

Abstract
Background
Chronic inflammation has been recognized as one of the hallmarks of cancer. We recently showed that parainflammation, a unique variant of inflammation between homeostasis and chronic inflammation, strongly promotes mouse gut tumorigenesis upon p53 loss. Here we explore the prevalence of parainflammation in human cancer and determine its relationship to certain molecular and clinical parameters affecting treatment and prognosis.
Results
We generated a transcriptome signature to identify parainflammation in many primary human tumors and carcinoma cell lines as distinct from their normal tissue counterparts and the tumor microenvironment and show that parainflammation-positive tumors are enriched for p53 mutations and associated with poor prognosis. Non-steroidal anti-inflammatory drug (NSAID) treatment suppresses parainflammation in both murine and human cancers, possibly explaining a protective effect of NSAIDs against cancer.
Conclusions
We conclude that parainflammation, a low-grade form of inflammation, is widely prevalent in human cancer, particularly in cancer types commonly harboring p53 mutations. Our data suggest that parainflammation may be a driver for p53 mutagenesis and a guide for cancer prevention by NSAID treatment.
Electronic supplementary material
The online version of this article (doi:10.1186/s13059-016-0995-z) contains supplementary material, which is available to authorized users.
Keywords: Inflammation, Parainflammation, p53 mutations, NSAID treatment, Cancer prevention, Genomics
Background
Inflammation is one of the enabling hallmarks of cancer [1] and it has been estimated that approximately 20 % of cancers are caused by chronic inflammation [2, 3]. Tumor-promoting inflammation can contribute to various stages of tumor development, from tumor initiation to metastasis [4]. We recently developed and characterized a mouse model of intestinal cancer based on tissue-specific ablation of CKIα [5, 6]. Inducible ablation of CKIα in the gut epithelium has several immediate consequences, Wnt activation due to stabilization of β-catenin, induction of DNA damage response with robust p53 activation, and elicitation of a low-grade inflammatory response in the epithelium. This inflammatory reaction, which is detected by mRNA and protein analysis of the gut epithelial cells, is an atypical one, having a relatively low secretory component and, conspicuously, is not accompanied by a typical inflammatory cell infiltrate in CKIα-deficient mucosa. We coined this low-grade inflammatory reaction “parainflammation” (PI) based on its relationship to a term defined by Medzhitov: a low-grade inflammatory response at an intermediate state between tissue homeostasis and classic inflammation which can be induced by persistent tissue stress, serving to restore tissue homeostasis [7]. It has been proposed that PI could play a role in several conditions, such as aging and obesity [8]. In contrast to “classic” inflammation, often ignited by an extrinsic assault such as bacterial infection, PI may erupt due to tissue-intrinsic assaults, such as DNA damage [6, 7], and cooperate with the tumor suppressor p53, contributing to tissue protective senescence and counteracting tumor progression. Upon p53 ablation, however, PI loses its beneficial role and, instead, contributes to carcinogenesis [6]. Here we constructed a PI gene signature based on analysis of the CKIα and CKIα-p53-deficient gut epithelium and, using existing databases, examined this signature and its implications in thousands of human tumors and cell lines. We noticed a striking occurrence of PI in several cancer types distinct from their normal tissue counterparts. Our study indicates that PI is an important factor in tumor development, with a significant influence on prognosis. Notably, we found that PI can be markedly attenuated in human samples by non-steroidal anti-inflammatory drug (NSAID) treatment, possibly alluding to a mechanism of cancer prevention.
Results
Deriving the PI gene signature from CKIα-deficient mice
Our strategy to derive a gene expression signature for PI consisted of two steps. First, we compiled a list of 840 inflammatory response genes. This list is based on three manually curated databases: the Ingenuity inflammatory response gene list (http://www.qiagen.com/ingenuity), InnateDB innate immunity genes [9], and the Immunogenetic Related Information Source (IRIS) [10] (Fig. 1a; Additional file 1). We then intersected the 840 genes with a list of genes found to be significantly upregulated in RNA-seq expression profiles of two mouse models featuring gut PI: CKIα-deficient and CKIα-p53-deficient gut epithelium (Additional file 2) [6]. This procedure generated a list of 40 inflammatory context genes which are upregulated in the PI mouse models, possibly representing a gain-of-function mechanism (Fig. 1b; Additional file 3: Table S1). Using Ingenuity’s upstream regulator analysis [11], we revealed a strong resemblance of the PI signature to the lipopolysaccharide (LPS) response (Fig. 1c; Additional file 4). This response is mediated via two major arms, the NF-κB and the interferon (IFN) signaling pathways [12], only the latter of which is found to be strongly associated with PI. The Ingenuity analysis also confirmed our observation of upregulation of the genes: 37 of the genes are known to be upregulated during the LPS/IFN response (based on manual curation of the literature). Nevertheless, no common regulator could be found for all PI genes, suggesting that PI is a unique inflammatory response whose origin and mechanism have yet to be described. It should be pointed out that while this signature includes innate immunity genes which are commonly expressed by leukocytes, in the mouse models these genes are explicitly expressed by epithelial cells [6]. Notably, the signature has a remarkable paucity of chemokines or other secreted factors, suggesting that PI is primarily cell-autonomous in the epithelial tissue and explaining why it doesn’t tend to initiate recruitment of immune cells.
Fig. 1.
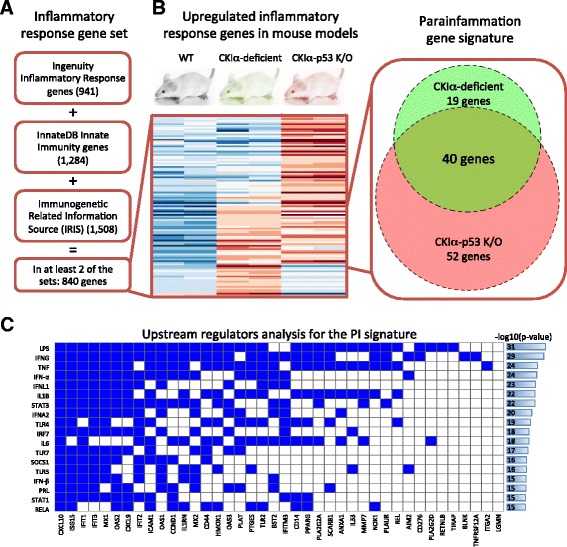
The parainflammation signature. a An inflammatory response gene set was generated by combining genes that are found in at least two of three manually curated databases: the Ingenuity inflammatory response gene list, InnateDB innate immunity genes, and the Immunogenetic Related Information Source (IRIS). b Differential expression analysis of CKIα-deficient and CKIα-p53-deficient mice against wild type (WT) revealed 59 and 92 upregulated inflammatory response genes in the single and double knock outs (K/O), respectively, and 40 genes upregulated in both. The 40 genes were designated as the parainflammation (PI) gene signature. The heatmap shows the expression levels of the 111 upregulated genes (red, high expression; blue, low expression). c Upstream regulatory analysis revealed a strong enrichment for genes regulated by lipopolysaccharide and immune pathways that are activated by it. The rows show the top enriched, non-chemical regulators, the columns are the 40 PI genes, and a blue cell represents regulation
PI is suppressed by NSAID treatment in mouse tumors
We next sought to validate the 40-gene signature for PI in an established model system. PI is associated with cellular senescence and is apparent in a pure primary epithelial tissue, such as gut epithelial organoids [6, 13]. APC-mutated human and mouse polyps represent early neoplastic lesions which mostly do not progress, possibly due to senescence-associated PI [14], thus representing a good source for testing the PI signature we characterized. We prepared organoid cultures from APC-mutated normal gut epithelium (MIN) and adenomatous polyps of APCmin/+ mice (adenoma) and analyzed them via RNA-seq (Additional file 5). Our analysis confirmed the overexpression of many of the PI genes in adenomatous polyps in comparison with organoids prepared from the adjacent normal tissue of APCmin/+ mouse tumors (MIN): 17 (42.5 %) of the PI genes showed significant upregulation in the tumor-derived organoids (false discovery rate < 1 %) compared with 17.5 % of all genes and 17.0 % of all inflammatory response genes (chi-square test p value = 4.9e-5) (Fig. 2a). Only two genes (5.0 %) showed significant, yet modest, downregulation. Quantitative PCR (qPCR) analysis confirmed these findings (Additional file 3: Figure S1).
Fig. 2.
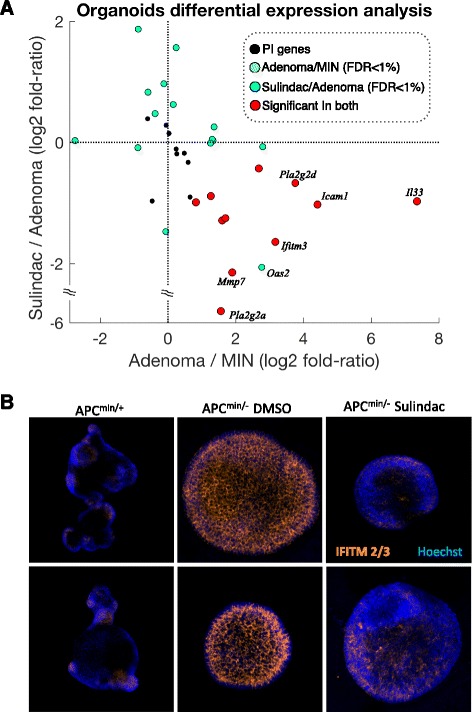
Parainflammation decreases in response to NSAID treatment in mouse organoids. a The adenoma/MIN log2 expression fold ratio of PI genes (x-axis) against the sulindac-treated adenomas/adenoma log2 expression fold ratio of PI genes (y-axis). Colors represent significance in the differential expression analyses (false discovery rate (FDR) <1 %). Along the x-axis it can be observed that 17 of 19 significant genes in adenoma/MIN are upregulated in the adenomas. Of those, 11 are significantly downregulated in the sulindac-treated samples and none are significantly upregulated. b IFITM2/3 immunofluorescence in APCmin/+, adenoma and Sulindac-treated adenoma organoids. Both rows show different fields of same experiment.
It is not yet known why NSAID treatment has an impact on cancer prevalence, progression, and mortality in human solid tumors [15–18]. We have previously shown that gut epithelium PI can be suppressed by NSAID treatment in the CKIα-deficient mouse model [6]. We therefore hypothesized that cancer cells may harbor tumor-promoting PI subject to suppression by NSAID treatment. To test this hypothesis, we generated a tumor model for examining NSAID treatment effects. We prepared organoids from normal gut and adenomatous polyps of APCmin/+ mice, treated them with the NSAID sulindac, and performed RNA-seq on three biological replicates (Additional file 6). Strikingly, of the 17 upregulated PI genes observed in the tumor organoids, 11 (64.7 %) were significantly downregulated in the sulindac-treated samples, compared with 33.2 % of all genes and 38.3 % of all inflammatory response genes (p value = 0.0053) (Fig. 2a). Immunofluorescence staining shows that the protein expression of PI gene Ifitm2/3 (Fig. 2b) is markedly elevated in the tumor organoids and is mostly suppressed by sulindac treatment. These data validate the PI signature in a new mouse tumor model, underscoring both the epithelial origin and the tumor specificity of this new signature, and show that NSAID treatment can reduce PI.
Analogously to the procedure described above, we also found 75 inflammatory context genes which are downregulated in the PI mouse models (Additional file 3: Table S2). This list of genes is again highly enriched with LPS and IFN γ response genes but also contains interleukin (IL)2/IL4 response genes. However, we didn’t observe expression changes of those 75 genes in the organoids model, suggesting that the downregulated genes are not part of the general PI mechanism. Therefore, while it is well established that activation of innate inflammation pathways leads to both up- and downregulation of genes involved in activation and inhibition of this highly regulated network [19–21], we chose to derive the PI signature only from the upregulated genes in the mouse and organoid PI models.
Human cancers display the PI signature discovered in mice
Observing PI in mouse tumors prompted us to look for a similar phenomenon in human cancers. To that end, we first analyzed the expression of 40 human homologs of the mouse PI genes in human cancers, utilizing data from the Cancer Cell Line Encyclopedia (CCLE) [22]. Whereas immune and inflammatory genes are normally expressed in hematopoietic cells, a wide range of carcinoma cell lines (n = 634) expressed the PI genes to a higher level than hematopoietic and lymphoid cancer cell lines (n = 180) (Fig. 3a; Additional file 3: Figure S2). Further investigation revealed that, compared with non-PI genes, many PI genes are broadly overexpressed in a subset of the cell lines. To quantify the distribution of PI genes in cell lines, we computed the expression distribution in all the carcinoma cell lines and identified the peak of the distribution. Then, by defining overexpression as twofold expression level over the peak (1 in log2 expression levels), we counted the number of cell lines overexpressing each gene (examples in Fig. 3b). Our analysis revealed that 74.4 % of the PI genes are overexpressed in more than 10 % of the carcinomas, significantly more than a non-discriminatory pool of random inflammatory response genes (40.5 %, chi-square test p value = 2.9e-5) and all genes (29.6 %, p value = 1.0e-9) (Fig. 3b). The median number of overexpressing cell lines was 125 (19.7 % of carcinoma samples) for the PI genes compared with 39 (6.2 %, U-test p value = 2.7e-5) across random inflammatory response genes and only 31 (4.9 %, p value = 2.6e-8) across random genes.
Fig. 3.
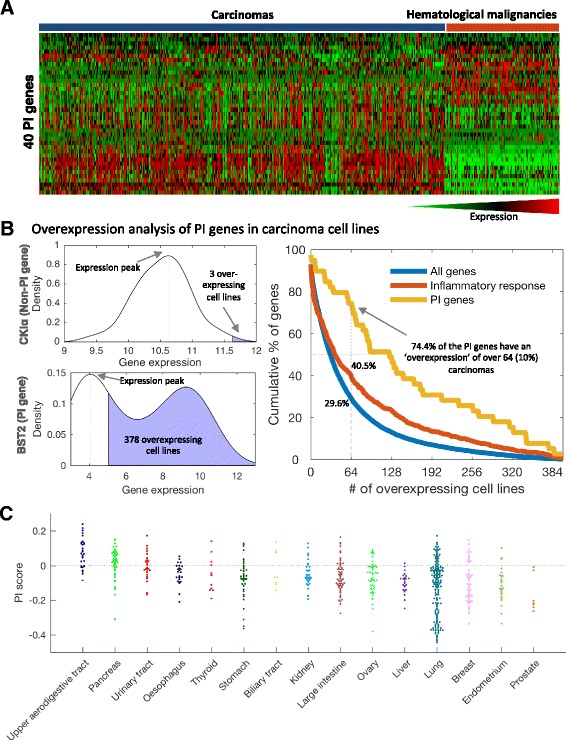
Parainflammation genes are overexpressed in carcinoma cell lines. a Heatmap of the expression of 39 PI genes in 634 carcinoma cell lines and 180 hematopoietic and lymphoid cancer cell lines from CCLE. One PI gene, IFITM3, is not represented in CCLE. Of the PI genes, 19 show significantly higher expression in carcinomas compared with cancers originated from immune cell types; 10 PI genes are more abundant in immune cancers. b Left: the distribution of expression of two representative genes across 634 carcinoma cell lines from CCLE. We detected the expression peak and counted the number of samples that express the gene twofold (1 in log2 scale) more than the peak. The top example, the gene CSNK1A1 (CKIα), which here represents a housekeeping gene, is a typical normally distributed gene with low “overexpression” rate and the bottom example, BST2, which is part of the PI signature, shows a gene with a bimodal expression pattern, corresponding to a high “overexpression” rate. Right: the cumulative overexpression rates for the PI genes, all inflammatory response genes, and all genes. Of the PI signature genes, 29 (74.4 %) are overexpressed in at least 10 % of the carcinoma cell lines (≥64 samples) compared with 29.6 % and 40.5 % of all and inflammatory response genes, respectively. The yellow curve (PI genes) shows remarkably higher levels of overexpression along the whole graph. c Spread plot of tThe PI score in 634 carcinoma cell lines grouped by tissue types. The dashed blue line differentiates PI+ and PI− samples as defined by the proportion of PI+ of tumor samples
We next computed a score for each cell line by performing single-sample gene set enrichment analysis (ssGSEA) [23] for the PI gene signature. We defined this score, which is an enrichment measure of the overexpression of the PI genes, as the PI score (Additional file 7). The PI score revealed major differences in PI both between cancer types and within cancer types, varying from high levels of PI in head and neck and pancreatic cancer to low levels in samples originating from prostate and liver (Fig. 3c). Thus, the PI expression and PI score patterns suggest that PI occurs across most cancer types, although not uniformly. It is important to note that while the PI score is based on a signature of 40 genes, the PI phenomenon is not restricted to these genes alone but has a wide effect on numerous genes over many different molecular functions (Additional file 8). Finally, we observed a high correlation between the PI score and scores derived from the downregulated PI genes (Spearman R = 0.654, p value <1e-20; Additional file 3: Figure S3). This once again confirms the validity of our gene sets in human and the relevance of the PI mouse models to a phenomenon that is also observed in human.
Next, we explored the representation of the PI signature in primary human cancers. As differences in PI expression between individual samples may be explained solely by differences in purity levels of the samples [24], we designed a simple adjustment procedure for removing inflammatory gene expression originating from immune infiltrations (Additional file 3: Figure S4). This adjustment procedure consists of two steps: first, utilizing expression data of normal tissues from the Genotype-Tissue Expression (GTEx) project [25], we learned the normal association of gene expression with immune infiltrations in each tissue type. We then used the expression level of PTPRC (CD45), a pan-hematopoietic exclusive marker expressed in all leukocyte cell types but not in other tissues, as an estimate for immune infiltrations. Finally, we applied the gene-specific, tissue-specific learned slopes to adjust the expression levels of The Cancer Genome Atlas (TCGA) tumor samples. This procedure diminishes expression differences among samples which are most likely explained by differences in purity.
We collected gene expression data for 6523 primary tumors and 582 patient-matched normal samples of 18 cancer types from TCGA (Additional file 3: Table S3; http://cancergenome.nih.gov) and performed the adjustment procedure. As in the cancer cell lines, we computed the scores for the PI gene signatures using the adjusted expression of each tumor sample (Additional file 9). Cancer samples demonstrated a wide range of PI scores regardless of CD45 expression (Fig. 4a; Additional file 3: Figure S5). Furthermore, malignant tumors demonstrated significantly higher levels than adjacent normal samples, both overall and across most tested cancer types (Fig. 4b; Additional file 3: Figure S6). As in cell lines, the PI score analyses revealed considerable differences between cancer types and within cancer types (Fig. 4c). We next defined the PI threshold as the score that appears in only 5 % of the adjacent normal samples; cancer samples over the threshold were designated as PI positive (PI+). Strikingly, over all cancer types 25.9 % of the tumor samples were PI+, compared with a null expectation of 5 %, with varying proportions among cancer types, from 77.7 % in pancreatic adenocarcinoma (PAAD) to none in kidney renal clear cell carcinoma (KIRC). The PI score of a sample is highly correlated with the number of upregulated genes in the sample (R = 0.649; Additional file 3: Figure S7). Accordingly, the median number of PI genes activated in PI+ samples is 17 (42.5 %; compared with eight in PI− samples), the same number we observed to be upregulated in the adenoma organoids. Interestingly, we observed that different sets of PI genes are activated in different cancer types (Fig. 4d). Finally, using the 25.9 % proportion of PI+ samples, we determined a threshold of PI+ samples in the cancer cell lines dataset (where there are no normal samples to determine the threshold). Remarkably, we found high concordance in the abundance of PI+ samples in cancer types between tumors and cell lines (Pearson coefficient R = 0.875, p value = 1.9e-4 (Fig. 4e). These results again suggest that the PI gene signature is being expressed by the cancer cells, distinct from immune infiltration.
Fig. 4.
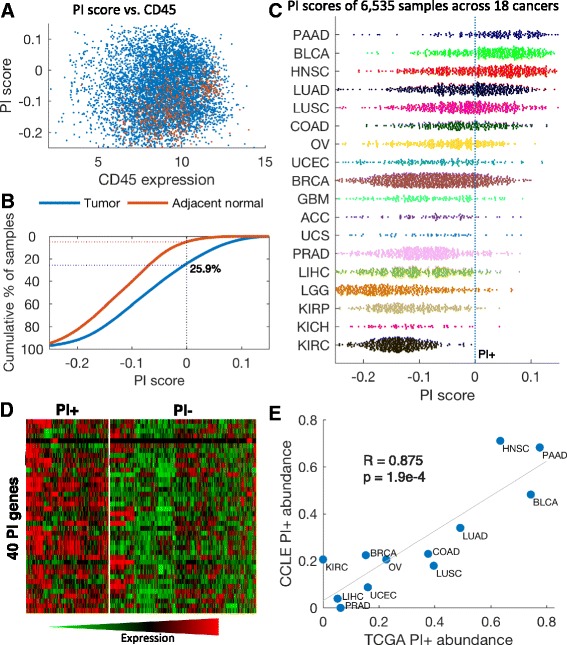
Pan-cancer parainflammation in human cancers. a PI scores (y-axis) of all 6535 tumor samples (blue) and 582 adjacent normal samples (red) versus the CD45 expression level (x-axis). No correlation is observed between the PI score and CD45 after the adjustment. b PI scores in the tumor samples (blue) and the adjacent normal samples (red). The y-axis is the cumulative percentage of samples over the score in x; 25.9 % of the tumor samples (dashed blue line) are over a threshold which only 5 % of the adjacent normal tissues pass (dashed red line). The PI score is shifted accordingly, so PI+ samples have positive scores. c Spread plot of the PI score in 6535 tumor samples from 18 cancer types. The dashed blue line differentiates PI+ and PI− samples. PAAD pancreatic adenocarcinoma, BLCA bladder carcinoma, HNSC head and neck squamous cell carcinoma, LUAD lung adenocarcinoma, LUSC lung squamous cell carcinoma, COAD colon adenocarcinoma, OV ovarian serous cystadenocarcinoma, UCEC uterine corpus endometrial carcinoma, BRCA breast carcinoma, GBM glioblastoma multiforme, ACC adrenocortical carcinoma, UCS uterine carcinosarcoma, PRAD prostate adenocarcinoma, LIHC liver hepatocellular carcinoma, LGG lower grade glioma, KIRP kidney renal papillary cell carcinoma, KICH kidney chromophobe, KIRC kidney renal clear cell carcinoma. d Heatmap of expression profiles of PI genes across TCGA samples: left, PI+ samples; right, PI− samples. Different subsets of PI genes are expressed in PI+ samples. The expression levels presented are after adjustment to immune infiltrations and are standardized across all samples. e Correlation of the fraction of PI+ samples in each tumor type from TCGA with corresponding tissue origin in CCLE. The same types of cancers have low or high PI+ levels in tumors and in cell lines. The Pearson coefficient is presented
We noted earlier that the PI gene signature is enriched in genes related to the IFN signaling pathway but not to the NF-κB pathway. To test this hypothesis further we first expanded the list of PI genes, identifying 215 genes with expression levels highly correlated with the PI score (R > 0.5) in the carcinoma cell lines (Additional file 8). We then performed transcription factor binding enrichment analysis using the ENCODE ChIP-Seq Significance Tool [26] (Additional file 3: Table S4). The analysis again affirmed our claim that PI has a distinct pattern of inflammation, where the top enriched transcription factors were STAT2, IRF1, STAT1 and STAT3, c-Fos, and PRDM1, but only modest enrichment for a key regulator of inflammation such as NF-κB. We further utilized enrichment scores of functional immune gene sets (Additional file 3: Table S5) and correlated them with the PI score in both tumors and cell lines. As expected, this analysis revealed high correlations in all datasets with the type I IFN response, which is a hallmark of PI, but a much weaker association with the tumor necrosis factor and NF-κB signaling pathways (Fig. 5a; Additional file 3: Table S6). We also did not detect any correlation with the immune cytolytic activity metric [27], which is a well-described anti-tumor immunity measure. It should be pointed out that the same correlations were also found in the CCLE dataset, again showing that PI is activated in the tumor and not in its microenvironment. These results again support our claim that PI is distinct from canonical chronic inflammation or other previously described inflammatory responses.
Fig. 5.
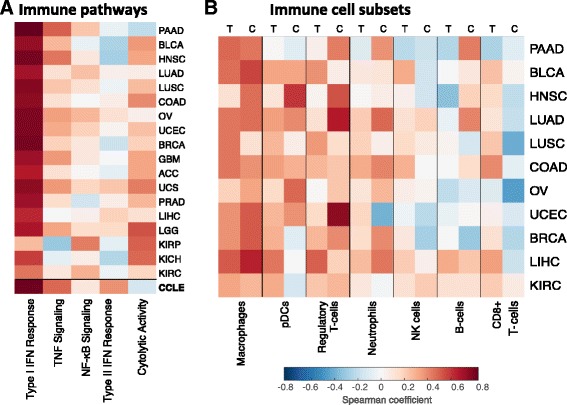
Cancer parainflammation resembles macrophage infiltration. a Heatmap of the Spearman correlations between the PI score and immune functional gene sets across different cancer types. Correlations in CCLE are shown as well (Additional file 3: Tables S5 and S6). b Heatmap of the Spearman correlations between the PI score and the immune subset enrichments calculated using gene sets (Additional files 6 and 9) across different cancer types derived from both TCGA (T) and CCLE (C). Similar correlation trends are observed for a cancer type whether the data were derived from TCGA or CCLE, suggesting that the correlation is due not to association of PI with immune subset presence but to shared functionality with the gene signatures. PAAD pancreatic adenocarcinoma, BLCA bladder carcinoma, HNSC head and neck squamous cell carcinoma, LUAD lung adenocarcinoma, LUSC lung squamous cell carcinoma, COAD colon adenocarcinoma, OV ovarian serous cystadenocarcinoma, UCEC uterine corpus endometrial carcinoma, BRCA breast carcinoma, GBM glioblastoma multiforme, ACC adrenocortical carcinoma, UCS uterine carcinosarcoma, PRAD prostate adenocarcinoma, LIHC liver hepatocellular carcinoma, LGG lower grade glioma, KIRP kidney renal papillary cell carcinoma, KICH kidney chromophobe, KIRC kidney renal clear cell carcinoma
Notably, certain PI genes are members of the Toll-like receptor (TLR) activation pathway (TLR2, CD14, and TIRAP) and, when upregulated, could have mediated an innate immune response to tissue-associated microbiota, igniting conventional inflammation with inflammatory infiltrate, secondary to PI. We therefore hypothesized that PI may enhance the recruitment of certain immune cell subsets to the tumors. To this end, we utilized hematoxylin and eosin (H&E) estimations provided by TCGA and gene signature enrichments of immune subset types from Rooney et al. [27] (Additional file 3: Table S5) and associated them with PI scores in tumor samples across cancer types. H&E estimations of major immune subtypes (lymphocytes, monocytes, and neutrophils) did not, however, show significant associations with PI scores across different cancer types (Additional file 3: Table S7). We further correlated PI scores of individual tumors with specific immune subsets based on gene signature enrichments in both TCGA and CCLE samples (Fig. 5b; Additional file 3: Table S8). Among the immune subsets the PI score demonstrated highest correlations across tumor types with the macrophage signature (average Spearman coefficient = 0.362). However, we observed the same trend of correlation between the PI score and macrophages in cell lines (average R = 0.407). This observation rules out the possibility that PI is dependent on macrophages infiltrating the tumor. Moreover, whereas the role of macrophages in orchestrating PI has been previously suggested [7], our finding supports this inference yet suggests that the tumors themselves may fulfill the macrophage inflammatory function in PI by expressing macrophage-relevant genes. Importantly, we did not observe any correlation with CD8+ enrichment, which is the main component of the “immunoscore” [28], thus suggesting that PI may represent a different immunotype, which, similarly to the immunoscore, may serve as a clinical parameter in evaluating tumorigenesis.
Thus, the PI signature is widely expressed in human tumors, distinguishing the tumor cells from adjacent normal tissue and the tumor microenvironment, with certain cancer types having stronger PI signatures than others. PI appears to be primarily a cancer cell-autonomous phenomenon, distinct from all other well-established cancer-promoting immune inflammatory responses.
PI is associated with poor prognosis and p53 mutations
Based on the analysis of the CKIα-deficient mouse models, PI itself may either serve as a tumor suppressor mechanism or help promote carcinogenesis [6]. To determine PI’s role in human cancer, we analyzed clinical data from TCGA, which allowed us to examine the association of PI with survival time and other clinical features available from TCGA database in 18 cancer types. Our analysis revealed higher mortality rates for patients with high PI scores in most cancer types (Additional file 3: Table S9). Prominent examples of high PI score tumors associated with bad prognosis are head and neck squamous cell carcinoma (HNCS; Cox regression p value = 1.4e-3), low-grade glioma (LGG; p value = 8.4e-4), lung adenocarcinoma (LUAD; p value = 9.4e-3), and pancreatic adenocarcinoma (PAAD; p value = 6.7e-5). Median time of mortality of PI+ compared with PI− patients was 1.54-, 3.50-, 1.33-, and 3.53-fold faster, respectively (Fig. 6a; Kaplan-Meier plots). A pan-cancer survival analysis of all samples confirmed a negative correlation of PI with survival, showing a consistent earlier mortality for the PI+ patients (p value = 7.1e-29; Fig. 6b). Notably, low-grade glioma has a low overall PI score; however the small subset of PI+ patients have a significantly lower survival rate, indicating a value of screening for PI even in tumor types with low PI. Analysis of other clinical features, including age, gender, race, and smoking, showed that the PI score is generally independent of these variables (Additional file 3: Table S10). Accordingly, multivariate regression analysis controlling for age and smoking did not change the power of the PI score in predicting survival (Additional file 3: Table S9).
Fig. 6.
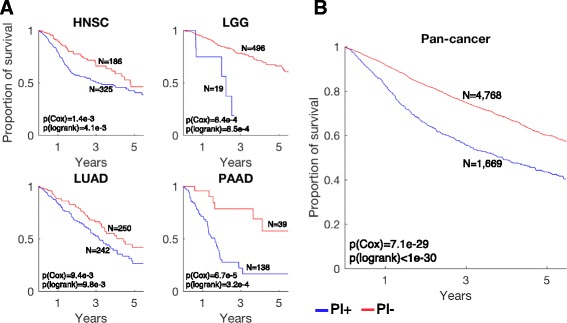
Parainflammation is associated with worse prognosis. a Kaplan-Meier plots for PI+ samples (blue) versus PI− samples (red) in head and neck squamous cell carcinoma (HNSC), lower grade glioma (LGG), lung adenocarcinoma (LUAD), and pancreatic adenocarcinoma (PAAD). The p values were calculated using Cox proportional hazard regression of the PI scores and the log-rank method between PI+ and PI− samples. b Pan-cancer survival analysis of 6437 patients with survival data and PI scores
In the CKIα-deficient mouse model, PI promotes carcinogenesis only after loss of p53, suggesting that PI+ samples will be enriched with p53 mutations. We therefore investigated the relationship of PI and the status of p53 in human cancer. By analyzing the mutation data from whole-exome sequencing provided by TCGA across all samples with expression data, we found 1309 samples with mutations in the TP53 gene versus 2363 with wild-type (WT) TP53. The PI score was significantly higher in the mutated p53 samples (U-test p value = 1.4e-47) and 35.4 % of the p53 mutated samples were PI+ compared with 15.2 % in WT p53 samples (Fig. 7a). This result was recapitulated in the carcinoma cell lines—29.3 % PI+ in mutated p53 and 20.6 % in WT—although less significant (p value = 0.016). p53 mutations are observed more frequently in cell lines than in primary tumors (59.8 % versus 35.7 %), possibly reflecting the growth advantage of p53 mutants in tissue culture, which may hinder a stronger association between p53 mutations and PI in cancer cell lines. Notably, cancer types with known, albeit unexplained low rates of p53 mutations, such as kidney cancer types and prostate adenocarcinoma (PRAD) [29], tend to show low rates of PI, whereas cancers with high p53 mutation rates, such as pancreatic adenocarcinoma (PAAD) and colon adenocarcinoma (COAD), show high rates of PI (R = 0.740, p value = 0.002) (Fig. 7b). Finally, it is well known that patients harboring tumors with p53 mutations have significantly worse prognosis [30] (in TCGA: Cox regression p value = 4.9e-15). We tested whether the association of PI with survival relates to p53 mutations. For 2954 patients with both mutations and clinical data, our analysis revealed only a small decrease in significance between controlled and uncontrolled Cox regression analyses (p value without controlling for p53 = 4.7e-17, p value with controlling for p53 = 2.4e-11), indicating that the poor survival associated with PI cannot entirely be attributed to p53 loss.
Fig. 7.
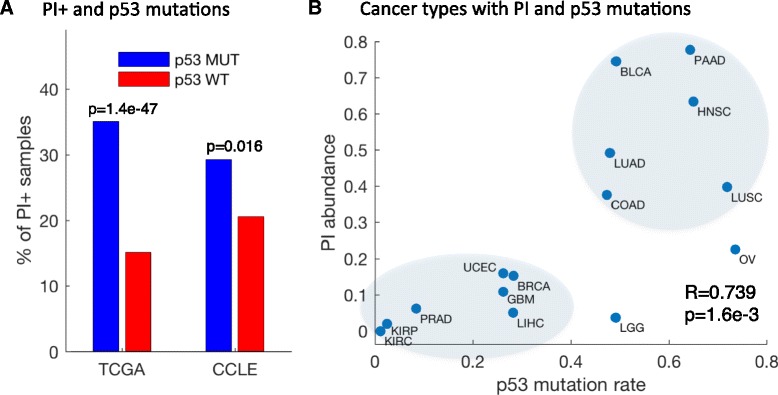
Parainflammation is associated with p53 mutations. a Percentage of p53 nonsense mutated PI+ samples (blue) versus percentage of p53 WT PI+ samples (red) in TCGA and in CCLE. b Abundance of p53 missense mutations (x-axis) versus the abundance of PI+ samples (y-axis) in 15 cancer types with sufficient information. Cancer types with higher rates of p53 nonsense mutations tend to have higher rates of PI. The Spearman coefficient is presented
PI response to NSAID treatment in human tumors
Our mouse tumor data indicate that tumor PI is suppressed by sulindac treatment (Fig. 2). Prolonged NSAID treatment was found to reduce both the incidence and the mortality of human cancer [15–17]. To study the relevance of PI in NSAID treatment in humans, we first searched for public expression data of NSAID treatment in cancer cell lines. While there are several such databases, notably the Broad Institute Connectivity Map (C-Map) [31], most of the data are limited to cell lines that show low PI levels before treatment and, therefore, no treatment effect is expected. Nevertheless, we observed one particular cell line with a high PI score, SCC-25, an oral cancer cell line treated with 2 mM of aspirin (GSE58162). This cell line has a high PI score, which ranks it 19th out of 634 CCLE carcinomas. We observed a sharp decrease in the expression of the PI genes in response to the treatment, and in two of the three replicates the PI gene set is significantly downregulated compared with all genes: U-test p value = 8.6e-4, 0.318, and 0.009 in the three replicates, respectively. Similarly, we found that the PI score decreased in all three replicates following aspirin treatment (Fig. 8a). We further noticed that the response to aspirin is significantly enhanced when examining a large set of genes that are upregulated in correlation with the PI score; 74.0 % of the top 215 correlated genes are downregulated in response to aspirin treatment (U-test p value = 8.1e-25; Additional file 3: Figure S8).
Fig. 8.
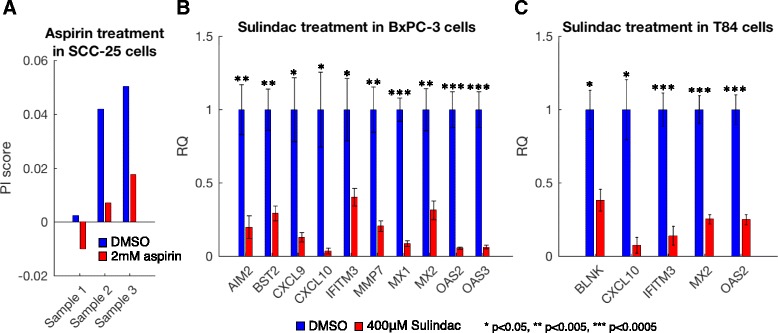
Parainflammation decreases in response to NSAID treatment in human cell lines. a average of the differential expression (in log2 scale) of three replicates each of control and aspirin-treated SCC-25 cells for the PI genes. A kernel smoothed regression curve for all genes is presented as reference (red). The p value presented was calculated using a Wilcoxon rank-sum test. b qPCR analysis of highly expressed PI genes in control and sulindac-treated BxPC3 cells. Error bars show the standard error of the mean (SEM) of triplicates. c qPCR analysis of highly expressed PI genes in control and sulindac-treated T84 cells. Error bars show the SEM of triplicates
To confirm these computational findings, we treated two human cancer cell lines, predicted to have high PI scores from the CCLE dataset analysis, with sulindac: the BxPC3 cell line, a pancreatic cancer cell line with a PI score which ranks it ninth out of the 634 CCLE carcinomas, and the T84 cell line, a colorectal cancer cell line ranked 18th. Sulindac treatment led to a decrease of more than 50 % in the expression of all tested PI genes which are highly expressed in BxPC3 and T84 cells, respectively (Fig. 8b, c). This, together with the analyses of the SCC-25 cell line and the mouse tumor organoids (Fig. 2), confirms the repressive effect of NSAID treatment on PI, suggesting that part of the cancer-prevention mechanism of NSAIDs may be attributed to PI suppression.
Discussion
Inflammation is emerging as one of the hallmarks of cancer [1] yet histologically detectable chronic inflammation characterizes early tumor development in only a minority of solid tumors [32]. In this respect it is surprising that NSAID treatment is remarkably effective in reducing mortality rates associated with major human solid tumors, albeit their mechanism of action in cancer is controversial and there is no common property distinguishing cancers in which NSAID treatment is beneficial. NSAIDs are non-selective Cox2 inhibitors and it is thus not surprising that they are effective in cancers where Cox2 is indeed elevated. However, NSAIDs are also effective in cancers where Cox2 is not activated, occurring through hereditary or stochastic mutations and not preceded by prolonged inflammation. This indicates that inflammation may have a covert course that plays a major unappreciated role in cancer. Accordingly, in spite of many studies addressing this question, the mechanism of NSAID action in cancer prevention remains mostly elusive. Medzhitov coined the term parainflammation for a low-grade inflammatory response, referring to an adaptive response due to tissue stress or malfunction [7]. Here we defined PI on the basis of analysis of existing databases and experimental data: an epithelial-autonomous inflammatory response observed in genetically modified mouse models. We then investigated the occurrence of PI in human cancer, asking (a) whether PI is a universal cancer phenomenon and (b) does PI have clinical/prognostic implications in human cancer. Using several large sets of human cancer samples, we detected frequent occurrence of PI in human cancers with several interesting features: PI is cancer cell-autonomous, readily detectable in human carcinoma cell lines; its repertoire is distinct from other common inflammatory signatures; and it doesn’t bear the phenotypic hallmarks nor some of the regulatory characteristics of common inflammatory reactions (e.g., NF-κB). PI is very common in certain types of cancer, including bladder, head and neck, cervical, and colorectal cancer, and conspicuously absent in other types, such as hepatocellular carcinoma, prostate and endometrial adenocarcinoma, and low grade glioma.
In the CKIα-deficient mouse model, PI promotes tumorigenesis only after loss of p53. We therefore investigated whether p53 loss/mutation in human tumors is associated with occurrence of PI. Indeed, the p53 status of the tumor displays a high association with PI in a variety of cancer types. A similar association of p53 mutation or loss with PI occurs across the entire spectrum of cancer types: those with low PI, like prostate, liver, thyroid, and melanoma, have few p53 mutations. Why certain cancer types have low p53 mutation frequencies with no evidence of p53 pathway suppression is an enigma. The strong association between PI and p53 mutations in cancer suggests that PI may be one of the major driving forces for inactivating the p53 pathway. This corresponds to the relationship between PI and p53 deficiency in mouse tumor models, showing a sharp PI switch from a tumor suppressor to a tumor promoter mechanism upon p53 loss [6]. Such a tumorigenesis switch mechanism may be a particularly powerful cancer driver mechanism and is thus possibly one of the main mechanisms enforcing p53 mutations in cancer, which cannot be substituted by loss of other tumor suppressor genes. While we are not aware yet of other means of switching PI from a cancer suppressor to a promoter, it is possible that certain other tumor-specific genetic aberrations may fulfill a function similar to p53 inactivation, pushing cancer progression. Indeed, PI is associated with worse prognosis, beyond the p53 status of the tumor. These retrospective analyses could indicate the value of screening individual tumors for PI manifestation as a prognostic or treatment/prevention tool. Particularly relevant in this aspect is NSAID treatment. Retrospective analysis of cancer patients who were routinely treated with aspirin, including low dose aspirin, for non-cancer indications showed a surprisingly beneficial effect in reducing cancer mortality following surgical removal of the tumor [17]. Nearly all the cancer types in which beneficial aspirin effects were noted (e.g., colorectal, pancreatic, lung, and breast) [17, 18, 33] are characterized by high PI, either throughout the entire cancer type or a significant subtype, calling for implementation of PI screening of tumor biopsy or resection samples. Supporting our hypothesis implicating PI suppression as an important anti-tumorigenic mechanism of NSAID is our study of a mouse tumor model and human cancer cell lines displaying high PI: we demonstrated effective sulindac repression of the majority of the tested PI genes upregulated in the tumor cells. Thus, whereas we are still far from understanding the origin and mechanisms of PI emergence in tumors, its recognition and monitoring may be of great value in the clinical care of cancer.
Conclusions
In this work, we have characterized a novel PI signature present in 25.9 % of all human tumors and in the vast majority of certain types of cancer, such as gastrointestinal, lung, bladder, and head and neck tumors. PI has a distinct signature, originating endogenously from the tumor cells, and does not coincide with canonical inflammation. Our data indicate that PI is linked to p53 mutations, suggesting it might be a major driving force for p53 mutation. As PI is suppressible by NSAID treatment in vitro and is particularly prominent in cancers in which aspirin treatment is beneficial, we propose that a tumor PI signature should be tested as a potential biomarker/clinical indication for NSAID chemoprevention and treatment of cancer.
Methods
Data analysis
PI gene signature
We obtained raw RNA-seq counts from Pribluda et al. [6] for WT, CKIα-deficient and CKIα-p53 doubly deficient gut epithelium, each with two replicates (Additional file 2). Using the DESeq2 package for R [34], we calculated p values and fold ratio changes against the WT counts. Upregulated genes were chosen as genes with fold ratio >2 and adjusted p value <0.01. Genes upregulated in the mouse models were then intersected with our list of inflammatory response genes. The downregulated gene signature was defined in the same manner but was more than twofold lower in the deficient mouse models compared with WT.
Upstream regulator analysis
The analysis was performed through the use of QIAGEN’s Ingenuity® Pathway Analysis (IPA®, QIAGEN Redwood City, http://www.qiagen.com/ingenuity). Figure 1c shows the top 18 non-chemical regulators; Additional file 4 presents all data.
Adjustment for blood admixture
Infiltration of immune cells into tumor samples may influence the analysis of gene expression profiles of the tumor cells and these should therefore be adjusted. The level of blood cells in tumor samples may be efficiently analyzed using leukocyte-specific expression profiles, e.g., by the ESTIMATE score [35]. We found tight correlation between the ESTIMATE measurement and CD45 expression levels over tumor samples across all cancer types (Additional file 3: Figure S4a). The ESTIMATE score, which is calculated using a gene signature of 141 genes, might be perturbed by our notion of PI; thus, we adjusted the expression levels of each gene according to CD45 expression alone. Using expression data of normal samples from the Genotype-Tissue Expression Project (GTEx) [25], we first fit a linear curve for each gene according to its expression level and the expression level of CD45 in each tissue type (Additional file 3: Figure S4b). The slope learned from this section represents the normal tissue-specific association between the gene expression levels and the immune infiltration levels. Using this slope we then adjust the expression levels of the gene for all TCGA samples in the corresponding cancer type (Additional file 3: Figure S4c). Finally, the expression of all samples is shifted so the minimal level for each gene is 0.
PI score
The PI score is the ssGSEA score of the 40 PI genes (Additional file 3: Table S1). A TCGA tumor sample was considered PI positive (PI+) if its PI score is over 0.2951. This threshold is found in less than 5 % of TCGA adjacent normal samples and corresponds to 25.9 % of the tumor samples overall. In the CCLE dataset only 9.5 % of the carcinoma cell lines were over this threshold. However, the ssGSEA method is highly sensitive to different expression platforms with different numbers of genes and is thus incomparable between these. Of the carcinoma cell lines, 25.9 % have a PI score over 0.1859. This notion is true for other datasets used in this article for calculating the PI scores: the scores should only be compared between samples from the same expression-measuring platform in the species. To allow some degree of comparison between the different datasets, for visualization the scores were shifted in each data set such that PI+ samples have positive scores.
Organoid culture
Organoids were produced from WT and APCmin/+ mice as previously described [13, 36]. For adenoma and APCmin/+ organoids, adenomas and normal adjacent tissue were separated and processed accordingly. All organoids were grown in Advanced DMEM/F12 culture medium (Gibco) supplemented with Noggin, EGF, bFGF (Peprotech, 1:1000), R-spondin1 (Peprotech, 1:500) and B27 (Gibco, 1:50). For staining, organoids were fixed in 4 % paraformaldehyde and incubated overnight with the primary antibody (IFITM2/3 - 1:200, Abcam). Secondary antibody was Alexa fluor-647 donkey anti-rabbit (Molecular Probes, 1:1000). Hoechst (1 μg/ml, Molecular Probes) was used to stain nuclei.
NSAID treatment of human cancer cell lines
BxPC3 cells were grown in RPMI medium. T84 cells were grown in 1:1 DMEM/F12 medium. For sulindac treatment, cells were incubated with 400 μM sulindac (Sigma) for 48 h before harvesting. Controls were treated with DMSO, the NSAID vehicle.
RNA extraction and qPCR analysis
RNA was extracted using the miRNeasy kit (Qiagen). cDNA was produced using qScript cDNA kit (Quanta). Primers used for qPCR analysis can be found in Additional file 3: Table S11.
Abbreviations
CCLE, Cancer Cell Line Encyclopedia; GEO, Gene Expression Omnibus; GTEx, Genotype-Tissue Expression; H&E, hematoxylin and eosin; IFN, interferon; IL, interleukin; KIRC, kidney renal clear cell carcinoma; LPS, lipopolysaccharide; NSAID, non-steroidal anti-inflammatory drug; PAAD, pancreatic adenocarcinoma; PI, parainflammation; qPCR, quantitative PCR; ssGSEA, single-sample gene set enrichment analysis; TCGA, The Cancer Genome Atlas; WT, wild type.
Acknowledgments
We thank Haya Hamza for technical assistance.
Funding
This work was supported by the Gruss Lipper Postdoctoral Fellowship to D.A., grants from the Israel Science Foundation (ISF)—Centers of Excellence and the European Research Council within the FP-7 to YB-N (294390 PICHO) and EP (281738 LIVERMICROENV), the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (AMRF), the I-CORE program of ISF (grant number 41/11) and the Israel Cancer Research fund to Y.B.N., and Israel Science Foundation grant number 1162/12, I-CORE Program of the Planning and Budgeting Committee and the Israel Science Foundation grant number 41/11, and the Rosetrees Trust grant to A.H and the National Cancer Institute of the National Institutes of Health U24 CA195858 to A.J.B.
Availability of data and materials
The results published here are in part based on data generated by TCGA Research Network (http://cancergenome.nih.gov). TCGA datasets were downloaded on 15 January 2015. The organoid RNA-seq data are available at the Gene Expression Omnibus (GEO) under accession GSE81836.
Tumor and adjacent normal samples: Level 3 RNA-seq data (RNAseqV2 normalized RSEM), clinical data, and Mutation Annotation Format (MAF) files for multiple human cancers and adjacent normal samples were downloaded from TCGA data portal on 15 January 2015 (http://cancergenome.nih.gov; Additional file 3: Table S3). Only primary tumors were included in the analyses.
Normal samples: Processed RNA-seq data were downloaded from the Genotype-Tissue Expression (GTEx) portal (http://www.gtexportal.org/home/).
Cancer cell lines: Normalized Affymetrix U133 + 2 array data were downloaded from the Cancer Cell Line Encyclopedia (CCLE) website [22].
Aspirin treatment: Illumina HumanHT-12 V4.0 expression beadchip of three replicates obtained from oral cancer cell line SCC25 treated with 2.0 mM of aspirin and untreated samples used as a control were downloaded from the NCBI GEO (accession GSE58162).
Software: All statistical analyses were performed using MATLAB®. The Deseq2 package for R [34] was used for differential expression re-analysis of the RNA-seq of the CKIα-deficient mice. The RSEM (RNA-Seq by Expectation-Maximization) software package [37] was used for the analysis of the organoid RNA-seq experiments. The GSVA package [38] was used for the single-set gene set enrichment analysis (ssGSEA).
Authors’ contributions
Conception and design: DA, AL, MB, EP, AH, YB-N. Development of methodology: DA, AL. Acquisition of data: DA, AL, AZ. Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): DA. Writing, review, and/or revision of the manuscript: DA, AL, EP, AH, YB-N. Study supervision: YB-N, EP, AH, AJB. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
All mouse experiments were conducted in accordance with guidelines of the Hebrew University animal ethics committee, accredited by AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care International).
Additional files
List of 840 inflammatory response genes based on three manually curated databases: inflammatory response gene lists curated by Ingenuity, InnateDB innate immunity genes, and the Immunogenetic Related Information Source (IRIS). (XLSX 74 kb)
Results from differential expression analyses of CKIα-deficient and CKIα-p53-deficient gut epithelium versus wild-type gut epithelium. Raw RNA-seq counts were obtained from Pribluda et al. [6]. Each set has two replicates. The analysis was performed using the DESeq2 package for R. (XLSX 4406 kb)
qPCR of PI genes in organoids. Figure S2. Expression of PI genes in cell lines. Figure S3. Correlation between the PI scores derived from up- and downregulated genes. Figure S4. Adjusting TCGA expression levels to immune infiltrations. Figure S5. Correlation of the PI score with CD45 expression. Figure S6. PI scores in TCGA cancer and adjacent normal samples. Figure S7. The PI score is correlated with the number of activated PI genes. Figure S8. PI-associated gene responses to aspirin treatment. Table S1. Parainflammation gene signature. Table S2. Down-regulated parainflammation genes. Table S3. List of samples acquired from TCGA. Table S4. Transcription factor binding enrichment analysis of PI genes. Table S5. Immune gene signatures. Table. S6. PI score and functional immune gene sets. Table S7. PI score and immune subsets estimated by hematoxylin and eosin staining. Table S8. PI score and immune subset estimate by gene signatures. Table S9. Survival and parainflammation. Table S10. Clinical features and parainflammation. Table S11. Primers used for qPCR analyses. (PDF 1608 kb)
Results from an Ingenuity upstream regulator analysis performed on the 40 PI genes. (XLSX 123 kb)
Results from a differential expression analysis of organoid cultures from APC-mutated normal gut epithelium (MIN) versus adenomatous polyps of APCmin/+ mice (Adenoma). See “Methods” for organoid culture. Each set has three biological replicates. Raw reads and counts are available at the GEO under accession GSE81836. The analysis was performed using the RSEM software package. (XLSX 773 kb)
Results from a differential expression analysis of organoid cultures from adenomatous polyps of APCmin/+ mice (Adenoma) versus adenomatous polyps of APCmin/+ mice treated with sulindac. See “Methods” for organoid culture and sulindac treatment. Each set has three biological replicates. Raw reads and counts are available at the GEO under accession GSE81836. The analysis was performed using the RSEM software package. (XLSX 731 kb)
Parainflammation (PI) scores for 634 carcinoma cell lines from the CCLE project. The PI score is calculated using ssGSEA for the 40 PI genes. (XLSX 69 kb)
Spearman coefficients of correlations between gene expression profiles and the PI scores across the carcinoma cell lines. Many genes not in part of the PI signature are highly correlated with the PI score, suggesting their role in PI. (XLSX 422 kb)
Parainflammation (PI) scores for 6523 primary tumors and 582 patient-matched normal samples of 18 cancer types from TCGA. The PI score is calculated using ssGSEA for the 40 PI genes using the immune-adjusted expression profiles. (XLSX 260 kb)
Contributor Information
Atul J. Butte, Email: atul.butte@uscf.edu
Yinon Ben-Neriah, Phone: +972-2- 6758718, Email: yinonb@ekmd.huji.ac.il.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal BB, Vijayalekshmi RV, Sung B. Targeting inflammatory pathways for prevention and therapy of cancer: short-term friend, long-term foe. Clin Cancer Res. 2009;15:425–30. doi: 10.1158/1078-0432.CCR-08-0149. [DOI] [PubMed] [Google Scholar]
- 3.Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol. 2012;22:33–40. doi: 10.1016/j.semcancer.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 4.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elyada E, Pribluda A, Goldstein RE, Morgenstern Y, Brachya G, Cojocaru G, Snir-Alkalay I, Burstain I, Haffner-Krausz R, Jung S, et al. CKIalpha ablation highlights a critical role for p53 in invasiveness control. Nature. 2011;470:409–13. [DOI] [PubMed]
- 6.Pribluda A, Elyada E, Wiener Z, Hamza H, Goldstein RE, Biton M, Burstain I, Morgenstern Y, Brachya G, Billauer H, et al. A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell. 2013;24:242–56. [DOI] [PubMed]
- 7.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–35. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 8.Chovatiya R, Medzhitov R. Stress, inflammation, and defense of homeostasis. Mol Cell. 2014;54:281–8. doi: 10.1016/j.molcel.2014.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Breuer K, Foroushani AK, Laird MR, Chen C, Sribnaia A, Lo R, Winsor GL, Hancock RE, Brinkman FS, Lynn DJ. InnateDB: systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res. 2013;41:D1228–33. [DOI] [PMC free article] [PubMed]
- 10.Kelley J, de Bono B, Trowsdale J. IRIS: a database surveying known human immune system genes. Genomics. 2005;85:503–11. doi: 10.1016/j.ygeno.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Kramer A, Green J, Pollard J, Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30:523–30. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doyle S, Vaidya S, O'Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, Cheng G . IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–63. [DOI] [PubMed]
- 13.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H . Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–5. [DOI] [PubMed]
- 14.Wolff WI, Shinya H. Definitive treatment of "malignant" polyps of the colon. Ann Surg. 1975;182:516–25. doi: 10.1097/00000658-197510000-00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burn J, Gerdes AM, Macrae F, Mecklin JP, Moeslein G, Olschwang S, Eccles D, Evans DG, Maher ER, Bertario L, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet. 2011;378:2081–7. [DOI] [PMC free article] [PubMed]
- 16.Lasry A, Zinger A, Ben-Neriah Y. Inflammatory networks underlying colorectal cancer. Nature Immunology. 2016;17:230–240. doi: 10.1038/ni.3384. [DOI] [PubMed] [Google Scholar]
- 17.Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. 2011;377:31–41. doi: 10.1016/S0140-6736(10)62110-1. [DOI] [PubMed] [Google Scholar]
- 18.Streicher SA, Yu H, Lu L, Kidd MS, Risch HA. Case–control study of aspirin use and risk of pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2014;23:1254–63. doi: 10.1158/1055-9965.EPI-13-1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hawiger J. Innate immunity and inflammation: a transcriptional paradigm. Immunol Res. 2001;23:99–109. doi: 10.1385/IR:23:2-3:099. [DOI] [PubMed] [Google Scholar]
- 20.Hashimoto S, Suzuki T, Dong HY, Nagai S, Yamazaki N, Matsushima K. Serial analysis of gene expression in human monocyte-derived dendritic cells. Blood. 1999;94:845–52. [PubMed] [Google Scholar]
- 21.Hashimoto S, Suzuki T, Dong HY, Yamazaki N, Matsushima K. Serial analysis of gene expression in human monocytes and macrophages. Blood. 1999;94:837–44. [PubMed] [Google Scholar]
- 22.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. [DOI] [PMC free article] [PubMed]
- 23.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–12. [DOI] [PMC free article] [PubMed]
- 24.Aran D, Sirota M, Butte AJ. Systematic pan-cancer analysis of tumour purity. Nat Commun. 2015;6:8971. doi: 10.1038/ncomms9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Consortium GT The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–5. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Auerbach RK, Chen B, Butte AJ. Relating genes to function: identifying enriched transcription factors using the ENCODE ChIP-Seq significance tool. Bioinformatics. 2013;29:1922–4. doi: 10.1093/bioinformatics/btt316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, Lugli A, Zlobec I, Hartmann A, Bifulco C, et al. Towards the introduction of the 'Immunoscore' in the classification of malignant tumours. J Pathol. 2014;232:199–209. [DOI] [PMC free article] [PubMed]
- 29.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. [DOI] [PMC free article] [PubMed]
- 30.Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26:2157–65. doi: 10.1038/sj.onc.1210302. [DOI] [PubMed] [Google Scholar]
- 31.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–35. [DOI] [PubMed]
- 32.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fraser DM, Sullivan FM, Thompson AM, McCowan C. Aspirin use and survival after the diagnosis of breast cancer: a population-based cohort study. Br J Cancer. 2014;111(3):623–7. doi: 10.1038/bjc.2014.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, Trevino V, Shen H, Laird PW, Levine DA, Van Houdt WJ, Pronk A, Van Gorp J, Siersema PD, Clevers H. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. [DOI] [PMC free article] [PubMed]
- 36.Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J, Siersema PD, Clevers H. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology. 2011;141:1762–72. [DOI] [PubMed]
- 37.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The results published here are in part based on data generated by TCGA Research Network (http://cancergenome.nih.gov). TCGA datasets were downloaded on 15 January 2015. The organoid RNA-seq data are available at the Gene Expression Omnibus (GEO) under accession GSE81836.
Tumor and adjacent normal samples: Level 3 RNA-seq data (RNAseqV2 normalized RSEM), clinical data, and Mutation Annotation Format (MAF) files for multiple human cancers and adjacent normal samples were downloaded from TCGA data portal on 15 January 2015 (http://cancergenome.nih.gov; Additional file 3: Table S3). Only primary tumors were included in the analyses.
Normal samples: Processed RNA-seq data were downloaded from the Genotype-Tissue Expression (GTEx) portal (http://www.gtexportal.org/home/).
Cancer cell lines: Normalized Affymetrix U133 + 2 array data were downloaded from the Cancer Cell Line Encyclopedia (CCLE) website [22].
Aspirin treatment: Illumina HumanHT-12 V4.0 expression beadchip of three replicates obtained from oral cancer cell line SCC25 treated with 2.0 mM of aspirin and untreated samples used as a control were downloaded from the NCBI GEO (accession GSE58162).
Software: All statistical analyses were performed using MATLAB®. The Deseq2 package for R [34] was used for differential expression re-analysis of the RNA-seq of the CKIα-deficient mice. The RSEM (RNA-Seq by Expectation-Maximization) software package [37] was used for the analysis of the organoid RNA-seq experiments. The GSVA package [38] was used for the single-set gene set enrichment analysis (ssGSEA).