

Abstract
Quorum sensing (QS) is a generic term used to describe cell-cell communication and collective decision making by bacterial and social insects to regulate the expression of specific genes in controlling cell density and other properties of the populations in response to nutrient supply or changes in the environment. QS mechanisms also have a role in higher organisms in maintaining homeostasis, regulation of the immune system and collective behavior of cancer cell populations. In the present study, we used a p190BCR-ABL driven pre-B acute lymphoblastic leukemia (ALL3) cell line derived from the pleural fluid of a terminally ill patient with ALL to test the QS hypothesis in leukemia. ALL3 cells don’t grow at low density (LD) in liquid media but grow progressively faster at increasingly high cell densities (HD) in contrast to other established leukemic cell lines that grow well at very low starting cell densities. The ALL3 cells at LD are poised to grow but shortly die without additional stimulation. Supernates of ALL3 cells (HDSN) and some other primary cells grown at HD stimulate the growth of the LD ALL3 cells without which they won’t survive. To get further insight into the activation processes we performed microarray analysis of the LD ALL3 cells after stimulation with ALL3 HDSN at days 1, 3, and 6. This screen identified several candidate genes, and we linked them to signaling networks and their functions. We observed that genes involved in lipid, cholesterol, fatty acid metabolism, and B cell activation are most up- or down-regulated upon stimulation of the LD ALL3 cells using HDSN. We also discuss other pathways that are differentially expressed upon stimulation of the LD ALL3 cells. Our findings suggest that the Ph+ ALL population achieves dominance by functioning as a collective aberrant ecosystem subject to defective quorum-sensing regulatory mechanisms.
Keywords: Acute lymphoblastic leukemia, quorum sensing, cholesterol metabolism, cancer cell population, cancer ecosystem, hallmark of cancer, FAM129C
Introduction
Collective behavior in higher organisms is an important characteristic regulating many biological processes and functions such as cell migration, stem-cell maintenance, maintenance of proper organ size, immune system regulations and regeneration. Individual cells employ ‘autocrine’ and/or ‘paracrine’ factors to coordinate these beneficial collective behaviors. Bacteria utilize secreted molecules to ‘count’ their population numbers to determine whether the conditions are appropriate to perform any simple or complex collective behavior. Intercellular communication between bacteria to execute community behavior is an example of quorum sensing (QS). Bacteria release, detect, and respond to the release of small chemical signaling molecules, termed autoinducers, to coordinate the various activities of cells in order to function like a multicellular organism such as formation of complex biofilms, antibiotic production, motility, sporulation, virulence, swarming, competence, conjugation, symbiosis, production of virulence factors, and to attract prey, avoid predators or find mates that would not be possible as individual cells [1-8]. Bacteria use autoinducers [3,4,6] or QS peptides [9-11] to modulate gene-expression and to coordinate intercellular and also interspecies communications [12,13]. Thus, QS allows bacteria to behave like a multicellular organism. Ants operate without central control and work collectively to perform tasks [14]. They establish their activities using interactions largely based on smell [15-17].
QS like mechanisms are also important in maintaining appropriate cell numbers in each organ and tissue for its optimum function in mammals [18-23]. The immune system uses QS mechanisms to control their populations, discriminate between self and non-self, maintain homeostasis of lymphocyte numbers, prevent uncontrolled lymphocyte proliferation during immune responses to accelerate T-cells activation when they encounter antigens, and maintain the size and diversity of the pool of T-memory cells [24-26]. Signals derived from differentiated cells of the same lineage and cells in the microenvironment or niche are also important in maintaining homeostasis of hematopoiesis and progenitor cell quiescence [27].
Various processes such as abnormal cell proliferation, replication, metastasis, invasion, defective immune responses, therapeutic resistance, resisting cell death and metabolic reprogramming have been described as hallmarks of cancer [28-33]. Cancer cells use various intrinsic and extrinsic factors to utilize these complex functions to enhance their excessive growth. Disruption of QS in breast cancer stem cells (CSC) triggers tumorigenesis [34]. According to mathematical modeling based on a QS hypothesis for CSC, their proliferation is regulated by negative feedback regulation and any approach to eliminate CSCs using differentiation therapies as a single anticancer treatment would be ineffectual [35]. The hallmarks of metastatic colonization include organ specific homing, attachment and interaction of cells with each other and with other cells, cell surface adhesion, colonization, and tumor cell-stromal cell interaction. The cancer cells accomplish these cooperatively to form complex and heterogeneous structures to support the formation of vascularized metastatic lesions to supply enough nutrients [36]. However, they still remain subclinical until sufficiently large cell populations are reached. The fundamental biochemical and biological QS mechanisms governing these processes in different types of cancers are still poorly understood.
Like normal adult stem cell populations, all human cancers contain variable numbers of quiescent stem/progenitor cells (S/P cells) or CSC that can become reactivated and reinitiate the cancer if the proliferating cancer cells are killed. Drugs designed to kill proliferating cells usually do not kill quiescent S/P cells, and their survival is thus a major reason for failure to cure even highly chemo-sensitive tumors. With rare exceptions it is not possible to cure any disseminated cancers without eradicating dormant CSC. This can sometimes be accomplished with irradiation or non-cell cycle specific drugs (NCCSD) in localized cancers or in disseminated cancers that are especially sensitive to irradiation and NCCSD (e.g. ALL, rapidly growing lymphomas, male germ cell tumors), but the mutant stem cells initiating most cancers do not have sufficient heightened differential sensitivity to cytotoxic drugs compared to normal stem cells and thereof cannot be eradicated without intolerable toxicity. Because human CSC are rare and largely ill-defined, there is little quantitative information in most cancers about their characterization such as their prevalence, phenotype, cell cycle kinetics, duration of dormancy, activating signaling pathways, or in identifying genomic, epigenetic, or biochemical differences between normal and cancer stem cells, especially differences between quiescent cancer and normal stem cells that might be selectively targeted [37]. Another important hallmark of many CSC is their failure to respond to normal regulatory mechanisms that curtail cell production in the bone marrow or at other sites when normal homeostatic cell density equilibrium is reached, balancing cell production and cell death. The magnitude of the excessive expansion of the cancer cells in some of the hematologic malignancies can be remarkable, often reaching 5-10 times or greater than the normal homeostatic levels. The leukemic cells not only expand in their sites of origin in the bone marrow or lymph nodes, but also may infiltrate and proliferate in the long bones, spleen, liver and other organs. It is less clear why the CSC continue to produce cells long after the normal homeostatic cell density is reached at which normal cells curtail production.
Chronic myelogenous leukemia (CML) is an excellent paradigm of neoplasms that characteristically undergo progression from a relatively benign and treatable phase to a more malignant and usually rapidly fatal phase; without effective treatment, blastic transformation consistently occurs in CML after a median chronic phase (CP) duration of ~3 years [38]. Imatinib and the newer TKIs have proven so effective in controlling the CP that its average duration is considerably extended, but if treatment is stopped too soon because of drug intolerance or development of resistance, the disease almost always relapses. CML patients who achieve durable deep molecular remissions (MR) on TKIs (4-4.5 MR) which are maintained for several years or longer in different trials performed better, and only about 50% or fewer have relapsed so far [39-43]. Once blastic transformation occurs it responds poorly to any available therapy and is usually rapidly fatal. Cytokinetic studies during the CP have shown that the CML cells’ proliferative kinetics are similar to normal cells at similar bone marrow or blood cell densities, but that proliferation progressively slows as the cell density increases [44-48]. In blastic phase (BP) CML the blasts proliferate even slower as in other types of acute leukemia, but they still inevitably replace the faster dividing CP CML cells. It is important to understand how cancer S/P cells and entire cancer cell populations far exceed normal homeostatic cell density limits due to the abnormalities in QS.
As a first step towards understanding the abnormalities in QS that permit cancer S/P cells and cancer populations to greatly overexpand, we have been studying the cytokinetics of a recently obtained p190BCR-ABL driven pre-B cell line (ALL3) derived from the pleural fluid of a patient who was terminally ill with widely disseminated Ph+ ALL. Largely in accord with hypotheses postulated by previous investigators [36,49,50] we have assumed that the Ph+ acute leukemia population functions as an interactive cell society in which the group dynamic governs overall behavior, and which further ignores or disobeys normal homeostatic cell density and other normal QS regulations. Thus, rather than clonal succession and faster growth of the most aggressive new mutant clones being the sole underlying cause responsible for malignant progression, increasingly defective QS may also have an important role in the progressive expansion of the Ph+ ALL cells. This may be also be the case in other hematological malignancies such as other types of acute leukemia and BP CML as well as solid tumors undergoing malignant progression. The ALL3 cell line used in this study provided a unique opportunity to investigate the mechanisms regulating the growth of these malignant cells that closely simulate the pleural fluid ecosystem in which they were growing in the patient.
ALL is a heterogeneous disease affecting ~6000 individuals in the United States each year, 60% of whom are children (www.cancer.gov). ALL is a clonal proliferation of mutated progenitors of B or T lymphocyte origin that arise in the bone marrow [51]. The disease has a bimodal age distribution, being most commonly seen in children [52,53]. With current available therapies pediatric ALL is one of the greatest therapeutic success stories, with overall long-term event-free survival rates exceeding 80%, but in adults only 30%-40% [54,55]. Ph+ ALL is one the most lethal types of ALL both in children and adults, and is often only curable with marrow ablation and allogeneic stem cell transplants and perhaps with CAR T-cell therapy. The usual driving mutation, p190BCR-ABL, is similar to the common driving mutation in CP and BP CML, p210BCR-ABL, and the proliferative kinetics of the leukemic blast cells are similar in Ph+ ALL and BP CML [45,56].
It is difficult to initiate new human acute leukemic cell lines. In August 2007 we were given a new p190BCR-ABL driven pre-B leukemic cell line named ALL3 by Drs. Renier Brentjens and Mark Frattini which they had recently established from the free-floating leukemic cells in the pleural fluid of an adult patient who was dying of Ph+ ALL that was no longer responsive to Imatinib or other BCR-ABL TKIs. They had already shown that the cells injected in large numbers (estimated several million) caused a rapidly fatal leukemia in immunodeficient mice and would also grow in the absence of any cytokines in liquid culture if started at very high cell concentrations (~0.5-1 × 106 cells/ml). We confirmed their initial findings in NOD-SCID mice. Mice injected S.C. with as few as 5 × 105 ALL3 cells developed tumors reaching 2 cm3 in size between 4 and 6 weeks after injection, and mice injected i.v. with 5 × 105 or more ALL3 cells all developed disseminated leukemia within 5 weeks after injection. However mice injected i.v. with 5 × 103, 2.5 × 104, and 5 × 104 ALL3 cells did not develop leukemia, unlike many other human established leukemic cell lines which cause disseminated leukemia in the majority of immunodeficient mice with much fewer cells. Also, in contrast to many other long-established human or murine BCR-ABL driven leukemic cell lines, ALL3 cells do not form colonies in methylcellulose, do not grow in liquid culture at low cell densities (~5000-10,000 cells/ml), and grow increasingly faster at progressively higher cell densities between 20,000 cells/ml and 3-4 × 105 cells/ml with doubling times varying between ~20-100 hr without stimulation by any growth factors (GFs). ALL3 cells grow almost equally well in ALL3 media, IMDM with 10% FCS without additives, and QBSF-60, but die rapidly in CellGro and all other protein-free medias that have been tested. We shortly observed that ALL3 cells do not proliferate at very low starting cell densities (LD) (104 cells/ml or less) but grow increasingly well in liquid culture at higher cell densities (HD) (2.5 × 104-3-4 × 105 cells/ml). We immediately froze 50 aliquots, each containing millions of the cells, after receiving the cells so they would remain as closely as possible to their conditions in the pleural fluid. Periodically when we were ready to do an experiment, an aliquot stored in liquid nitrogen or at -80°C was thawed and regrown in ALL3 media or QBSF-60 media and about half of the aliquot was promptly refrozen for future use. It usually took several weeks before cells surviving the freeze/thaw procedure would begin growing again at HD with their maximum doubling times of ~20-24 hr which they would usually maintain for the next 4-8 months or so. During this period they would not grow at low cell densities at ~104 cells/ml or lower, but later they often began to adapt to the culture conditions and began to grow at progressively lower cell densities in liquid culture, but still never formed any colonies in soft agar or methyl cellulose. All the experiments shown were performed during the initial period when the cells behaved as when first received when they would only grow at HDs and not at LD.
The ALL3 cells are unresponsive to any known hematopoietic cytokines, produce no clones in semi-solid media, not even tiny ones, and don’t grow as a single cell in 60-well single cell cloning plates. The cell-free supernates from ALL3 cells grown at high starting cell densities (HDSN) were found to stimulate the growth of the ALL3 cells at LD at which they otherwise don’t grow. The ALL3 cells shortly enter apoptosis and die at LD, but the apoptosis of LD ALL3 cells can be repressed in the presence of the HDSN. Labeling studies with Ki67, BrdU or EdU showed that the LD ALL3 cells are poised to begin proliferating but cannot do so without being triggered by HDSN from ALL3 cells or some other normal or leukemic cells growing at HD.
Microarray gene expression analysis was performed to try to identify critical differentially regulated genes between non-stimulated and HDSN stimulated LD ALL3 cells. The gene expression studies were performed at days 1, 3, and 6, representing early, intermediate, and late stages of activation of the LD ALL3 in the presence of HDSN. A large number of genes were differentially expressed and they were analyzed and grouped according to their functions using gene set enrichment analysis (GSEA). We found enrichment of genes involved in various pathways such as cholesterol, lipid, and sterol metabolism. We found that B cell activation, signaling pathways, and anti-apoptotic pathways were also differentially expressed. These results reveal some clues about the complex underlying mechanisms responsible for the defective QS regulatory networks in ALL that may also be involved in other hematologic malignancies and solid cancers. They also provide strong evidence that the great majority of individual ALL3 cells by themselves have very limited self renewal and proliferative potential, and are only able to proliferate continuously and achieve dominance in vitro and presumably in vivo in animals or humans because they have learned to function collectively as a semi-independent closely interactive tumor ecosystem.
Materials and methods
ALL3 cells
The human p190BCR-ABL driven ALL cells line (ALL3) was derived from the rapidly growing Ph+ ALL leukemic cells growing in ascitic form in the pleural fluid of a patient with widely disseminated Ph+ ALL who died shortly thereafter. Multiple aliquots of ALL3 cells were frozen to preserve the cells’ condition as closely as possible to their status in the pleural fluid. When experiments were planned, an aliquot was thawed about a month or so ahead of time as it took a few months for the majority of cells surviving the freeze/thaw procedures to resume growing at about their original rate in the pleural fluid and immediately after collection of the thoracentisis fluids in vitro. A portion of the thawed cells was refrozen for future use. During the course of a series of experiments the cells were passaged serially for about 4-8 months during which they usually maintained their original growth characteristics but with longer passage, they sometimes began to adapt to the liquid culture conditions and started to grow at lower cell densities at which they would not grow originally. During this optimal experimental period the cells were maintained in Iscove’s modified Dulbecco’s medium (IMDM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Hyclone Laboratories, Logan, Utah, USA), 1% penicillin-streptomycin solution (PSN; 10,000 I.U. penicillin and 10,000 μg/ml streptomycin) (Corning #30-001-CI, Fisher Scientific, Pittsburgh, PA, USA), 1% sodium pyruvate (Mediatech Inc, Manassas, VA, USA #25-000-CI), 1% HEPES buffer (N-2-hydroxyethylpiperazine-N’-2-ethanesulphonic acid) (Mediatech #25-060-CI), 1% non-essential amino acids (NEAA; Mediatech #25-025) and 0.1% β-mercaptoethanol (BME) (Life Technologies, Grand Island, NY, USA #21985-023). Hereafter, we will use the term ALL3 media to describe the above culture medium used to culture the ALL3 cells.
Cell lines
R10(-) negative and R10(+) positive are two subclones of Mo7/p210BCR-ABL, a cell line originally established from an infant with acute megakaryoblastic leukemia and transfected with p210BCR-ABL by Dr. Brian Druker who kindly gave us the line. These cells were grown in IMDM containing 10% FBS supplemented with 1% PSN and are described more fully in [57]. The Ph-positive cell line, RWLeu4, derived from individual patients in the acute phase of CML was maintained in Roswell Park Memorial Institute medium (RPMI 1640) supplemented with 10% FBS and 1% PSN as described [58]. The SKL7 (cell line derived from the peripheral blood of a child with acute myelomonocytic leukemia) was grown as described [59]. The CML cell line K562 was grown in IMDM supplemented with 10% FBS and 1% PSN. The murine BM185 cell line was originally obtained from bone marrow cells of BALB/C mice transduced with a retroviral vector encoding the human BCR-ABL fusion protein. BM185 is a BCR-ABL driven acute lymphoblastic mouse cell line originally established by Kochling et al [60] and subsequently given to us by Dr. David Scheinberg (MSKCC, NY). The BM185 cell line was maintained in RPMI 1640 supplemented with 10% FBS, 2 mM L-glutamine, 100 units/liter PSN and 10-5 M BME.
Growth factors
The Granulocyte-colony stimulating factor (G-CSF), granulocyte-macrophage-colony stimulating factor (GM-CSF), and interleukin-3 (IL-3) were obtained as gifts from Kirin Brewery Co., Gunma, Japan. The kit ligand (KL or stem cell factor (SCF)), FLT3 (Flt3 ligand), TPO (Thrombopoietin), IL-6 (interleukin-6), and EPO (Erythropoietin) were purchased from R&D systems, Inc. (Minneapolis, MN, USA). Other growth factors such as IL-2, hTGF-β, IL-21, IL-1, IL-7, Leptin, IGF-I (Insulin Growth Factor I), IGF-II (Insulin Growth Factor-II), and IL-4 were obtained from MSKCC labs.
Collection and processing of supernatant (condition media) from cell lines
ALL3 cells were grown in ALL3 media at starting cell densities of 0.5-1 × 106 cells/ml and cultured for 72-96 hr at 37°C in a 5% CO2 atmosphere in incubator. The cell cultures were harvested after 3-4 days of growth and the cells were centrifuged at 1,200 rpm for 7-10 min. The cell-free supernate was collected carefully and filtered through 50-ml conical vacuum filter unit with 0.2 μm filters (Millipore, Billerica, Massachusetts, USA). The cell-free filtered-supernate was used for stimulating cells. All the other cell lines, K562, BM185, RWLeu4, R10-, R10+, and SKL7 were grown at the starting cell densities of 0.2-0.5 × 106 cells/ml for 72-96 hr at 37°C in an incubator and supernates were collected and processed as described above.
[3H]-thymidine incorporation assay
The supernates from cells growing at high densities were collected as described above. For growth assays, cells were counted using the trypan blue dye exclusion method and cells equivalent to 5,000-10,000 cells/ml (LD ALL3 cells) were placed into 15-ml falcon tubes. The adjusted ALL3 cells at the LD were washed with fresh ALL3 media once, and replaced with HDSN collected from different cell lines or with plain ALL3 media (as negative control). In each experiment the cell suspension volume was 6-7 ml. Then, the LD cells were resuspended thoroughly and dispensed into two 6-well flat bottom plates with 3 ml cell suspension per well in each plate and kept at 37°C, 5% CO2 in an incubator until harvesting them for assay. The day before harvesting the cells on a filter plate, 200 μl of cell suspensions were taken from the 6-well plate and seeded in triplicate in 96-well round bottom plates. Then, these cells were incubated with 20 μl (for each well) of 0.3 μCi of [3H]-thymidine (PerkinElmer Life Sciences, Shelton, CT, USA), and the cells were incubated for 18 hr at 37°C and 5% CO2. Next day, cells were harvested on Unifilter GF/C 96-well plates (Perkin Elmer Life Sciences #1450-521) using Unifilter-96 cell harvester (Perkin Elmer Life Sciences #961961) as described by manufacturer. The plates were allowed to air dry, and 20 μl Microscint-20 fluid (Perkin Elmer Life Sciences #6013621) was added and plate was covered with transparent top-seal and opaque back-seal, and [3H]-thymidine radioactivity was measured in TopCount Microplate Scintillation Counter (Perkin Elmer Sciences). This assay system was used to determine stimulatory activity of different sources of supernates on the growth of the LD ALL3 cells.
Trypan-blue dye exclusion method
To determine the cell number and cell viability, the trypan blue exclusion method was used [61]. Cell number and viability was determined by hemocytometer cell counts but were inaccurate at low cell densities of ~104 cells/ml or lower. Cells stained with trypan blue were considered as dead cells. We used a 0.4% solution of trypan blue (Sigma Aldrich, St. Louis, MO, USA #T8154) in buffered isotonic salt solution, pH 7.2 to 7.3 (i.e., phosphate-buffered saline; PBS).
Patient samples
CML blood samples were obtained from patients newly diagnosed with CP CML. The CML samples, AML, and BP CML were all obtained from patients hospitalized at MSKCC and given to us by our clinical colleagues as left-over cells obtained from peripheral blood or leukapheresis samples drawn for laboratory tests or therapeutic purposes (e.g., leukapheresis). After informed consent on MSKCC Institutional Review Board-approved protocols, peripheral blood mononuclear cells (PBMCs) from patients were obtained by Ficoll density centrifugation for research purpose. The CML blood samples were processed to obtain enriched PBMCs and frozen until further use. Defrosted PBMC from the patient samples were individually used to isolate the CD34+ fraction using the midiMACS immune-magnetic separation Kit from Miltenyi (Miltenyi Biotec, Bergisch Gladbach, Germany, Cat# 130-0460701) [37,56]. The cord blood samples were purchased from New York Blood Bank Center, NY.
CD34+ cells enrichment
Peripheral blood mononuclear cells (PBMCs) were isolated on a Ficoll gradient (Ficoll-Paque PLUS, GEHealthcare, Cat# 17-1440-03) from the total nucleated cells of peripheral blood of leukemic patients. In the case of the patient samples, PBMCs after Ficoll-gradient were frozen in liquid nitrogen in RPMI plus 10% dimethyl sulfoxide and 10% FBS and stored until used for experiments. CD34+ cells were positively selected using a midiMACS immune-magnetic separation Kit after one round of purification, the recovered cells were passed through another round of purification using a second column [37].
Co-culture experiments in transwells and adding HDSN
To study the effect of diffusible and soluble factors from HD ALL3 cells or any other test cells, we used permeable transparent 6-well transwells (Corning, Tewskbury, MA, USA) with the top-chamber (insert) having a 0.4 μm permeable polycarbonate membrane and a bottom well that only allows diffusion of soluble factors and prevent exchange of cells. The effect of ALL3 cells other test cells growing at different HD in the upper inserts on the LD ALL3 cells in the lower wells was observed. This method was not suitable for long-term culture since the overgrowth of cells beyond plateau phase releases unwanted materials like metabolites and release proteins from dying cells that hampers growth of ALL3 cells at LD. To circumvent these problems, we cultured HD cells for 3-4 days, and filtered HDSN were used to stimulate the LD ALL3 cells, and the growth was monitored using the [3H]-thymidine uptake assay.
Ki67 labeling
ALL3 cells were kept at LD and HD, and collected and stained with MIB-1 Ki67 antibody and nuclei were stained with DAPI. The Ki67 positive cells were counted and imaged using an immunofluorescence microscope with help from the Molecular Cytology Core Facility at Memorial Sloan-Kettering Cancer Center (MSKCC), NY.
EdU (5-ethynyl-2’-deoxy uridine) labeling
The Click-iT EdU Imaging Kit (Invitrogen, Carlsbad, CA, USA) was used. In this method, cells were collected, centrifuged, and washed twice with cold PBS. Cells were pulsed with EdU for 1 hr, fixed, permeabilised and treated with the reagent as described in the product manual provided by the manufacturer. Cells were also counter-stained with DAPI (4’,6-Diamidino-2-phenylindole dihydrochloride) (Sigma, St.Loius, MO, USA #D9542). The EdU positive cells were counted and imaged using immunofluorescence microscope with help from Molecular Cytology Core Facility at MSKCC, NY. BrdU was also used to determine the percentage of cells in S phase in some experiments, and using both methods simultaneously the results were almost identical.
Caspase 3/7 assay
The Caspase-Glo3/7 assay system (Promega, Madison, WI, USA #G8091) was used to monitor cell apoptosis as described in the product manual. Briefly, equal number of cells growing at LD and HD were seeded in white-walled 96-plate in triplicate. 100 μl of Caspase-Glo3/7 reagent was added to each well, mixed using a plate shaker and incubated at room temperature (RT) for 1 hr. After incubation luminescence was measured using a luminometer.
TUNEL (TdT-mediated dUTO Nick-End labeling) assay
Dead EndTM Fluorometric TUNEL assay system from Promega was used following manufacturer’s instructions (Promega #G3250).
AnnexinV-FITC/propidum iodide apoptotic assay
The LD ALL3 cells were stimulated with or without the HDSN from the same ALL3 cells growing at HD for 3-4 days. The non-stimulated and stimulated cells were collected, spun down, and then the supernate was discarded and cells were washed twice with cold PBS. Then, the cells were suspended in 100 μl binding buffer (PBS+2% bovine serum albumin (BSA)). 100 μl cell suspensions were treated using annexin V-FITC (BD Biosciences, San Jose, CA, USA) and 0.5 μl of 100 μg/ml PI (Propium Iodide) solution, and incubated at RT for 15 min in the dark. After the incubation period, 400 μl of binding buffer was added, mixed gently, kept on ice, and analyzed using BD FACS Calibur (BD Biosciences, San Jose, CA, USA).
RNA isolation, labeling, hybridization, and microarray
For the microarray study, the LD ALL3 cells with or without stimulation from HDSN were used. Cells from both groups were monitored for growth and viability using the trypan-blue exclusion method and the [3H]-thymidine uptake assay at different time points as described earlier. On each day, the cells were collected, spun down and media was removed, suspended in 1-1.5 ml of TRIzol reagent (Invitrogen, Carlsbad, CA, USA #15596-026) and rapidly frozen and stored at -80°C until further processing. Total RNA was isolated from each group of cells following the manufacturer’s method for TRIzol reagent. RNA quality was ensured before labeling by analyzing 5 pg of each individual sample using the RNA 6000 picoAssay and a Bioanalyzer 2100 (Agilent Technologies Inc., Santa Clara, CA, USA). For each sample meeting the standards, 20 ng of total RNA from the samples were labeled using the GeneChip two-cycle target labeling kit (Affymetrix, Inc., Santa Clara, CA, USA). Ten micrograms of labeled and fragmented cRNA were then individually hybridized to the Human Genome U133 plus 2.0 array (Affymetrix) at 45°C for 16 hr. Automated washing and staining were performed using the Affymetrix Fluidics Station 400 according to the manufacturer’s protocols. Finally, chips were scanned with a high-numerical Aperture and flying objective lens (FOL) in the GS3000 scanner (Affymetrix). In summary, RNAs were extracted from each group (stimulated or non-stimulated LD ALL3 cells), and separate gene expression profile and the changes in gene expression were derived from the statistical analysis in which we compared non-stimulated and stimulated LD ALL3 cells with HDSN collected from HD ALL3 at day 1, 3, and 6 in two independent separate experiments.
Microarray data analysis
Differential gene expression analysis comparing ALL3 HDSN stimulated and non-stimulated LD ALL3 cells was performed based on a model utilizing the LIMMA (linear models for microarray data) [62]. Microarray data analysis was performed with help from the Bioinformatics core facility at MSKCC, NY. Genes with FDR<1%, fold change larger than 2 were treated as differentially expressed genes.
Gene set enrichment analysis
A series of analyses were undertaken with the differentially expressed genes. EnrichR was employed for functional annotation analysis to understand the localization, biological significance and signaling pathways associated with large lists of differentially expressed genes [63].
Results
Growth of ALL3 cells and other CML cell lines at different starting cell densities
To evaluate the growth of the ALL3 cell line compared to other leukemic cell lines like RWLeu4, R10+, R10-, SKL7, BM185 and K562, we followed their growth at different starting cell densities over different time periods using the trypan-blue dye exclusion method (Figure 1). It is important to note that at very low cell densities hemocytometer cell counts are increasingly inaccurate. We found that the ALL3 cell line and other leukemic cell lines grew faster at progressively higher starting cell densities between 5 × 104 and 3 × 105 cells/ml (Figure 1A-D) with doubling times (DTs) of ~31-39 hr. ALL3 cells did not grow in liquid culture at LDs of 0.5 × 104-1 × 104 cells/ml but other long established human or murine BCR/ABL driven leukemic cells grew very well at these (Figure 1E-K) and even lower cell densities (Figure 1L-O).
Figure 1.
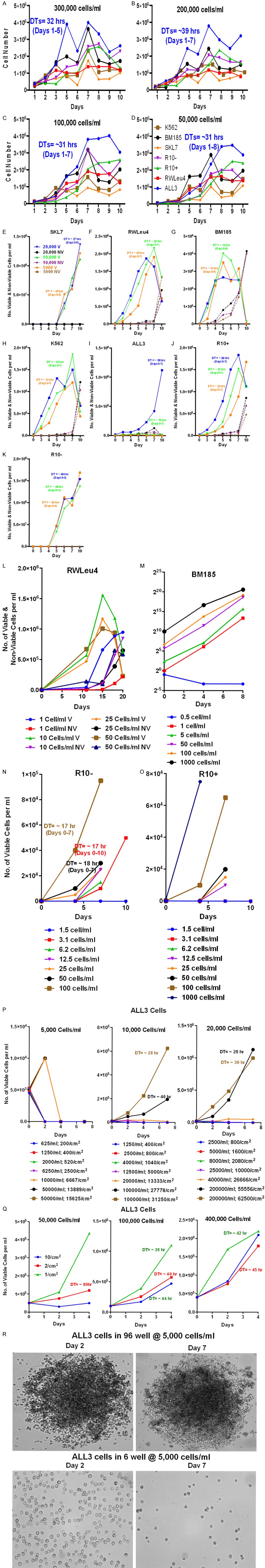
Comparative growth of various leukemic cell lines at different starting cell densities. Growth of leukemic cell lines at HD of (A) 3 × 105 cells/ml (B) 2 × 105 cells/ml (C) 1 × 105 cells/ml and (D) 5 × 104 cells/ml. The DT for ALL3 cells are color coded and shown in figures. Comparison of the growth of leukemic cell lines (E) SKL7 (F) RWLeu4 (G) BM185 (H) K562 (I) ALL3 (J) R10+ and (K) R10- at low starting cell densities of 0.5 × 104, 1 × 104, 2 × 104 cells/ml. The doubling time (DTs) for each cell lines at different cell densities are shown in graph and color-coded. BM185 was fastest growing cell line with DTs of 13-14 hr. Other cell lines SKL7, RWLue4, K562 and R10+ were growing with DTs of ~20-26 hr. The R10- cell line at starting cell densities of 1 × 104-2 × 104 and 0.5 × 104 cells/ml was growing with DTs of ~48 hr and ~64 hr, respectively. ALL3 cells at starting cell densities of 2 × 104 cells/ml grew with DTs of ~38 hr, at 1 × 104 cells/ml they had slight growth with DTs of ~75 hr and they did not grow at all at 0.5 × 104 cells/ml. (L) RWLeu4 was growing with DTs of ~21-25 hr at very low cell densities (1-50 cells/ml in 5 ml in 6-well plates) with little cell death. (M) Growth of BM185 cells in liquid culture (RPMI-1640) at 6 low cell densities. The cells grow equally well at cell densities as low as 1 cell/ml (5 cells/well) and as high as 105 cells/ml with DT of 11-15 hr, but rapidly became overgrown at higher starting densities. (N) Growth of R10- and (O) R10+ cells in liquid culture at cell concentrations from 1.5 cell/ml to 1000 cells/ml. R10- cells grew with similar DTs at cell densities as low as 3 cells/ml (15 cells/tube) and R10+ as low as 12.5 cells/ml (62 cells/tube). (P and Q) Growth or failure to grow of ALL3 cells at the same total starting cell numbers but at different cell densities per ml and per cm2 of growing surface area. (P) 5,000 cells failed to grow at all except for initially doubling once and then promptly dying at the 3 highest cell densities (104-5 × 104 cells/ml) and most tightly packed surface growing areas (6667 -15625 cells/cm2) (left panel). 10,000 cells grow fairly slowly with a DT of ~28 hr at a starting cell density of 105 cells/ml and a growing area cell concentration of 31250 cells/cm2 and even more at 105 cells/ml with a DTs of ~52 hr with fewer cells per growing area (27778/cm2) (middle panel). There was no growth at LD or with lower number of cells/cm2. Starting with a total of 20,000 cells, the cells again grew best at the two highest cell densities per ml and per growing areas with DTs of ~29 hr and hardly at all at lower cell concentration (right panel). (Q) At starting cell densities of 5 × 104 and 105 cells/ml as shown in the left and middle panels the cells grow faster and to higher cell densities on the smallest surface area (1 cm2), whereas at very high starting cell densities (4 × 105 cells/ml), the growing surface area is less important (right panel). (R) Photos of ALL3 cells starting with a total of 5000 cells growing in 6 well and 96 well plates at 2 and 7 days. The cells only doubled transiently once in first 2 days in the small 96-wells and then died but much more slowly than the cells in the larger 6-well plates. They didn’t grow at all in the larger 6-well plates; most were still viable at 2 days since the LD ALL3 cells were prepared by dilution of HD cells, but all were dead by 7 days and only a few corpses remained. These photos are shown to illustrate the contrast between the wide dispersal of the cells in the larger wells compared to their tight packing together in the small 96-well plates.
We also found that ALL3 cells don’t grow at all in liquid media at starting cell densities at or below ~5000 cells/ml unless they are tightly packed together as in round-bottom 96 well plates. The ALL3 cells don’t adhere or are only very loosely attach to the bottom surface or growing area of the culture vehicle. Proximity or close contact are clearly important as even at HD the ALL3 cells (5 × 104 and 105 cells/ml) grow faster on the smallest surface area (1 cm2), whereas at very high starting cell densities (4 × 105 cells/ml), the growing surface area is less important (Figure 1P and 1Q). As shown in Figure 1R, the LD ALL3 cells at 5000 cells/ml did not grow at all in the larger 6-well plates and almost all were dead by 7 days, The cells doubled only once by 2 days in the smaller 96-well plates, but then mostly stopped growing but didn’t die as rapidly as the same cells in the larger 6-well plates. All of these observations are consistent with the conclusion that the ALL3 cells need adequate numbers of cells and close proximity in order to communicate with each other and thereby enable them to proliferate.
ALL3 cells growing at low or high densities don’t respond to growth factors
We made several attempts to stimulate the growth of ALL3 cells using different growth factors (GFs) namely KL (SCF), GM-SCF, G-SCF, IL-3, IL-6, TPO, FLT3, EPO, IL-2, hTGF-β, IL-21, IL-1, IL7, and IL-4. We also used 3 GFs combination (KL+FLT3+TPO; data not shown) or 7 GFs combination (KL+FLT3+TPO+IL-3+IL-6+G-CSF+GM-CSF) to maximally stimulate the growth of ALL3 cells at different densities. ALL3 cells at starting densities of 2.5 & 5 × 104 cells/ml did not respond to 7 GFs (Figure 2A and 2B). None of the GFs tested further stimulated the growth of ALL3 cells at HD of 2.5 × 105 cells/ml and some such as hTGF-β, IGF, EGF, BLys and combination of these GFs (Figure 2C). Other set of GFs tested at starting densities of 0.5 × 104, 1 × 104, 2 × 104, and 5 × 104 cells/ml in the presence of IL-2, IL-4, IL-6, IL-7, and 7 GFs combination also didn’t stimulate their growth (Figure 2D-G) and some combinations actually inhibited growth of both LD and intermediate cell densities (Figure 2H-K). As shown in Figure 2H-K, Leptin, KL, FLT3 (FL), TPO, IGF 1, and IGF II also did not stimulate the growth of the ALL3 cells at starting densities of 0.5 × 104, 1 × 104, 2 × 104, and 5 × 104 cells/ml. These cell count results were also confirmed using a [3H]-thymidine uptake assay which also showed that both LD and intermediate density ALL3 cells were not stimulated to grow in the presence of any single GF alone or any combination of GFs tested (not shown).
Figure 2.
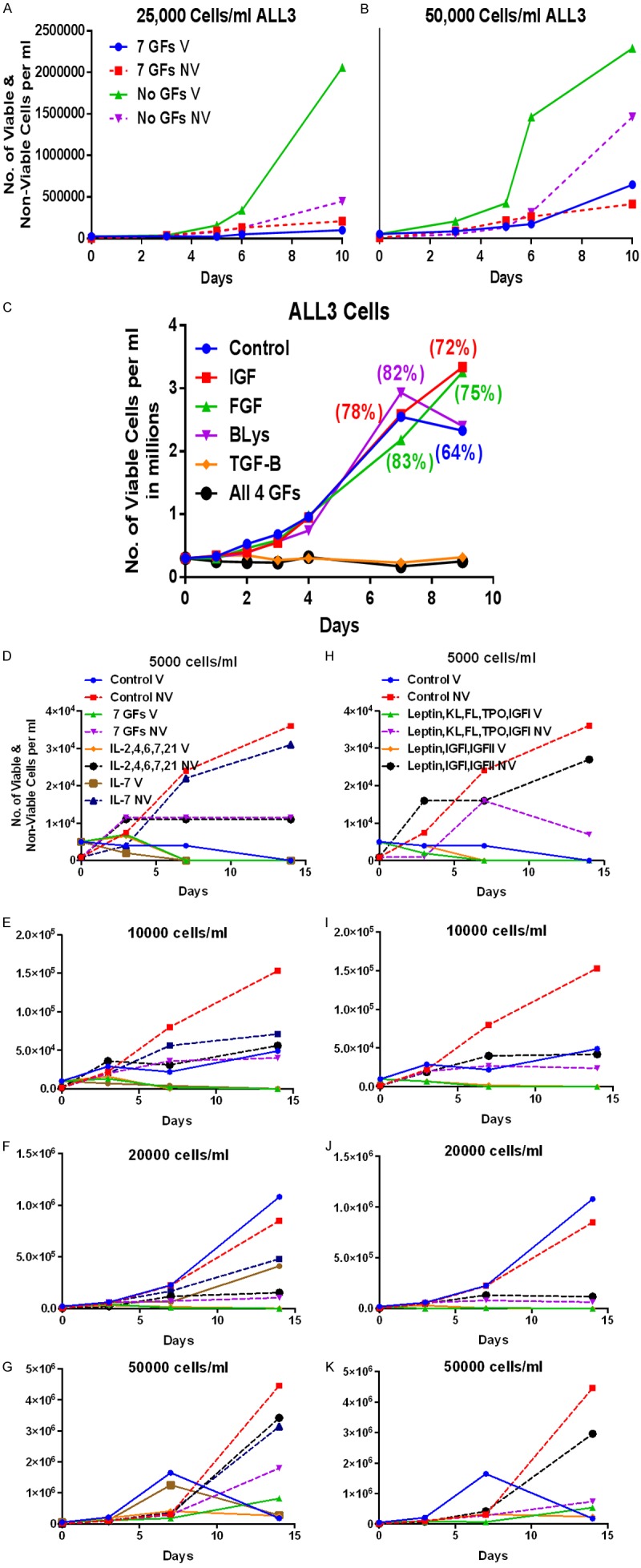
Comparison of the growth of the ALL3 cells at different starting cell densities upon growth factor stimulation. 7 GFs= (KL+FLT3+TPO+IL-3+IL-6+G-CSF+GM-CSF). Unless otherwise stated the GFs concentrations in all experiments were: KL, FLT3, TPO each 50 ng/ml; G-CSF, GM-CSF, IL-3, IL-6, IL-2, hTGF-β, IL-21, IL-1, IL-7, IL-4, IGF-I, IGF-II each 10 ng/ml; and EPO at 1 IU/ml. (KL+FL+TPO@50 ng/ml+G-CSF+GM-CSF+IL-S+IL-6@10 ng/ml) Leptin (100 ng/ml), Note: cell number (y-axis) refers to viable number of cells/ml unless otherwise stated. A, B. Growth of ALL3 cells in liquid culture at starting cell concentrations of 50,000 and 25,000 cells/ml with and without 7 GFs. C. Effect of IGF, FGF, BLys, TGF-β, and all 4 on the growh of ALL3 cells in liquid culture at starting cell concentration of 250,000 cells/ml. D-K. Lack of stimulatory activity of various GFs as shown in legends on the growth of ALL3 cells at 5 × 103, 104, 2 × 104, and 5 × 104 cells/ml.
Comparison of the growth of the ALL3 cells and other CML cell lines at single cell levels
We also used 60-well single cell cloning plates to study the growth behavior of the ALL3 and other leukemic cell lines when they are dispensed at either 1, 2, or up to 20 cells per well. Wells with 1 cell or 2-6 cells did not grow, but in a few wells with 7-20 cells they grew to >100 cells/well but then died rapidly in the tiny wells (Figure 3A and 3B). In contrast cell lines like RWLeu4, K562, and R10- (Figure 3C-E) grow significantly better than ALL3 cells (Figure 3A and 3B) as single or a few cells. R10- cells were less capable of growing as single a few cells than RWLeu4 or K562 but more efficient than ALL3 cells (Figure 3C-E).
Figure 3.
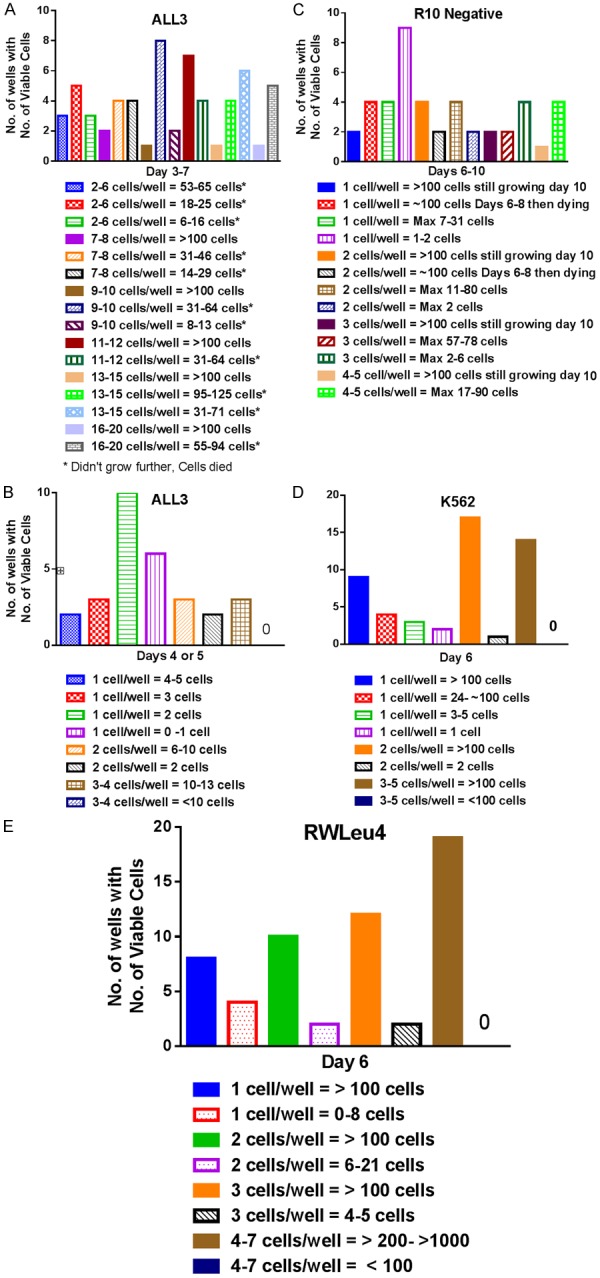
Single cell cloning of the ALL3 cells and other leukemic cell lines using 60-well single cell cloning plates. A. Cloning of ~10 (2 to 20) ALL3 cells in ALL3 media in 60-well single cell cloning plates. No wells with 1 cell (not shown) or 2-6 ALL 3 cells grew to >100 cell/well (Solid bars) and continued growing, and only a few wells starting with 7-20 cells did so. Some wells started with 13-20 cells grew to >90-125 cells, but then the cells died. The maximum cell count per well at 3-7 days is shown. B. Cloning of 1 to 4 ALL3 cells in ALL3 media in 60-well single cell cloning plates. C. Cloning of single or a few R10- cells in 60 well single cell cloning plates. Only 10% of single cells and 20-27% of 2-5 cells continued growing to >100 cells at 10 days. D, E. Cloning of single or a few K562 cells in 60-well plates. 67% of single and 83% of two RW-Leu4 cells and >50% of single and 94% of two K562 cells per well grew to >100 cells and continued growing as did 96% of the cells in wells containing 3 or more cells of either cell line. Cell counts greater than 100 cells per well are inaccurate because of crowding.
In all the single or few cell experiments in single cell cloning plates the cells were counted individually microscopically. When only a few cells were initially present or even 10-20, they were rarely in direct contact with each other. Thus the observations that the wells containing greater than 7-20 ALL3 cells sometimes grew transiently to >100 cells (Figure 3A) suggest that even such small number of cells in the tiny wells were sometimes capable of communicating with each other without being in direct contact in order to initiate proliferation at least transiently, although their growth was not sustained when the cells were transferred to larger wells. In contrast, RwLeu4 and K562 even when started at 1 or 2 cells often grew rapidly in the first 6 days or less to >100 or even 1000 cells in the tiny wells (Figure 3D and 3E), and if then transferred to slightly larger wells would usually continue to grow indefinitely on serial passage. These observations again confirm that a certain minimum number of ALL3 cells are necessary to initiate even transient growth, but only if the cells are in fairly close proximity but not necessarily in direct contact in the tiny wells in the 60-well plates they are able to communicate closely with each other. This behavior is quite unlike that of the long established RwLeu4 and K562 BP CML cell lines in which most of the blasts can sustain proliferation indefinitely starting with only one or two cells.
Growth of the cord blood CD34+ cells in liquid culture and as single cells
Enriched CD34+ from cord blood (CB) cells were stimulated to grow in the presence of GFs when grown at starting cell densities of 400, 1000, 5000, and 9000 cells/ml, reaching their fastest DTs of ~20-30 hr which was independent of starting cell densities; the cells failed to grow without GFs except slightly at 9000 cells/ml (Figure 4A). As shown more clearly using a different scale in Figure 4B, the enriched CD34+ CB cells grew transiently at a starting cell density of 9000 cells/ml without any GFs while at still lower densities they failed to grow. The enriched CD34+ CB cells hardly grew at all without GFs as single or 2 cells, but with 7 or 8 GFs most of the wells with single cells or 2 or 3 cells grew extremely well (Figure 4C and 4D). The experimental observations are described in more detail in the legends. The fact that enriched primitive CB CD34+ cells grew even transiently at 9000 cells/ml but not at lower cell densities with no GFs suggests that they, like ALL3 cells, may also be behaving collectively and communicating with each other at intermediate densities. However, unlike ALL3 cells which don’t respond at all to GFs, the CB CD34+ S/P cells require additional stimuli such as multiple GFs to maintain growth or to initiate cell division at very LD. We have made similar observations with some but not all acute leukemic and BP CML blasts. The blasts obtained from newly diagnosed and untreated patients generally grow much better than those from pre-treated patients or those with very advanced disease.
Figure 4.

Single cell cloning of the enriched CD34+ cord blood (CB) cells using 60-well single cell cloning plates. (A) Growth of enriched CD34+ cord blood cells in liquid culture (QBSF-60) starting at 400, 1000, 5000, & 9000 cells/ml without GF and with 7 or 8 GFs (KL, FLT3, TPO, G-CSF, GM-CSF, IL-3, IL-6, +/- EPO); MFE= maximum fold expansion. The cells didn’t grow at all without GFs except at the highest starting cell density of 9000 cells/ml when they grew slightly. When stimulated with 7 or 8 GFs the cells grew rapidly at all starting cell densities for 13 days, and, depending on the starting density, they underwent 154x to 2500x MFE after which they began to die rapidly as most of the cells matured. The cells usually grew slightly faster with the addition of EPO and sometimes to higher numbers, but also usually with a higher death rate (NV cells). (B) Growth or no growth of same CD34+ CB cells as in (A) without any GFs plotted on a different scale to show transient proliferation at a starting cell density of 9000 cells/ml but not at lower cell densities. The graph shows the initial rapid proliferation of the CD34+ CB cells starting at 9000 cells/ml without any GF with a DT of ~22 hr during the first 48 hr, which then slowed abruptly and almost all the cells were dead by day 15. The cells started at the LD without GFs died promptly. (C) Growth of highly enriched CD34+ CB cells in 60-well single cell cloning plates without GFs and with 7 or 8 GFs. CD34+ CB cells hardly grew at all without GFs as single or 2 cells, but with 7 or 8 GFs respectively 79 and 72% of single cells, 60-86% of 2 cells, and 100% of 3 cells continued growing to >100 cells within 6-10 days. (D) Growth of enriched CB CD34+ cells in 60-well single cell plates when stimulated by 8 GFs, and representative examples of proliferative fate of individual single cells. The growth of representative single or two CD34+ CB cells shown in (C) in the right panel with 8 GFs is shown here in more detail: 20/28 (72%) of single cells grew to >100 viable cells within 6-10 days (blue symbols) while one didn’t grow at all and 6 produced the numbers of cells shown in the (C) and then died (right Panel). Some of the 20 single cells that reached >100 cells kept growing after day 13 until they packed the small well while others began dying after ~10 days. Cell counts >100 or 200 cells/well are only rough estimates because of extreme crowding in the tiny wells.
Comparison of the growth BP CML and AML blasts at single cell levels
The BP CML total blasts or enriched CD34+ blasts didn’t grow at all as single or a few cells without GFs but grew better with 7 GFs (Figure 5A). The BP CML enriched CD34+ blasts and total blasts cells grew similarly when stimulated by 7 GFs (Figure 5B). In contrast, the blasts obtained from AML patients did not grow well at all with or without GFs as single or a few cells (Figure 5C). The AML blasts cells also did not grow in liquid culture when started at HD of 1-5 × 105 cells/ml without GFs but in the presence of 7 GFs they grew very slowly at these relatively high starting cell densities, but died rapidly after about 10 days (Figure 5D).
Figure 5.

Single cell cloning with and without 7 GFs of enriched CD34+ blasts from transformed BP CML and AML patients. A and B. Growth in single cell cloning plates of total blasts and enriched CD34+ blasts obtained from a 32 year old woman with CP CML treated with Imatinib whose disease had undergone blast transformation after about a year. Her WBC was over 70,000/mm3 with 70% blasts. A. leukapheresis was done before resuming treatment and multiple aliquots of the total mononuclear cells (including all blasts) were frozen and later thawed for phenotyping and other studies. Flow Cytometry showed that only 6% of the blasts were CD34+ and these were included with the Total Blasts. Only 6% of the 70% PB blasts obtained by leukapheresis were CD34+ by flow cytometry and only ~0.1% of the CD34+ blasts were recovered after enrichment. Presumably the 6% CD34+ blasts were residual CP CML blasts. A. None of the single or 2 or 3 total blasts per well grew to more than 3-4 cells and most didn’t grow at all without GFs. They grew much better with 7 GFs: 15%, 39%, and 50% respectively of the cells starting at 1, 2, or 3 cells/well grew to >100 cells, and while 44% and 21% of the single and 2 cells didn’t grow at all, most of the wells starting with 3 cells/well that failed to reach >100 cells grew to at least 48-92 cells by Day 9. B. Growth of the 6% enriched CD34+ blasts from the total blasts in single cell cloning plates. Enriched CD34+ blasts and total blasts grew similarly when stimulated by 7 GFs with the latter slightly better. Respectively 11, 16, and 30% of the single, 2, and 3-4 enriched CD34+ blasts grew to >100 cells (solid color) compared to 15, 39, and 50% of the single, 2, and 3 total blasts. C and D. The patient (J.A.) from whom these blast cells were obtained by leukapheresis was a 42 year old man diagnosed with MDS which rapidly progressed to AML with a high number of circulating blasts. He was leukapheresed at another hospital and placed on hydroxyurea, but his WBC rose rapidly again. He had no other chemotherapy. On admission to Memorial Hospital the WBC was 71,000/mm3 with 84% blasts. Leukapheresis was repeated and numerous aliquots of the MNCs (mostly blasts) were frozen for later experiments. 30% of the blasts were CD34+ which were used for this experiment. The 7 GFs and their concentrations were the same as in previous experiments. C. Single cell cloning in QBSF-60 with and without 7 GFs of enriched CD34+ AML blasts from patients J.A. with AML progressed from MDS. All the blasts appeared viable and excluded trypan blue on day 0, but none of the single or few cells grew in single cell cloning experiments and all were dead after 2 days. D. Attempted growth of JA’s AML enriched CD34+ blasts in QBSF at HD (105 and 5 × 105 cells/ml) without and with 7 GFs. There was no growth at 105 or 5 × 105 cells/ml without GFs and the cells were all dead by 2 days. With 7 GFs after an initial sharp decline the surviving cells grew very slowly at starting densities of 105 and 5 × 105 cells/ml for 9 days with DTs of 120-135 hr, but almost all were dead by 2 weeks. The AML blasts were also cloned in methylcellulose, but no colonies were produced (not shown).
LD ALL3 cells can be stimulated to grow in the presence of HD ALL3 cells in transwells
Transwells with polycarbonate membranes with 0.4 µm pore size were used for co-culture experiments (Figure 6A). For this method we used 6-well plates; the bottom wells have a growth area of 9.5 cm2 and hold 2.6 ml while the upper insert growth area is 4.67 cm2 and hold 1.5 ml (Figure 6A). We observed that ALL3 cells growing at 3-4 × 105 cells/ml or higher in the upper insert consistently stimulated growth of the LD ALL3 cells in the lower wells at 0.5 × 104 cells/ml whereas plain media or ALL3 cells at LD in upper inserts failed to do so (Figure 6B). Thus, ALL3 cells did not grow spontaneously at LD but were stimulated to grow by soluble, diffusible factors produced by the ALL3 cells growing exponentially at HD in the upper inserts of the transwells. Our data show that ALL3 cells growing at starting cell concentrations of 3-4 × 105 cells/ml provide full stimulation of LD ALL3 cells in the lower wells, but with a DT of ~24-30 hr they rapidly reach saturation density within a few days, begin to die and release toxic factors that inhibit rather than stimulate growth of cells in the lower wells. If started at still higher cell density in the upper inserts they reach saturation density so quickly that stimulation of the LD ALL3 cells is less than at starting cell densities of ~3-4 × 105 cells/ml.
Figure 6.
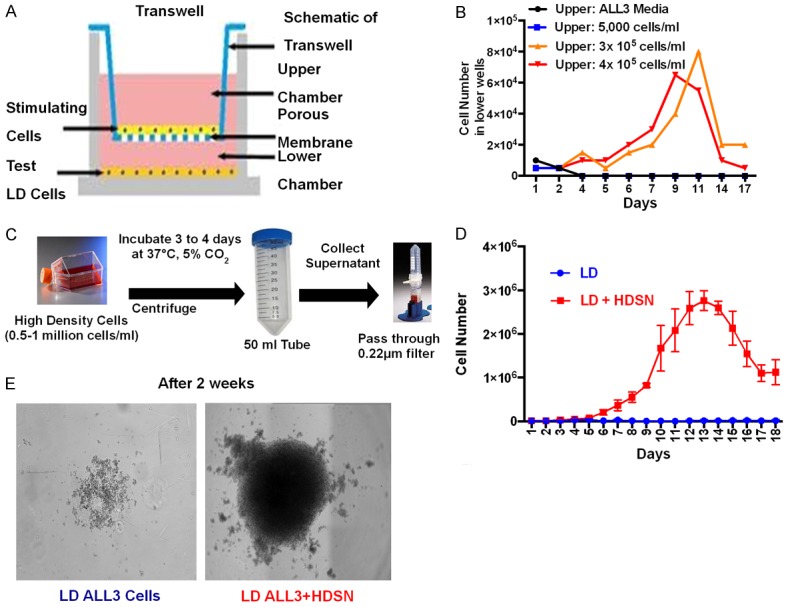
Effect of HD ALL3 supernatants on growth of LD ALL3 cells. A. Schematics of transwell experiment. The upper insert can hold 1.5 ml and lower well can hold 2.6 ml. Upper insert and lower well are separated by a membrane with 0.4 μm pore size (growth area of insert is 44 cm2 for 100 mm dish in 75 mm transwell). Stimulating cells to be tested were kept in upper inserts and test cells in lower wells. B. Transwell experiments demonstrated stimulation of the LD ALL3 cells in the lower wells by HD ALL3 cells growing exponentially in the upper inserts. The graph shows total number of viable cells per ml in lower wells at different conditions as shown in legends. The ALL3 cells growing at HD of 3 × 105-4 × 105 cells/ml in the upper inserts stimulated the growth of the LD ALL3 cells at 5,000 cells/ml in lower wells shown as orange or red color lines, respectively. LD ALL3 cells in lower wells did not grow in the presence of either ALL3 media only or LD ALL3 cells (5,000 cells/ml) in the upper inserts shown as black and blue lines, respectively. The growth of the viable LD ALL3 cells in lower wells was determined using the trypan-blue exclusion assay method. C. Scheme showing collection of filtered HDSN. The HD ALL3 cells were grown at 0.5-1 million cells/ml in ALL3 media for 3 or 4 days. Then, the ALL3 cells were centrifuged and HDSN was filtered using 0.2 μm 50-ml Millipore tubes. Filtered HDSN was used for various assays. D. Comparative growth of the LD ALL3 cells in the presence (red line) and absence (blue line) of filtered HDSN. The growth was followed using the trypan blue exclusion method for 18 days. The y-axis represents the total number of viable cells per ml. E. Light microscopy images showing the lack of growth of the LD ALL3 cells in the absence (left panel) and presence of HDSN (right panel) after two weeks.
Filtered HDSN stimulated growth of LD ALL3 cells but not of HD ALL3 cells
As described above, toxic factors released by continuously growing HD ALL3 cells in upper inserts of transwells as they approached saturation density hampered growth of the LD ALL3 cells when kept in the lower wells for too long. Thus, it was necessary to develop an alternative method to observe the stimulatory effect of diffusible factor(s) secreted by the HD ALL3 cells on the LD ALL3 cells. To do so, the ALL3 cells were grown at starting HD of 0.5-1 × 106 cells/ml in ALL3 media. After 3 or 4 days of growth cell suspensions were centrifuged and supernates were carefully filtered using a 0.2 µm filter 50-ml tube leaving the cell pellets behind. The detailed scheme for obtaining filtered HDSN is shown in Figure 6C. We then used the filtered HDSN to observe its effect on the LD ALL3 cells. In the presence of HDSN collected from HD ALL3 cells, the LD ALL3 at 0.5-1 × 104 cells/ml grew nicely with a DT of ~24-26 hr whereas LD ALL3 cells did not grow at all in plain ALL3 media without HDSN (Figure 6D). The trypan blue exclusion method was used to observe the growth of the cells. As shown in Figure 6E, after two weeks of growth in 6-well plates the LD ALL3 cells were growing excellently in the presence of filtered HDSN but hardly at all in plain ALL3 media.
The [3H]-thymidine uptake assay was also used to confirm our findings. The scheme for the [3H]-thymidine uptake assay is described in detail in the experimental method section and the scheme is also shown in Figure 7A. The LD ALL3 cells were kept in HDSN collected from the HD ALL3 cells after 3 days of growth, and followed for up to 18 days using the [3H]-thymidine uptake assay. The LD ALL3 cells grow exponentially in the presence of filtered HDSN providing maximum stimulatory activity around ~10-15 days and decline in activity after ~2 weeks of growth (Figure 7B). It is important to note that the timing of maximum stimulatory activity of the HSDN on the LD ALL3 cells depends on the starting cell density of HD ALL3 cells and also on the timings when the HDSN has been collected (either 3, 4, or 5 days of HD ALL3 cells’ growth). In most of our experiments, we grew cells at high densities of 0.5-1 × 106 cells/ml for 3-4 days.
Figure 7.
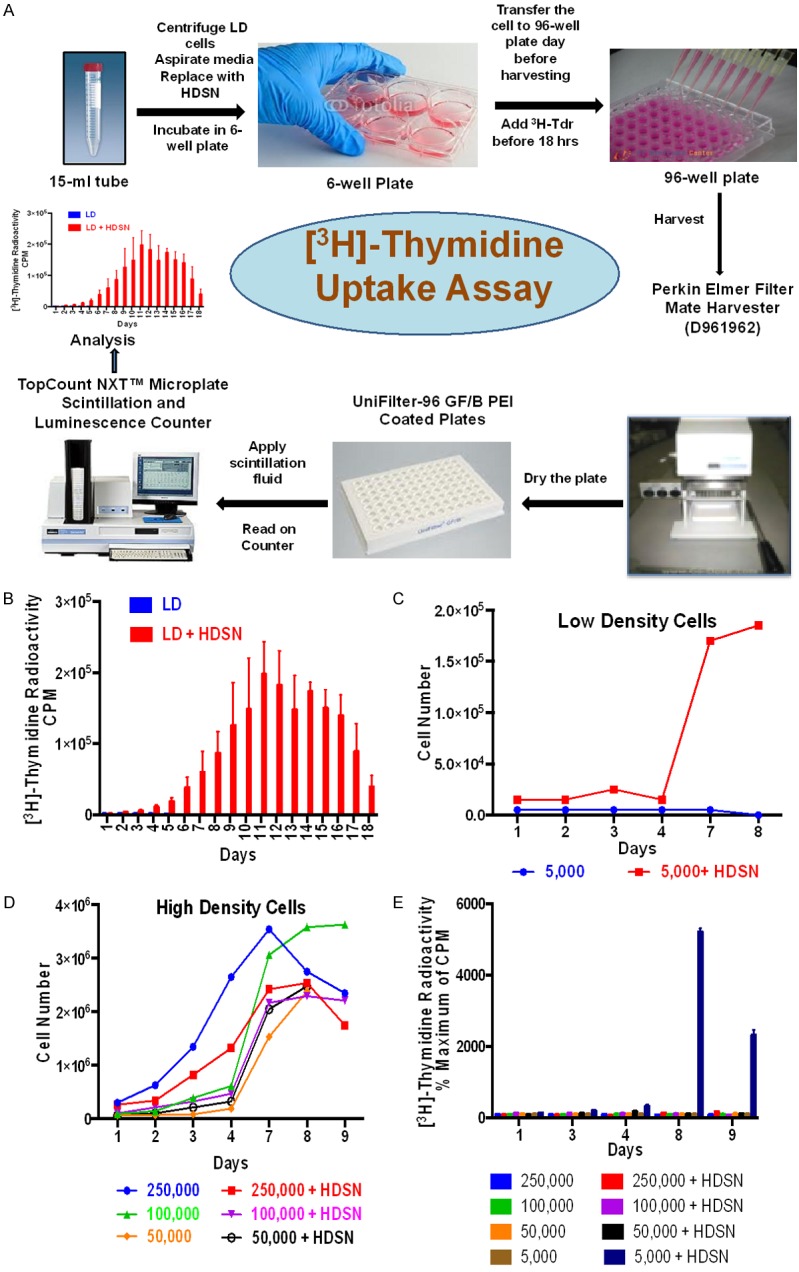
Effect of supernates on the growth of LD and HD ALL3 cells using trypan-blue exclusion method and [3H]-thymidine uptake assay. (A) Scheme showing steps used for performing the [3H]-thymidine uptake assay. Refer to experimental method section for more detail. (B) The filtered HDSN were collected and mixed with LD ALL3 cells. Growth of the LD ALL3 cells in the presence or absence of HDSN was followed using the [3H]-thymidine uptake assay. The y-axis represents radioactive counts per minute (CPM). Blue bars represent LD ALL3 cells alone in media without HDSN and red bars represent LD ALL3 cells growing in HDSN. (C) Comparative growth of the LD ALL3 cells in absence (blue line) or presence (red line) of HDSN as followed using trypan blue exclusion. (D) Comparative stimulatory activity of the same HDSN used in (C) on the growth of HD ALL3 cells at starting cell densities of 2.5 × 105, 105 and 0.5 × 105 cells/ml followed using the trypan-blue exclusion method. The HDSN did not stimulate further growth of the ALL3 cells grown at HD at starting cell densities of 2.5 × 105 (red line), 105 (pink line) or 0.5 × 105 cells/ml (black line) and was moderately inhibitory. (E) The effect of the HDSN on the growth of HD and LD ALL3 cells using [3H]-thymidine uptake assay. The y-axis represents percentage CPM (% of maximum CPM) comparing stimulated and non-stimulated ALL3 cells on that particular day. As in (C) only the LD ALL3 cells at 5,000 cells/ml were stimulated by HDSN to grow.
Filtered HDSN stimulated growth of the LD ALL3 cells but not of the HD ALL3 cells
We also investigated the effect of HDSN on the HD ALL3 cells to see if there was any beneficial effect on the growth of the HD ALL3 cells using both the trypan-blue exclusion method and [3H]-thymidine uptake assay. The filtered HDSNs were collected as described before and stimulated ALL3 cells at different HD and LD. As expected, profound stimulatory activity of the LD ALL3 cells was observed at 0.5 × 104 cells/ml in the presence of HDSN (Figure 7C and 7E), whereas there was no growth advantage or stimulation of HD cells at 2.5 × 105, 105, or 0.5 × 105 cells/ml in the presence of HDSN (Figure 7D and 7E). Growth of the HD ALL3 cells was actually diminished in the presence of HDSN (Figure 7D). These data suggest that ALL3 cells below a certain cell density, in this case <10,000 cells/ml, require additional stimulus without which they do not survive, whereas ALL3 cells growing at HD either don’t need more stimulation or the presence of the diffusible factor generates an abnormal signal for HD ALL3 cells that are already growing vigorously leading to their diminished growth. Another probable contributing cause may be that the HDSN was obtained from very HD ALL3 cells starting at 0.5-1 × 106 cells/ml and grown for 3 days before harvesting their HDSN. Thus the cells were approaching saturation density and had undoubtedly exhausted more of their nutrients than the test LD ALL3 cells starting at lower cell densities (0.5-2.5 × 105 cells/ml) in fresh media.
Similar findings were observed in which CB MNCs had the greatest stimulatory effect on the LD ALL3 cells at 0.5-1 × 104 cells/ml but which failed to stimulate ALL3 cells started at 5 × 104 cells/ml.
Supernatants from some but not other leukemic cell lines also stimulate growth of LD ALL3 cells
We also used other established leukemic cell lines to test whether the supernates collected from these cell lines can also stimulate the growth of the LD ALL3 cells or not. Figure 8A shows the growth of the established leukemic cell lines tested at a starting cell density of 3.5 × 105 cells/ml; they all grew but with different DTs and all with more than ~90% cell viability (Figure 8B). As shown in Figure 8C, the supernatants collected from the cell lines HD ALL3, R10+, R10-, and BM185 stimulated the growth of the LD ALL3 cells whereas LD ALL3, RwLeu4, SKL7 and K562 had no effect on the growth of the LD ALL3 cells (Figure 8C). The data also indicates that the stimulation kinetics on the LD ALL3 cells are different for different leukemic cell line supernates, suggesting there may be different secreted stimulatory factors or different levels of the same diffusible factor(s) as secreted by the HD ALL3 cells.
Figure 8.
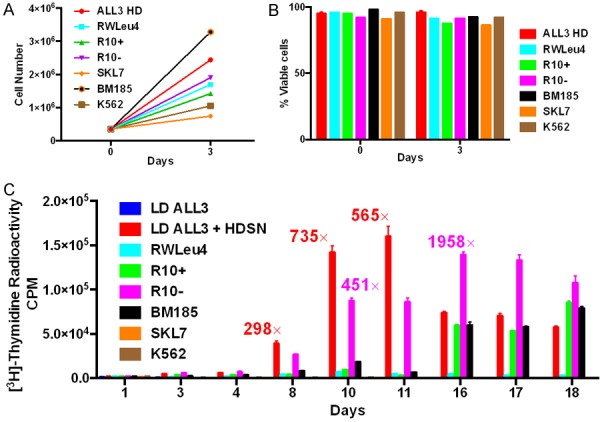
Effect of supernates from established leukemic cell lines on the growth of the LD ALL3 using [3H]-thymidine uptake assay. A. Growth of the established leukemic cell lines started at HD of 3.5 × 105 cells/ml. B. The viability of all the HD cell lines was 90% or more at both 1 and 3 days determined using the trypan blue exclusion method. C. The effect of supernates collected from the established leukemic cell lines started at HD of 3.5 × 105 cells/ml on the LD ALL3 cells using [3H]-thymidine uptake assay. As expected HD ALL3 SNs were the most stimulatory for the first 11 days while the unstimulated LD ALL3 cells didn’t grow at all. The next most stimulatory was R10- cells which peaked later at day 16. The recorded fold increases of the stimulated over the unstimulated ALL3 cells measures the difference on each day so 1958 on day 16 is not necessarily more stimulatory than 565 on day 11 as more of the control unstimulated LD ALL3 cells have died so their CPM are lower on day 16 than day 11. The SN of HD R10+ and BM185 cells were the next most stimulatory but this occurred still later (Days 16-18). SN of RWLeu4, SKL7, and K562 were not stimulatory at all. The y-axis represents radioactive CPM.
Presumably the non-stimulatory supernates of long established BP CML lines, RWLeu4 and K562, which usually grow vigorously as single cells as shown earlier, have fully adapted over many years of continuous passage to surviving and growing as single or a few cells, and no longer have a need for collective communications in order to grow. This observation leads to an important distinction because many conclusions derived from experiments on such long passaged cell lines are not necessarily valid if too readily extrapolated to reach similar conclusions regarding leukemic or other neoplastic cells recently derived directly from patients. In particular this distinction may apply to cells that were immediately frozen after collection from patients such as the ALL3 cell line.
Effect of cord blood, CP and BP CML CD34+ and MNC on the growth of the LD ALL3 cells
Next, we used pooled CB MNCs and highly enriched CB CD34+ cells to test whether they would also stimulate the growth of the LD ALL3 cells (Refer to [37] for enrichment protocol). In all cases, transwell systems were used to assess the stimulatory effect of test cells on the LD ALL3 cells. Growth of the CB MNCs and CD34+ cells with or without GF stimulation, and ALL3 cells in upper inserts are shown in Figure 9A. Neither the CB MNC or CD34+ cells grew without GFs and the more primitive enriched CD34+ cells grew faster to higher maximum cell numbers. As shown in Figure 9B, in addition to the HD ALL3 cells both CB MNCs and enriched CB CD34+ cells stimulated the growth of the LD ALL3 cells in the lower wells. But the CB MNCs growing in the presence of 7 GFs were more stimulatory than CB MNCs grown with 0 and 3 GFs, and CD34+ cells with 0, 3, and 7 GFs despite the latters’ faster growth and to higher numbers in the upper inserts as shown in Figure 9A. In another set of experiments as shown in Figure 9C and 9D, we observed similar stimulatory effect of GFs stimulated CB CD34+ and MNCs on the LD ALL3 cells. As shown in Figure 9E, CB MNCs cells stimulated by 3 or 8 GFs stimulated the growth of the ALL3 cells at LD of 0.5-1 × 104 cells/ml, but had no effect on ALL3 cells started at 5 × 104 cells/ml since they grow fine at this intermediate cell density without need for additional stimulation. The experimental observations are described in more detail in the Figure 9 legends. The most surprising observation was the late stimulatory effect of the primitive CB CD34+ cells with no GFs on the LD ALL3 cells in the lower wells (Figure 9B, left panel) since the CD34+ cells without GFs didn’t grow at all in the upper inserts and were all non-viable by ~ day 5, at least as estimated by trypan blue staining (Figure 9A, middle panel). This unexpected finding illustrates the complexity of trying to understand the stimulatory effects and interactions of different cells, here suggesting that even dying or apoptotic [64-68] or senescent [69-72] normal primitive cells might release factors that revitalize and stimulate growth of leukemic cells that were themselves almost completely dead, in this case after not growing for the first 16 days (Figure 9A).
Figure 9.

Experiments showing the stimulatory effect of pooled cord blood cells growing at high cell densities in the upper inserts of transwells on the ALL3 cells in the lower wells at a LD at which they will not grow without stimulation. (A) Comparative growth of HD and LD ALL3 cells, and pooled CB MNCs and enriched CD34+ cells starting at a HD (4 × 105 cells/ml) in the upper inserts of transwells. Pooled enriched CB CD34+ cells starting at a HD (4 × 105 cells/ml) without GFs and with stimulation by 3 and 7 GFs in the upper inserts of transwells. The CD34+ cells grow faster and to higher cell densities (to ~9-10,000,000 cells/ml) than the MNCs when stimulated by 3 or 7 GFs (to ~1.4-2.4 × 105 cells/ml, and neither grew with no GFs. The HD ALL3 cells grew faster then the CB CD34+ cells, but the latter reached about double the HD ALL3 ultimate highest cell density a few days later. (B) Comparative growth of ALL3 cells at a LD (5000 cells/ml) in the lower wells of transwells when stimulated by the following cells growing in the upper inserts: Left Panel: LD (5000/ml) alone and HD ALL3 cells (3 × 105 cells/ml)); enriched CB CD34+ cells (3 × 105/ml) without GFs and with 3 or 7 GFs, and Right Panel: CB MNCs with and without the same 3 and 7 GFs. The CB MNCs grew slower and to lower maximum cell densities in the upper inserts than the enriched CD34+ cells (A), but the former were more stimulatory to the LD ALL3 cells which grew to >2 × 106 cells/ml in the lower wells when stimulated by the MNCs in the upper inserts with 7 GFs (B, right panel). As shown in the left panel of (C), the CD34+ cells with 3 GFs were more stimulatory to the ALL3 cells in the lower wells than the CD34+ cells with 7 GFs, probably because the latter were approaching saturation density sooner. Unexpectedly the CB CD34+ cells with no GFs which didn’t grow in the upper insert (A) had minimal stimulatory effect on the LD ALL3 cells until after day 16 when the ALL3 began to grow with a DTs of ~24 hr during the next 6 days (B, Left Panel). The CB MNCs without GFs may also have begun to stimulate growth of the ALL3 cells by day 16 (B, right panel). (C) Growth of CB MNCs and CD34+ cells with and without 7 GFs in upper inserts of transwells to test the HD CB cells’ ability to stimulate the growth of the LD ALL3 cells in lower wells. This was another pooled CB transwell experiment in which the CB CD34+ cells without GFs at a high starting cell density in the upper insert grew slowly in the upper inserts but the CB MNCs did not (left panel). With 7 GFs the HD CB CD34+ cells after an initial decline, grew faster but had a high death rate as many cells differentiated and died, while the CB MNC barely grew (right panel). (D) Stimulatory effect of these same CB CD34+ and MNCs with 7 GFs in the upper inserts on the LD ALL3 cells in the lower wells. Despite the failure of the MNCs to grow in the upper insert, both the MNCs and CD34+ cells stimulated the LD ALL3 cells to grow with a DT of ~26 hr. Note the earlier growth but the much higher death rate of the ALL3 cells stimulated by the HD CB CD34+ cells (left panel) than by the HD MNCs (right panel). (E) Stimulatory effect of HD CB MNCs with no, 3 and 8 GFs on the ALL3 cells starting at three different cell densities in the lower wells. As expected based on the other experiments, the CB MNCs had the greatest stimulatory effect on the LD ALL3 cells started at 0.5-1 × 104 cells/ml, and negligible effect on ALL3 cells started at 50,000 cells/ml since they grow well by themselves at this intermediate cell density. The GFs used in all these experiments have no stimulatory effect on the ALL3 cells themselves (see Figure 2), only on the CB cells in the upper inserts.
Next, we investigated whether or not CP CML MNCs and enriched CD34+ cells can stimulate growth of the LD ALL3 cells. The growth of the CP CML CD34+, MNCs and ALL3 cells in the upper inserts of transwells are shown in Figure 10A and 10C. As expected, the CML CD34+ cells grew very slowly without GFs in the upper inserts and progressively better with 3 or 7 GFs. However these same slowly growing CP CML CD34+ cells in the absence of GFs in the upper in the upper inserts were more stimulatory to the LD ALL3 cells than the same cells stimulated with 3 or 7 GFs (Figure 10A and 10B). This is presumably because although they only grew slowly without GFs, they remained viable as primitive cells and were not forced to differentiate and die as the GF stimulated cells were in the 2 right panels of Figure 10A. The growth of the CP CML MNCs with or without stimulation by 3 or 7 GFs are shown in Figure 10C and are also described in the legends of Figure 10. Despite their slow growth, they were all stimulatory to the LD ALL3 cells growing in lower wells of transwells (Figure 10D).
Figure 10.
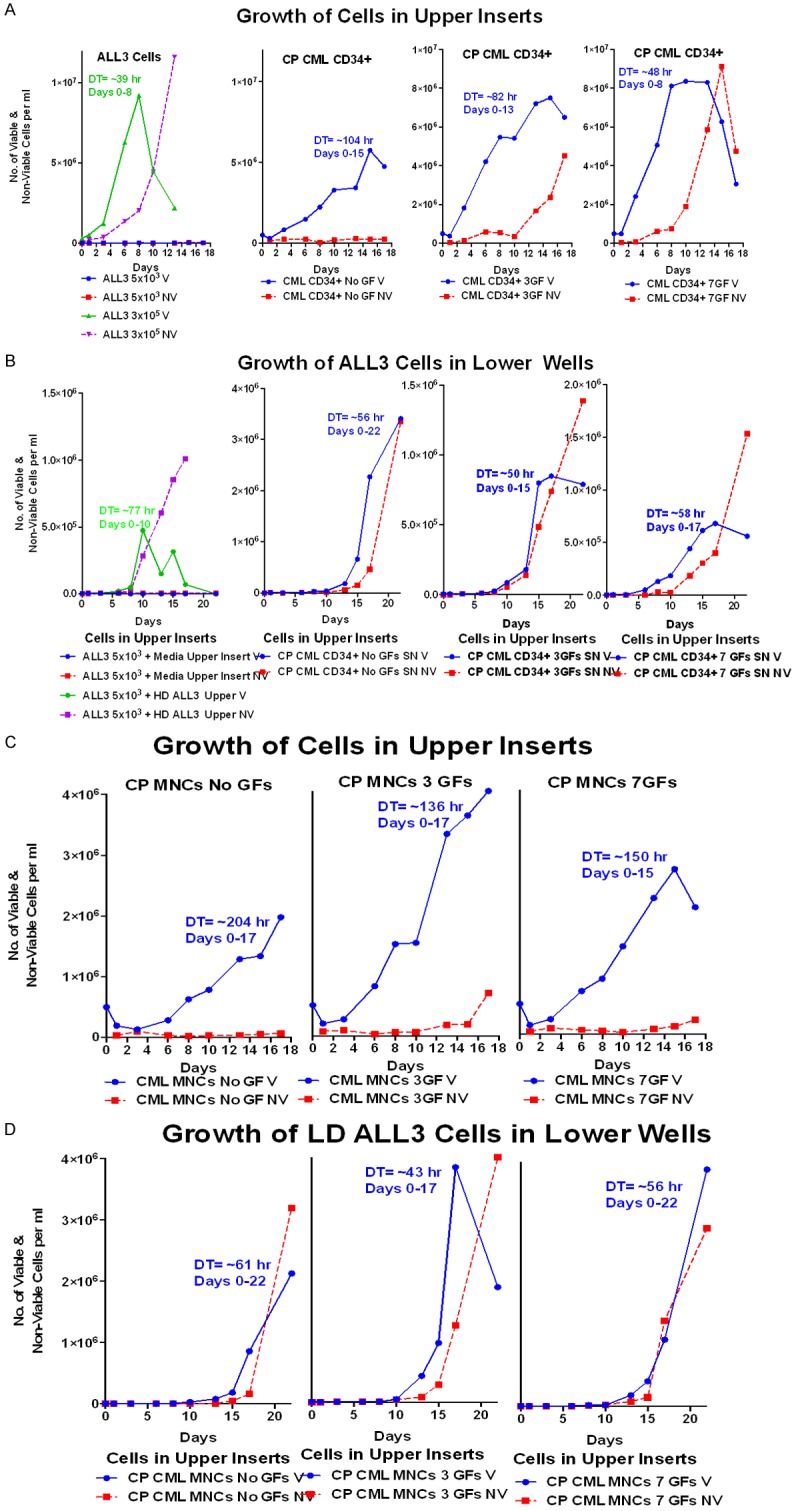
The stimulatory effects of CP CML MNC and enriched CD34+ cells on the growth of ALL3 cells at LD. (A) Growth of the ALL3 cells starting at 5,000-3 × 105 cells/ml and enriched CP CML CD34+ cells, all without any GFs, in upper inserts of transwells. The ALL3 cells didn’t grow at all starting at 5000 cells/ml but grew rapidly from 3 × 105 to saturation density ~9 × 106 cells/ml at about 8 days when they begin to die rapidly. The HD CP CML CD34+ cells grew slowly for about 15 days without GFs, meanwhile maintaining good viability. The HD CP CML CD34+ cells grew progressively faster and reached higher saturation densities with GFs, but this was accompanied by increasing cell death. (B) Comparison of the growth stimulatory activities of diffusible factors secreted by HD ALL3 cells (5 × 105 cells/ml) and HD CP CML CD34+ cells growing in the upper inserts, both without any GFs, on LD ALL3 cells (5000 cells/ml) in the lower wells. The HD ALL3 cells stimulated growth a bit faster, but the DTs was slower, the LD ALL3 cells died faster and didn’t reach nearly as high a maximum cell density as did the LD ALL3 cells stimulated by the HD CP CML CD34+ cells, even though the latter were only growing slowly in the upper insert in the absence of GFs stimulation. Growth of LD ALL3 cells (5000 cells/ml) in lower wells stimulated with enriched CP CML CD34+ cells in the upper inserts of transwells with 0, 3 and 7 GFs. The HD CP CML CD34+ cells in the upper insert without any GFs is clearly secreting more stimulatory diffusible factors to enhance growth of LD ALL3 cells than the same CD34+ cells stimulated themselves with 3 or 7 GFs, but in all cases there is a high loss of viability accompanying the growth of the LD ALL3 cells. The greater stimulation without GFs is probably because many of the stimulated HD CD CD34+ cells are forced to differentiate and begin dying whereas the CD34+ cells without cytokines remain viable longer as more primitive S/P cells as shown in (A). (C) Growth of CP CML MNCs in upper inserts of transwells without or with 3 and 7 GFs. (D) Growth of the LD ALL3 cells in lower wells with CP CML MNCs in upper inserts of transwells with 0, 3 and 7 GFs (same cells from C). The CP CML MNCs, which are far less enriched for S/P cells than the CD34+ cells, usually grow slower than the latter, with or without stimulation by the same GFs and are also usually less stimulatory to the LD ALL3 cells.
We also used the [3H]-thymidine uptake assay to confirm the stimulatory activity of CP CML MNCs and enriched CD34+ cells growing with or without GFs stimulation. The CP CML MNCs and CD34+ cells were grown with or without stimulation by GFs (Figure 11A and 11C). After 6 days of growth supernates were collected to test their stimulatory activity for the LD ALL3 cells. The slow growing CP CD34+ cells without GFs were by far the most stimulatory to the LD ALL3 cells rather than CP CD34+ cells grown with 3 and/or 7 GFs as confirmed in two separate experiments (Figure 11A-D). Again this is because the slow growing CD34+ cells without GFs remained largely as primitive cells and thus were far more stimulatory than the same cells forced to differentiate by the GFs. The CML MNCs grew poorly and their SNs were not stimulatory to the LD ALL3 cells or at best only very slightly on day 2 in the 1st experiment (Figure 11B) These observations were further confirmed with another set of experiments as shown in Figure 11E-H.
Figure 11.

Stimulatory effects of CP CML MNC and enriched CD34+ cells on the growth of ALL3 cells at LD using [3H]-thymidine uptake assay. (A and C) Comparison of the growth of CP CML MNCs and enriched CD34+ cells which were obtained from fresh/frozen leukapheresis sample from a newly diagnosed patient with CP CML presenting with a WBC of 700,000/mm3. Aliquots of the frozen MNC were thawed, highly enriched for CD34+ cells, and the growth of the MNCs and CD34+ cells at a HD (5 × 105/ml) in QBSF-60 with and without GFs stimulation was compared. The CD34+ cells grew about twice as fast with GFs stimulation as those without GFs while the MNCs hardly grew at all. (B and D) The supernates from these two HD cultures (A and C) were collected after 6 days to determine if they would stimulate the growth of LD ALL3 cells (5000 cells/ml) at which they wouldn’t grow without stimulation as measured by [3H]-thymidine uptake assay. The representative numbers indicate the fold increase in uptake on that particular day of the cells growing with added HDSN compared to the control LD ALL3 cells with no HDSN. The very slow growing HD CD34+ without GFs were by far the most stimulatory with the maximum increased fold difference in CPM between the control and stimulated ALL3 cells (2059x) occurring on day 17. The greater stimulatory activity of the HDSN of the CD34+ cells without GFs was probably because the GFs induced the majority of the CP CML cells to begin differentiating and the differentiated cells were no longer producing the same proliferation triggering factors secreted by the more primitive CML CD34+ S/P cells. The MNCs hardly grew and their HDSN didn’t stimulate the LD ALL3 cells except minimally on day 2. (E-H) Repeat experiment similar to (A-D). Because of the LD ALL3 cells stimulated by the HDSN of the CD34+ cells with no GFs were strikingly stimulated, this experiment was repeated using the same protocol and the same CML cells obtained from another frozen-thawed aliquot from the same patient except the CD34+ and MNCs cultures were started at an even higher cell density (9 × 105 cells/ml). The results were similar except that the maximum fold increase in CPM of the CD34+ cells with no GFs occurred sooner on day 11 and was lower (2247x control) than in the first experiment. The same CD34+ cells starting at almost twice the cell density as in the first experiment grew more slowly, and neared saturation density sooner so they probably were producing less stimulatory factors by day 6 when the HDSNs were collected.
Next, we used blastic phase (BP) CML MNCs and enriched CD34+ cells to test their effect on stimulation of the LD ALL3 cells. The BP CML MNCs and enriched CD34+ cells were grown with or without stimulation by GFs for 16 days (Figure 12A). As usual, the BP CML CD34+ blasts grew only slowly and transiently after an initial sharp decline with 7 GFs and not at all with no GFs in liquid culture. The BP CML MNCs didn’t grow at all (Figure 12A, right panel). Supernates from each group from Figure 12A were collected to observe if they had any stimulatory activity to the LD ALL3 cells as measured by the [3H]-thymidine uptake assay (Figure 12B), but none of the BP CML supernates stimulated uptake of [3H]-thymidine by the LD ALL3 cells (Figure 12B and 12C). We also used transwell experiments to test BP CML cells obtained directly from patients. Whereas the BP CML CD34+ cells didn’t grow in the upper inserts in the presence of GFs (Figure 12D), they did stimulate late and slow growth of the LD ALL3 cells to some degree (Figure 12D, right panel). When GFs were added multiple times to maximally stimulate the BP CML CD34+ blasts freshly obtained from a newly diagnosed patient with BP CML with a rapidly rising blast cell count in the peripheral blood, the blasts grew quite well in the upper inserts of transwells with 7 GFs and more slowly with 3 GFs or none (Figure 12E). These same BP CML CD34+ blasts also stimulated the LD ALL3 cells to proliferate (Figure 12F, right panel), but later than HD ALL3 cells (Figure 12F, left panel). The late stimulation of the LD ALL3 cells shown in the right panel by the BP CML CD34+ cells without any GFs is at first glance surprising since these cells only grew quite slowly in the upper insert (Figure 12E). However as discussed earlier, similar late stimulation of growth of the LD ALL3 cells by poorly or non-growing CB and CP CML CD34+ cells in the absence of GFs was previously observed repeatedly. In all of these and other transwell experiments growth stimulation of LD ALL3 cells was accompanied by high rates of cell death, presumably either due to exhaustion of the media in the small volume transwell chambers, or differentiation and terminal maturation in the case of GF stimulation.
Figure 12.
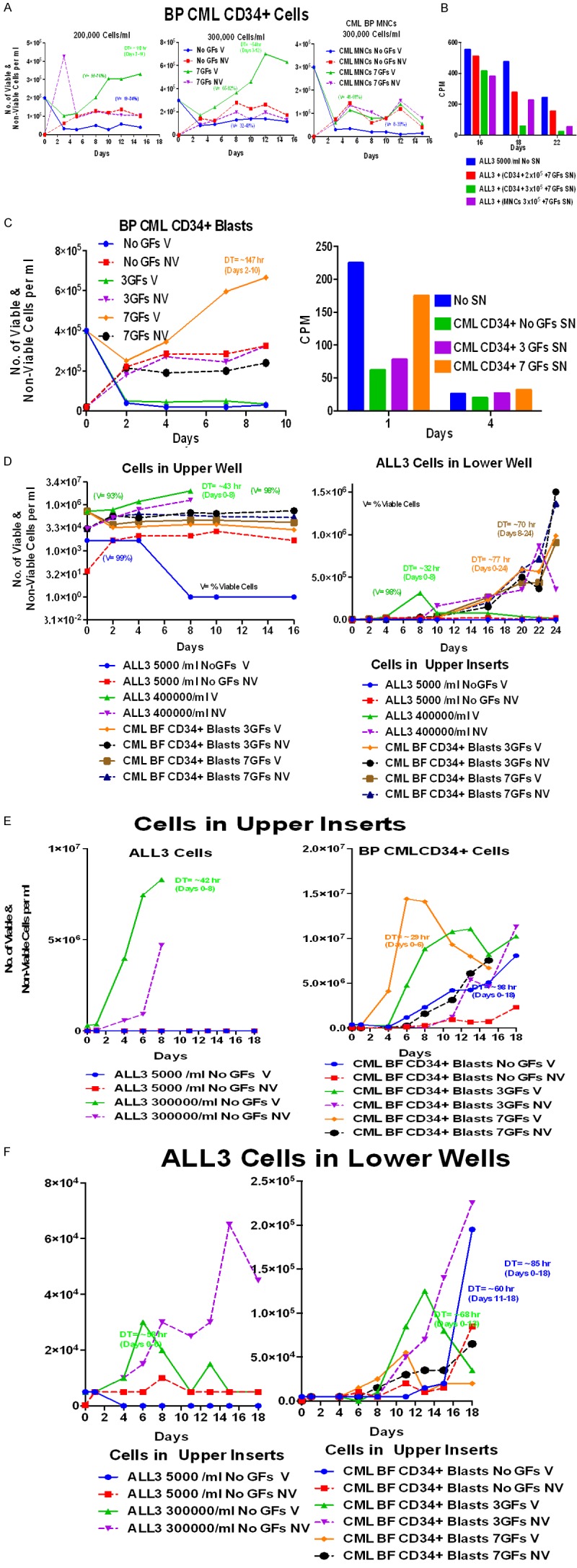
Effect of BP CML MNCs or enriched CD34+ cells on stimulation of the LD ALL3 cells. These experiments were only possible using BP CML cells from occasional BP CML patients because the blasts from most patients with advanced BP disease blasts died too rapidly to obtain meaningful results. (A) Growth of BP CML MNC and enriched CD34+ cells in QBSF medium without and with 7 GFs. After a sharp decline the surviving CD34+ cells from a BP CML grew slowly in QBSF-60 at HD for about 12-14 days when stimulated with 7 GFs, but not without; the MNCs hardly grew at all. (B) Stimulatory activity of BP CML MNCs and CD34+ cells stimulated with 7 GFs in (A). The MNCs and CD34+ culture supernates were collected on day 16 to replace the media of freshly prepared LD ALL3 cells to see if any of the SNs would stimulate the LD ALL3 cells to proliferate, but none of the SN were stimulatory; only the CD34+ SN plus 7 GFs is shown. (C) Growth of BP CML enriched CD34+ blasts at HD from another BP CMP patient grown in QBSF-60. All the cells from the cultures without or with only 3 GFs died promptly but some surviving CD34+ blasts stimulated with 7 GFs grew very slowly for about 10 days (DT= ~147 h) and then died (left panel). None of the supernates from these BP CML HD CD34+ cultures was stimulatory to the LD ALL3 cells (right panel, C). (D) Right Panel: The lack of growth and rapid loss of viability of the enriched CD34+ blasts from another BP CML patient even when stimulated by 3 or 7 GFs. LD ALL3 cells (5000 cells/ml) also in the upper inserts of transwells were 50% non-viable by the day 2 and 100% by day 8. As a control, ALL3 cells at HD (4 × 105 cells/ml) grew well without any GFs in the upper inserts. Left Panel: Stimulatory effects of these cells in the upper inserts on the LD ALL3 cells in the lower wells. The HD ALL3 cells stimulated rapid growth of the LD cells for the first 8 days, but the cells then died quickly, probably because the HD media was exhausted; as expected the LD ALL3 cells in the upper inserts were not stimulatory. Unexpectedly, although the HD CML BP CD34+ cells didn’t grow at all in the upper inserts (D), they did stimulate some of the LD ALL3 cells to proliferate slowly beginning around days 8-10, albeit with a continuing high loss of viability. (E) Growth of LD and HD ALL3 cells without GFs and HD enriched CD34+ BP CML blasts from a patient with newly diagnosed, untreated BP CML in upper inserts of tranwells stimulated with 0, 3, and 7 GFs added on days 0, 4, and 6. Enriched CD34+ cells were obtained from a newly diagnosed untreated patient with BP CML with over 80,000 circulating blasts/mm3. The CD34+ blasts started at 4 × 105 cells/ml in the upper transwell inserts grew rapidly to over 14 and 10 million cells/ml respectively when stimulated by 7 and 3 GFs, and even the blasts without GFs grew slowly for 18 days. The left panel shows that as usual the LD ALL3 cells didn’t grow at all while the HD grew with a DT of 42 hr accompanied by a progressive increase in non-viable cells. (F) Growth of the LD ALL3 cells (5000 cells/ml) in lower wells in transwells stimulated by cells in upper inserts as shown. As usually observed, the HD ALL3 in the left panel stimulated the LD ALL3 cells to proliferate sooner than the BP CML CD34+ cells, but not to as high an ultimate cell peak.
Diffusible factors secreted by all the primary cells tested were more or less stimulatory to the LD ALL3 cells, but the degree of stimulation was quite variable, probably for many reasons, including different types of normal and tumor cells, cells from different patients and the quality of cord blood and other samples. The mechanisms responsible for stimulation of the test LD ALL3 cells by the same and other normal (cord blood) and CP and BP CML and AML cells are not known and undoubtedly are highly complex and varied.
ALL3 cells had no quiescent cells and their proliferative capacity is not affected by HDSN
Next we tested whether non-growing LD ALL3 become quiescent or not using various proliferation markers. We found that non-growing LD ALL3 cells were poised to begin proliferating since 98-100% were Ki67+, similar to growing HD ALL3 cells (Figure 13A). This is as expected because the LD ALL3 cells were obtained by dilution of rapidly growing HD cells. We also used EdU staining as a proliferation marker to determine whether ALL3 cells at HD and LD have different proliferative profiles. After 24 hr, there were ~28% and ~41% of HD and LD EdU positive cells respectively (Figure 13C, left and middle upper panels) and on day 2 there were ~25% and ~40% EdU positive cells present in HD and LD ALL3 cells respectively (Figure 13B). Results of staining with BrdU were almost identical to EdU staining (not shown).
Figure 13.
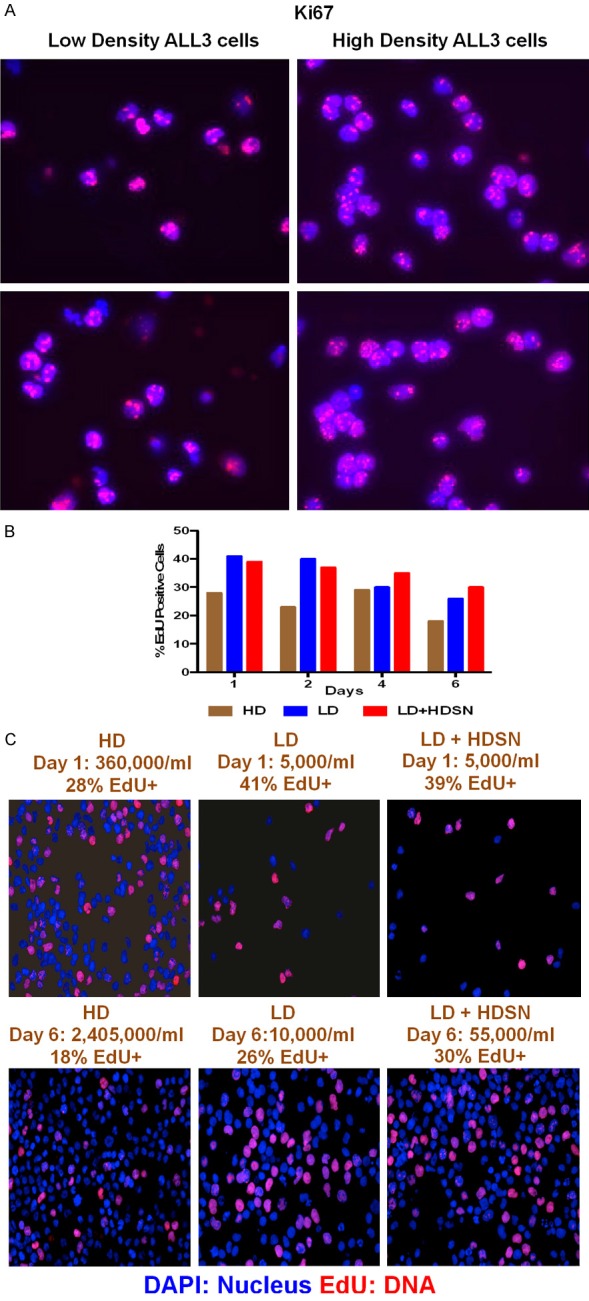
Effect of ALL3 HDSN on proliferation of LD ALL3 cells. A. Pictures of LD and HD ALL3 cells on day 2 that were stained with the MIB-1 Ki67 antibody and counter stained with nuclei stain DAPI. The LD cells hadn’t grown at all by day 2, their viability was only 50%, but >90% of the viable cells were Ki67+. The HD cells had grown to 4.85 × 105 cells by day 2, were 84% viable, and 100% were Ki67+. B. Bar graph of the HD and LD ALL3 cells started on day 0 at 20 × 104 and 0.5 × 104 cells/ml and cells were stimulated using HDSN were pulse labeled with EdU at days 1, 2, 4, and 6. Percent of EdU+ cells are shown on each day. C. Representative pictures of the HD and LD ALL3 cells started on day 0 at 2 × 105 and 5 × 103 cells/ml that were pulse labeled with EdU at days 1 and 6. Serial pulse labeling with EdU for ALL3 cells growing for 6 days in ALL3 media at both HD of 2 × 105 and LD of 5 × 103 cells/ml, the latter with and without addition of HDSN from another culture of ALL3 cells growing exponentially for 3 days starting at 3 × 105 cells/ml. Despite their poor growth, the LD cells alone initially had a higher percentage of cells (41%) incorporating EdU (middle panel) than the proliferating HD cells (left panel; 28%), but the poorly growing LD cells in S phase dropped to 26% by 6 days (middle panel) whereas the percentage of LD cells in S phase stimulated to grow by the HDSN (right panel) declined less rapidly (39% to 30%). The cell counts/ml and percent EdU + cells are shown on each day. The lower percent in S phase of the HD cells at 6 days (18%) is probably because the HD ALL3 cells were entering saturation phase. The higher percentage of LD ALL3 cells in S phase than the HD cells on day 1 (41% vs 28%) was because the LD ALL3 cells were prepared by dilution of proliferating HD ALL3 cells were still mostly viable by day 1 and some continued to enter S phase which they were unable to complete before dying (middle panel). The LD cells rescued by HDSN (right panel) were growing nicely by day 6 and had about the same percentage of cells in S phase (30%) as the HD cells on day 1 (28%).
We also determined the proliferation profiles of the LD ALL3 cells that were not stimulated and stimulated with HDSN, but found that the LD ALL3 cells growing in the presence of stimulatory factor did not show much difference in the percentage of cells in S phase between the non-growing and growing cells as estimated by EdU or BrdU staining (Figure 13B and 13C). Thus, LD ALL3 cells are not truly quiescent cells but rather are poised to begin dividing. Their non-growth or very slow growth is not because they are quiescent, but because they require additional stimulatory factors to begin proliferating without which they have a progressive spontaneous death rate. In interpreting the results of Ki67 and EdU/BrdU staining it should be noted that in all experiments the LD cells were always obtained by cell dilution of HD cell cultures just prior to beginning the experiment. So on day 0 the LD cells were usually >90% viable by the trypan blue exclusion. Thus, like the proliferating HD cells, the LD cells were almost all Ki67 positive and their higher labeling of EdU or BrdU during the first 2 days surely indicates that many of the cells continued cycling for the first day or so and continued to enter S phase, but were unable to complete DNA synthesis, became arrested and died without further stimulation within a few more days.
The HDSN rescue the LD ALL3 cells from apoptosis
To estimate the cell death rate upon population depletion, we used several apoptosis assays. The ALL3 cells at different LD and HD cell concentrations were collected after 24 hr of growth. Figure 14A shows a typical comparison of the rapid growth of HD and non-growth of LD ALL3 using the trypan-blue dye exclusion method, and during the same time period there was a substantial increase in apoptosis in the LD ALL3 cells compared to the HD ALL3 cells using the caspase 3/7 assay (Figure 14B). A similar increase in the death rate of the non-growing LD ALL3 cells compared to the growing HD ALL3 cells was found using a TUNEL assay (Figure 14C).
Figure 14.
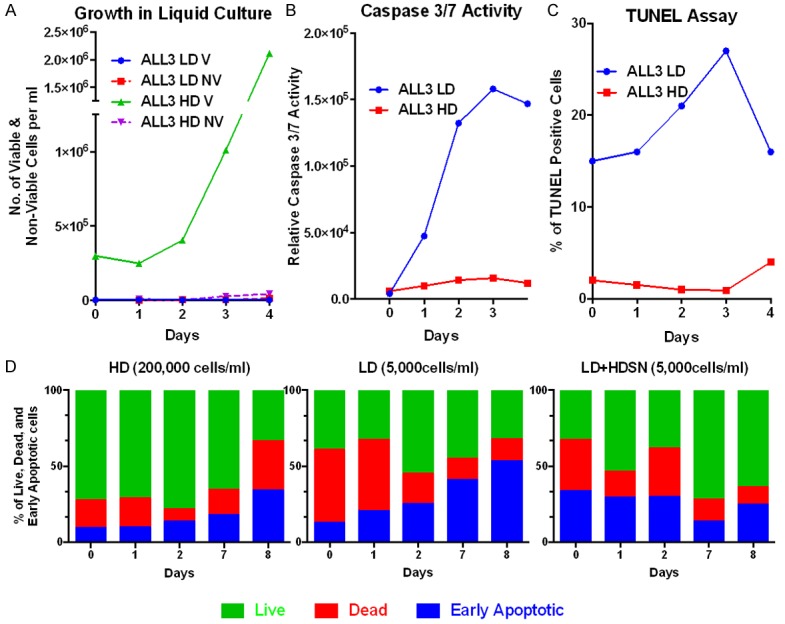
Various apoptosis assays during growth of the HD ALL3 and no growth of the LD ALL3 cells in ALL3 media. A. Total number of viable and non-viable LD and HD ALL3 cells using the trypan blue exclusion method. B. Caspase 3/7 activity (Promega Technical Bulletins: Caspase Glo 3/7 assay, 2012) remained very low in rapidly proliferating cells starting at 3 × 105 cells/ml, but rose rapidly in non-growing cells starting at 5000 cells/ml. C. Percent of TUNEL positive cells (Dead EndTM Fluorometric TUNEL System) remained very low in rapidly proliferating cells starting at 3 × 105 cells/ml, but rose rapidly in non-growing cells starting at 5000 cells/ml. D. Daily flow cytometric AnnexinV-FITC/PI assay was conducted for the HD and LD ALL3 cells stained with Propidium iodide (PI) and AnnexinV-FITC. The HD ALL3 cells showed a rapid increase in early and late apoptosis as they approached saturation density on Day 8. The poorly growing LD cells alone without HDSN had an early increase in dead cells and progressive increase in early apoptotic cells with annexin and PI staining. Even though the LD ALL3 cells plus HDSN were immediately stimulated to grow rapidly with a DTs of 26 hr during the first 3 days, the majority of the LD+HDSN cells were estimated by FACS to still be in the early or late stages of apoptosis and by day 2 only about ~20% of the cells were alive. However, by days 7 and 8 the surviving cells had recovered and about 60-70% were ascertained to be live by FACS and the cells continued to grow well with a DTs of 35 hr. The Bar graphs represent the percent of total cells in each of the below defined gating categories. Green bar represents live cells (AnnexinV-FITC Neg/PI Neg), red bar represents dead cell population (AnnexinV-FITC Neg/PI Pos), and blue bar represents early apoptotic cell populations (AnnexinV-FITC Pos/PI Neg).
We also evaluated apoptotic levels of the LD ALL3 cells in the presence or absence of filtered HDSN. The AnnexinV-FITC/PI based flow cytometry based approach was used to evaluate apoptosis (Figure 14D). As also seen with the caspase 3/7 assay, we observed a dramatic increase in early apoptosis (annexin V-FITC positive cells; blue bars, lower right quadrant) of ALL3 cells at LD compared to HD on day 7 than day 0 (Figure 14D). In addition to the increase in apoptosis over time, we also found that there was a marked initial increase in dead cells on days 0 and 1 (red bars) in the LD compared to the HD ALL3 cells. The LD ALL3 cells in the presence of HDSN had a striking increase in live cells at days 7 & 8 (annexin V (-), PI (-); green bars, lower left quadrant) and a decrease in early apoptotic and dead cells (blue and red bars) compared to the LD ALL3 without HDSN. Thus, the HDSN not only stimulates the growth of still viable surviving LD ALL3 cells but also inhibits apoptosis and death of marginally viable or near dying cells (Figure 14D).
Gene-expression data analysis
We used microarray gene-expression to study the genes activated upon stimulation of the LD ALL3 cells with diffusible HDSN compared to non-stimulated LD ALL3 population. We collected filtered HDSN as described before and stimulated LD ALL3 cells, and monitored the growth of the LD ALL3 cells using the trypan-blue exclusion method as well as the [3H] thymidine uptake assay method over time in two independent experiments conducted in duplicate, and also collected cells on each day for RNA extraction. We selected three time-points, days 1, 3 and 6, that are representative of ‘early’, ‘intermediate’ and ‘late’ stages of stimulation. The method used for RNA extraction and gene-expression data analyses are described in detail in the method section of this paper.
Using bioinformatics analysis we found that there were 165 genes up-regulated and 122 genes down-regulated on the 1st day, 153 genes up-regulated and 251 genes down-regulated on the 3rd day, and 573 genes up-regulated and 268 genes down-regulated on the 6th day for stimulated LD ALL3 cells compared to non-stimulated LD ALL3 cells (Figure 15A and 15B). We also found that there were 92 common genes between day 1, 3, and 6, 50 of which were up-regulated and 42 genes which were down-regulated, differentially expressed between stimulated to non-stimulated LD ALL3 cells (Figure 15A and 15B). Most of these differentially expressed genes are shown in Table 1.
Figure 15.
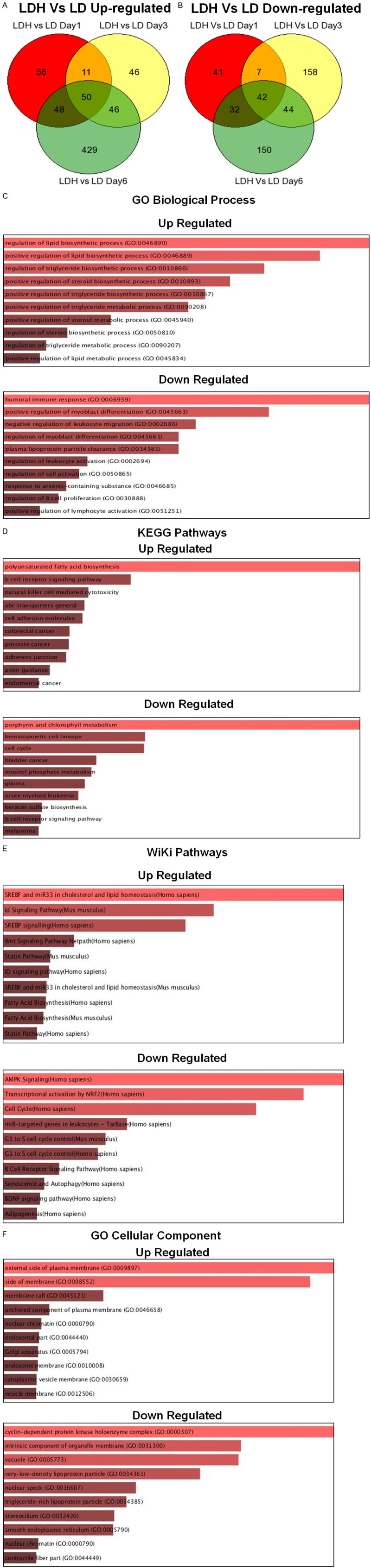
Gene expression analysis comparing the LD ALL3 in presence and absence of HDSN. (A) Venn-diagram showing up-regulated genes on days 1, 3, and 6. There are 50 genes up-regulated commonly in LDH comparing LD at day1, 3, and 6. Here, LD= LD ALL3 cells (non-stimulated cells) and LDH= LD+HDSN (stimulated cells). (B) Venn-diagram showing down-regulated genes on days 1, 3, and 6. There are 42 genes down-regulated commonly in LDH comparing LD at days 1, 3, and 6. The visualization for the bar graphs (C-F) have benn made according to combined score (Combined score= p-value computed using the Fisher exact test, and the z-score computed to assess the deviation from the expected rank). The length of the bar represents the significance of that specific gene-set or term. In addition, the brighter the color, the more significant that term is (adapted from and refer to [63] for more details).(C and D) show Gene ontology (GO) biological process and cellular component analsis of differentially expressed common 92 genes using EnrichR. (E and F) show KEGG and and WiKi pathway analysis of differentially expressed common 92 genes using EnrichR.
Table 1.
List of some of the common genes with significantly differential expression in HDSN stimulated LD ALL3 cells (LDH) compared to non-stimulated LD ALL3 (LD) cells
| Gene Symbol | Fold Change (LDH Vs LD) | ||
|---|---|---|---|
|
|
|||
| Day 1 | Day 3 | Day 6 | |
| Cholesterol, lipid, steroid, triglyceride metabolism and biosynthesis pathways | |||
| ABCG1 | 27.8 | 49.3 | 57.7 |
| ABCA1 | 9.2 | 18.5 | 21.1 |
| MYLIP | 8.8 | 7.8 | 10.9 |
| SREBF1 | 8.7 | 8.4 | 12.1 |
| INSIG1 | 8.3 | 2.5 | 4.0 |
| SCD | 8.1 | 3.6 | 4.3 |
| LDLR | 5.6 | 2.6 | 3.4 |
| SNTB1 | 4.8 | 2.2 | 2.6 |
| FASN | 3.3 | 3.0 | 2.2 |
| PIP5K1B | 3.1 | 2.3 | 2.8 |
| IGF1R | 3.0 | 3.5 | 4.6 |
| FADS1 | 2.9 | 2.1 | 2.0 |
| TCF7L2 | 2.3 | 4.7 | 4.0 |
| ELOVL2 | 2.2 | 2.1 | 3.8 |
| VLDLR | -2.1 | -2.5 | -4.3 |
| B cell proliferation/Signaling pathways | |||
| FAM129C | 27.4 | 49.2 | 121.2 |
| IFITM1 | 10.7 | 26.4 | 26.4 |
| CD22 | 4.1 | 2.7 | 4.1 |
| CD48 | 3.4 | 3.0 | 2.4 |
| NFATC1 | 2.7 | 2.2 | 2.2 |
| CD40 | 2.6 | 5.1 | 3.3 |
| TCF3 | 2.3 | 2.2 | 3.6 |
| HLA-E | 2.3 | 2.1 | 2.9 |
| BLNK | -2.7 | -2.2 | -2.4 |
| MEF2C | -2.9 | -2.3 | -3.2 |
| CDKN1A | -3.4 | -4.1 | -4.4 |
| PRDM1 | -4.1 | -3.4 | -7.5 |
| MS4A1 | -5.5 | -4.4 | -4.3 |
| CD1C | -6.4 | -3.1 | -8.2 |
| Cell cycle regulation/G1 to S cell cycle control | |||
| CTGF | 4.7 | 4.4 | 7.2 |
| TCF3 | 2.3 | 2.2 | 3.6 |
| PLCB1 | -2.5 | -3.5 | -2.4 |
| CDKN1A | -3.4 | -4.1 | -4.4 |
| CCPG1 | -3.6 | -7.9 | -5.8 |
| CCNA1 | -4.5 | -4.7 | -8.5 |
| Apoptosis | |||
| FAIM3 | 20.9 | 14.2 | 6.9 |
| BACH2 | 10.1 | 9.1 | 17.8 |
| TNFRSF19 | -3.7 | -4.5 | -4.4 |
| HMOX1 | -14.4 | -6.7 | -19.4 |
| Cell Secretion | |||
| CTGF | 4.7 | 4.4 | 7.2 |
| RAB27A | 3.2 | 2.6 | 4.1 |
| SRGN | -2.0 | -3.9 | -5.5 |
| LAMP2 | -2.7 | -3.0 | -3.8 |
| Transporter activity | |||
| SLC25A23 | 2.0 | 2.4 | 2.2 |
| VLDLR | -2.1 | -2.5 | -4.3 |
| SLC35B1 | -2.5 | -2.1 | -2.0 |
| SLC16A6 | -5.4 | -4.9 | -10.0 |
| SLC7A11 | -8.0 | -7.6 | -9.3 |
| Other Cancer related genes | |||
| ADARB1 | 10.0 | 5.2 | 12.8 |
| CHI3L2 | 7.6 | 5.3 | 14.0 |
| FAM69B | 7.5 | 6.3 | 10.2 |
| GPR18 | 4.8 | 5.4 | 4.3 |
| MAML3 | 4.6 | 7.1 | 15.2 |
| VWA5A | -1.1 | -5.4 | -10.3 |
| DOCK4 | -4.7 | -3.2 | -6.8 |
| SAMD9L | -5.1 | -4.3 | -3.4 |
| CHST2 | -5.6 | -2.5 | -10.7 |
| FTH1 | -7.4 | -3.7 | -8.6 |
| EGR3 | -9.3 | -3.4 | -6.9 |
| THSD7A | -12.1 | -5.0 | -9.0 |
| LOC284801 | -4.8 | -5.7 | -37.4 |
| CHST2 | -5.6 | -2.5 | -10.7 |
| MPEG1 | -5.8 | -6.3 | -11.2 |
The fold change is linear and positive values mean genes more expressed in LDH and negative values vice versa. In the case of single genes where more than one set of probes is significantly differentially expressed, the highest value amongst them is shown.
GSEA for common differentially expressed genes
For the 50 consistently up-regulated genes, there was enrichment of pathways related to lipid biosynthesis, triglyceride biosynthesis, steroid biosynthesis, poly-unsaturated fatty acid biosynthesis, cholesterol and lipid homeostasis, SREBP signaling, Wnt signaling, ID signaling, and B-cell receptor signaling for up-regulated genes. In the case of 42 consistently down-regulated genes, we found enrichment of pathways related to AMPK signaling, NRF2-dependent transcription activation, G1 to S cell cycle, B-cell receptor signaling, adipogenesis, BDNF signaling, leukocyte activation and B-cell proliferation signaling and activation pathways as shown in Figure 15C-E.
The cellular components of the up-regulated genes include the external side of plasma membranes, membrane rafts, nuclear chromatin, Golgi apparatus, and receptor complex. In the case of down-regulated genes they are components of cyclic-dependent protein kinase holoenzyme complexes, organelle membranes, and vacuoles (Figure 15F).
We focused on these 92 consistently differentially expressed genes to extend the analysis to look for genes that could corroborate our conclusions. Genes are grouped according to their functions, and their possible significance are discussed in hematopoiesis, leukemia and perhaps other cancers. Some of the genes are discussed for their possible role in regulating cancer cell proliferative behavior.
Cholesterol, lipid, steroid, triglyceride metabolism and biosynthesis related genes
Among the most significant differentially expressed genes between stimulated and non-stimulated LD ALL3 cells many of them are associated with lipid, cholesterol, steroid, and triglyceride homeostasis, metabolism and biosynthetic processes. Common differentially expressed genes from this group includes: ABCG1, ABCG1, MYLIP, SREBF1, INSIG1, SCD, LDLR, SNTB1, FASN, PIPK1B, IGF1R, FADS1, TCF7L2, ELOVL2, and VLDLR. These are the first genes shown in Table 1 but only the first 4 progressively increase on days 1-6 and only 1 continues to decrease (VLDLR).
Abnormal cholesterol homeostasis has been observed in several hematological malignancies. The ATP-binding cassette transporters ABCA1 and ABCG1 are involved in cholesterol efflux and their suppression affects hematopoietic stem cell proliferation [73]. Mutation or inactivation of ABCA1 and/or ABCG1 might prevent cholesterol efflux and disable anti-tumor activity [74-76]. Change in the cholesterol efflux pathways is associated with an increase in hematopoietic stem and multipotential progenitor cell mobilization and extramedullary hematopoiesis, favoring the granulocyte rather than the macrophage lineages [77]. Cholesterol-modulating agents have also been shown to kill AML cells [78], urothelial cancer cells [79], and chronic lymphocytic leukemia [80]. MYLIP (also termed IDOL “Inducible degrader of the LDL receptor”) is a ubiquitin ligase that ubiquinates LDL receptors [81]. LDLR is also up-regulated. We also found upregulation of SREBP1 (also termed SREBF1), a well known master transcription factor, that controls lipid metabolism and has been shown to be involved in cancer [74]. Another up-regulated transcription factor was TCF7L2 which is involved in regulation of a member of the Wnt signaling pathways and has also been implicated in several cancers [82-84]. Still another up-regulated gene, SNTB1, has been found to interact with ABCA1 and also regulates cholesterol efflux [85] but its direct role in cancer is still unknown. SCD, FASN, ELOVL2, and INSIG1 have been implicated in breast cancer [86].
Differential expression of the above genes in these pathways provide evidence that up-regulation of the cholesterol and lipid metabolism related pathways are probably necessary to initiate and sustain the growth of non-growing LD ALL3 cells. Cancer cells are known to undergo extensive metabolic reprogramming. Whereas cholesterol and lipid biosynthesis are normally restricted to defined tissues, cancer cells can become independent of systemic regulation and reactivate the lipogenic machinery. Thus, cancer cells are able to fulfill their increased requirement for energy, membrane proliferation, and signaling molecules by rewiring the metabolic pathways [87-91].
In conclusion, it seems that alterations in cholesterol, lipid, sterol, and fatty acid metabolism are important factors in enabling non-growing LD ALL3 cells to initiate and continue proliferation when triggered to do so by stimulatory factors in the HDSN. It also provides an opportunity to consider possible new therapeutic strategies to intervene in the above involved signaling pathways to decrease the growth of leukemic cells and maybe other cancers [74,92].
B cell proliferation and signaling pathways related genes
The most significantly up-regulated genes that we found between the stimulated and non-stimulated LD ALL3 are those that belong to the B cell proliferation and signaling pathways group: FAM129C (BCNP1), IFITM1, CD22, CD48, NFATC1, CD40, TCF3, HLA-E (Table 1).
FAM129C (BCNP1; B-cell novel protein 1)
FAM129C is by far the most up-regulated gene in HDSN stimulated ALL3 cells (121 fold at day 6), but its role in leukemia or cancer is still poorly understood. Boyd and colleagues discovered BCNP1 in their study of the B cell-surface plasma membrane proteins [93]. In their study, they cloned the entire BCNP1 gene from normal spleen and cDNA library of the Daudi cell line and also found that BCNP1 was highly expressed in multiple leukemia and lymphoma patient samples and cell lines compared to normal tissues. BCNP1 expression was found to be restricted to B-cell rich normal tissues, being particularly high in lymph nodes, spleen and the thymus [93]. It has been more than a decade since BCNP1 was first identified, but the role of BCNP1 in B-cell development and malignancies is still uncertain. Our combined gene-expression and biological data suggest that it may well have an important role in activating leukemic Ph+ ALL cells that are already poised to proliferate to do so.
IFITM1 was also highly up-regulated (~26 fold at days 3 and 6) (Table 1). IFITM1 is a component of a multimeric complex involved in the transduction of antiproliferative and cell adhesion signals [94]. CD22 was over-expressed ~4 fold. CD22 is a B-lineage differentiation antigen, which is expressed on the surface of B-lineage cells from the early progenitor state (pro-B cells) to terminally differentiated plasma cells [95,96]. CD48 was also upregulated but only 2.4-3.4 fold. It is expressed on differentiated hematopoietic cells but not on quiescent HSC [97,98]. It also regulates hematopoietic S/P cells and suppresses tumor formation and also acts as an environmental sensor to regulate progenitor cell number [99]. NFATC1 is a transcription factor playing a critical role during immune responses; it regulates both normal homeostasis and differentiation of B cells [100] and also T cells [101]. CD40 receptor is expressed on the B-cell lineage ALL [102] and its high expression on B-cell precursor ALL blasts has been proposed as a marker indicating superior relapse-free survival [103]. The TCF3 gene is a member of the helix-loop-helix transcription factor family. It is involved in several chromosomal translocations and other abnormalities in B-lineage acute leukemia [104-106].
Among the genes in the B cell proliferation and signaling pathways related group that are down-regulated upon stimulation of LD ALL3 cells are BLNK, MEF2C, CDKN1A, PRDM1, MS4A1 (CD20), and CD1C (Table 1). The BLNK expression in B-lineage ALL is higher than in B cells [107], and BLNK acts as tumor suppressor in pre-B cell ALL [108]. MEF2C, a transcription factor, has been suggested to be a poor-risk marker in some subtypes of AML [109], and its abnormal expression due to chromosomal rearrangements is also linked to leukemia [110,111]. PRDM1 is overexpressed in some cases of B and T-cell lymphoma [112] and acts as a tumor suppressor in human colon cancer [113]. CD20 (also termed MS4A1) is commonly expressed on leukemic B cells [114]; It is not expressed in HSC or pro-B cells but is expressed in pre-B cells and also on mature B cells in the bone marrow and blood [115]. The ALL3 cells are ~100% positive for the CD20 (MS4A1) marker but it is down-regulated in the HDSN stimulated LD ALL3 cells. Anolik et al, found that CD40 activation leads to down-regulation of CD20 on the normal B cells [116]. In our gene-expression data we found that CD40 is up-regulated (~2-5 fold) in the stimulated LD ALL3 cells. This suggests that the stimulatory factors up-regulate CD40 which leads to down-regulation of CD20 upon the stimulation of the LD ALL3 cells and thereof permits survival and growth of LD ALL3 cells. CD1C is also down-regulated in B cell CLL [117].
All these genes that are differentially expressed in stimulated LD ALL3 cells compared to non-stimulated LD ALL3 cells belong to the B-cell proliferation, activation, and signaling pathways group shown in Table 1. The ALL3 cell line used in this study is of pre-B lineage as determined by surface cell antigen markers using flow cytometry: CD19, CD20, CD38, CD45RA are all 95-100% Positive, and IgG1, CD34, CD117 & CD3 are all negative providing additional evidence that the stimulatory factor(s) are stimulating leukemic pre-B cells. The role of the above genes in regulating cell-cell communication and cell number maintenance is presently unclear, but appears to be highly complex and additional studies will be required to determine their functional roles.
Cell cycle regulation/G1 to S cell cycle control
Of the genes linked to cell cycle regulation, the differentially expressed genes include: CTGF, TCF3 PLCB1, CDKN1A, CCPG1, and CCNA1 (Table 1). The failure of LD ALL3 cells to grow is not because they are quiescent, but rather because although they are already primed to proliferate they require additional stimulatory factors to begin to do so without which they all die within about a week. In the presence of HDSN stimulatory factors many of the LD ALL3 cells which are arrested in S phase resume DNA synthesis and all the surviving cells in other phases of the cell cycle again begin cycling (Figure 13) (depending on the timing of adding HDSN).
Apoptosis pathways
We found the following differentially expressed genes that have been reported to be associated with apoptosis: FAIM3, BACH2, HMOX1, TNFRSF19 (Table 1).
FAIM3 (Fas apoptotic inhibitory molecules) protects cells from Fas-, TNF-alpha-, and FADD- induced apoptosis [118,119] and is highly expressed on CLL B cells [120]. BACH2 is a transcription factor involved in apoptosis by repressing HMOX1 in Ph+ CML through the PI3K/S6 kinase pathways [121]. TNFRSF19, a member of the TNF receptor superfamily, is overexpressed in glioblastoma, and its knockdown results in prolonged survival of an in vivo xenograft of glioblastoma cells in mice [122].
In our study, FAIM3 and BACH2 were up-regulated and HMOX1 was down-regulated upon stimulation of the LD ALL3 cells. As noted earlier, the LD ALL3 cell died rapidly without growth stimulation, and in the presence of the stimulatory factors in HD ALL3 SN there was a decrease in apoptosis and an increase in live cells (Figure 14). This of course suggests that the stimulatory factors suppress apoptosis by upregulating the two anti-apoptotic genes FAIM3 and BACH2 and by downregulating pro-apoptotic genes HMOX1 and TNFRSF19.
Gene involved in cell secretion
In the genes linked to cell secretory pathways two genes, CTGF and RAB27A were upregulated, and two genes, SRGN and LAMP2, downregulated in stimulated compared to non-stimulated LD ALL3 cells (Table 1). CTGF (also termed CCN3) is present in cellular compartments. The tyrosine-phosphorylated BCR-ABL kinase activity leads to an increase in CCN3 secretion and a decrease in cellular CCN3 at the protein level that can be reversed by using BCR-ABL kinase inhibitors such as Imatinib. Overexpression of CCN3 in BCR-ABL positive cells also led to a decrease in proliferation and clonogenic potential [123]. CCN3 is an important player in stem cell regulation, hematopoiesis and bone marrow niche maintenance [124]. In our study the CCN3 (CTGF) gene is up-regulated at the mRNA level ~4.5-7 fold in stimulated LD ALL3 cells. It is plausible that the stimulatory factors induce expression of endogenous CCN3 at the mRNA level and increase CCN3 secretion, allowing the stimulated LD ALL3 cells to commence and sustain proliferation.
Another up-regulated gene, Rab27A controls the exosome secretion pathways [125] and is highly expressed in melanocytes and hematopoietic and other secretory cells [126]. Its expression has been clinically related to hepatocellular carcinoma [127] and pancreatic cancer [128]. Exosomes have been shown to carry proteins, lipids, RNAs and DNAs [129]. Raimondo et al have shown that CML-derived exosomes promote the proliferation and survival of tumor cells in an autocrine fashion by activating anti-apoptotic pathways [130]. The up-regulation of RAB27A (~2-4 fold) in the HDSN stimulated LD ALL3 cells suggests that this induces release and secretion of exosomes which contain factors that can stimulate the non-growing LD ALL3 cells to survive and resume proliferation.
Down-regulated SRGN is the ligand for CD44 involved in in the adherence and activation of lymphoid cells [131]. It is actively secreted in a functional form by various hematopoietic cells [132]. The SRGN deficient mice shows enlargement of multiple lymphoid organs [133]. Another down-regulated gene, LAMP2, has been linked to autophagy. It is also involved in lysosome protection, maintenance and adhesion. Its knockdown had been shown to reduce viability of AML cells [134].
It is plausible that the differential expressions of the above genes activate or repress certain secretory pathways so the LD ALL3 cells are enabled to grow in the presence of stimulatory factors released in the secretome or exosomes of the HD ALL3, CB or other proliferating cells. Much further experimental evidence is of course needed to define the precise roles of the above genes or others in communications between ALL3 cells as well as with other cells. However, these genes are good candidates to begin unraveling the extreme complexity of how cells communicate with each other and perhaps to identify differences in the means of communication whereby leukemic or other malignant cells can function as semi-autonomous collective ecosystems that proliferate according to their own rules, disregarding normal restraining regulatory mechanisms that tightly maintain homeostasis in normal organs and tissues.
Genes related to solute carrier transporter activity
Membrane transporters are involved in the transfer of exogenous and endogenous metabolites and small molecules. The genes that encode these transporter family members constitute approximately 4% of genes in the human genome [135]. The genes differentially expressed in this group in our study include: SLC25A23, VLDLR, SLC35B1, SLC16A6, and SLC7A11.
The SLC25A23 plays an important role in mitochondrial matrix Ca2+ influx [136], SLC35B1 is an UDP-galactose translocator, SLC16A6 is a monocarboxylate transporter, and SLC7A11 is a cysteine/glutamate transporter. The SLC7A11 is also overexpressed in human cancer patients [137] and also involved in chemoresistance [138].
The numerous differentially expressed genes in the stimulated LD ALL3 cells also suggests that the leukemic cells are undergoing profound metabolic reprogramming to sustain their growth and continue supplying building blocks necessary for synthesis for DNA, RNA, lipids, and proteins. The solute carrier family member proteins are involved in transport of several small molecules such as metabolites and are dysregulated in cancers [139]. Their inhibition could be one of the ways to block tumor progression.
There are recent reports that in addition to communicating by chemical autoinducers, bacteria can also communicate with each other via electrical signaling which is also used in the brain to convey information between neurons. Prindle and colleagues have shown that ion channels that are present on the surface of bacteria control the transfer of charged particles and that this opening and closing changes the charges of neighboring cells; the process continues on and on to coordinate the collective behavior of the bacterial cells [140]. Bacteria also use an electrical signaling mechanism for biofilm formation [141]. Various prokaryotes such as cyanobacteria Oscillatoria terebriformis, Geitlerinema sp. and Halothece sp., the unicellular eukaryotes: yeast Saccharomyces cerevisiae and ciliates Paramecium caudatum, and H. lucorum have also shown some electrical activity when measured individually at single cell levels [142].
There is no direct evidence as yet that ALL3 cells use electrical signaling to communicate. However, the differential expression of so many transporters and solute family member proteins suggest that it is possible that the LD ALL3 cells might use a similar electrical system to that in bacteria or other simpler organisms to communicate with each other.
Other differentially expressed cancer related genes (Table 1)
ADARB1, an enzyme involved in RNA editing, has been shown to be abnormally expressed in acute leukemias [143] and is also involved in malignant reprogramming of myeloid progenitors in CML [144]. GPR18 is an orphan G-protein receptor and has been implicated in tumor cell survival but its detailed mechanism and role in cancer has not been studied [145].
VWA5A is implicated as tumor suppressor in breast cancer [146], and in nasopharyngeal carcinoma [147]. DOCK4 is a GTPase exchange factor which is down-regulated in MDS. It is also hyeprmethylated and its expression is reduced in bone marrow stem cells of MDS [148]. Its depletion leads to dysplastic morphology in erythroid cells [149] and it is also involved in AML [150]. Inactivation of SAMD9L promotes cell proliferation in hepatocellular carcinoma [151] and it also acts as a tumor growth suppressor in bladder cancer [152].
CHST2, a sulfotransferase, has been implicated to play role in pre-B ALL [153]. FTH1 regulates intracellular iron storage and its level varies at disease conditions and interacts with NCOA4 to regulate ferrintophagy and plays an essential role in erythropoiesis [154].
EGR3, a transcription factor, has been shown to regulate proliferative potential of HSCs [155]. It has also been shown as “a strong limiting factor” in HSC quiescence [156]. It is also hypermethylated in adult T-cell leukemia [157] and is also dysregulated in other cancers such as breast [158], prostate [159], and gastric cancers [160]. THS7DA is a soluble N-glycoprotein that increases endothelial cell migration and tube formation in angiogenesis [151,161], but its role in cancer is not understood. LOC284801 seems to be part of a long non-coding RNA. It is highly down-regulated (~37 fold) upon stimulation of the LD ALL3 cells. It is found to be up-regulated in non-small cell lung cancer [162] and might have some role in mesothelioma [163] and hepatoma [164].
With regard to other genes in the “other cancer related genes category”, the affected gene products cover a wide range of functions including proteins involved in cell signaling, RNA editing, migration, angiogenesis and transcriptional control.
Discussion
The Ph+ ALL3 cell line was used as a model system to try to understand how cells communicate with each other and with other cells and the abnormalities in quorum sensing that permit these S/P cells to far exceed normal homeostatic cell densities. The ALL3 cells line is somewhat unique because the cells were frozen in multiple aliquots shortly after they were obtained and thus more closely simulate the behavior of the same cells in a patient’s pleural fluid in which they were growing as free-floating ascitic cells in the terminally ill patient with Ph+ ALL than do many established leukemic cell lines. We found that ALL3 cells behave very differently when grown in various cell culture systems at low and high starting cell densities. ALL3 cells fail to grow at LD but grow progressively faster at increasing cell densities. Using various proliferation markers, we also found LD ALL3 cells are not quiescent but rather are poised to begin proliferating but are unable to do so without stimulatory factors secreted or provided in exosomes from ALL3 or some other cells growing at HD. In addition, we found that LD ALL3 cells undergo progressive apoptosis and death as measured by various apoptosis assays unless they are rescued by the stimulatory factors. These observations that changes in cell density can have a dramatic growth effect on the proliferation, survival, and growth of ALL3 cells strongly suggest that these characteristics or due to collective behavior of the cells which are regulated by complex intercellular communications and intracellular signaling pathways.
Many other studies have also suggested a relationship between changes in cell density and changes in survival, death, apoptosis, stem-cell maintenance, proliferation, and metastasis in cancer. As one example, Dolnick et al demonstrated that the rTS gene regulates the level of rTSbeta protein in a cell density dependent manner and is controlled by secretion of lipophilic metabolites [20]. The authors of this work found that diffusible signaling molecules generated growth variations upon increase in rTSbeta protein levels. In another study, Falk et al demonstrated that cysteine was required for survival of Burkitt lymphoma cells when seeded at low density [165]. Within any cancer cell population the cells are very heterogeneous and consist of diverse subpopulations or clones of cancer cells with different functional properties, such as growth rate, ability to metastatize, ability to develop therapeutic resistance, and survival. Heppner et al showed that tumor subpopulations do not behave independently of each other but instead they form a society of cells in which they impact each other’s growth and support the maintenance of tumor heterogeneity. Thus cancer cell behavior is an overall outcome of the behavior of the cancer cell society and its mixed interactions [31]. In a similar line of research, Seton-Rogers et al demonstrated that co-operation between lineage-restricted subclones is the result of aberrant expression of secreted signaling molecules and thus maintains tumor cell heterogeneity, tumor progression and subclonal malignant diversities [166]. Agur et al, showed the importance of QS in the regulation of breast CSCs proliferation in in vitro experiments [34]. Melanoma cells grown at different starting cell numbers demonstrated different metastatic properties [167], and Welch et al demonstrated that the relative number of metastases formed by rat mammary adenocarcinoma clones depends on paracrine factors secreted by donor cells growing at different cell densities [168].
Our study was initiated in attempt to understand the fundamental molecular mechanisms responsible for the differential growth of the ALL3 cells when grown at LD or HD. We observed a number of interesting findings that correlate nicely with some of the biological and functional abnormalities that have previously been observed in other systems. These findings include the following.
(i) The ALL3 cells do not grow at low starting cell densities but grow very well with good viability at high starting cell densities. This observation is nicely coordinate with older observations in bacteria in which light emission was determined to occur only at a high cell population density [4]. Chen et al found that plucking hair at different densities leads to a regeneration of up to five times more neighboring, unplucked resting hairs by a two-step mechanism. Injured hair cells release CCL2 leading to recruitment of TNF-α secreting macrophages which activate regeneration of hair, indicating a collective decision-making process [169].
(ii) The non-growing LD and HD ALL3 cells are similar in their proliferation marker profiles, but the latter grows progressively faster at increasing cell densities while the LD ALL3 cells don’t grow at all without additional stimulatory factors provided by supernates of the same or other cells growing at HD to support their growth.
(iii) At low starting cell density ALL3 cells fail to survive; they shortly undergo profound apoptosis compared to cells growing at high starting cell densities. Challa et al suggested that Burkitt-like lymphoma cells which are directly obtained from a patient sustained their growth by secreting CD40 ligand when the cell number fell below a certain threshold [170]. However, in our study LD ALL3 cells died rapidly and were unable to secret any stimulatory factors that could sustain their growth.
(iv) The LD ALL3 cells are unresponsive to stimulation by any of a large number of cytokines tested alone or in various combinations but they are stimulated to grow by fresh supernates of ALL3 cells growing at HD. Thus, an ‘autocrine’ factor(s) is sufficient to stimulate growth of LD ALL3 cells. This observation suggests that ALL3 cells are simultaneously able to secrete and sense the same signaling molecule(s) for a variety of biological functions that allows them to achieve versatile social behaviors. Youk et al used budding yeast to study synthetic secrete-and-sense circuit motif to understand complex social and asocial behaviors [171].
(v) The LD ALL3 cells can also be stimulated to grow in the presence of supernate collected from some but not all leukemic cell lines and also from primary MNCs or enriched CD34+ cells obtained from cord blood or CML patients. Thus, ‘paracrine’ factors can also stimulate growth of the LD ALL3 cells suggesting that in some cases cells can also respond to other external environmental cues.
(vi) The HDSN from HD ALL3 cells that have intense stimulatory activity on the LD ALL3 cells do not have any additional stimulatory effect on HD ALL3 cells which are already growing well. Similarly supernates from enriched CD34+ normal or CML cells that vigorously stimulate growth of LD ALL3 cells fail to stimulate growth of HD ALL3 cells. This suggest the following three possibilities. First, the ALL3 cells at high starting cell density are self-stimulatory or secrete enough stimulatory factor to support their own growth. Second, the same stimulatory factor(s) that stimulate growth of LD ALL3 cells inhibit growth of HD ALL3 cells. Third, the HDSN might contain both stimulatory and inhibitory factor(s); the former is only effective when the cell numbers are low, and the latter when the cell numbers are increasing to establish a limiting threshold. It is also possible that the same factors might have paradoxical effects depending on the cell population density. Paradoxical signaling has been observed in various immune systems [172]. As an example, IL-2 can increase both proliferation and death rate of T cells [173]. What the benefits are of having such paradoxical signaling still remains unclear.
(vii) Once the LD ALL3 cells are stimulated by HDSN, the gene expression data clearly shows that they have marked overexpression of genes associated with cholesterol, lipid and sterol regulatory pathways. Thus, the LD ALL3 cells rapidly change their metabolic requirements and signaling machinary to support their resumption of growth after stimulation.
(viii) FAM129C was highly upregulated (~27, ~49 and ~121 fold on days 1, 3, and 6, respectively) by HDSN stimulated LD ALL3 cells. FAM129A [174] and FAM129B [175] are other members of the family. Yuki et al showed that FAM129A is involved in cell density dependent cell proliferation by a ZNF777-mediated pathway [176]. The findings by Yuki et al on FAM129A and strikingly high up-regulation of FAM129C in stimulated LD ALL3 cells indicates that it probably has an important role in cell density-dependent cell proliferation at least in B-cell specific development. Our study also shows BCNP1 phosphorylation at serine residues are linked to the PI3K and p38 MAPK signaling pathways and might have role in cancers (Patel SJ, In submission).
(ix) Upon stimulation of the LD ALL3 cells in the presence of HDSN other genes involved in B-cell activation, apoptosis, exosome release and secretion are also differentially expressed. Thus, the LD ALL3 cells clearly use complex regulatory gene networks to support their growth after stimulation. ALL3 cells produce no clones in semi-solid media, don’t grow as a single cell(s) in single cell cloning plates in liquid media, and grow significantly better at the some cell densities per ml in liquid culture if the culture vessel or well has a smaller rather than a large bottom growing area. Thus as might be expected close cell proximity is also an important factor enhancing cell communication and collective behavior.
(x) LD and HD ALL3 cells are un-responsive to growth stimulation by any of the single or multiple combinations of cytokines tested that are essential for growth of normal hematopoietic S/P cells. Instead the growth of HD or intermediate density ALL3 cells is inhibited by several of the same single or combinations of cytokines that are absolutely required by the normal S/P cells. Another striking difference is that while a high percentage of normal cord blood or other normal S/P cells will grew with stimulation by multiple cytokines as single cells either in liquid culture in single cell cloning plates or in soft agar or methyl cellulose (MC), the ALL3 cells will not grow as single cells nor produce any colonies at all in agar or MC. These are striking differences. If the misregulatory mechanisms and aberrant signaling pathways were better understood, it is quite possible that ALL3 cells and other acute leukemia cells could be found to be preferentially vulnerable to different forms of therapies then the normal S/P cells.
We also performed a biophysical study using atomic force microscopy to compare non-growing LD and proliferating HD ALL3 cells. We found that there are substantial differences in the biophysical parameters such as Young’s modulus and the pericellular brush length between HD and LD ALL3 cells. The HD ALL3 cells showed ~3.6x increase in the pericellular brush length and also ~2.5x increase in effective brush size compared to the LD ALL3 cells while the LD ALL3 cells showed ~6x increase in the Young’s elastic modulus of the cell body compared to HD cells (Nataliia V. Guz, Sapan J. Patel et al. unpublished work). These observations indicate that the proliferating HD ALL3 cells were softer than the non-growing LD ALL3 cells which were stiffer, probably because they were undergoing considerable cytoskeletal reorganization.
Normal organ systems including hematopoiesis behave collectively as interactive cell societies in which homeostasis is rigidly maintained by complex regulatory networks and QS mechanisms. Leukemia and other cancer ecosystems also behave collectively and their component cells interact with each other and with neighboring or distant normal cells, but as the malignant transformation proceeds the neoplastic cells further ignore or disobey normal homeostatic cell density and other normal QS regulations. Thus, rather than clonal succession and faster growth of the most aggressive new mutant clones, increasingly defective QS may be the predominant biological functional defect responsible for the progressive expansion and domination by the Ph+ ALL3 and other acute leukemic cells and the transformed BP clones in CML, and this may also be the case in other hematological malignancies and solid tumors undergoing malignant progression.
In summary, we have demonstrated that diffusible factors secreted or released by proliferating cells are capable of initiating and sustaining growth of Ph+ ALL3 cells at low cell densities at which they do not grow without these stimulatory factors but rather succumb to apoptosis, and death. So far, there have not been any reports attempting to directly study quorum sensing in ALL. In the presence of diffusible, soluble factor(s) obtained from the supernates of ALL3 or other cells growing at HD, extensive changes take place in the LD ALL3 cells including metabolic reprogramming, suppression of apoptosis, and reactivation of B cell pathways that sustain the propagation of non-growing LD ALL3 cells. Whether the abnormalities described here in Ph+ ALL3 cells can be reproduced to find similar or parallel disorders in other malignant tumors needs to be determined, but if defective QS and collective behavior of cancer cells can be shown to be common abnormalities of many cancers they should be included among the other hallmarks of cancer. Activation of the ALL3 cells at LD following stimulation appears to be highly complex and involves the coordinated and selective induction of expression and repression of hundreds of genes. Considering the astounding complexity of all of the stimulatory factor(s), other regulatory factors and cellular interactions that determine whether non-growing LD ALL3 cells are going to proliferate or die, it is hardly surprising that our present understanding of the mechanisms regulating cancer cell collective behavior is inadequate. In our analysis, it appears that in many cases, clusters of presumably related genes that are differentially expressed are all associated with a particular stage of development or function, suggesting that their common dysfunction in collective behavior of ALL3 cells may involve aberrant co-regulation. The alterations in gene expression described here must be confirmed in other human cell culture systems or directly in cancer patients using advanced technologies. As the search proceeds, the significance of some of the differences in gene expression reported here may become clearer and eventually lead to discovery of new ways to selectively disrupt aberrant cell-cell communication.
Acknowledgements
This work was supported in part by The Enid A Haupt Charitable Trust, the MeadRock Foundation, the Albert C. Bostwick Foundation (Bessemer Trust), and the E./S. Sindina Lymphoma Research Fund. This research was funded in part through the NIH/NCI cancer center support grant P30 CA008748 and friends of the senior author, especially Nancy Taylor, Judy Murray and Alexander McLanahan. We would like to thank Drs. Renier Brentjens and Mark Frattini for giving us the ALL3 cell line, Ms. Caryl Lambek and Dr. Chong-Yuan Liu for performing some of the early experiments with the ALL3 cells, Dr. Katia Manova (Molecular cytology core facility head) and Mesruh Turkekul for their help with EdU immunofluroscence experiments, and Dr. Lilian Reich for providing left-over leukopheresis samples. We also want to thank Dr. Agnes Viale, head of functional genomics initiative core, for help with designing the microarray experiments, Drs. Nicholas Socci (Bioinformatics core), Raya Khanin, and Hui Zhao for help with the gene-expression data analysis and Drs. Raju Chaganti, Stephen Larson for their support and Dr. Darren Veach for helpful scientific discussion. We also thank research volunteers Lauren Bellavia and Gaurang Trivedi for their help with experiments and Ms. Lucinda Lewis for her great assistance. Numerous colleagues at MSKCC have provided helpful support, advice and use of technical instruments and facilities in their laboratories without which many of the experiments could not have been performed. We are especially grateful to Drs. Minkui Luo, Ronald Hendrickson, Alex Kentsis, David Spriggs, Michael Kharas, Jan Hendricks, Elisa DeStanchiana and Hediye Erdjument-Bromage for invaluable assistance. Finally we want to thank Drs. David Scheinberg, George Bosl, Jose Baselga, Craig Thompson and Ms. Carol Slattery for support and encouragement without which it would not have been possible to complete this work.
Disclosure of conflict of interest
None.
References
- 1.Hastings JW, Nealson KH. Bacterial bioluminescence. Annu Rev Microbiol. 1977;31:549–595. doi: 10.1146/annurev.mi.31.100177.003001. [DOI] [PubMed] [Google Scholar]
- 2.Hastings JW. Bacterial bioluminescence light emission in the mixed function oxidation of reduced flavin and fatty aldehyde. CRC Crit Rev Biochem. 1978;5:163–184. doi: 10.3109/10409237809177143. [DOI] [PubMed] [Google Scholar]
- 3.Miller MB, Bassler BL. Quorum sensing in bacteria. Annu Rev Microbiol. 2001;55:165–199. doi: 10.1146/annurev.micro.55.1.165. [DOI] [PubMed] [Google Scholar]
- 4.Nealson KH, Hastings JW. Bacterial bioluminescence: its control and ecological significance. Microbiol Rev. 1979;43:496–518. doi: 10.1128/mr.43.4.496-518.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruby EG. Lessons from a cooperative, bacterial-animal association: the Vibrio fischeri-Euprymna scolopes light organ symbiosis. Annu Rev Microbiol. 1996;50:591–624. doi: 10.1146/annurev.micro.50.1.591. [DOI] [PubMed] [Google Scholar]
- 6.Sifri CD. Healthcare epidemiology: quorum sensing: bacteria talk sense. Clin Infect Dis. 2008;47:1070–1076. doi: 10.1086/592072. [DOI] [PubMed] [Google Scholar]
- 7.Sperandio V. Striking a balance: inter-kingdom cell-to-cell signaling, friendship or war? Trends Immunol. 2004;25:505–507. doi: 10.1016/j.it.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Visick KL, McFall-Ngai MJ. An exclusive contract: specificity in the Vibrio fischeri-Euprymna scolopes partnership. J Bacteriol. 2000;182:1779–1787. doi: 10.1128/jb.182.7.1779-1787.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kolodkin-Gal I, Engelberg-Kulka H. The extracellular death factor: physiological and genetic factors influencing its production and response in Escherichia coli. J Bacteriol. 2008;190:3169–3175. doi: 10.1128/JB.01918-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolodkin-Gal I, Hazan R, Gaathon A, Carmeli S, Engelberg-Kulka H. A linear pentapeptide is a quorum-sensing factor required for mazEF-mediated cell death in Escherichia coli. Science. 2007;318:652–655. doi: 10.1126/science.1147248. [DOI] [PubMed] [Google Scholar]
- 11.Wynendaele E, Bronselaer A, Nielandt J, D’Hondt M, Stalmans S, Bracke N, Verbeke F, Van De Wiele C, De Tre G, De Spiegeleer B. Quorumpeps database: chemical space, microbial origin and functionality of quorum sensing peptides. Nucleic Acids Res. 2013;41:D655–659. doi: 10.1093/nar/gks1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar S, Kolodkin-Gal I, Engelberg-Kulka H. Novel quorum-sensing peptides mediating interspecies bacterial cell death. MBio. 2013;4:e00314–00313. doi: 10.1128/mBio.00314-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ackermann M. A functional perspective on phenotypic heterogeneity in microorganisms. Nat Rev Microbiol. 2015;13:497–508. doi: 10.1038/nrmicro3491. [DOI] [PubMed] [Google Scholar]
- 14.Gordon D. Ant encounters: interaction networks and colony behavior. Princeton, N.J.: Princeton University Press; 2010. [Google Scholar]
- 15.Franks NR, Stuttard JP, Doran C, Esposito JC, Master MC, Sendova-Franks AB, Masuda N, Britton NF. How ants use quorum sensing to estimate the average quality of a fluctuating resource. Sci Rep. 2015;5:11890. doi: 10.1038/srep11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greene MJ, Pinter-Wollman N, Gordon DM. Interactions with combined chemical cues inform harvester ant foragers’ decisions to leave the nest in search of food. PLoS One. 2013;8:e52219. doi: 10.1371/journal.pone.0052219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sturgis SJ, Greene MJ, Gordon DM. Hydrocarbons on harvester ant (Pogonomyrmex barbatus) middens guide foragers to the nest. J Chem Ecol. 2011;37:514–524. doi: 10.1007/s10886-011-9947-y. [DOI] [PubMed] [Google Scholar]
- 18.Altschuler SJ, Wu LF. Cellular heterogeneity: do differences make a difference? Cell. 2010;141:559–563. doi: 10.1016/j.cell.2010.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balazsi G, van Oudenaarden A, Collins JJ. Cellular decision making and biological noise: from microbes to mammals. Cell. 2011;144:910–925. doi: 10.1016/j.cell.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dolnick BJ, Angelino NJ, Dolnick R, Sufrin JR. A novel function for the rTS gene. Cancer Biol Ther. 2003;2:364–369. doi: 10.4161/cbt.2.4.424. [DOI] [PubMed] [Google Scholar]
- 21.Foster KR. The sociobiology of molecular systems. Nat Rev Genet. 2011;12:193–203. doi: 10.1038/nrg2903. [DOI] [PubMed] [Google Scholar]
- 22.Guttal V, Couzin ID. Social interactions, information use, and the evolution of collective migration. Proc Natl Acad Sci U S A. 2010;107:16172–16177. doi: 10.1073/pnas.1006874107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perkins TJ, Swain PS. Strategies for cellular decision-making. Mol Syst Biol. 2009;5:326. doi: 10.1038/msb.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Almeida AR, Amado IF, Reynolds J, Berges J, Lythe G, Molina-Paris C, Freitas AA. Quorum-Sensing in CD4(+) T Cell Homeostasis: A Hypothesis and a Model. Front Immunol. 2012;3:125. doi: 10.3389/fimmu.2012.00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Callard RE, Stark J, Yates AJ. Fratricide: a mechanism for T memory-cell homeostasis. Trends Immunol. 2003;24:370–375. doi: 10.1016/s1471-4906(03)00164-9. [DOI] [PubMed] [Google Scholar]
- 26.Sabatos CA, Doh J, Chakravarti S, Friedman RS, Pandurangi PG, Tooley AJ, Krummel MF. A synaptic basis for paracrine interleukin-2 signaling during homotypic T cell interaction. Immunity. 2008;29:238–248. doi: 10.1016/j.immuni.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mondal BC, Mukherjee T, Mandal L, Evans CJ, Sinenko SA, Martinez-Agosto JA, Banerjee U. Interaction between differentiating cell- and niche-derived signals in hematopoietic progenitor maintenance. Cell. 2011;147:1589–1600. doi: 10.1016/j.cell.2011.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 29.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 30.Ben-Jacob E, Coffey DS, Levine H. Bacterial survival strategies suggest rethinking cancer cooperativity. Trends Microbiol. 2012;20:403–410. doi: 10.1016/j.tim.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 31.Heppner GH. Cancer cell societies and tumor progression. Stem Cells. 1993;11:199–203. doi: 10.1002/stem.5530110306. [DOI] [PubMed] [Google Scholar]
- 32.Deisboeck TS, Couzin ID. Collective behavior in cancer cell populations. Bioessays. 2009;31:190–197. doi: 10.1002/bies.200800084. [DOI] [PubMed] [Google Scholar]
- 33.Lambert G, Estevez-Salmeron L, Oh S, Liao D, Emerson BM, Tlsty TD, Austin RH. An analogy between the evolution of drug resistance in bacterial communities and malignant tissues. Nat Rev Cancer. 2011;11:375–382. doi: 10.1038/nrc3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agur Z, Kogan Y, Levi L, Harrison H, Lamb R, Kirnasovsky OU, Clarke RB. Disruption of a Quorum Sensing mechanism triggers tumorigenesis: a simple discrete model corroborated by experiments in mammary cancer stem cells. Biol Direct. 2010;5:20. doi: 10.1186/1745-6150-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vainstein V, Kirnasovsky OU, Kogan Y, Agur Z. Strategies for cancer stem cell elimination: insights from mathematical modeling. J Theor Biol. 2012;298:32–41. doi: 10.1016/j.jtbi.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 36.Hickson J, Diane Yamada S, Berger J, Alverdy J, O’Keefe J, Bassler B, Rinker-Schaeffer C. Societal interactions in ovarian cancer metastasis: a quorum-sensing hypothesis. Clin Exp Metastasis. 2009;26:67–76. doi: 10.1007/s10585-008-9177-z. [DOI] [PubMed] [Google Scholar]
- 37.Affer M, Dao S, Liu C, Olshen AB, Mo Q, Viale A, Lambek CL, Marr TG, Clarkson BD. Gene Expression Differences between Enriched Normal and Chronic Myelogenous Leukemia Quiescent Stem/Progenitor Cells and Correlations with Biological Abnormalities. J Oncol. 2011;2011:798592. doi: 10.1155/2011/798592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Monfardini S, Gee T, Fried J, Clarkson B. Survival in chronic myelogenous leukemia: influence of treatment and extent of disease at diagnosis. Cancer. 1973;31:492–501. doi: 10.1002/1097-0142(197303)31:3<492::aid-cncr2820310302>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 39. http://www.cmladvocates.net/3-news/newsflash/421-ash-report-1-stopping-cml-treatment-for-therapy-free-remission.
- 40.Rea D, Nicolini FE, Tulliez M, Rousselot P, Guilhot F, Gardembas M, Guerci A, Coiteux V, Legros L, Guillerm G, Pignon JM, Giraudier S, Etienne G, Villemagne B, Escoffre M, Charbonnier A, Mahon F-X. 56th ASH Annual Meeting and Exposition. San Francisco, CA: 2014. Dasatinib or Nilotinib Discontinuation in Chronic Phase (CP)-Chronic Myeloid Leukemia (CML) Patients (pts) with Durably Undetectable BCR-ABL Transcripts: Interim Analysis of the STOP 2G-TKI Study with a Minimum Follow-up of 12 Months - on Behalf of the French CML Group. [Google Scholar]
- 41.Mahon FX, Richter J, Guilhot J, Muller MC, Dietz C, Porkka K, Hjorth-Hansen H, Gruber F, Panagoitidis P, Ossenkoppele GJ, Mayer J, Almeida A, Polakova KM, Ehrencrona H, Kairisto V, Berger MG, stromberg UO, Mustjoki S, Hochhaus A, Pfirrmann M, Saussele S. 56th ASH Annual Meeting and Exposition. San Francisco, CA: 2014. Interim Analysis of a Pan European Stop Tyrosine Kinase Inhibitor Trial in Chronic Myeloid Leukemia: The EURO-SKI study. [Google Scholar]
- 42.Branford S, Ross DM, Yeung DT, Braley J, Hughes TP. 56th ASH Annual Meeting and Exposition. San Francisco, CA: 2014. Achieving the Deep Molecular Response Levels Required for an Imatinib Discontinuation Trial Is Strongly Associated with the BCR-ABL Level at the First Qualifying Timepoint. [Google Scholar]
- 43.Yhim HY, Lee NR, Song EK, Yim CY, Jeon SY, Lee B, Kim JA, Kim HS, Cho EH, Kwak JY. 56th ASH Annual Meeting and Exposition. San Francisco, CA: 2014. Long-Term Follow-up Results after Imatinib Discontinuation in Chronic Myeloid Leukemia Patients with Undetectable Minimal Residual Disease. [DOI] [PubMed] [Google Scholar]
- 44.Clarkson B, Fried J, Strife A, Sakai Y, Ota K, Okita T. Studies of cellular proliferation in human leukemia. 3. Behavior of leukemic cells in three adults with acute leukemia given continuous infusions of 3H-thymidine for 8 or 10 days. Cancer. 1970;25:1237–1260. doi: 10.1002/1097-0142(197006)25:6<1237::aid-cncr2820250602>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 45.Clarkson B, Strife A. Linkage of proliferative and maturational abnormalities in chronic myelogenous leukemia and relevance to treatment. Leukemia. 1993;7:1683–1721. [PubMed] [Google Scholar]
- 46.Clarkson BD, Strife A, Wisniewski D, Lambek C, Carpino N. New understanding of the pathogenesis of CML: a prototype of early neoplasia. Leukemia. 1997;11:1404–1428. doi: 10.1038/sj.leu.2400751. [DOI] [PubMed] [Google Scholar]
- 47.Stryckmans P, Debusscher L, Socquet M. Regulation of bone marrow myeloblast proliferation in chronic myeloid leukemia. Cancer Res. 1976;36:3034–3038. [PubMed] [Google Scholar]
- 48.Ogawa M, Fried J, Sakai Y, Strife A, Clarkson BD. Studies of cellular proliferation in human leukemia. VI. The proliferative activity, generation time, and emergence time of neutrophilic granulocytes in chronic granulocytic leukemia. Cancer. 1970;25:1031–1049. doi: 10.1002/1097-0142(197005)25:5<1031::aid-cncr2820250507>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 49.Axelrod R, Axelrod DE, Pienta KJ. Evolution of cooperation among tumor cells. Proc Natl Acad Sci U S A. 2006;103:13474–13479. doi: 10.1073/pnas.0606053103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brock A, Chang H, Huang S. Non-genetic heterogeneity--a mutation-independent driving force for the somatic evolution of tumours. Nat Rev Genet. 2009;10:336–342. doi: 10.1038/nrg2556. [DOI] [PubMed] [Google Scholar]
- 51.Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet. 2013;381:1943–1955. doi: 10.1016/S0140-6736(12)62187-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Faderl S, O’Brien S, Pui CH, Stock W, Wetzler M, Hoelzer D, Kantarjian HM. Adult acute lymphoblastic leukemia: concepts and strategies. Cancer. 2010;116:1165–1176. doi: 10.1002/cncr.24862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jabbour EJ, Faderl S, Kantarjian HM. Adult acute lymphoblastic leukemia. Mayo Clin Proc. 2005;80:1517–1527. doi: 10.4065/80.11.1517. [DOI] [PubMed] [Google Scholar]
- 54.Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, Behm FG, Pui CH, Downing JR, Gilliland DG, Lander ES, Golub TR, Look AT. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002;1:75–87. doi: 10.1016/s1535-6108(02)00018-1. [DOI] [PubMed] [Google Scholar]
- 55.Kantarjian H, O’Brien S, Cortes J, Wierda W, Faderl S, Garcia-Manero G, Issa JP, Estey E, Keating M, Freireich EJ. Therapeutic advances in leukemia and myelodysplastic syndrome over the past 40 years. Cancer. 2008;113:1933–1952. doi: 10.1002/cncr.23655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clarkson B, Strife A, Wisniewski D, Lambek CL, Liu C. Chronic myelogenous leukemia as a paradigm of early cancer and possible curative strategies. Leukemia. 2003;17:1211–1262. doi: 10.1038/sj.leu.2402912. [DOI] [PubMed] [Google Scholar]
- 57.Berman E, Jhanwar S, McBride M, Strife A, Wisniewski D, Lambek C, Clarkson B. Characterization of two novel sublines established from a human megakaryoblastic leukemia cell line transfected with p210(BCR-ABL) Leuk Res. 2000;24:289–297. doi: 10.1016/s0145-2126(99)00179-4. [DOI] [PubMed] [Google Scholar]
- 58.Huhn RD, Posner MR, Rayter SI, Foulkes JG, Frackelton AR Jr. Cell lines and peripheral blood leukocytes derived from individuals with chronic myelogenous leukemia display virtually identical proteins phosphorylated on tyrosine residues. Proc Natl Acad Sci U S A. 1987;84:4408–4412. doi: 10.1073/pnas.84.13.4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clarkson B, Strife A, De Harven E. Continuous culture of seven new cell lines (SK-L1 to 7) from patients with acute leukemia. Cancer. 1967;20:926–947. doi: 10.1002/1097-0142(196706)20:6<926::aid-cncr2820200603>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 60.Kochling J, Konig-Merediz SA, Stripecke R, Buchwald D, Korte A, Von Einsiedel HG, Sack F, Henze G, Seeger K, Wittig B, Schmidt M. Protection of mice against Philadelphia chromosome-positive acute lymphoblastic leukemia by cell-based vaccination using nonviral, minimalistic expression vectors and immunomodulatory oligonucleotides. Clin Cancer Res. 2003;9:3142–3149. [PubMed] [Google Scholar]
- 61.Weisenthal LM, Dill PL, Kurnick NB, Lippman ME. Comparison of dye exclusion assays with a clonogenic assay in the determination of drug-induced cytotoxicity. Cancer Res. 1983;43:258–264. [PubMed] [Google Scholar]
- 62.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 63.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beer L, Zimmermann M, Mitterbauer A, Ellinger A, Gruber F, Narzt MS, Zellner M, Gyongyosi M, Madlener S, Simader E, Gabriel C, Mildner M, Ankersmit HJ. Analysis of the Secretome of Apoptotic Peripheral Blood Mononuclear Cells: Impact of Released Proteins and Exosomes for Tissue Regeneration. Sci Rep. 2015;5:16662. doi: 10.1038/srep16662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haider T, Hoftberger R, Ruger B, Mildner M, Blumer R, Mitterbauer A, Buchacher T, Sherif C, Altmann P, Redl H, Gabriel C, Gyongyosi M, Fischer MB, Lubec G, Ankersmit HJ. The secretome of apoptotic human peripheral blood mononuclear cells attenuates secondary damage following spinal cord injury in rats. Exp Neurol. 2015;267:230–242. doi: 10.1016/j.expneurol.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 66.Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, Xu Y, Autenrieth IB, Schulze-Osthoff K, Belka C, Stuhler G, Wesselborg S. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. 2003;113:717–730. doi: 10.1016/s0092-8674(03)00422-7. [DOI] [PubMed] [Google Scholar]
- 67.Mildner M, Hacker S, Haider T, Gschwandtner M, Werba G, Barresi C, Zimmermann M, Golabi B, Tschachler E, Ankersmit HJ. Secretome of peripheral blood mononuclear cells enhances wound healing. PLoS One. 2013;8:e60103. doi: 10.1371/journal.pone.0060103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pavo N, Zimmermann M, Pils D, Mildner M, Petrasi Z, Petnehazy O, Fuzik J, Jakab A, Gabriel C, Sipos W, Maurer G, Gyongyosi M, Ankersmit HJ. Long-acting beneficial effect of percutaneously intramyocardially delivered secretome of apoptotic peripheral blood cells on porcine chronic ischemic left ventricular dysfunction. Biomaterials. 2014;35:3541–3550. doi: 10.1016/j.biomaterials.2013.12.071. [DOI] [PubMed] [Google Scholar]
- 69.Han NK, Kim BC, Lee HC, Lee YJ, Park MJ, Chi SG, Ko YG, Lee JS. Secretome analysis of ionizing radiation-induced senescent cancer cells reveals that secreted RKIP plays a critical role in neighboring cell migration. Proteomics. 2012;12:2822–2832. doi: 10.1002/pmic.201100419. [DOI] [PubMed] [Google Scholar]
- 70.Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009;9:81–94. doi: 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- 71.Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, Honda K, Ishikawa Y, Hara E, Ohtani N. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- 72.Zullo J, Matsumoto K, Xavier S, Ratliff B, Goligorsky MS. The cell secretome, a mediator of cell-to-cell communication. Prostaglandins Other Lipid Mediat. 2015;120:17–20. doi: 10.1016/j.prostaglandins.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, Welch CL, Wang N, Randolph GJ, Snoeck HW, Tall AR. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010;328:1689–1693. doi: 10.1126/science.1189731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guo D, Bell EH, Mischel P, Chakravarti A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr Pharm Des. 2014;20:2619–2626. doi: 10.2174/13816128113199990486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Peeters SD, van der Kolk DM, de Haan G, Bystrykh L, Kuipers F, de Vries EG, Vellenga E. Selective expression of cholesterol metabolism genes in normal CD34+CD38- cells with a heterogeneous expression pattern in AML cells. Exp Hematol. 2006;34:622–630. doi: 10.1016/j.exphem.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 76.Raaijmakers MH. ATP-binding-cassette transporters in hematopoietic stem cells and their utility as therapeutical targets in acute and chronic myeloid leukemia. Leukemia. 2007;21:2094–2102. doi: 10.1038/sj.leu.2404859. [DOI] [PubMed] [Google Scholar]
- 77.Westerterp M, Gourion-Arsiquaud S, Murphy AJ, Shih A, Cremers S, Levine RL, Tall AR, Yvan-Charvet L. Regulation of hematopoietic stem and progenitor cell mobilization by cholesterol efflux pathways. Cell Stem Cell. 2012;11:195–206. doi: 10.1016/j.stem.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li HY, Appelbaum FR, Willman CL, Zager RA, Banker DE. Cholesterol-modulating agents kill acute myeloid leukemia cells and sensitize them to therapeutics by blocking adaptive cholesterol responses. Blood. 2003;101:3628–3634. doi: 10.1182/blood-2002-07-2283. [DOI] [PubMed] [Google Scholar]
- 79.Resnik N, Repnik U, Kreft ME, Sepcic K, Macek P, Turk B, Veranic P. Highly Selective Anti-Cancer Activity of Cholesterol-Interacting Agents Methyl-beta-Cyclodextrin and Ostreolysin A/Pleurotolysin B Protein Complex on Urothelial Cancer Cells. PLoS One. 2015;10:e0137878. doi: 10.1371/journal.pone.0137878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pallasch CP, Schwamb J, Konigs S, Schulz A, Debey S, Kofler D, Schultze JL, Hallek M, Ultsch A, Wendtner CM. Targeting lipid metabolism by the lipoprotein lipase inhibitor orlistat results in apoptosis of B-cell chronic lymphocytic leukemia cells. Leukemia. 2008;22:585–592. doi: 10.1038/sj.leu.2405058. [DOI] [PubMed] [Google Scholar]
- 81.Zelcer N, Hong C, Boyadjian R, Tontonoz P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. 2009;325:100–104. doi: 10.1126/science.1168974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Roose J, Clevers H. TCF transcription factors: molecular switches in carcinogenesis. Biochim Biophys Acta. 1999;1424:M23–37. doi: 10.1016/s0304-419x(99)00026-8. [DOI] [PubMed] [Google Scholar]
- 83.Hazra A, Fuchs CS, Chan AT, Giovannucci EL, Hunter DJ. Association of the TCF7L2 polymorphism with colorectal cancer and adenoma risk. Cancer Causes Control. 2008;19:975–980. doi: 10.1007/s10552-008-9164-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Slattery ML, Folsom AR, Wolff R, Herrick J, Caan BJ, Potter JD. Transcription factor 7-like 2 polymorphism and colon cancer. Cancer Epidemiol Biomarkers Prev. 2008;17:978–982. doi: 10.1158/1055-9965.EPI-07-2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Okuhira K, Fitzgerald ML, Sarracino DA, Manning JJ, Bell SA, Goss JL, Freeman MW. Purification of ATP-binding cassette transporter A1 and associated binding proteins reveals the importance of beta1-syntrophin in cholesterol efflux. J Biol Chem. 2005;280:39653–39664. doi: 10.1074/jbc.M510187200. [DOI] [PubMed] [Google Scholar]
- 86.Hilvo M, Denkert C, Lehtinen L, Muller B, Brockmoller S, Seppanen-Laakso T, Budczies J, Bucher E, Yetukuri L, Castillo S, Berg E, Nygren H, Sysi-Aho M, Griffin JL, Fiehn O, Loibl S, Richter-Ehrenstein C, Radke C, Hyotylainen T, Kallioniemi O, Iljin K, Oresic M. Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Res. 2011;71:3236–3245. doi: 10.1158/0008-5472.CAN-10-3894. [DOI] [PubMed] [Google Scholar]
- 87.Baenke F, Peck B, Miess H, Schulze A. Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech. 2013;6:1353–1363. doi: 10.1242/dmm.011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cruz PM, Mo H, McConathy WJ, Sabnis N, Lacko AG. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: a review of scientific findings, relevant to future cancer therapeutics. Front Pharmacol. 2013;4:119. doi: 10.3389/fphar.2013.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18:153–161. doi: 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim S, Lee Y, Koo JS. Differential expression of lipid metabolism-related proteins in different breast cancer subtypes. PLoS One. 2015;10:e0119473. doi: 10.1371/journal.pone.0119473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang F, Du G. Dysregulated lipid metabolism in cancer. World J Biol Chem. 2012;3:167–174. doi: 10.4331/wjbc.v3.i8.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tirado-Velez JM, Joumady I, Saez-Benito A, Cozar-Castellano I, Perdomo G. Inhibition of fatty acid metabolism reduces human myeloma cells proliferation. PLoS One. 2012;7:e46484. doi: 10.1371/journal.pone.0046484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Boyd RS, Adam PJ, Patel S, Loader JA, Berry J, Redpath NT, Poyser HR, Fletcher GC, Burgess NA, Stamps AC, Hudson L, Smith P, Griffiths M, Willis TG, Karran EL, Oscier DG, Catovsky D, Terrett JA, Dyer MJ. Proteomic analysis of the cell-surface membrane in chronic lymphocytic leukemia: identification of two novel proteins, BCNP1 and MIG2B. Leukemia. 2003;17:1605–1612. doi: 10.1038/sj.leu.2402993. [DOI] [PubMed] [Google Scholar]
- 94.Akyerli CB, Beksac M, Holko M, Frevel M, Dalva K, Ozbek U, Soydan E, Ozcan M, Ozet G, Ilhan O, Gurman G, Akan H, Williams BR, Ozcelik T. Expression of IFITM1 in chronic myeloid leukemia patients. Leuk Res. 2005;29:283–286. doi: 10.1016/j.leukres.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 95.Tedder TF, Poe JC, Haas KM. CD22: a multifunctional receptor that regulates B lymphocyte survival and signal transduction. Adv Immunol. 2005;88:1–50. doi: 10.1016/S0065-2776(05)88001-0. [DOI] [PubMed] [Google Scholar]
- 96.Tedder TF, Tuscano J, Sato S, Kehrl JH. CD22, a B lymphocyte-specific adhesion molecule that regulates antigen receptor signaling. Annu Rev Immunol. 1997;15:481–504. doi: 10.1146/annurev.immunol.15.1.481. [DOI] [PubMed] [Google Scholar]
- 97.Chambers SM, Boles NC, Lin KY, Tierney MP, Bowman TV, Bradfute SB, Chen AJ, Merchant AA, Sirin O, Weksberg DC, Merchant MG, Fisk CJ, Shaw CA, Goodell MA. Hematopoietic fingerprints: an expression database of stem cells and their progeny. Cell Stem Cell. 2007;1:578–591. doi: 10.1016/j.stem.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 99.Boles NC, Lin KK, Lukov GL, Bowman TV, Baldridge MT, Goodell MA. CD48 on hematopoietic progenitors regulates stem cells and suppresses tumor formation. Blood. 2011;118:80–87. doi: 10.1182/blood-2010-12-322339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Peng SL, Gerth AJ, Ranger AM, Glimcher LH. NFATc1 and NFATc2 together control both T and B cell activation and differentiation. Immunity. 2001;14:13–20. doi: 10.1016/s1074-7613(01)00085-1. [DOI] [PubMed] [Google Scholar]
- 101.Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472–484. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- 102.Law CL, Wormann B, LeBien TW. Analysis of expression and function of CD40 on normal and leukemic human B cell precursors. Leukemia. 1990;4:732–738. [PubMed] [Google Scholar]
- 103.Troeger A, Glouchkova L, Ackermann B, Escherich G, Meisel R, Hanenberg H, den Boer ML, Pieters R, Janka-Schaub GE, Goebel U, Laws HJ, Dilloo D. High expression of CD40 on B-cell precursor acute lymphoblastic leukemia blasts is an independent risk factor associated with improved survival and enhanced capacity to up-regulate the death receptor CD95. Blood. 2008;112:1028–1034. doi: 10.1182/blood-2007-11-123315. [DOI] [PubMed] [Google Scholar]
- 104.Kamps MP, Murre C, Sun XH, Baltimore D. A new homeobox gene contributes the DNA binding domain of the t(1;19) translocation protein in pre-B ALL. Cell. 1990;60:547–555. doi: 10.1016/0092-8674(90)90658-2. [DOI] [PubMed] [Google Scholar]
- 105.Brambillasca F, Mosna G, Colombo M, Rivolta A, Caslini C, Minuzzo M, Giudici G, Mizzi L, Biondi A, Privitera E. Identification of a novel molecular partner of the E2A gene in childhood leukemia. Leukemia. 1999;13:369–375. doi: 10.1038/sj.leu.2401338. [DOI] [PubMed] [Google Scholar]
- 106.Inaba T, Roberts WM, Shapiro LH, Jolly KW, Raimondi SC, Smith SD, Look AT. Fusion of the leucine zipper gene HLF to the E2A gene in human acute B-lineage leukemia. Science. 1992;257:531–534. doi: 10.1126/science.1386162. [DOI] [PubMed] [Google Scholar]
- 107.Imai C, Ross ME, Reid G, Coustan-Smith E, Schultz KR, Pui CH, Downing JR, Campana D. Expression of the adaptor protein BLNK/SLP-65 in childhood acute lymphoblastic leukemia. Leukemia. 2004;18:922–925. doi: 10.1038/sj.leu.2403349. [DOI] [PubMed] [Google Scholar]
- 108.Marin R, Ruiz-Cabello F, Pedrinaci S, Mendez R, Jimenez P, Geraghty DE, Garrido F. Analysis of HLA-E expression in human tumors. Immunogenetics. 2003;54:767–775. doi: 10.1007/s00251-002-0526-9. [DOI] [PubMed] [Google Scholar]
- 109.Laszlo GS, Alonzo TA, Gudgeon CJ, Harrington KH, Kentsis A, Gerbing RB, Wang YC, Ries RE, Raimondi SC, Hirsch BA, Gamis AS, Meshinchi S, Walter RB. High expression of myocyte enhancer factor 2C (MEF2C) is associated with adverse-risk features and poor outcome in pediatric acute myeloid leukemia: a report from the Children’s Oncology Group. J Hematol Oncol. 2015;8:115. doi: 10.1186/s13045-015-0215-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cante-Barrett K, Pieters R, Meijerink JP. Myocyte enhancer factor 2C in hematopoiesis and leukemia. Oncogene. 2014;33:403–410. doi: 10.1038/onc.2013.56. [DOI] [PubMed] [Google Scholar]
- 111.Nagel S, Meyer C, Quentmeier H, Kaufmann M, Drexler HG, MacLeod RA. MEF2C is activated by multiple mechanisms in a subset of T-acute lymphoblastic leukemia cell lines. Leukemia. 2008;22:600–607. doi: 10.1038/sj.leu.2405067. [DOI] [PubMed] [Google Scholar]
- 112.Garcia JF, Roncador G, Garcia JF, Sanz AI, Maestre L, Lucas E, Montes-Moreno S, Fernandez Victoria R, Martinez-Torrecuadrara JL, Marafioti T, Mason DY, Piris MA. PRDM1/BLIMP-1 expression in multiple B and T-cell lymphoma. Haematologica. 2006;91:467–474. [PubMed] [Google Scholar]
- 113.Kang HB, Lee HR, Jee da J, Shin SH, Nah SS, Yoon SY, Kim JW. PRDM1, a Tumor-Suppressor Gene, is Induced by Genkwadaphnin in Human Colon Cancer SW620 Cells. J Cell Biochem. 2016;117:172–179. doi: 10.1002/jcb.25262. [DOI] [PubMed] [Google Scholar]
- 114.Ai J, Advani A. Current status of antibody therapy in ALL. Br J Haematol. 2015;168:471–480. doi: 10.1111/bjh.13205. [DOI] [PubMed] [Google Scholar]
- 115.Nadler LM, Ritz J, Hardy R, Pesando JM, Schlossman SF, Stashenko P. A unique cell surface antigen identifying lymphoid malignancies of B cell origin. J Clin Invest. 1981;67:134–140. doi: 10.1172/JCI110005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Anolik J, Looney RJ, Bottaro A, Sanz I, Young F. Down-regulation of CD20 on B cells upon CD40 activation. Eur J Immunol. 2003;33:2398–2409. doi: 10.1002/eji.200323515. [DOI] [PubMed] [Google Scholar]
- 117.Zheng Z, Venkatapathy S, Rao G, Harrington CA. Expression profiling of B cell chronic lymphocytic leukemia suggests deficient CD1-mediated immunity, polarized cytokine response, altered adhesion and increased intracellular protein transport and processing of leukemic cells. Leukemia. 2002;16:2429–2437. doi: 10.1038/sj.leu.2402711. [DOI] [PubMed] [Google Scholar]
- 118.Hancer VS, Diz-Kucukkaya R, Aktan M. Overexpression of Fc mu receptor (FCMR, TOSO) gene in chronic lymphocytic leukemia patients. Med Oncol. 2012;29:1068–1072. doi: 10.1007/s12032-011-9821-3. [DOI] [PubMed] [Google Scholar]
- 119.Yu Z, Zhou KS, Li ZJ, Yi SH, Hao M, Li CH, Qi JY, Qiu LG. [Expression and prognostic significance of TOSO in CD19+ B cells from Chinese CLL patients] . Zhonghua Yi Xue Za Zhi. 2011;91:1371–1374. [PubMed] [Google Scholar]
- 120.Vire B, David A, Wiestner A. TOSO, the Fcmicro receptor, is highly expressed on chronic lymphocytic leukemia B cells, internalizes upon IgM binding, shuttles to the lysosome, and is downregulated in response to TLR activation. J Immunol. 2011;187:4040–4050. doi: 10.4049/jimmunol.1100532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yoshida C, Yoshida F, Sears DE, Hart SM, Ikebe D, Muto A, Basu S, Igarashi K, Melo JV. Bcr-Abl signaling through the PI-3/S6 kinase pathway inhibits nuclear translocation of the transcription factor Bach2, which represses the antiapoptotic factor heme oxygenase-1. Blood. 2007;109:1211–1219. doi: 10.1182/blood-2005-12-040972. [DOI] [PubMed] [Google Scholar]
- 122.Loftus JC, Dhruv H, Tuncali S, Kloss J, Yang Z, Schumacher CA, Cao B, Williams BO, Eschbacher JM, Ross JT, Tran NL. TROY (TNFRSF19) promotes glioblastoma survival signaling and therapeutic resistance. Mol Cancer Res. 2013;11:865–874. doi: 10.1158/1541-7786.MCR-13-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.McCallum L, Price S, Planque N, Perbal B, Pierce A, Whetton AD, Irvine AE. A novel mechanism for BCR-ABL action: stimulated secretion of CCN3 is involved in growth and differentiation regulation. Blood. 2006;108:1716–1723. doi: 10.1182/blood-2006-04-016113. [DOI] [PubMed] [Google Scholar]
- 124.McCallum L, Irvine AE. CCN3--a key regulator of the hematopoietic compartment. Blood Rev. 2009;23:79–85. doi: 10.1016/j.blre.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 125.Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, Moita CF, Schauer K, Hume AN, Freitas RP, Goud B, Benaroch P, Hacohen N, Fukuda M, Desnos C, Seabra MC, Darchen F, Amigorena S, Moita LF, Thery C. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. 2010;12:19–30. doi: 10.1038/ncb2000. sup pp 1-13. [DOI] [PubMed] [Google Scholar]
- 126.Tolmachova T, Anders R, Stinchcombe J, Bossi G, Griffiths GM, Huxley C, Seabra MC. A general role for Rab27a in secretory cells. Mol Biol Cell. 2004;15:332–344. doi: 10.1091/mbc.E03-07-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Spiegelberg HL, Fishkin BG. Human myeloma IgA half-molecules. J Clin Invest. 1976;58:1259–1265. doi: 10.1172/JCI108580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang Q, Ni Q, Wang X, Zhu H, Wang Z, Huang J. High expression of RAB27A and TP53 in pancreatic cancer predicts poor survival. Med Oncol. 2015;32:372. doi: 10.1007/s12032-014-0372-2. [DOI] [PubMed] [Google Scholar]
- 129.Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569–579. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 130.Raimondo S, Saieva L, Corrado C, Fontana S, Flugy A, Rizzo A, De Leo G, Alessandro R. Chronic myeloid leukemia-derived exosomes promote tumor growth through an autocrine mechanism. Cell Commun Signal. 2015;13:8. doi: 10.1186/s12964-015-0086-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Toyama-Sorimachi N, Sorimachi H, Tobita Y, Kitamura F, Yagita H, Suzuki K, Miyasaka M. A novel ligand for CD44 is serglycin, a hematopoietic cell lineage-specific proteoglycan. Possible involvement in lymphoid cell adherence and activation. J Biol Chem. 1995;270:7437–7444. doi: 10.1074/jbc.270.13.7437. [DOI] [PubMed] [Google Scholar]
- 132.Toyama-Sorimachi N, Kitamura F, Habuchi H, Tobita Y, Kimata K, Miyasaka M. Widespread expression of chondroitin sulfate-type serglycins with CD44 binding ability in hematopoietic cells. J Biol Chem. 1997;272:26714–26719. doi: 10.1074/jbc.272.42.26714. [DOI] [PubMed] [Google Scholar]
- 133.Wernersson S, Braga T, Sawesi O, Waern I, Nilsson KE, Pejler G, Abrink M. Age-related enlargement of lymphoid tissue and altered leukocyte composition in serglycin-deficient mice. J Leukoc Biol. 2009;85:401–408. doi: 10.1189/jlb.1008670. [DOI] [PubMed] [Google Scholar]
- 134.Sukhai MA, Prabha S, Hurren R, Rutledge AC, Lee AY, Sriskanthadevan S, Sun H, Wang X, Skrtic M, Seneviratne A, Cusimano M, Jhas B, Gronda M, MacLean N, Cho EE, Spagnuolo PA, Sharmeen S, Gebbia M, Urbanus M, Eppert K, Dissanayake D, Jonet A, Dassonville-Klimpt A, Li X, Datti A, Ohashi PS, Wrana J, Rogers I, Sonnet P, Ellis WY, Corey SJ, Eaves C, Minden MD, Wang JC, Dick JE, Nislow C, Giaever G, Schimmer AD. Lysosomal disruption preferentially targets acute myeloid leukemia cells and progenitors. J Clin Invest. 2013;123:315–328. doi: 10.1172/JCI64180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, Yao A, Ye J, Zhan M, Zhang W, Zhang H, Zhao Q, Zheng L, Zhong F, Zhong W, Zhu S, Zhao S, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An H, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigo R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi H, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays A, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu X, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen M, Wu D, Wu M, Xia A, Zandieh A, Zhu X. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 136.Hoffman NE, Chandramoorthy HC, Shanmughapriya S, Zhang XQ, Vallem S, Doonan PJ, Malliankaraman K, Guo S, Rajan S, Elrod JW, Koch WJ, Cheung JY, Madesh M. SLC25A23 augments mitochondrial Ca(2)(+) uptake, interacts with MCU, and induces oxidative stress-mediated cell death. Mol Biol Cell. 2014;25:936–947. doi: 10.1091/mbc.E13-08-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Huang Y, Dai Z, Barbacioru C, Sadee W. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 2005;65:7446–7454. doi: 10.1158/0008-5472.CAN-04-4267. [DOI] [PubMed] [Google Scholar]
- 139.El-Gebali S, Bentz S, Hediger MA, Anderle P. Solute carriers (SLCs) in cancer. Mol Aspects Med. 2013;34:719–734. doi: 10.1016/j.mam.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 140.Prindle A, Liu J, Asally M, Ly S, Garcia-Ojalvo J, Suel GM. Ion channels enable electrical communication in bacterial communities. Nature. 2015;527:59–63. doi: 10.1038/nature15709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Masi E, Ciszak M, Santopolo L, Frascella A, Giovannetti L, Marchi E, Viti C, Mancuso S. Electrical spiking in bacterial biofilms. J R Soc Interface. 2015;12:20141036. doi: 10.1098/rsif.2014.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Grechenko TN KAN, Zhegallo AV. Evolutionary paths of electric oscillators. Eksperimental’naâ psihologiâ. Experimental Psychology (Russia) 2015;8:105–118. [Google Scholar]
- 143.Ma CH, Chong JH, Guo Y, Zeng HM, Liu SY, Xu LL, Wei J, Lin YM, Zhu XF, Zheng GG. Abnormal expression of ADAR1 isoforms in Chinese pediatric acute leukemias. Biochem Biophys Res Commun. 2011;406:245–251. doi: 10.1016/j.bbrc.2011.02.025. [DOI] [PubMed] [Google Scholar]
- 144.Jiang Q, Crews LA, Barrett CL, Chun HJ, Court AC, Isquith JM, Zipeto MA, Goff DJ, Minden M, Sadarangani A, Rusert JM, Dao KH, Morris SR, Goldstein LS, Marra MA, Frazer KA, Jamieson CH. ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc Natl Acad Sci U S A. 2013;110:1041–1046. doi: 10.1073/pnas.1213021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Qin Y, Verdegaal EM, Siderius M, Bebelman JP, Smit MJ, Leurs R, Willemze R, Tensen CP, Osanto S. Quantitative expression profiling of G-protein-coupled receptors (GPCRs) in metastatic melanoma: the constitutively active orphan GPCR GPR18 as novel drug target. Pigment Cell Melanoma Res. 2011;24:207–218. doi: 10.1111/j.1755-148X.2010.00781.x. [DOI] [PubMed] [Google Scholar]
- 146.Anghel SI, Correa-Rocha R, Budinska E, Boligan KF, Abraham S, Colombetti S, Fontao L, Mariotti A, Rimoldi D, Ghanem GE, Fisher DE, Levy F, Delorenzi M, Piguet V. Breast cancer suppressor candidate-1 (BCSC-1) is a melanoma tumor suppressor that down regulates MITF. Pigment Cell Melanoma Res. 2012;25:482–487. doi: 10.1111/j.1755-148X.2012.01018.x. [DOI] [PubMed] [Google Scholar]
- 147.Zhou YQ, Chen SL, Ju JY, Shen L, Liu Y, Zhen S, Lv N, He ZG, Zhu LP. Tumor suppressor function of BCSC-1 in nasopharyngeal carcinoma. Cancer Sci. 2009;100:1817–1822. doi: 10.1111/j.1349-7006.2009.01261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhou L, Opalinska J, Sohal D, Yu Y, Mo Y, Bhagat T, Abdel-Wahab O, Fazzari M, Figueroa M, Alencar C, Zhang J, Kambhampati S, Parmar S, Nischal S, Hueck C, Suzuki M, Freidman E, Pellagatti A, Boultwood J, Steidl U, Sauthararajah Y, Yajnik V, McMahon C, Gore SD, Platanias LC, Levine R, Melnick A, Wickrema A, Greally JM, Verma A. Aberrant epigenetic and genetic marks are seen in myelodysplastic leukocytes and reveal Dock4 as a candidate pathogenic gene on chromosome 7q. J Biol Chem. 2011;286:25211–25223. doi: 10.1074/jbc.M111.235028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sundaravel S, Duggan R, Bhagat T, Ebenezer DL, Liu H, Yu Y, Bartenstein M, Unnikrishnan M, Karmakar S, Liu TC, Torregroza I, Quenon T, Anastasi J, McGraw KL, Pellagatti A, Boultwood J, Yajnik V, Artz A, Le Beau MM, Steidl U, List AF, Evans T, Verma A, Wickrema A. Reduced DOCK4 expression leads to erythroid dysplasia in myelodysplastic syndromes. Proc Natl Acad Sci U S A. 2015;112:E6359–6368. doi: 10.1073/pnas.1516394112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kjeldsen E, Veigaard C. DOCK4 deletion at 7q31.1 in a de novo acute myeloid leukemia with a normal karyotype. Cell Oncol (Dordr) 2013;36:395–403. doi: 10.1007/s13402-013-0145-5. [DOI] [PubMed] [Google Scholar]
- 151.Wang CH, Su PT, Du XY, Kuo MW, Lin CY, Yang CC, Chan HS, Chang SJ, Kuo C, Seo K, Leung LL, Chuang YJ. Thrombospondin type I domain containing 7A (THSD7A) mediates endothelial cell migration and tube formation. J Cell Physiol. 2010;222:685–694. doi: 10.1002/jcp.21990. [DOI] [PubMed] [Google Scholar]
- 152.Hensel J, Duex JE, Owens C, Dancik GM, Edwards MG, Frierson HF, Theodorescu D. Patient Mutation Directed shRNA Screen Uncovers Novel Bladder Tumor Growth Suppressors. Mol Cancer Res. 2015;13:1306–1315. doi: 10.1158/1541-7786.MCR-15-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Harvey RC, Mullighan CG, Wang X, Dobbin KK, Davidson GS, Bedrick EJ, Chen IM, Atlas SR, Kang H, Ar K, Wilson CS, Wharton W, Murphy M, Devidas M, Carroll AJ, Borowitz MJ, Bowman WP, Downing JR, Relling M, Yang J, Bhojwani D, Carroll WL, Camitta B, Reaman GH, Smith M, Hunger SP, Willman CL. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010;116:4874–4884. doi: 10.1182/blood-2009-08-239681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mancias JD, Pontano Vaites L, Nissim S, Biancur DE, Kim AJ, Wang X, Liu Y, Goessling W, Kimmelman AC, Harper JW. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife. 2015;4 doi: 10.7554/eLife.10308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cheng H, Hao S, Liu Y, Pang Y, Ma S, Dong F, Xu J, Zheng G, Li S, Yuan W, Cheng T. Leukemic marrow infiltration reveals a novel role for Egr3 as a potent inhibitor of normal hematopoietic stem cell proliferation. Blood. 2015;126:1302–1313. doi: 10.1182/blood-2015-01-623645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mukherjee S. “Blood feuds”. Blood. 2015;126:1264–1265. doi: 10.1182/blood-2015-07-659540. [DOI] [PubMed] [Google Scholar]
- 157.Yasunaga J, Taniguchi Y, Nosaka K, Yoshida M, Satou Y, Sakai T, Mitsuya H, Matsuoka M. Identification of aberrantly methylated genes in association with adult T-cell leukemia. Cancer Res. 2004;64:6002–6009. doi: 10.1158/0008-5472.CAN-04-1422. [DOI] [PubMed] [Google Scholar]
- 158.Lee SE, Kim SJ, Yoon HJ, Yu SY, Yang H, Jeong SI, Hwang SY, Park CS, Park YS. Genome-wide profiling in melatonin-exposed human breast cancer cell lines identifies differentially methylated genes involved in the anticancer effect of melatonin. J Pineal Res. 2013;54:80–88. doi: 10.1111/j.1600-079X.2012.01027.x. [DOI] [PubMed] [Google Scholar]
- 159.Pio R, Jia Z, Baron VT, Mercola D UCI NCI SPECS Consortium of the Strategic Partners for the Evaluation of Cancer Signatures-Prostate Cancer. Early growth response 3 (Egr3) is highly over-expressed in non-relapsing prostate cancer but not in relapsing prostate cancer. PLoS One. 2013;8:e54096. doi: 10.1371/journal.pone.0054096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Liao F, Ji MY, Shen L, Qiu S, Guo XF, Dong WG. Decreased EGR3 expression is related to poor prognosis in patients with gastric cancer. J Mol Histol. 2013;44:463–468. doi: 10.1007/s10735-013-9493-8. [DOI] [PubMed] [Google Scholar]
- 161.Kuo MW, Wang CH, Wu HC, Chang SJ, Chuang YJ. Soluble THSD7A is an N-glycoprotein that promotes endothelial cell migration and tube formation in angiogenesis. PLoS One. 2011;6:e29000. doi: 10.1371/journal.pone.0029000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Li P, Li J, Yang R, Zhang F, Wang H, Chu H, Lu Y, Dun S, Wang Y, Zang W, Du Y, Chen X, Zhao G, Zhang G. Study on expression of lncRNA RGMB-AS1 and repulsive guidance molecule b in non-small cell lung cancer. Diagn Pathol. 2015;10:63. doi: 10.1186/s13000-015-0297-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ivanova AV, Ivanov SV, Prudkin L, Nonaka D, Liu Z, Tsao A, Wistuba I, Roth J, Pass HI. Mechanisms of FUS1/TUSC2 deficiency in mesothelioma and its tumorigenic transcriptional effects. Mol Cancer. 2009;8:91. doi: 10.1186/1476-4598-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Shan J, Lopez MC, Baker HV, Kilberg MS. Expression profiling after activation of amino acid deprivation response in HepG2 human hepatoma cells. Physiol Genomics. 2010;41:315–327. doi: 10.1152/physiolgenomics.00217.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Falk MH, Meier T, Issels RD, Brielmeier M, Scheffer B, Bornkamm GW. Apoptosis in Burkitt lymphoma cells is prevented by promotion of cysteine uptake. Int J Cancer. 1998;75:620–625. doi: 10.1002/(sici)1097-0215(19980209)75:4<620::aid-ijc21>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 166.Seton-Rogers S. Tumour heterogeneity: A cooperative tumour cell community. Nat Rev Cancer. 2014;14:294. doi: 10.1038/nrc3732. [DOI] [PubMed] [Google Scholar]
- 167.Hill RP, Chambers AF, Ling V, Harris JF. Dynamic heterogeneity: rapid generation of metastatic variants in mouse B16 melanoma cells. Science. 1984;224:998–1001. doi: 10.1126/science.6719130. [DOI] [PubMed] [Google Scholar]
- 168.Welch DR, Aeed PA, Earhart RH, McClure SA. Evidence for paracrine regulation of experimental metastasis in 13762NF rat mammary adenocarcinoma cell clones. Anticancer Res. 1994;14:1743–1751. [PubMed] [Google Scholar]
- 169.Chen CC, Wang L, Plikus MV, Jiang TX, Murray PJ, Ramos R, Guerrero-Juarez CF, Hughes MW, Lee OK, Shi S, Widelitz RB, Lander AD, Chuong CM. Organ-level quorum sensing directs regeneration in hair stem cell populations. Cell. 2015;161:277–290. doi: 10.1016/j.cell.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Challa A, Eliopoulos AG, Holder MJ, Burguete AS, Pound JD, Chamba A, Grafton G, Armitage RJ, Gregory CD, Martinez-Valdez H, Young L, Gordon J. Population depletion activates autonomous CD154-dependent survival in biopsylike Burkitt lymphoma cells. Blood. 2002;99:3411–3418. doi: 10.1182/blood.v99.9.3411. [DOI] [PubMed] [Google Scholar]
- 171.Youk H, Lim WA. Secreting and sensing the same molecule allows cells to achieve versatile social behaviors. Science. 2014;343:1242782. doi: 10.1126/science.1242782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.You L, Cox RS 3rd, Weiss R, Arnold FH. Programmed population control by cell-cell communication and regulated killing. Nature. 2004;428:868–871. doi: 10.1038/nature02491. [DOI] [PubMed] [Google Scholar]
- 173.Hart Y, Reich-Zeliger S, Antebi YE, Zaretsky I, Mayo AE, Alon U, Friedman N. Paradoxical signaling by a secreted molecule leads to homeostasis of cell levels. Cell. 2014;158:1022–1032. doi: 10.1016/j.cell.2014.07.033. [DOI] [PubMed] [Google Scholar]
- 174.Majima S, Kajino K, Fukuda T, Otsuka F, Hino O. A novel gene “Niban” upregulated in renal carcinogenesis: cloning by the cDNA-amplified fragment length polymorphism approach. Jpn J Cancer Res. 2000;91:869–874. doi: 10.1111/j.1349-7006.2000.tb01027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Old WM, Shabb JB, Houel S, Wang H, Couts KL, Yen CY, Litman ES, Croy CH, Meyer-Arendt K, Miranda JG, Brown RA, Witze ES, Schweppe RE, Resing KA, Ahn NG. Functional proteomics identifies targets of phosphorylation by B-Raf signaling in melanoma. Mol Cell. 2009;34:115–131. doi: 10.1016/j.molcel.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Yuki R, Aoyama K, Kubota S, Yamaguchi N, Kubota S, Hasegawa H, Morii M, Huang X, Liu K, Williams R, Fukuda MN, Yamaguchi N. Overexpression of zinc-finger protein 777 (ZNF777) inhibits proliferation at low cell density through down-regulation of FAM129A. J Cell Biochem. 2015;116:954–968. doi: 10.1002/jcb.25046. [DOI] [PubMed] [Google Scholar]