SUZ12 in WT and Ezh2Δ/Δ cells and quantifying the amount of crosslinked RNA. To control for potential differences in UV crosslinking efficiency and RNA abundance, we also performed CLIP for FUS in parallel. We found that deletion of Ezh2 increased SUZ12 RNA crosslinking by an average of sixfold, even though the contribution of EZH2 to RNA binding had been lost (Fig. 5C; Supplemental Fig. S8A). In contrast, FUS exhibited equal RNA crosslinking efficiency in the presence or absence of EZH2. This increase in SUZ12 RNA interaction does not reflect ectopic RNA binding events caused by Ezh2 deletion because the positions of the RNA crosslink sites remain identical in Ezh2Δ/Δ cells (Fig. 4).
Deletion of Ezh2 disrupts core PRC2, so the increase in SUZ12 RNA binding may be a consequence of this. To determine whether release of intact core PRC2 from chromatin also increased RNA binding, we repeated the experiment in WT and Jarid2−/− cells, in which PRC2 is depleted from chromatin (Supplemental Fig. S7C; Landeira et al. 2010) but the core complex remains intact (Supplemental Fig. S5D). We found that SUZ12/EZH2 RNA crosslinking was increased approximately threefold in Jarid2−/− cells compared with their WT counterparts, with FUS crosslinking remaining equal (Fig. 5D; Supplemental Fig. S8B). These results show that release from chromatin increases the availability of PRC2 for RNA binding, and we conclude that PRC2 chromatin association antagonizes its interaction with RNA.
RNA degradation increases interaction of PRC2 with chromatin
We next considered that if the relationship between RNA binding and chromatin association is antagonistic, then removal of RNA in cells should increase the recruitment of PRC2 to chromatin. To test this, we permeabilized ES cells, performed a mock or RNase A treatment, and generated cytoplasmic/nucleoplasmic and chromatin fractions (Zoabi et al. 2014). We then blotted the fractions for SUZ12, EZH2, and other chromatin-associated proteins (Fig. 6A,B; Supplemental Fig. S8C,D). We found that treatment with RNase A increased association of SUZ12 and EZH2 with the chromatin fraction by approximately twofold (P < 0.05, paired t-test). In contrast, no significant changes were observed in the association of histone H3, RNA Pol II, or BRD4 with the chromatin fraction. Opposite to that observed for PRC2, RNase treatment reduced the association of FUS with chromatin, consistent with this being mediated by RNA. Confirming that the increase in PRC2 chromatin association was due to loss of RNA rather than an indirect effect on chromosomal architecture, washing away RNase A and titrating in yeast tRNA restored the lower level of PRC2 chromatin association (Supplemental Fig. S9A). Furthermore, no changes in chromatin accessibility were observed upon RNaseA treatment (Supplemental Fig. S9B), and deletion of the Cohesin subunit Rad21, which results in loss of chromosomal architecture (Sofueva et al. 2013), had no effect on PRC2 chromatin association (Supplemental Fig. S9C).
Figure 6.

Interaction of PRC2 with chromatin is increased upon RNA degradation. (A) Immunoblots of SUZ12, EZH2, RNA Pol II (POLR2A), FUS, BRD4, histone H3, beta-actin, and alpha-tubulin in cytoplasmic plus nucleoplasmic (C + N) and chromatin (Chr) fractions from mock- or RNase A–treated cells. (B) Ratio of the amount of protein present in the chromatin fraction between RNase A–treated and mock-treated cells (mean and SD, n = 3). (*) P < 0.05 (paired Student's t-test). (C) ChIP-qPCR for SUZ12, EZH2, and histone H3 in mock-treated and RNase A–treated cells (mean and SD, n = 3). (D) Average SUZ12 chromatin binding profiles around gene TSS in mock- (black) and RNase A–treated (red) ESCs (mean and SE). Genes are separated into active (nuclear and total RNA RPKM > 1) and inactive (RPKM = 0) and further divided by the presence or absence of a CpG island (CGI) at their TSS. Data are the average of duplicate experiments. (E) SUZ12 ChIP-seq read density (reads per million) at Arf6 (top) and Gmeb2 (bottom) following mock (black) or RNase A treatment (red) (two replicates). CpG islands are marked in green. (F) Percentage of genes that are occupied by PRC2 in differentiated cells (Yue et al. 2014). Genes are divided into those that gain PRC2 upon RNase A treatment and those that exhibit no change. (G) Average change in PRC2 binding upon RNase A treatment at genes at which PRC2 is gained (red), lost (purple), or unchanged (gray) upon inhibition of RNA Pol II with triptolide.
To verify whether RNA degradation increased PRC2 chromatin association using a different method, we performed ChIP for SUZ12, EZH2, and histone H3 in mock- or RNase-treated ESCs. Measurement of DNA enrichment by qPCR confirmed that RNA degradation caused an increase in PRC2 chromatin association (Fig. 6C).
We next considered that if binding to nascent RNA antagonizes PRC2 chromatin association, the increase in chromatin occupancy upon RNase treatment should be localized to transcriptionally active genes. To test this, we performed ChIP-seq for SUZ12 in mock- and RNase-treated ESCs and plotted SUZ12 chromatin occupancy at transcriptionally active and inactive genes. This revealed that the increase in PRC2 chromatin association upon RNA degradation was indeed concentrated at active genes (Fig. 6D; Supplemental Table S3). The increase in PRC2 binding was much more pronounced at genes with CpG islands, revealing that RNA degradation maintained PRC2 preference for CpG islands but now allowed binding to these elements at active genes (Fig. 6D,E). Furthermore, these sites were normally occupied by PRC2 in differentiated cells (Fig. 6F), confirming that RNA degradation induces PRC2 binding to physiological target sites at which recruitment is normally prevented in ESCs. In contrast to the effect on PRC2 chromatin binding, RNase A treatment did not alter binding of EP300, further demonstrating that the effect was specific for PRC2 (Supplemental Fig. S9E).
To confirm whether these changes were directly caused by loss of RNA, we asked whether RNA Pol II inhibition caused similar changes to PRC2 chromatin association. We found that RNase A treatment mimicked the changes in PRC2 binding that occurred upon inhibition of RNA Pol II with triptolide, with genes that exhibit an increase in PRC2 binding upon triptolide treatment also exhibiting an increase in PRC2 binding upon RNase A treatment (P = 1.4 × 10−10, Wilcoxon signed-rank test) (Fig. 6G). We conclude that RNA antagonizes recruitment of PRC2 to otherwise permissive CpG islands at active genes. Taken together with the corresponding increase in RNA binding upon release from chromatin, these results show that the interaction of PRC2 with RNA or chromatin is mutually antagonistic in cells.
RNA and nucleosomes compete for PRC2 binding in vitro
To verify whether RNA antagonized PRC2 chromatin association by an independent method, we measured the effect of RNA on the interaction of recombinant PRC2 with biotinylated nucleosomes (Fig. 7A). We found that titration of purified mouse nuclear RNA inhibited the interaction of PRC2 with nucleosomes, consistent with the result of RNA degradation in cells. Furthermore, this effect was also observed with yeast tRNA, consistent with the promiscuous binding of PRC2 in cells revealed by iCLIP. To confirm that PRC2 chromatin association could reciprocally antagonize RNA binding, we tested whether nucleosomes could compete with biotinylated nascent RNA for PRC2 binding. Titration of nucleosomes strongly reduced pull-down of PRC2 by biotinylated RNA (Fig. 7B). These results support the findings from our iCLIP and functional experiments in cells that the binding of PRC2 to RNA and chromatin is mutually antagonistic.
Figure 7.

RNA and chromatin compete for PRC2 binding in vitro. (A) Pull-down of PRC2 (13 ng/µL, 49 nM) by biotinylated nucleosomes (21 ng/µL, 100 nM) in the absence and presence of purified nuclear RNA or tRNA (both titrated from 0.5–65 ng/µL). BSA at 330 ng/µL (5 µM) was used to control for nonspecific occlusion effects. (B, top) Pull-down of PRC2 (13 ng/µL, 49 nM) by 2.5 µg of biotinylated nascent RNA (bio-RNA) in the absence and presence of reconstituted nucleosomes (titrated from 0.024–2.4 µM). 5-Ethynyl uridinylated RNA (EU-RNA), the precursor in the biotinylation reaction, was used as control. (Bottom) Dot blot of input bio- and EU-RNA with streptavidin-HRP. (C) Our data support a model in which PRC2 can bind either to chromatin or to RNA in a mutually antagonistic fashion. At lowly expressed genes, there is active competition between chromatin and RNA for PRC2 binding (center). At highly active genes (right), repeated rounds of nascent RNA synthesis outcompete chromatin for PRC2 binding, protecting genes from inappropriate silencing. The loss of RNA upon gene repression allows PRC2 recruitment to chromatin (left). Positive feedback mediated by EED binding to H3K27me3, and between PRC1 and PRC2, allows for stable chromatin association that resists the loss of PRC2 to RNA caused by low level or stochastic transcriptional activation. This bistable state therefore buffers changes due to transcription noise and may ensure that only robust changes in transcriptional activity are propagated as changes in epigenetic state.
Discussion
We have discovered that PRC2 interacts with nascent RNA at essentially all active genes and that RNA binding and chromatin association are mutually antagonistic in cells and in vitro. SUZ12 directly binds RNA in cells and is sufficient for PRC2 RNA binding activity. RNA degradation in cells induces recruitment of PRC2 to chromatin at active genes. Correspondingly, release of PRC2 from chromatin in cells increases RNA binding. Consistent with this, RNA and nucleosomes compete for PRC2 interaction in vitro.
Taken together, our data support a new model in which PRC2 RNA binding and chromatin association are mutually antagonistic, helping to explain the observed pattern with which PRC2 is recruited to chromatin (Fig. 7C). In this model, RNA and chromatin compete for PRC2 interaction, and it is the balance between RNA level and chromatin state that dictates the extent to which PRC2 is recruited to chromatin at CpG islands. At lowly expressed genes, there is active competition between chromatin and RNA for PRC2 binding. At highly active genes, repeated rounds of nascent RNA synthesis constantly outcompete chromatin for PRC2 binding. When a gene is silenced during differentiation, RNA synthesis is down-regulated, shifting the balance toward PRC2 chromatin interaction. Positive feedback mediated by EED binding to H3K27me3 (Margueron et al. 2009) and between PRC1 and PRC2 (Blackledge et al. 2014; Cooper et al. 2014; Kalb et al. 2014) then allows for the formation of a stable repressive complex. We propose that this stable chromatin association is able to resist loss of PRC2 to RNA upon stochastic gene activation. The mutual antagonism between RNA and chromatin could therefore constitute the bistable switch previously suggested to exist between Polycomb repressed and active gene states (Pietersen and van Lohuizen 2008; Angel et al. 2011; Klose et al. 2013; Steffen and Ringrose 2014). This would ensure that stochastic changes in transcriptional activity are buffered with the important implication that only robust changes in transcriptional activity, occurring in response to developmental signals, are propagated as changes in epigenetic state.
Our model differs from those previously postulated from RIP-seq or PAR-CLIP data generated in the absence of experimental modulation of RNA and chromatin binding (Davidovich et al. 2013; Kaneko et al. 2013). In contrast to the data shown here, Kaneko et al. (2013) reported that RNA inhibited EZH2 methyltransferase activity at a specific set of genes without affecting PRC2 association with chromatin, leaving PRC2 in a poised state. Although allowing for the possibility of RNA acting to inhibit PRC2 chromatin association under certain circumstances, the “junk mail” model of Davidovich and colleagues is centered on the proposition that promiscuous RNA binding aids PRC2 recruitment to chromatin at Polycomb target genes that have escaped repression (Davidovich et al. 2013; Davidovich and Cech 2015). In contrast, we find that RNA antagonizes the association of PRC2 with its target genes.
Our data are consistent with the increase in PRC2 chromatin occupancy at genes silenced by the inhibition of RNA Pol II (Riising et al. 2014), but the recapitulation of this effect by RNA degradation (Fig. 6G) argues that this is mediated by loss of RNA rather than loss of RNA Pol II transit through chromatin, as originally envisaged. This antagonistic effect of RNA may also contribute to the observed loss of PRC2 binding at CpG islands when positioned next to active promoters and enhancers (Lynch et al. 2011; Jermann et al. 2014). The results of our in vitro RNA–nucleosome competition assay are consistent with the repression of PRC2 H3K27 methyltransferase activity observed upon addition of RNA in vitro (Cifuentes-Rojas et al. 2014; Herzog et al. 2014; Kaneko et al. 2014b) but suggest this is because RNA competes with nucleosomes for PRC2 binding rather than serving as an allosteric inhibitor of methyltransferase activity. Although PcG recruitment mechanisms differ between mammals and Drosophila, it has previously been suggested that the process of transcription through Polycomb response elements (PREs) removes PcG complexes from chromatin (Schmitt et al. 2005). At the vestigial PRE, it was reported that the direction of transcription dictates whether the element acts as a silencer or activator but that both RNA strands could inhibit PRC2 methyltransferase activity in vitro, potentially indicating that PRC2 binding to certain RNAs is prevented by other RNA binding proteins in vivo (Herzog et al. 2014). However, we do not find evidence for a population of RNAs that are depleted for PRC2 binding in mammalian ESCs. The formation of R loops upstream of RNA Pol II has also been found to inhibit PRC2 chromatin association (Chen et al. 2015), although the lack of PRC2 binding to RNA–DNA hybrids (Zhao et al. 2008, 2010; Kanhere et al. 2010) and differences between PRC2 chromatin binding upon RNase A and RNase H treatment (Supplemental Fig. S9F) indicate this is a separate phenomenon.
Our results do not rule out that some lincRNAs function to target PRC2 to chromatin, but they do constrain such models. Rather than having any particular propensity for binding PRC2, some lincRNAs may instead be able to target the complex to specific genomic loci due to their particular localization on chromatin. The ability of chromatin to compete with RNA for PRC2 binding that we have identified here may then allow PRC2 to be transferred from the lincRNA to the locus with which the RNA is associated. Alternatively, although lincRNAs interact with PRC2, this interaction may not be required for Polycomb recruitment, with the lincRNA instead repressing gene expression by another mechanism, which then allows for Polycomb recruitment (Brockdorff 2013). For example, a number of studies recently determined that Xist RNA interacts with the transcriptional repressor SPEN (also known as SHARP) and that this is required for gene silencing (Chu et al. 2015; McHugh et al. 2015; Minajigi et al. 2015; Moindrot et al. 2015; Monfort et al. 2015).
Whether PRC2 displayed a preference for certain RNAs or RNA sequences has been uncertain. We found that PRC2 interacts with nascent RNA at essentially all active genes, showing it binds RNA promiscuously in cells. Consistent with this, yeast tRNA can compete with nucleosomes for PRC2 binding (Fig. 7A), and tRNA can reverse the increase in PRC2 chromatin association that is caused by RNase A treatment in cells (Supplemental Fig. S9A). However, although promiscuous in terms of the RNAs with which it interacts, we found that PRC2 was enriched at exon–intron boundaries and the 3′ UTR. PRC2 thus binds nascent RNA nonselectively but with enrichment at defined locations that are related to gene structure.
Recombinant SUZ12 has been shown to interact with RNA in vitro (Kanhere et al. 2010; Cifuentes-Rojas et al. 2014), but whether it directly interacted with RNA in cells was unknown. Although antibodies to SUZ12 have previously been used in native RNA IP, this method does not discriminate between direct SUZ12 RNA binding and RNA binding by other PRC2 subunits or coprecipitating proteins. Our results demonstrate that not only does SUZ12 directly interact with RNA in cells but also it is able to recapitulate the PRC2 RNA binding profile in the absence of other PRC2 subunits. The invariance in PRC2 RNA binding upon Ezh2 deletion suggests either that SUZ12 determines PRC2 RNA binding or that SUZ12 and EZH2 RNA binding activities are redundant. It is not possible to perform iCLIP for EZH2 in SUZ12-deficient cells to distinguish between these possibilities because EZH2 is degraded in the absence of SUZ12 (Fig. 3A; Pasini et al. 2007). In either case, it can be concluded that SUZ12 is sufficient to establish the characteristic PRC2 RNA binding profile in cells. This discovery will likely be important for future structural studies seeking to understand how PRC2 interacts with RNA.
In summary, we have determined that PRC2 interacts with nascent RNA at essentially all active genes in ESCs and that mutual antagonism between RNA binding and chromatin association helps shape the pattern of PRC2 recruitment to chromatin. This mutual antagonism may also allow for the formation of a bistable switch between active and Polycomb-repressed states that ensures that only robust transcriptional changes are propagated as changes in epigenetic state. We propose that RNA may play a similar role in the regulation of other epigenetic modifications during development.
Methods
Mouse ESC culture
Ezh2fl/fl (Kanhere et al. 2010), JM8 (Jarid2+/+), Jarid2−/− (Landeira et al. 2010), and Suz12−/− ESCs (Pasini et al. 2007) were maintained on 0.1% gelatin, as previously described (culture conditions detailed in Supplemental Methods). Ezh2fl/fl cells were treated with 4-hydroxy-tamoxifen for 96 h (800 nM, Sigma) to induce Cre-mediated deletion of the SET domain, generating Ezh2Δ/Δ cells. The cKO Eed mESC clone 4D2 was cultured as previously described (Ura et al. 2008) and treated with doxycycline (10 μg/mL, Sigma) for 96 h to repress the MYC-tagged Eed transgene.
CLIP, PAR-CLIP, and iCLIP
CLIP, PAR-CLIP, and iCLIP were performed as previously described (Hafner et al. 2010; Huppertz et al. 2014) with the variations described in the Supplemental Methods. We used 2 × 108 cells for SUZ12, IgG, and GFP iCLIP and used 2.5 × 107 cells for FUS and HNRNPC iCLIP. Five microliters of α-SUZ12 (Cell Signaling) or 5 μg of α-EZH2 (Cell Signaling), α-Flag (Sigma), α-MYC (Cell Signaling), α-FUS (Novus), nonspecific IgG (Santa Cruz), or α-GFP (Abcam) antibody were used per experiment. Crosslinked RNPs running 10–30 (SUZ12), 10–45 (FUS), 10–40 (HNRNPC), and 5–30 (GFP) kDa above the sizes of their respective proteins were isolated. For IgG, we isolated RNPs of the same molecular weight as for SUZ12. Libraries were sequenced using an Illumina HiSeq 2500 (50-bp single-end reads). Crosslinked RNA was quantified using a Typhoon phosphorimager (GE) and ImageQuantTL (GE). EZH2 PAR-CLIP data (GSE49435) were processed as previously described (Kaneko et al. 2013), retaining only those reads containing a T > C transition. A list of ezRNA genes was obtained from Kaneko et al. (2013).
iCLIP data processing
iCLIP data were processed with iCount (http://icount.biolab.si/) and filtered to remove any crosslinks overlapping a RepeatMasker feature, ncRNAs under 200 nt in length, or snoRNAs. High-confidence clusters of crosslink sites were identified in iCount (FDR < 0.05) (Konig et al. 2010). Where a crosslink could map to multiple segments, it was assigned as either intron, UTR, or CDS, in that order, with protein-coding genes taking precedence over lincRNA. TopHat2 (Kim et al. 2013) was used to identify reads at spliced and unspliced exon junctions. DESeq2 (Love et al. 2014) was used to identify genes with significantly more PRC2 crosslinks compared with FUS, HNRNPC, or RNA-seq (FDR < 0.05). Further details on the bioinformatics pipeline can be found in the Supplemental Methods.
RNA-seq
Total RNA was purified with TRIzol and treated with DNase-Turbo (Ambion). Ribosomal RNA was depleted with Ribo-zero gold (EpiCentre). Libraries were constructed using the Illumina directional mRNA-seq sample prep and the NEBNext multiplex small RNA library prep kits and sequenced on an Illumina HiSeq 2500 (50 bp paired-end). Total and nuclear (GSE57092) (Bulut-Karslioglu et al. 2014) RNA-seq reads were trimmed to remove adaptors and low-quality bases and aligned with TopHat2.
RNase A treatment and cell fractionation
RNase A treatment and cell fractionation were performed as previously described (Zoabi et al. 2014), with variations described in the Supplemental Methods. ESCs were trypsinized, permeabilized with 0.05% Tween-20 in PBS for 10 min on ice, and mock-treated or treated with 1 mg/mL RNase A (Sigma) for 30 min at RT. Cells were either crosslinked for ChIP or fractionated into cytoplasmic, nucleosplasmic, and chromatin fractions for immunoblotting.
ChIP
ChIP was performed for SUZ12 (Abcam), EZH2 (Cell Signaling), JARID2 (Cell Signalling), H3K27me3 (Abcam), total H3 (Abcam), and EP300 (Bethyl) as previously described (Kanhere et al. 2012), with variations described in the Supplemental Methods. Enrichment of specific gene sequences was measured by qPCR (Applied Biosystems). Libraries were sequenced using an Illumina HiSeq 2500 (50 bp single-end). ChIP-seq reads were trimmed to remove adaptors and low-quality bases and aligned with Bowtie 2 (Langmead and Salzberg 2012). MACS14 (Zhang et al. 2008) and MAnorm (Shao et al. 2012) were used to identify sites at which SUZ12 binding was significantly changed by RNase treatment (P < 0.05).
Nucleosome and RNA pull-downs
Nucleosomes incorporating biotinylated and unmodified DNA were reconstituted as previously described (Bartke et al. 2010). Nascent RNA was biotinylated using the Click-iT nascent RNA capture kit (Thermo). Recombinant PRC2 was from Active Motif (no. 31387). Binding reactions were performed as previously described (Schmitges et al. 2011), except for the inclusion of 0.05% IGEPAL CA-630, 1 mM DTT, and 33 ng/µL BSA for 3 h at 4°C.
Data access
All iCLIP, RNA-seq, and ChIP-seq data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE66831.
Acknowledgments
We thank Neil Brockdorff and Hiroshi Koide for Eed cKO cells, Amanda Fisher for Ezh2fl/fl cells, Amanda Fisher and Bill Skarnes for JM8 and Jarid2−/− cells, Diego Pasini for Suz12−/− cells, and Suzana Hadjur for Rad21fl/fl NSC. We thank Deborah Hughes at the Institute for Neurology and the UCL Cancer Institute Genomics Core Facility (supported by the Cancer Research UK–UCL Centre) for performing the sequencing runs and Tomaž Curk and Igor Ruiz de los Mozos for submission of data to iCount. This work was funded by a European Research Council (ERC) Starting Grant (ChromatinRNA, 311704), a Worldwide Cancer Research project grant (13-0256), a Fast-track Award from the UCLH Clinical Research and Development Committee, and a European Molecular Biology Organization (EMBO) Long term Fellowship (54-2011) to M.B.
Author contributions: M.B. and R.G.J. initiated the research program. M.B., C.M.Y., L.S., C.R.S., T.B., J.U., and R.G.J. designed experiments. M.B., L.S., M.D., F.P.R., and C.L.F. performed experiments. M.B., K.V., C.L.F., B.M.F., and T.B. provided reagents. C.M.Y. performed bioinformatics analyses. All authors interpreted data. R.G.J. wrote the paper, with input from all authors.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.197632.115.
References
- Angel A, Song J, Dean C, Howard M. 2011. A Polycomb-based switch underlying quantitative epigenetic memory. Nature 476: 105–108. [DOI] [PubMed] [Google Scholar]
- Bartke T, Vermeulen M, Xhemalce B, Robson SC, Mann M, Kouzarides T. 2010. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell 143: 470–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackledge NP, Farcas AM, Kondo T, King HW, McGouran JF, Hanssen LL, Ito S, Cooper S, Kondo K, Koseki Y, et al. 2014. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and Polycomb domain formation. Cell 157: 1445–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockdorff N. 2013. Noncoding RNA and Polycomb recruitment. RNA 19: 429–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulut-Karslioglu A, De La Rosa-Velazquez IA, Ramirez F, Barenboim M, Onishi-Seebacher M, Arand J, Galan C, Winter GE, Engist B, Gerle B, et al. 2014. Suv39h-dependent H3K9me3 marks intact retrotransposons and silences LINE elements in mouse embryonic stem cells. Mol Cell 55: 277–290. [DOI] [PubMed] [Google Scholar]
- Chen PB, Chen HV, Acharya D, Rando OJ, Fazzio TG. 2015. R loops regulate promoter-proximal chromatin architecture and cellular differentiation. Nat Struct Mol Biol. 22: 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C, Zhang QC, da Rocha ST, Flynn RA, Bharadwaj M, Calabrese JM, Magnuson T, Heard E, Chang HY. 2015. Systematic discovery of Xist RNA binding proteins. Cell 161: 404–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Rojas C, Hernandez AJ, Sarma K, Lee JT. 2014. Regulatory interactions between RNA and Polycomb repressive complex 2. Mol Cell 55: 171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper S, Dienstbier M, Hassan R, Schermelleh L, Sharif J, Blackledge NP, De Marco V, Elderkin S, Koseki H, Klose R, et al. 2014. Targeting Polycomb to pericentric heterochromatin in embryonic stem cells reveals a role for H2AK119u1 in PRC2 recruitment. Cell Rep 7: 1456–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidovich C, Cech TR. 2015. The recruitment of chromatin modifiers by long noncoding RNAs: lessons from PRC2. RNA 21: 2007–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidovich C, Zheng L, Goodrich KJ, Cech TR. 2013. Promiscuous RNA binding by Polycomb repressive complex 2. Nat Struct Mol Biol 20: 1250–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidovich C, Wang X, Cifuentes-Rojas C, Goodrich KJ, Gooding AR, Lee JT, Cech TR. 2015. Toward a consensus on the binding specificity and promiscuity of PRC2 for RNA. Mol Cell 57: 552–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Croce L, Helin K. 2013. Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol 20: 1147–1155. [DOI] [PubMed] [Google Scholar]
- Guil S, Soler M, Portela A, Carrere J, Fonalleras E, Gomez A, Villanueva A, Esteller M. 2012. Intronic RNAs mediate EZH2 regulation of epigenetic targets. Nat Struct Mol Biol 19: 664–670. [DOI] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M, et al. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141: 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog VA, Lempradl A, Trupke J, Okulski H, Altmutter C, Ruge F, Boidol B, Kubicek S, Schmauss G, Aumayr K, et al. 2014. A strand-specific switch in noncoding transcription switches the function of a Polycomb/Trithorax response element. Nat Genet 46: 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppertz I, Attig J, D'Ambrogio A, Easton LE, Sibley CR, Sugimoto Y, Tajnik M, Konig J, Ule J. 2014. iCLIP: protein–RNA interactions at nucleotide resolution. Methods 65: 274–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jermann P, Hoerner L, Burger L, Schubeler D. 2014. Short sequences can efficiently recruit histone H3 lysine 27 trimethylation in the absence of enhancer activity and DNA methylation. Proc Natl Acad Sci 111: E3415–E3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalb R, Latwiel S, Baymaz HI, Jansen PW, Muller CW, Vermeulen M, Muller J. 2014. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat Struct Mol Biol 21: 569–571. [DOI] [PubMed] [Google Scholar]
- Kaneko S, Son J, Shen SS, Reinberg D, Bonasio R. 2013. PRC2 binds active promoters and contacts nascent RNAs in embryonic stem cells. Nat Struct Mol Biol 20: 1258–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Bonasio R, Saldana-Meyer R, Yoshida T, Son J, Nishino K, Umezawa A, Reinberg D. 2014a. Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol Cell 53: 290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Son J, Bonasio R, Shen SS, Reinberg D. 2014b. Nascent RNA interaction keeps PRC2 activity poised and in check. Genes Dev 28: 1983–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanhere A, Viiri K, Araujo CC, Rasaiyaah J, Bouwman RD, Whyte WA, Pereira CF, Brookes E, Walker K, Bell GW, et al. 2010. Short RNAs are transcribed from repressed Polycomb target genes and interact with Polycomb repressive complex-2. Mol Cell 38: 675–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanhere A, Hertweck A, Bhatia U, Gökmen MR, Perucha E, Jackson I, Lord GM, Jenner RG. 2012. T-bet and GATA3 orchestrate Th1 and Th2 differentiation through lineage-specific targeting of distal regulatory elements. Nat Commun 3: 1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, et al. 2009. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci 106: 11667–11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Cooper S, Farcas AM, Blackledge NP, Brockdorff N. 2013. Chromatin sampling: an emerging perspective on targeting Polycomb repressor proteins. PLoS Genet 9: e1003717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J. 2010. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol 17: 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, et al. 2008. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 4: e1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landeira D, Sauer S, Poot R, Dvorkina M, Mazzarella L, Jorgensen HF, Pereira CF, Leleu M, Piccolo FM, Spivakov M, et al. 2010. Jarid2 is a PRC2 component in embryonic stem cells required for multi-lineage differentiation and recruitment of PRC1 and RNA Polymerase II to developmental regulators. Nat Cell Biol 12: 618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg S. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT. 2012. Epigenetic regulation by long noncoding RNAs. Science 338: 1435–1439. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MD, Smith AJ, De Gobbi M, Flenley M, Hughes JR, Vernimmen D, Ayyub H, Sharpe JA, Sloane-Stanley JA, Sutherland L, et al. 2011. An interspecies analysis reveals a key role for unmethylated CpG dinucleotides in vertebrate Polycomb complex recruitment. EMBO J 31: 317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. 2011. The Polycomb complex PRC2 and its mark in life. Nature 469: 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ III, Voigt P, Martin SR, Taylor WR, De Marco V, et al. 2009. Role of the Polycomb protein EED in the propagation of repressive histone marks. Nature 461: 762–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh CA, Chen CK, Chow A, Surka CF, Tran C, McDonel P, Pandya-Jones A, Blanco M, Burghard C, Moradian A, et al. 2015. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 521: 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendenhall EM, Koche RP, Truong T, Zhou VW, Issac B, Chi AS, Ku M, Bernstein BE. 2010. GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet 6: e1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minajigi A, Froberg JE, Wei C, Sunwoo H, Kesner B, Colognori D, Lessing D, Payer B, Boukhali M, Haas W, et al. 2015. Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 349: pii:aab2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moindrot B, Cerase A, Coker H, Masui O, Grijzenhout A, Pintacuda G, Schermelleh L, Nesterova TB, Brockdorff N. 2015. A pooled shRNA screen identifies Rbm15, Spen, and Wtap as factors required for Xist RNA-mediated silencing. Cell Rep 12: 562–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monfort A, Di Minin G, Postlmayr A, Freimann R, Arieti F, Thore S, Wutz A. 2015. Identification of Spen as a crucial factor for Xist function through forward genetic screening in haploid embryonic stem cells. Cell Rep 12: 554–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini D, Bracken AP, Hansen JB, Capillo M, Helin K. 2007. The Polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol Cell Biol 27: 3769–3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietersen AM, van Lohuizen M. 2008. Stem cell regulation by Polycomb repressors: postponing commitment. Curr Opin Cell Biol 20: 201–207. [DOI] [PubMed] [Google Scholar]
- Riising EM, Comet I, Leblanc B, Wu X, Johansen JV, Helin K. 2014. Gene silencing triggers Polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol Cell 55: 347–360. [DOI] [PubMed] [Google Scholar]
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, et al. 2007. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129: 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogelj B, Easton LE, Bogu GK, Stanton LW, Rot G, Curk T, Zupan B, Sugimoto Y, Modic M, Haberman N, et al. 2012. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci Rep 2: 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitges FW, Prusty AB, Faty M, Stutzer A, Lingaraju GM, Aiwazian J, Sack R, Hess D, Li L, Zhou S, et al. 2011. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol Cell 42: 330–341. [DOI] [PubMed] [Google Scholar]
- Schmitt S, Prestel M, Paro R. 2005. Intergenic transcription through a Polycomb group response element counteracts silencing. Genes Dev 19: 697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Zhang Y, Yuan GC, Orkin SH, Waxman DJ. 2012. MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol 13: R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofueva S, Yaffe E, Chan WC, Georgopoulou D, Vietri Rudan M, Mira-Bontenbal H, Pollard SM, Schroth GP, Tanay A, Hadjur S. 2013. Cohesin-mediated interactions organize chromosomal domain architecture. EMBO J 32: 3119–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen PA, Ringrose L. 2014. What are memories made of? How Polycomb and Trithorax proteins mediate epigenetic memory. Nat Rev Mol Cell Biol 1: 340–356. [DOI] [PubMed] [Google Scholar]
- Tanay A, O'Donnell AH, Damelin M, Bestor TH. 2007. Hyperconserved CpG domains underlie Polycomb-binding sites. Proc Natl Acad Sci 104: 5521–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ura H, Usuda M, Kinoshita K, Sun C, Mori K, Akagi T, Matsuda T, Koide H, Yokota T. 2008. STAT3 and Oct-3/4 control histone modification through induction of Eed in embryonic stem cells. J Biol Chem 283: 9713–9723. [DOI] [PubMed] [Google Scholar]
- Wang KC, Chang HY. 2011. Molecular mechanisms of long noncoding RNAs. Mol Cell 43: 904–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Xu M, Huang C, Liu N, Chen S, Zhu B. 2011. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J Biol Chem 286: 7983–7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD, et al. 2014. A comparative encyclopedia of DNA elements in the mouse genome. Nature 515: 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. 2008. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322: 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, Lee JT. 2010. Genome-wide identification of Polycomb-associated RNAs by RIP-seq. Mol Cell 40: 939–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoabi M, Nadar-Ponniah PT, Khoury-Haddad H, Usaj M, Budowski-Tal I, Haran T, Henn A, Mandel-Gutfreund Y, Ayoub N. 2014. RNA-dependent chromatin localization of KDM4D lysine demethylase promotes H3K9me3 demethylation. Nucleic Acids Res 42: 13026–13038. [DOI] [PMC free article] [PubMed] [Google Scholar]