

Abstract
Copy number variants (CNVs) are a class of structural variants that may involve complex genomic rearrangements (CGRs) and are hypothesized to have additional mutations around their breakpoints. Understanding the mechanisms underlying CNV formation is fundamental for understanding the repair and mutation mechanisms in cells, thereby shedding light on evolution, genomic disorders, cancer, and complex human traits. In this study, we used data from the 1000 Genomes Project to analyze hundreds of loci harboring heterozygous germline deletions in the subjects NA12878 and NA19240. By utilizing synthetic long-read data (longer than 2 kbp) in combination with high coverage short-read data and, in parallel, by comparing with parental genomes, we interrogated the phasing of these deletions with the flanking tens of thousands of heterozygous SNPs and indels. We found that the density of SNPs/indels flanking the breakpoints of deletions (in-phase variants) is approximately twice as high as the corresponding density for the variants on the haplotype without deletion (out-of-phase variants). This fold change was even larger for the subset of deletions with signatures of replication-based mechanism of formation. The allele frequency (AF) spectrum for deletions is enriched for rare events; and the AF spectrum for in-phase SNPs is shifted toward this deletion spectrum, thus offering evidence consistent with the concomitance of the in-phase SNPs/indels with the deletion events. These findings therefore lend support to the hypothesis that the mutational mechanisms underlying CNV formation are error prone. Our results could also be relevant for resolving mutation-rate discrepancies in human and to explain kataegis.
CNVs (these include insertions, deletions, and duplications) are a class of structural variants (SVs) that are widely prevalent in the human population and can be benign. However, they are increasingly being implicated in a variety of disease phenotypes. CNVs can cause loss of function of genes (e.g., Nathans et al. 1986); alter the copy numbers of dosage-sensitive genes (Lupski and Stankiewicz 2005; Lee et al. 2007; Lupski 2009; Carvalho et al. 2013); and are also associated with complex human traits, such as schizophrenia, autism, mental retardation, Alzheimer’s and Parkinson’s diseases, susceptibility to HIV, Crohn’s disease, and pancreatitis (for review, see Zhang et al. 2009b).
CNVs occur as a result of changes in chromosome structure, which can occur due to homologous recombination (HR) or nonhomologous (NH) mechanisms (Hastings et al. 2009b). Specifically, nonallelic homologous recombination (NAHR) involves ectopic crossover between interacting strands of DNA mediated by paralogous low copy repeat (LCR) substrates (Stankiewicz and Lupski 2002; Liu et al. 2011). Nonhomologous (NH) mechanisms can be further subdivided into nonreplicative (e.g., NHEJ) and replicative processes (Hastings et al. 2009a). With the advent of NGS technologies, NH CNVs have often revealed complex genomic rearrangements (CGRs), nonblunt breakpoints, and additional mutations flanking the breakpoints. Multiple studies have reported small sequence insertions and microhomologies at SV breakpoints as well as SNVs/indels in the regions flanking the breakpoint junctions (Conrad et al. 2010; Kidd et al. 2010; Lam et al. 2010; Mills et al. 2011; Carvalho et al. 2013; Pang et al. 2013; Abyzov et al. 2015; Wang et al. 2015). CGRs are characterized by the presence of two or more breakpoint junctions and combinations of multiple simple rearrangements, such as deletions, duplications, inversions, and also triplications (Zhang et al. 2009a). Two replicative mechanisms have been proposed to explain the observed sequence features around NH CNVs: Fork Stalling and Template Switching (Slack et al. 2006; Lee et al. 2007); and Microhomology-Mediated Break Induced Replication (Hastings et al. 2009a). These replication-based mechanisms are hypothesized to be highly error prone (Carvalho et al. 2013) because they utilize a low-fidelity polymerase enzyme, leading to an increased mutation load around the breakpoints. Other studies (Deem et al. 2011; Arlt et al. 2012) exploring replication mechanisms in organisms, such as mouse and yeast, also found high error rates and mutations.
Although several studies have explored breakpoint complexity by working with data from patient cohorts (Lee et al. 2007; Carvalho et al. 2013; Wang et al. 2015), here we analyzed cell lines derived from presumably normal individuals (a Caucasian and Yoruban trio) from the 1000 Genomes Project Consortium (2015), to search for evidences in the germline lineage of a higher mutational load associated with complex deletion events. Specifically, we looked for the suggested signatures of the aforementioned error-prone replication mechanisms by asking the following questions: (1) Is there an elevated density of SNPs and indels around deletion breakpoints? and (2) Do the SNPs and indels occur concomitantly with the deletion events? To answer these questions, we analyzed, local to deletions, densities of SNP and indels that are in phase and out of phase with the deletions (Fig. 1). Unlike previous studies (Lee et al. 2007; Deem et al. 2011; Carvalho et al. 2013; Wang et al. 2015), our analysis examined deletions in human rather than in model organisms, and throughout the genome with the loci not necessarily associated with particular genes.
Figure 1.

Schematic and results of the analysis. (A) Personal real haplotypes with deletion (red) and without deletion (pure blue) are shown along with flanking SNPs/indels depicted by filled circles/rectangles. TruSeq data allows resolving SNPs/indels on the haplotype with deletion (the set of in-phase variants). The GATK variant calls (black) include SNPs/indels for both haplotypes and also provide genotype information. The reconstructed haplotype without deletion (the set of out-of-phase variants) is obtained through complement of GATK to TruSeq calls. Only heterozygous SNPs/indels are further counted. The dashed pink/green lines highlight error cases that increase the count for the haplotype with/without deletion, respectively. The pink errors are highly unlikely (see text), and the green cases can only contribute against our result. (B) Histogram showing the count of heterozygous SNPs on both haplotypes with respect to distance from the deletions’ breakpoints. A total of 262 loci with heterozygous deletions are considered. Statistically significant differences (by Z-test) in SNP counts in bins marked by star are observed in 6-kbp flanking windows. (C) Densities of heterozygous SNPs on haplotypes with and without deletions are roughly constant and higher than personal genome-wide average per haplotype. The density with deletions is twice as high as without. In-phase indel density is 33% higher than out-of-phase (not shown in figure).
Results
Deletion set selection and haplotype reconstruction
From the data resource provided by the 1000 Genomes Project Consortium (2015), we selected 376 large deletions genotyped as present in NA12878 and with breakpoints known at base pair resolution. From this set, we selected heterozygous deletions only, with consistent genotypes in parents (Methods), so that we could compare the occurrence of SNPs and indels between haplotypes, with and without the deletion. A few more deletions were eliminated from our analysis because alignment of TruSeq long-reads, using AGE (Abyzov and Gerstein 2011), did not agree with the breakpoints for these deletions. The final set consists of 262 deletions, with a median length of 1963 bp, a minimum length of 298 bp, and a maximum length of 79,598 bp (Supplemental File S1).
We classified these deletions by their likely mechanism of origin, using the BreakSeq pipeline (Lam et al. 2010). According to this pipeline, SVs have an NAHR mechanism of origin if among other criteria, there is extensive sequence identity (at least 85%, with a minimum homology of 50 bp) around the breakpoints. Further, NH events are those that cannot be classified as NAHR or transposable element insertions (TEIs). For these NH events, typical microhomology (MH) is of length smaller than 10 bp. Based on this classification scheme, 215 (82%) deletions are from NH events, 29 (11%) are NAHR, 14 (5%) are TEIs, and the remaining 4 (2%) are classified as unsure. Of the 215 NH-generated deletion events, 94 (44%) show evidence of being generated through replication-based mechanisms, i.e., they contained, around deletions’ breakpoints, sequence identity (MH) longer than 2 bp or a microinsertion (MI) longer than 10 bp. Although it is established (Hastings et al. 2009a,b) that MH is involved with replicative mechanisms, structural variants created by microhomology-mediated end joining (MMEJ) also have MH around their breakpoints (McVey and Lee 2008). Therefore, it is possible that some (likely a minor fraction) of the 94 deletions, e.g., those with MH longer than 5 bp, where generated by MMEJ.
Next, we reconstructed two local haplotypes around the selected heterozygous deletions in the genome of the subject NA12878: one with the deletion event, and the other without (Fig. 1A; Methods). To reconstruct a haplotype with deletion, we aligned Illumina TruSeq synthetic long-reads (McCoy et al. 2014) around deletions using AGE. From this alignment, we were able to find SNPs/indels in phase with each deletion. Of these in-phase TruSeq variants, we selected those that are present in the GATK (The 1000 Genomes Project Consortium 2015) derived unphased personal set of heterozygous variants, which is obtained from the analysis of deep-coverage, whole genome, short-read Illumina data. Variants deemed homozygous by GATK were excluded from our analysis because they are uninformative for the comparison of local haplotypes around deletions. SNPs/indels that are out of phase with the deletions were determined by subtracting the in-phase TruSeq variants from the aforementioned GATK personal variant set and by removing any remaining homozygous variants from this out-of-phase set. We provide the in-phase and out-of-phase variants in a VCF file that also contains their positions relative to the deletion breakpoints and the breakpoint coordinates (Supplemental File S2).
Count and density of SNPs/indels around deletion breakpoints
Our analysis yielded 2185 in-phase and 1065 out-of-phase SNPs, indicating a roughly doubled density of heterozygous SNPs on haplotypes with deletions (Fig. 1B). Similarly, we counted 359 in-phase and 270 out-of-phase indels, a 33% higher density of heterozygous indels on haplotypes with deletions. For both SNPs and indels, the differences in counts are statistically significant (P-value <4 × 10−4, by Z-test). The densities of in-phase and out-of-phase SNPs, in 6-kbp windows on either side of the breakpoints (Fig. 1C), are roughly constant (1.02 × 10−3 and 0.51 × 10−3 SNPs per bp per haplotype, respectively); and the density of the former is twice as high as the density of the latter (P-value <2.2 × 10−16, by paired t-test). This effect is driven by a number of deletions (rather than by one or just a few) (Supplemental Fig. S1). Both densities are higher than genome-wide per haplotype average heterozygous SNP density of 0.35 × 10−3 per bp. Higher SNP density around breakpoints has been previously observed and explained by deletion and SNP/indel co-occurrence in regions of relaxed selection (Abyzov et al. 2015). This is a likely explanation for the higher density of out-of-phase SNPs observed in this analysis. However, an even higher density of in-phase SNPs has not been previously observed.
We broke down the counts for in-phase and out-of-phase SNPs into those associated with the 94 NH deletions that are likely generated by replication-based mechanisms, and the remaining 168 (of the total 262) deletions. Within the 6-kbp windows, we counted 955 in-phase and 328 out-of-phase SNPs associated with the 94 deletions, which correspond to an almost threefold increase between the two haplotypes. The remaining 168 deletions gave corresponding counts of 1166 (in-phase) and 689 (out-of-phase) SNPs, i.e., only a 69% increase in the count of in-phase SNPs. The analysis of in-phase and out-of-phase SNP densities in relation to MH length did not reveal an obvious correlation (Supplemental Fig. S2). Note that some of the 168 deletions may have been generated by replication-based mechanisms, although they have not been classified so. This could happen because, for example, MH around breakpoints may be erased by another variant or not be detected due to sequencing errors. Additionally, deletions with an MH of 2 bp may also be generated by replication-based mechanisms. Besides, misclassification of replication-based deletions as NAHR or TEI is a possibility.
For indels, we counted 129 in-phase and 102 out-of-phase, flanking 10-kbp windows around the 94 NH deletions. The corresponding counts around the 168 deletions were 230 in-phase and 168 out-of-phase. However, a statistical test shows that the difference in proportions between the two groups (the 94 NH deletions and the remaining 168) is not statistically significant (P-value = 0.63, by the two-sample proportion test). Therefore, we did not observe a difference in the density of indels when compared with the deletions possibly generated by replicative mechanisms and those that are not.
Consideration of sequencing and calling errors
We carefully considered possible sequencing and genotyping errors to make sure that their effect on our results is negligible (Fig. 1A, dashed lines). Green dashed lines in Figure 1 represent error cases that contribute to increasing the count of out-of-phase heterozygous variants. Such errors can happen when TruSeq either misses a true variant or there is a false call or genotyping error in HiSeq data. However, these erroneous contributions only count against our result (hence, the green color).
Pink dashed lines represent error cases that contribute to increasing the count of in-phase heterozygous variants. Such errors can be a result of false variant call(s) from TruSeq/HiSeq data or incorrect genotyping for a true variant. Although the expected error rate of variant calling and genotyping is of the order of a few percent and so that such errors are unlikely to explain the observed difference in variant density, we replicated our analysis using a very high stringency variant set derived from a three-way consensus of variants in the GATK call set (instead of just the GATK call set), in the Complete Genomics (Drmanac et al. 2010) call set (derived from independent sequencing), and in the high confidence call set of Illumina Platinum Genomes (http://www.illumina.com/platinumgenomes/), derived from independent sequencing and elaborate pedigree analysis to minimize technical biases. Variants in the three-way consensus set were required to have a heterozygous genotype and matches in reference and alternate bases across the three sets.
The three-way consensus set consisted of 2008 (92% of the original set) in-phase and 849 (80% of the original set) out-of-phase SNPs, indicating a more than doubled density of heterozygous SNPs on haplotypes with deletions. The corresponding results for indels were 192 (54%) in-phase and 108 (40%) out-of-phase indel counts, respectively, with a 78% higher density of heterozygous indels on haplotypes with deletions. For both SNPs and indels, the differences in counts are statistically significant (P-value <2.4 × 10−6, by Z-test). Therefore, analysis with a more stringent set continues to show a marked, and an even larger and more significant, increase in the in-phase over the out-of-phase variant counts.
Variant counts with phasing by using family
The parents of NA12878 (i.e., NA12891 and NA12892) were also sequenced by the 1000 Genomes Project Consortium, and their personal SNPs/indels are also available. This knowledge of parental genomes allows for an independent (of TruSeq data) phasing of variants in a child. Using genotypes of the variants in this familial trio, we derived a phased set of heterozygous variants (Methods). Such a phasing spans across the entire genome, but is not complete because variants heterozygous in both parents and the child cannot be phased.
Next, using genotype information obtained from CNVnator (Abyzov et al. 2011; Methods) and the same trio-based method as above, we phased 220 heterozygous deletions (of the 262 selected above). For each of these deletions, we assigned the flanking variants to reconstruct the two haplotypes, with/without deletion, according to the phased genotypes of the deletion and the variants.
To compare with the analysis based on TruSeq phasing, we considered the same flanking regions around the deletions’ breakpoints, as obtained from TruSeq fragment data. Of the 1868 SNPs designated as in-phase by the trio analysis, 106 do not match the phasing assigned by TruSeq (Supplemental Table S1). For the out-of-phase SNPs, two of the 664 do not match the phasing assigned by TruSeq. Based on this, we calculated a high concordance of 95.7%. It is likely that the small discordance between trio and TruSeq-based phasing is due to errors in TruSeq synthetic reads. Evidence for this is provided by the high agreement between trio phasing and phased genotypes assigned by Illumina Platinum Genomes (Supplemental Table S1); however, errors due to recombination in cell lines (for parents or child) used to extract DNA cannot be completely ruled out. The ratio of in-phase to out-of-phase SNPs, from trio phasing, was 2.81, consistent, but higher than that from TruSeq phasing (a ratio of 2.18).
A larger value of the ratio could be a result of the bias inherent in the analysis with trio phasing when selection of phased deletions for haplotype reconstruction preferentially selects phased in-phase variants due to local linkage disequilibrium (LD), thereby inflating the count of in-phase SNPs. Specifically, a reconstructed haplotype around a given arbitrary locus will only contain SNPs that can be phased, which is a fraction (α) of all SNPs on the haplotype around the locus. Let N1 be the total number of SNPs in a region flanking a phased deletion, and n is the number of SNPs in LD with the deletion. The number of SNPs that contribute to an in-phase count is (N1 − n)α1 + n, where α1 is the fraction of SNPs that can be phased. Similarly, the number of SNPs that contribute to the out-of-phase count is N2α2, where N2 is the total number of SNPs flanking the same locus around the other haplotype (with no deletion), and α2 is the fraction of SNPs that can be phased. We now have the ratio as
Values of α1 and α2 can be different at each individual locus, but on an average will be the same and equal to a genome-wide average fraction of α = 0.74 of phased heterozygous SNPs. Thus, average overestimation of the true ratio N1/N2 over many sites will be
We carried out an empirical verification and estimation of this bias. For the entire deletion set, percentages of in-phase and out-of-phase SNPs that could not be phased by trio were within statistical error (Supplemental Table S2). However, for the subset of 220 deletions that could be phased by trio, the percentage of SNPs that could not be phased by trio was lower by 4.7% for in-phase SNPs (Supplemental Table S3), thus demonstrating the bias. Although this bias can explain some of the undercounting of out-of-phase SNPs in trio phasing, the major discrepancy in analysis between TruSeq and trio phasing is due to the differences in phasing of some SNPs to reconstruct the haplotypes. Although giving different quantitative results (i.e., different ratio values), both approaches agree qualitatively (i.e., show large excess of in-phase over out-of-phase SNPs).
Trio phasing
Since we just demonstrated that systematic bias in ratio estimation from trio phasing is small, we used this approach on a larger set of deletions (including smaller Pindel calls), for two familial trios—Caucasian (NA12878, NA12891, NA12892) and Yoruban (NA19239, NA19238, NA19240)—and over longer, 100-kbp flanking regions (Fig. 2). For NA12878, we phased 300 deletion events and counted 32,956 in-phase and 22,841 out-of-phase SNPs, for a total of 55,797. For NA19240, we phased 457 deletion events, with 56,392 in-phase and 43,282 out-of-phase SNPs, for a total of 99,674. For each individual, counts were distributed equally proportionally between deletions on maternal and paternal haplotypes (Table 1).
Figure 2.

Densities of in-phase (red) and out-of-phase (blue) heterozygous SNPs/indels flanking deletions in (A,C) Caucasian (NA12878) and (B,D) Yoruban (NA19240) individuals. Densities are obtained from trio-based haplotype reconstruction. Average out-of-phase densities for the displayed interval are shown by dashed lines (blue). Genome-wide average densities of heterozygous SNPs per haplotype are shown by black dashed lines. In the Yoruban trio, average out-of-phase and genome-wide densities are almost the same.
Table 1.
Breakdown of SNPs around deletions from trio phasing for maternal and paternal alleles
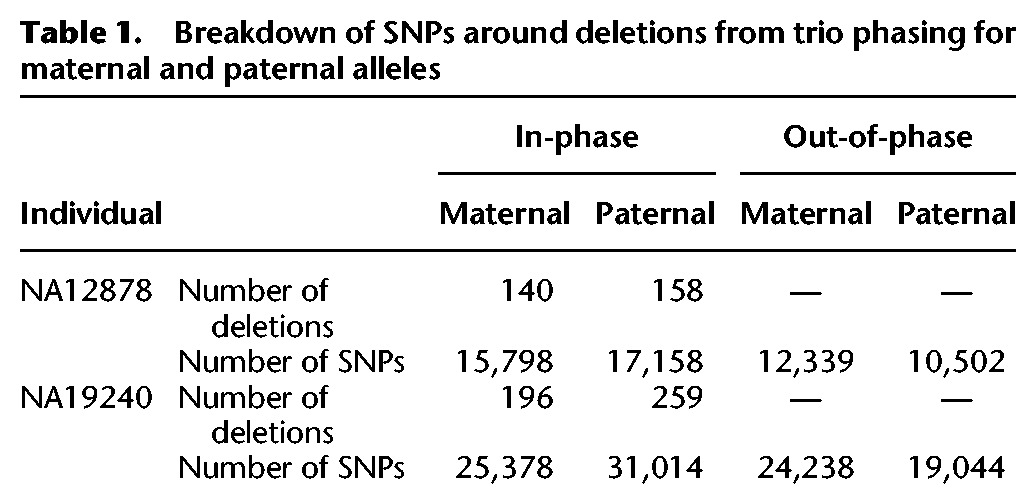
Qualitatively consistent with our result obtained from TruSeq analysis, we observed a higher density of in-phase SNPs than out-of-phase SNPs, and the latter was higher than the genome-wide average heterozygous SNP density per haplotype (Fig. 2A,B). The increase in the density of in-phase SNPs is apparent in a 50-kbp window around the deletions and is particularly pronounced in the 10-kbp window. In-phase average densities for each individual, in 10-kbp windows away from breakpoints, are 0.84 × 10−3 (for NA12878) and 0.89 × 10−3 for (NA19240). Corresponding out-of-phase densities are 0.39 × 10−3 and 0.45 × 10−3 (all in units of SNPs per haplotype per bp). The in-phase density tapers off to meet the out-of-phase density at a distance of ∼100 kbp from the deletions’ loci. Similarly, density of in-phase indels is higher than that of out-of-phase (Fig. 2C,D): 0.18 × 10−3 (for both NA12878 and NA19240) versus 0.08 × 10−3 (for NA12878) and 0.10 × 10−3 (for NA19240).
As we did with the TruSeq analysis above, we classified the trio-phased deletions according to their likely mechanism of origin and stratified in-phase and out-of-phase SNPs/indels into those associated with possibly replication-based NH deletions and the rest (Supplemental Table S4). For NA12878, we find qualitative agreement with the TruSeq results for SNPs and also statistically significant results for indels (likely a consequence of larger counts), with more than threefold ratios associated with possibly replication-based NH events.
Similar analyses with duplications are currently not feasible because there are only eight duplications in NA12878 and NA19240 with base pair resolution.
Allele frequency distribution of deletions and SNPs
To explore the hypothesis that in-phase SNPs occur concomitantly with the deletions, we analyzed the distribution of allele frequencies (AF) for deletions and in-phase and out-of-phase SNPs, phased with TruSeq, for NA12878. AF for 95.4% of deletions and 96.5% of SNPs in our set were provided by the 1000 Genomes Project Consortium based on the analysis of personal genomes for 2535 individuals (Sudmant et al. 2015; The 1000 Genomes Project Consortium 2015).
The normalized AF distributions for in-phase SNPs, out-of-phase SNPs, and deletions are different (Fig. 3A). AF distribution for deletions is shifted toward smaller values, i.e., deletions are enriched for rare events compared to SNPs. In contrast, AF distribution for out-of-phase SNPs is shifted toward more common events, whereas AF distribution for in-phase SNPs is between the two. Therefore, AF distribution for in-phase SNPs can be considered as a superposition of the background distribution (i.e., out-of-phase SNPs) and the one for additional SNPs associated with the introduction of a deletion into a haplotype. This is consistent with the hypothesis that SNPs are generated concomitantly with deletions.
Figure 3.
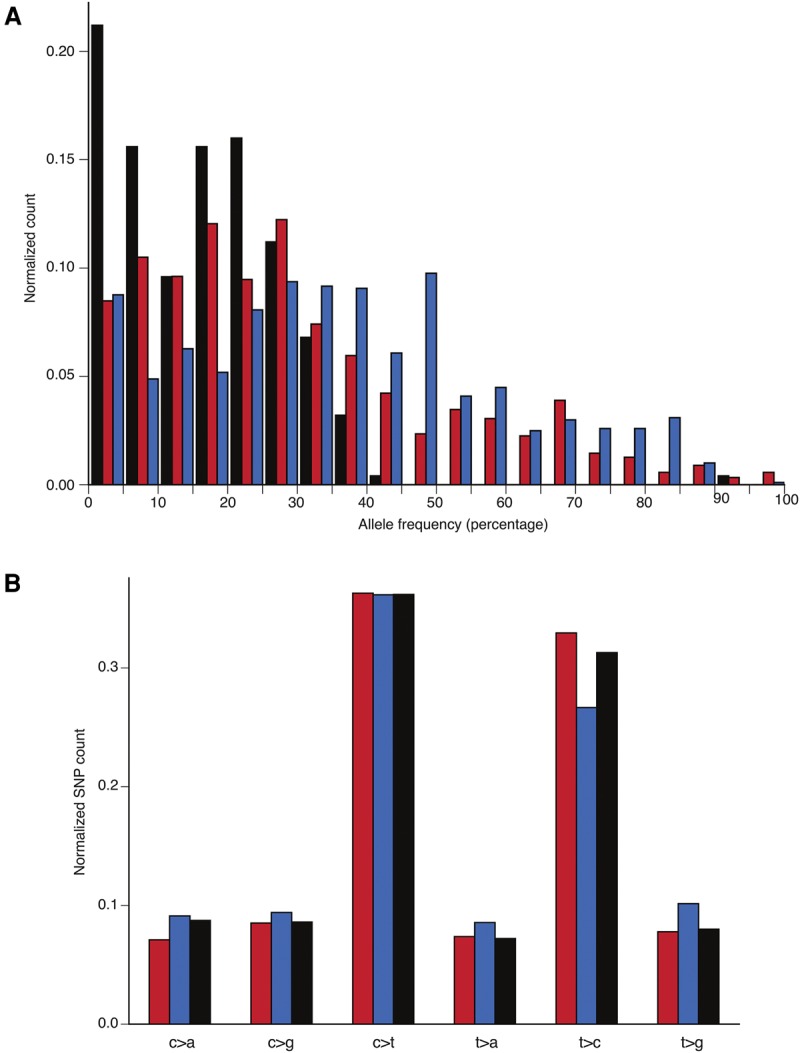
Histograms of normalized allele frequency distributions and normalized mutational profile for NA12878. (A) AF distribution for in-phase TruSeq SNPs (red) is between the distribution for deletions (black) and the distribution for out-of-phase TruSeq SNPs (blue), suggesting that it is a superposition of the two. This pattern is consistent with the hypothesis of SNPs being generated simultaneously with the deletions. (B) Mutational profile for in-phase (red) and out-of-phase (blue) TruSeq SNPs. The six possible transversions and transitions are shown, with the normalized count on the vertical axis. The third bar (black) represents personal genome-wide profile based on heterozygous SNPs.
These distributions for the set of 94 NH deletions (that are likely generated by replication-based mechanisms) and the remaining 168 deletions revealed the same trend (Supplemental Fig. S3). This is perhaps not surprising, given our previous observation of a higher in-phase versus out-of-phase SNP density for each of the sets and the corresponding discussion that the latter set may also contain deletions generated by replication-based mechanisms.
Mutational profile of SNPs
We further explored the mutational profile of TruSeq-derived in-phase and out-of-phase SNPs for NA12878 (Fig. 3B). In-phase SNPs had a higher frequency of transitions, with a Ti/Tv ratio of 2.25, than out-of-phase SNPs, with a Ti/Tv ratio of 1.69 (P-value = 2.5 × 10−4, by the two sample proportion test). The same analysis with the larger set of trio-phased SNPs revealed no differences in the ratios, with these being close to 2.08, which is the value obtained from personal heterozygous SNPs across the entire genome (Supplemental Table S5; Supplemental Fig. S4). Since we know that there is a discordance between TruSeq and trio analyses, we recalculated the preceding ratios, with the phase for 108 discordant SNPs assigned from trio analysis, and obtained statistically marginal significance (P-value = 0.05, by the two sample proportion test) for the difference in values of the ratio: 2.14 and 1.83 for in-phase and out-of-phase SNPs, respectively. Based on these results, we conclude that we do not see any special signature in the mutational profile of in-phase SNPs.
Discussion
In this study, we described an analysis of the density of SNPs/indels around deletion breakpoints in a germline lineage. Selection of heterozygous deletions and reconstruction of local haplotypes allowed us to compare the density, AF distribution, and mutational profile of SNPs that are in phase and out of phase with deletions. Because we are comparing haplotypes with exactly the same sequence content, other factors besides the presence/absence of the deletion are likely to have the same effect on the density and frequency of variants on both haplotypes and are unlikely to introduce any bias in our analysis. Therefore, the observed differences are (1) a higher density (at least, twofold increase in the case of SNPs) of in-phase variants; and (2) a shift in AF spectrum of in-phase variants toward the corresponding spectrum for deletions ought to be attributed to the presence of these deletions and provide evidence for the co-occurrence of SNPs/indels with structural variations, presumably during error-prone replication.
One can suggest an alternative hypothesis, that the introduction of a deletion into a haplotype increases its mutagenesis, resulting in a higher variant density and an increase in the fraction of rare variants (causing a shift in AF distribution of the variants toward lower values). From our analysis of SNPs around the subset of NH deletions carrying signatures of replication-based mechanisms of origin, we found an even higher (almost threefold) increase in the count of in-phase versus out-of-phase SNPs. Although this provides evidence against this possibility of higher mutagenesis, we cannot completely rule out this alternative hypothesis because the complementary set of deletions (those with none or insufficient evidence of replication-based origin) had a relatively weaker, but significant difference (∼70%–90%) in the counts. However, this difference can also be accounted for by the possibility that some of the deletions in the complementary set have a replication-based mechanism of origin, although not categorized as such, because we used fairly stringent criteria for this classification. The similarity in the shifts in AF spectrum of in-phase variants toward the corresponding spectrum for NH deletions (carrying signatures of replication-based mechanisms), and for the complementary set of deletions, could also be accounted for by the same reason. In order to resolve between the two hypotheses, we suggest that further analysis with a larger high-quality breakpoint set will likely be necessary because it will allow for finer stratification of variant densities by likely mutational mechanism of origin.
Also, previous studies (Carvalho et al. 2013; Wang et al. 2015) analyzed variants in a flanking distance of ∼40–50 bp from the SV breakpoints. We examined a 10-kbp flanking region using TruSeq long-reads and a 100-kbp flanking region using trio phasing. Although the numerical values of the densities of the in-phase, out-of-phase SNPs/indels, and the corresponding ratios were different, qualitatively, the results obtained (i.e., significantly higher density of in-phase SNPs/indels) from the two phasing approaches were in agreement. Quantitative differences can be explained by a few factors: (1) utilization of a different set of deletions (not all deletions phased by TruSeq can be phased by trio); (2) discordance in phasing of some SNPs; (3) bias in trio analysis; and (4) accessibility of uniformly longer flanks with trio phasing. The bias is due to LD between SNPs and the deletions, which enhances the ratio between in-phase and out-of-phase SNPs. We, however, demonstrated that the bias is small, and subsequently with trio-based phasing, observed a strong persistence of signal in the 50-kbp window flanking the deletions’ breakpoints, with the in-phase and out-of-phase densities merging further away around 100 kbp.
Additionally, trio analysis also allowed us to examine more than one subject, and we observed a qualitatively similar increase in the density of in-phase SNPs/indels compared with the out-of-phase SNPs/indels in both NA12878 (Caucasian) and NA19240 (Yoruba). Although with TruSeq phasing we did observe differences in the mutation profile and Ti/Tv ratio of in-phase compared to out-of-phase SNPs, these were not confirmed by trio analysis.
In a broader context, the observed dramatic increase of variant density on haplotypes with deletion may be a reason that, at least partially, can explain differences in mutation clock (Callaway 2015) and could underlie some clustered and phased mutations observed in cancer (Alexandrov et al. 2013).
Methods
Detailed instructions (README file) and Python scripts (to reproduce our analysis as described below) are provided in the Supplemental Scripts File.
Initial selection of Illumina TruSeq synthetic long-reads
From the 1000 Genomes Project Consortium, a confident set of precise deletion breakpoints is available for the subject NA12878 (ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/phase3/integrated_sv_map/supporting). There are a total of 376 available loci. Aligned Illumina TruSeq synthetic long-reads (McCoy et al. 2014) in BAM format are also available for this subject. We extracted reads in regions of interest by using SAMtools (Li et al. 2009). A region of interest is the chromosomal coordinate; and coordinate intervals [L − 10, L + 10], [R − 10, R + 10], where L and R refer to the left and right breakpoint coordinates in bp units. The chromosomal coordinate, L, and R are obtained from the breakpoints’ confident set. Using “SAMtools view” command, we extract several unique reads that overlap a region of interest. Of these, we only selected reads, longer than 2 kbp, for further analysis.
AGE alignment and further selection of long-reads
The above selected reads were realigned to the reference genome around the deletion breakpoints, using AGE (Abyzov and Gerstein 2011). The AGE options were “-coor1 = (L − 10000) − (R + 10000) -indel –go = −10 –mismatch = −10,” which specify alignment to the specified deletion, with breakpoints extended by 10 kbp downstream and upstream; indels are expected in the read sequence; gap open penalty is −10; and mismatch penalty is −10. For each of the 376 loci (deletions), we obtained the AGE output for the aligned reads in a text file, along with AGE-determined deletion breakpoint coordinates. A Python script, written by us, selected reads that aligned exactly with the specified deletion breakpoint coordinates. Of these, one read with the most balanced left and right flank lengths was output to another text file by our script. We were able to select 316 of the original 376 loci.
Determining heterozygous deletions using CNVnator
We genotyped deletions in the NA12878 and corresponding parents using 2 × 250 bp read Illumina data with approximately 60× coverage (ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/phase3/data/NA12878). The confident set of deletion breakpoint coordinates were passed to CNVnator (Abyzov et al. 2011), which was invoked with the “-genotype” option. Since we have a trio, we were able to check for the consistency of the heterozygosity of the deletion event, as determined by CNVnator, which returned the normalized read depth signal for each member of the trio.
The condition we used is: (0.5 ≤ rdSNA12878 ≤ 1.5) and (0.5 ≤ rdSNA12891 or 0.5 ≤ rdSNA12892) and (rdSNA12891 ≤ 1.5 or rdSNA12892 ≤ 1.5).
Applying this condition, we obtained 262 heterozygous loci of the 316 selected.
Counting heterozygous SNPs/indels
Our script wrote out the TruSeq long-reads SNPs/indels, flanking the 262 heterozygous deletions, into separate VCF files. For each selected read, we also kept track of the coordinates of the ends of the left and the right flanks of the deletion. Of course, the flanks are not of equal length.
To obtain a confident set of SNPs/indels, we carried out an intersection with the GATK confident set of variants for the trio (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/working/20131015_p3_high_cov_calls/HaplotypeCaller/CEU.wgs.Haplotype Caller.20131118.snps_indels.high_coverage_pcr_free.genotypes.vcf.gz). We used the vcf-isec command of the VCFtools (Danecek et al. 2011). The options were “-f –n = 2,” which specify that differences in sample numbers, etc., should be ignored; and positions that are present in exactly two files should be output. The two files we intersected were the SNPs/indels VCF and GATK VCF file.
To obtain the SNPs/indels on the haplotype without the deletion event, we subtracted the TruSeq SNPs/indels from the GATK confident set. We used vcf-isec ‘-f –c –r file,’ on the GATK and SNPs/indels VCF files. ‘–c’ implies taking the complement; and ‘–r file’ provides the chromosomal regions to which the complement should be limited. As mentioned before, we recorded the coordinates for each flank around the deletion: for the left flank, [start, L]; and for the right, [R, end], where L and R are the left and right breakpoint coordinates.
From the intersection and complement VCF files, we counted the heterozygous variants. For SNPs, we used the GATK genotype “0/1”; For indels, we use the GATK genotype, 0/x, where x can be 1, 2, … . This way we only counted the heterozygous SNPs/indels, for both haplotypes.
Density of heterozygous SNPs with TruSeq analysis
To obtain the density of heterozygous SNPs, we used a bin of size 250 and added up the SNPs in these bins. As mentioned above, not all of the 262 loci that contain the deletion events are of equal length. For each bin, we kept track of the number of flank fragments that contribute to it. To obtain the SNP density, we divided the total number of SNPs in a bin by the sum of the flank count in that bin. By normalizing this way, we were able to account for the differences in flank lengths.
Three-way consensus analysis
For the haplotype with the deletion event, we used, “vcf-isec –f –n = 4 –r file,” where “-r file” is as explained above; “-n = 4” specifies intersection of positions that are present in exactly four files, these being the GATK VCF (see above for download information), Complete Genomics VCF (ftp://ftp2.completegenomics.com/vcf_files/Build37_2.0.0/vcfBeta-NA12878-200-37-ASM.vcf.bz2), Illumina Platinum VCF (ftp://platgene_ro@ussd-ftp.illumina.com/hg19/older_releases/IlluminaPlatinumGenomes_v7.0/merged_ platinum/NA12878.vcf.gz), with these three providing the three-way consensus VCF file; and the Illumina TruSeq VCF file obtained by our script above.
Our three-way consensus Python script checked for agreement in chromosomal positions, the reference, and alternate bases among the files. We checked the heterozygosity of the variants using both the GATK and Platinum genotypes.
For the haplotype without the deletion event, we used, “vcf-isec –f –n = 3 –r file.” Only the “-n = 3” option needs to be explained. Here, the intersection is between exactly three files, these being the Illumina TruSeq complement VCF (obtained above), the Complete Genomics VCF, and the Illumina Platinum VCF. Note that (by simple set theory) this is the same as subtracting the Illumina TruSeq variants from the three-way consensus VCF. Once again, we checked for agreement as mentioned above.
Trio phasing
To phase the variants we used the genotype information available for the trio in the GATK call set mentioned above in the methods for counting of SNPs/indels. For NA12878, we used CEU.wgs.HaplotypeCaller.20131118.snps_indels.high_coverage_pcr_free.genotypes.vcf.gz and for NA19240, YRI.wgs.HaplotypeCaller.20131118.snps_indels.high_coverage_pcr_free.genotypes.vcf.gz. We only phased heterozygous variants for our analysis that had consistent genotypes in the trio.
To phase the deletion events, confident sets of deletion breakpoints (for NA12878 and NA19240) were passed to CNVnator invoked with the “-genotype” option. Following a logic very similar to that used to determine the heterozygosity of the deletions (methods above for determining heterozygous deletions using CNVnator), we determined the genotype of a deletion for the trio. The read depth satisfying the condition 0.5 ≤ rdSNA12878 ≤ 1.5 gives a heterozygous deletion event. Then, as an example, a condition of the form 1.5 < rdSNA12891 helps determine the paternal genotype as 0/0. If the maternal read depth satisfies rdSNA12892 ≤ 1.5, then we have the corresponding genotype as 0/1 or 1/1. Accordingly, for this example, we obtain the phased genotype for the deletion event as 0|1. Once again, inconsistent genotypes were discarded, and deletions with genotype 0/1, 0/1, 0/1 could not be phased.
Allele frequency (AF) analysis
For this analysis, we created two VCF files that contain the heterozygous SNPs for the two haplotypes. We used, “vcf-sec –f –n = 2 –r file” with the intersection being carried out between the VCF file containing the aggregated AF information for all the 2353 1000 Genomes Project samples and the VCF file produced by our analysis.
For the deletion events, we obtained the AF information for the 262 heterozygous loci (identified above) from the VCF file containing the merged calls for SVs for the 1000 Genomes Project samples.
We then bin (bin size of 5%) the counts, normalizing by the total count for each category (deletion; in-phase, out-of-phase SNPs) to make the histogram (Fig. 3).
Acknowledgments
We thank Pallavi Chhabra for her immense help with artwork for the figures and Aditya Bhagwate and Taejeong Bae for helpful discussions about various tools. We acknowledge support from the Center for Individualized Medicine at Mayo Clinic.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.205484.116.
References
- The 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abyzov A, Gerstein M. 2011. AGE: defining breakpoints of genomic structural variants at single-nucleotide resolution, through optimal alignments with gap excision. Bioinformatics 27: 595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abyzov A, Urban AE, Snyder M, Gerstein M. 2011. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 21: 974–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abyzov A, Li S, Kim DR, Mohiyuddin M, Stütz AM, Parrish NF, Mu XJ, Clark W, Chen K, Hurles M, et al. 2015. Analysis of deletion breakpoints from 1,092 humans reveals details of mutation mechanisms. Nat Commun 6: 7256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. 2013. Signatures of mutational processes in human cancer. Nature 500: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlt MF, Rajendran S, Birkeland SR, Wilson TE, Glover TW. 2012. De novo CNV formation in mouse embryonic stem cells occurs in the absence of Xrcc4-dependent nonhomologous end joining. PLoS Genet 8: e1002981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway E. 2015. DNA clock proves tough to set. Nature 519: 139–140. [DOI] [PubMed] [Google Scholar]
- Carvalho CM, Pehlivan D, Ramocki MB, Fang P, Alleva B, Franco LM, Belmont JW, Hastings PJ, Lupski JR. 2013. Replicative mechanisms for CNV formation are error prone. Nat Genet 45: 1319–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Bird C, Blackburne B, Lindsay S, Mamanova L, Lee C, Turner DJ, Hurles ME. 2010. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat Genet 42: 385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. 2011. The variant call format and VCFtools. Bioinformatics 27: 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deem A, Keszthelyi A, Blackgrove T, Vayl A, Coffey B, Mathur R, Chabes A, Malkova A. 2011. Break-induced replication is highly inaccurate. PLoS Biol 9: e1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, et al. 2010. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327: 78–81. [DOI] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR. 2009a. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 5: e1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Lupski JR, Rosenberg SM, Ira G. 2009b. Mechanisms of change in gene copy number. Nat Rev Genet 10: 551–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Graves T, Newman TL, Fulton R, Hayden HS, Malig M, Kallicki J, Kaul R, Wilson RK, Eichler EE. 2010. A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell 143: 837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam HY, Mu XJ, Stütz AM, Tanzer A, Cayting PD, Snyder M, Kim PM, Korbel JO, Gerstein MB. 2010. Nucleotide-resolution analysis of structural variants using BreakSeq and a breakpoint library. Nat Biotechnol 28: 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR. 2007. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131: 1235–1247. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Lacaria M, Zhang F, Withers M, Hastings PJ, Lupski JR. 2011. Frequency of nonallelic homologous recombination is correlated with length of homology: evidence that ectopic synapsis precedes ectopic crossing-over. Am J Hum Genet 89: 580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR. 2009. Genomic disorders ten years on. Genome Med 1: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, Stankiewicz P. 2005. Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet 1: 0627–0633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy RC, Taylor RW, Blauwkamp TA, Kelley JL, Kertesz M, Pushkarev D, Petrov DA, Fiston-Lavier AS. 2014. Illumina TruSeq synthetic long-reads empower de novo assembly and resolve complex, highly-repetitive transposable elements. PLoS One 9: e106689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVey M, Lee SE. 2008. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet 24: 529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, et al. 2011. Mapping copy number variation by population-scale genome sequencing. Nature 470: 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathans J, Piantanida TP, Eddy RL, Shows TB, Hogness DS. 1986. Molecular genetics of inherited variation in human color vision. Science 232: 203–210. [DOI] [PubMed] [Google Scholar]
- Pang AW, Migita O, Macdonald JR, Feuk L, Scherer SW. 2013. Mechanisms of formation of structural variation in a fully sequenced human genome. Hum Mutat 34: 345–354. [DOI] [PubMed] [Google Scholar]
- Slack A, Thornton PC, Magner DB, Rosenberg SM, Hastings PJ. 2006. On the mechanism of gene amplification induced under stress in Escherichia coli. PLoS Genet 2: 385–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. 2002. Genome architecture, rearrangements and genomic disorders. Trends Genet 18: 74–82. [DOI] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Hsi-Yang Fritz M, et al. 2015. An integrated map of structural variation in 2,504 human genomes. Nature 526: 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Su P, Hu B, Zhu W, Li Q, Yuan P, Li J, Guan X, Li F, Jing X, et al. 2015. Characterization of 26 deletion CNVs reveals the frequent occurrence of micro-mutations within the breakpoint-flanking regions and frequent repair of double-strand breaks by templated insertions derived from remote genomic regions. Hum Genet 134: 589–603. [DOI] [PubMed] [Google Scholar]
- Zhang F, Carvalho CM, Lupski JR. 2009a. Complex human chromosomal and genomic rearrangements. Trends Genet 25: 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Gu W, Hurles ME, Lupski JR. 2009b. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet 10: 451–481. [DOI] [PMC free article] [PubMed] [Google Scholar]