

Abstract
The purpose of this paper was to better understand the role of polyamine transport in pancreatic cancers.This paper identifies potential biomarkers for assessing the relative tumor commitment to polyamine biosynthesis or transport. Cell lines with low polyamine import activity and low ATP13A3 protein levels appear committed to polyamine biosynthesis and required high concentrations of the polyamine biosynthesis inhibitor, difluoromethylornithine (DFMO) to inhibit their growth (e.g., AsPC-1 and Capan 1). In contrast, cell lines with high polyamine import activity and high ATP13A3 protein expression (e.g., L3.6pl) demonstrated a commitment to polyamine transport and required lower DFMO concentrations to inhibit their growth. Pancreatic cancer cell lines which were most sensitive to DFMO also gave the highest EC50 values for the polyamine transport inhibitors (PTIs) tested indicating that more PTI was needed to inhibit the active polyamine transport systems of these cell lines. Most significant is that the combination therapy of DFMO+PTI was efficacious against both cell types with the PTI showing low efficacy in cell lines with low polyamine transport activity and high efficacy in cell lines with high polyamine transport activity. High ATP13A3 protein expression and moderate to low Cav-1 protein expression was shown to be predictive of tumors which effectively escape DFMO via polyamine import. In summary, this report demonstrates for the first time the role of ATP13A3 in polyamine transport and its use as a potential biomarker along with Cav-1 to select tumors most susceptible to DFMO. These findings may help stratify patients in the ongoing clinical trials with DFMO-based therapies and help predict tumor response.
Keywords: Difluoromethylornithine (DFMO), polyamines, pancreatic cancer, ATP13A3, polyamine transport
Introduction
The native polyamines, putrescine (Put), spermidine (Spd) and spermine (Spm) are key resources required by mammalian cells for growth and proliferation. These low molecular weight aliphatic amines play critical roles in chromatin remodeling, translation, eIF-5A biosynthesis, and transcription [1]. Polyamines exist primarily as polycations at physiological pH and interact strongly with biological polyanions such as RNA and DNA [2]. Unlike inorganic cations (e.g., Mg+2) the native polyamines can be biosynthesized. In addition, they can be imported from the extracellular milieu. The connection between increased polyamine metabolic flux, neoplasia and tumor spread is well established [3-5].
Compounds that disrupt polyamine homeostasis have been shown to be clinically useful for both the chemoprevention and treatment of human cancers [6,7]. Biomass generation is a critical requirement for rapidly-proliferating cancer cells and the altered metabolism of specific cancers make them especially sensitive to polyamine-targeting therapies [1,8]. Polyamine metabolites are needed to replicate the cancer cell’s contents. For example, Spd is required for the formation of the novel aminoacid hypusine, a critical residue in the formation of the required initiation factor eIF-5A [1]. In short, certain cancers seem to be ‘addicted’ to polyamines and rely on a combination of intracellular polyamine biosynthesis and import to maintain high levels of intracellular polyamines. The import process involves the polyamine transport system (PTS) which is used to scavenge polyamines from the extracellular milieu. This addiction to polyamine metabolites provides an opportunity to selectively target cancers via their upregulated PTS [9-11].
In principle, cancer cells exclusively committed to polyamine biosynthesis (Case A) could be targeted by α-difluoromethylornithine (DFMO), a compound which irreversibly inhibits ornithine decarboxylase (ODC), a key enzyme involved in the first step of polyamine biosynthesis. In practice, however, DFMO as a single therapy has been more challenging because DFMO-treated cancer cells often respond by upregulating polyamine import to circumvent DFMO’s blockade of polyamine biosynthesis. Alternatively, cells exclusively committed to polyamine import via high utilization of the PTS (Case B) could be targeted by cytotoxic polyamine-based compounds [9-11], which selectively enter and kill cells via the PTS [10] or via polyamine transport inhibitors (PTIs) [12-14]. As most cancers are expected to lie along the continuum of these two extremes (Cases A and B), combinations of these agents (e.g., DFMO+PTI) are expected to show promise even in the treatment of cancers with high tumor heterogeneity [6,15]. In addition, polyamine transport has been shown to be increased in hypoxic tumors [16], and the combination therapy of DFMO+PTI holds promise for the treatment of recalcitrant solid tumor types with aberrant hypoxia signaling such as pancreatic cancers [13,14,17-19].
The DFMO+PTI combination therapy may provide a ‘catch-all’ technology for cell populations with different commitment levels to polyamine biosynthesis and transport and this approach may be especially relevant in pancreatic cancers, where multiple cell populations with the ability to form tumors and self-renew have been identified [6,15,20]. Moreover, a recent report by Rao et al. demonstrating the critical roles that polyamines play in developing pancreatic cancers provides further impetus for this study [21]. Combination therapies involving DFMO+PTI are expected to be more efficacious than DFMO alone because they block an important cellular escape mechanism (i.e., polyamine import) and can thereby potentiate the in vivo effects of DFMO [13,14]. This report describes our progress in identifying the role of ATP13A3 and caveolin-1 (Cav-1) as biomarkers which indicate which cell types will best respond to the DFMO+PTI combination therapy.
Materials and methods
Reagents
DFMO was kindly provided by Dr. Patrick Woster (MUSC). Aminoguanidine was acquired from Sigma-Aldrich (St. Louis, MO), spermidine (Spd) from Acros Organics (Geel, Belgium), and 3H-Spd from Perkin-Elmer Inc, (Boston, MA). The primary rabbit polyclonal antibodies against ATP13A3 and caveolin-1 were purchased from Abgent (San Diego, CA) and from Santa Cruz Biotechnology (Dallas, TX), respectively. The primary rabbit monoclonal antibodies against c-Myc, MTAP, and p16 were purchased from Abcam (Cambridge, MA). The primary mouse monoclonal antibody against β-actin was obtained from Sigma-Aldrich. The secondary antibodies used included goat anti-rabbit and goat anti-mouse antibodies from Santa Cruz Biotechnology. All cell culture media and reagents were purchased from Life Technologies (Grand Island, NY). FBS was purchased from Atlanta Biologicals (Norcross, GA). All siRNA reagents, including ATP13A3 siRNA, scrambled siRNA and the Viromer Blue transfection agent were obtained from OriGene (Rockville, MD).
Cell culture
The L3.6pl pancreatic cell line was a gift from Dr. Isaiah Fidler at MD Anderson Cancer Center at Houston TX. hTERT-HPNE cells were a generous gift from Dr. Cheryl Baker at BioCurity Holdings, Inc. in Orlando, FL. CHO-K1 cells and CHO-MG cell lines were a gift from Dr. Wayne Flintoff at the University of Western Ontario, Canada. Other cell lines (AsPC-1, BxPC-3, Capan-1, MiaPaca-2, Pan02, Panc-1, and Su86.86) were purchased from ATCC (Manassas, VA), first cultured in the growth media suggested by ATCC and then shifted to RPMI 1640 media with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin, to allow testing of all cell lines under the same conditions. In the human cancer cell lines, 250 µM aminoguanidine was added to the media to prevent serum oxidase activity on polyamine-based compounds. For CHO and CHO-MG cells, 1 mM aminoguanidine was used. The media contained L-proline (2 μg/mL) for proper growth of the CHO-MG cells. Cells in early to mid-log phase were used for all experiments. All cells were grown at 37°C under a humidified 5% CO2 atmosphere.
Cell treatments
Cells were grown to 70-80% confluence. The cells were then trypsinized and re-plated at the appropriate cell density depending on the length of drug treatment. Prior to drug addition, the cells were incubated for 24 h (to facilitate re-attachment) before adding the appropriate compound or vehicle in PBS. DFMO dosage was adapted according to the length of incubation and cell line while Spd was used at 1 µM for all experiments. The cells were incubated for either 48 h or 72 h depending upon the experiment, with aminoguanidine present in the media.
IC50 and IC10 determinations
Cell growth was assayed in sterile 96-well microtiter plates (Costar 3599, Corning, NY). CHO and CHO-MG cells were plated at 1000 cells/well. All the pancreatic cell lines were plated at 500 cells/well, except Su86.86 which was plated at 1500 cells/well and incubated overnight. As these experiments required three additives, cells were plated in 70 µL volumes. DFMO, Spd or polyamine drug solutions (10 μL/well) of appropriate concentration in phosphate-buffered saline (PBS) were added. When needed, additional PBS was added to ensure that each well had a total volume of 100 μL for the experiment. After incubation with the compound(s) for 72 h, cell growth was determined by measuring formazan formation from the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfenyl)-2H tetrazolium, inner salt (MTS) using a SynergyMxBiotek microplate reader for absorbance (490 nm) measurements. All experiments were performed in triplicate. IC50 and IC10 values (i.e., the concentration of the compound needed to maintain 50% and 90% viability, respectively), were calculated. Thus, the IC10 value defines the maximum concentration of the compound which a cell line can tolerate with minimal toxic effects to the cell (≤ 10% toxicity).
Rescue assay and EC50 determination
Cells were seeded in 96 well plates as above and were exposed to the respective IC50 DFMO dose for that cell line, with or without a rescuing amount of Spd (1 µM) for 72 h. This rescue from DFMO treatment with exogenous Spd was cell-line dependent. For example, in L3.6pl cells the 72 h DFMO IC50 value was 4.2 mM and gave 50% viability. Spd addition to these DFMO treated L3.6pl cells resulted in 90% viability and defined a window spanning from DFMO only (50%) to DFMO+Spd (90% viability). The EC50 was defined as the concentration of the PTI needed to traverse halfway between these two endpoints (in this case the [PTI] needed to achieve 70% viability in the presence of DFMO+Spd). This approach allows ranking of the PTIs in different cell lines since it considers the different response to a fixed concentration of Spd (1 µM) in each of the DFMO-treated cell lines.
siRNA treatment and ATP13A3 knockdown
Three siRNAs against ATP13A3 along with a scrambled siRNA were purchased from OriGene. The effect on ATP13A3 expression as measured by immunoblot are as follows: ATP13A3 siRNA #1 (Cat# SR312433A) - poor knockdown, ATP13A3 siRNA #2 (Cat# SR312433B) - maximal knockdown, ATP13A3 siRNA #3 (Cat# SR312433C) - moderate knockdown. ATP13A3 siRNA #2 demonstrated the best knockdown of all three siRNAs tested and was used for all subsequent experimentation. For the experiments, L3.6pl cells were grown to 70-80% confluence in the presence of either ATP13A3 siRNA #2 or scrambled siRNA complexed with the Viromer Blue transfection agent. After 48 h incubation with the appropriate siRNA/transfection agent complexes, cells were washed with PBS, harvested by trypsinization, and re-plated into sterile 96-well plates at 500 cells/well. As described above, the rescue assay (48 h) was performed on the siRNA-treated cells to determine rescue efficiency with and without ATP13A3 knockdown and used DFMO (8 mM), Spd (1 µM) and aminoguanidine (250 µM). ATP13A3 knockdown was confirmed by western blotting. Since the scrambled siRNA treated cells behaved like non-siRNA treated cells in terms of their Spd rescue profile, we concluded that the reduced viability observed with the ATP13A3 siRNA treated cells was not due to general siRNA cell stress but due to the specific reduction in ATP13A3 expression.
Vmax determination
The kinetic profiles for Spd uptake and the ability of the PTIs to inhibit Spd uptake in the cell lines were determined using the protocols of Weeks et al. [19] Briefly, the respective cells (100,000/well) were seeded into a 24-well plate for log phase growth and incubated for 24 h at 37°C. The media was then replaced with preheated Hanks Balanced Salt Solution (HBSS, containing Ca+2 and Mg+2) at 37°C. For Km and Vmax determinations 3H-Spd was added at a range of 0-3 µM/well. For Ki determinations (Table 1 footnote) 3H-Spd (1 μM final concentration) was added with the respective PTI at different PTI concentrations (0, 0.1, 0.3, 1, 2 or 3 μM). Cells were incubated at 37°C for 15 min. The cells were then washed with cold HBSS and lysed with 0.1% sodium dodecyl sulfate (SDS) in water (500 μL). Each cell lysate was then transferred to an Eppendorf tube and centrifuged at 15,000 rpm for 15 min. A sample of each supernatant (200 μL) was transferred into a scintillation vial containing 2 mL of Scintiverse BD (Fisher Chemical, Pittsburgh, PA), and the resulting scintillation counts were measured. The amount of protein was determined using the Pierce BCA protein assay kit (Pierce, Rockford, IL) from the remaining lysate volume (approximately 300 μL) to normalize the radioactive counts obtained and the data reported as nmol 3H-Spd/mg protein. Ki and Km values were determined using double reciprocal Lineweaver-Burk plots. The Ki value was determined from the equation Ki = IC50/(1 + (L + Km)), where IC50 is the concentration of PTI required to block 50% of the relative uptake of 3H-Spd and L is the concentration of 3H-Spd used in the assay (1 μM). The Km and Vmax for Spd was calculated by plotting the inverse of [3H-Spd] versus the inverse of the nmol Spd/mg protein/min. The Km = −1/x intercept, and Vmax = 1/Y intercept.
Table 1.
Kinetic profiles (Vmax and Km) of human pancreatic cancer cell lines, murine (PanO2) and Chinese hamster ovary (CHO) cells
| Cell line | Vmax (3H Spd) (nmoles/mg protein/min) | Km for Spd (μM) |
|---|---|---|
| HPNE | 1.8 | 0.19 ± .02 |
| L3.6pl | 24 | 0.67 ± .07 |
| Panc-1 | 13 | 0.31 ± .01 |
| Su86.86 | 7.0 | 0.29 ± .01 |
| BxPC-3 | 9.0 | 0.18 ± .01 |
| AsPC-1 | 2.3 | 0.12 ± .03 |
| Capan-1 | 0.8 | 0.18 ± .02 |
| PanO2 | 9.0 | 0.36 ± .05 |
| CHO | 3.0 | 0.24 ± .06 |
| CHOMG | 0 | ND |
Legend: Aminoguanidine (AG, 250 µM) was added to all pancreatic and HPNE cell lines and AG (at 1 mM) was added for CHO cell experiments to prevent amine oxidase activity present in the fetal bovine serum used for cell culture. These AG concentrations were non-toxic to the respective cell lines in separate experiments (not shown). The Ki values for the PTIs, trimer44 (5a), trimer44NMe (5b) and Lys-Spm (6) in L3.6pl human pancreatic cancer cells, were 36, 55, and 26 nM, respectively.
Western blot analysis
Cells were grown to 70-80% confluence, washed with cold PBS, and harvested in PBS after centrifugation. Cell lysates were prepared in standard RIPA buffer. Protein concentration was measured with the Pierce BCA Assay kit. For western blotting, equal quantities of protein (50-75 µg) were loaded onto SDS-PAGE gels. Protein was transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA) and blocked with 5% non-fat dry milk in PBS with 0.1% Tween-20 (PBS-T). Membranes were incubated with primary antibodies overnight at 4°C at respective dilutions [ATP13A3 (1:500), Cav-1 (1:500), c-Myc (1:1000), MTAP (1:1000), p16 (1:500), β-actin (1:25,000)]. The next morning, the membrane was washed with phosphate buffer saline containing 0.1% Tween-20 (PBS-T). The membrane was then incubated for 1 h at room temperature in the respective secondary antibodies (1:10,000) conjugated to horseradish peroxidase. Protein bands were visualized using the SuperSignal West Pico ECL kit (Pierce) and subsequent exposure to X-ray films. For re-probing, the membranes were stripped with Restore PLUS stripping buffer (Life Technologies) and re-probed with the β-actin antibody as a loading control. The intensity of the protein exposures on the X-ray film was evaluated by densitometry using ImageJ software (National Institutes of Health, Bethesda, MD). The data is presented as intensity of protein of interest divided by intensity of β-actin in each respective lane.
ATP13A3 and Cav-1 mRNA expression analysis in public human cancer datasets
Genome-wide mRNA expression profiles of 238 different datasets containing human cancer samples were retrieved from the public Gene Expression Omnibus (GEO) dataset at the NCBI website http://www.ncbi.nlm.nih.gov/geo/ [22], from the EBI Express website http://www.ebi.ac.uk/arrayexpress/, and from the TCGA consortium website https://tcga-data.nci.nih.gov/tcga/, as annotated in Table S4. The data for the GSK-950 and Sadanandam-47 cell line sets used in Table S1 were from http://cbiit.nci.nih.gov and NCBI GEO (GSE17891), respectively. The large majority of studies were on the Affymetrix Gene Chip Human Genome U133A and Plus 2.0 platforms (U133A/U133P2; Affymetrix, Santa Clara, CA). CEL data were downloaded, and analyzed as described [23]. Briefly, gene transcript levels were determined from data image files using GeneChip operating software (MAS5.0 and GCOS1.0, from Affymetrix). Samples were scaled setting the average intensity of the middle 96% of all probe-set signals to a fixed value of 100 for every sample in the dataset, to allow transcript level comparison between samples and between studies. We determined significant present (“present call”) and absent expression using this software. The TranscriptView genomic analysis and visualization tool (http://bioinfo.amc.uva.nl/human-genetics/transcriptview/) was used to select probe-sets, except Agilent G4502 array probes, for which no sequence data were available. Probes had to show unique mapping in an anti-sense position within (late) coding exons and/or the 3’ UTR of the gene. When multiple correct probe-sets were available for a gene, the probe-set with the highest average expression and the highest amount of present calls for that dataset were used. The probe-sets selected for analysis of U133A/P2 arrays were 204069_at and 223282_at, for Illumina WG-6 v3.0 arrays 1663684 and 2149226, and for Agilent HG 4x44K arrays 24_P183094 and 23_P134454, for ATP13A3 and Cav-1, respectively. Analyses were performed using R2; a genomics analysis and visualization platform developed in the Department of Oncogenomics, Academic Medical Center, Amsterdam, The Netherlands (http://r2.amc.nl). Website standard settings were used for all tests on Oncomine (http://www.oncomine.org).
Statistical analysis of public human cancer datasets
Correlations between ATP13A3 and Cav-1 mRNA expression (Figures 8 and 9, Table S4) were calculated using a Pearson test on 2log-transformed expression values (with the significance of a correlation determined by t = R/sqrt((1-r^2)/(n-2)), where R is the correlation value and n is the number of samples, and distribution measure is approximately as t with n-2 degrees of freedom). Significant correlations were only calculated for datasets when ≥ 10% of samples had a present call for both genes. The Statistical Package for the Social Sciences software package for Windows (Version 13.0) was used for all statistical analyses. Numerical results (Table S1) are expressed as the mean value ± SEM. When values are shown as 2log-median centered (Figure 9A-D, Tables S2 and S3), statistically significant differences were determined by t-testing. Results were considered significant when p < 0.05.
Figure 8.
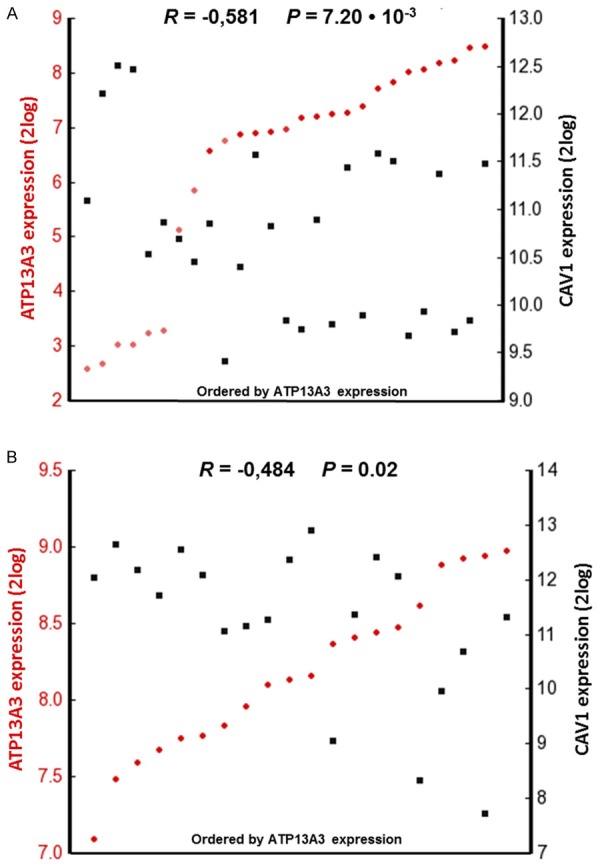
Inverse ATP13A3 versus CAV1 mRNA expression in two pancreatic cell line sets. Visual representation of ATP13A3 and CAV1 mRNA expression correlations calculated over (A) 27 pancreas cell lines in the GSK-950 dataset and (B) 20 pancreatic cancer cell lines in the Sadanandam-47 dataset as analyzed using R2. The tumors are ranked horizontally from left to right according to their ATP13A3 mRNA expression as determined by Affymetrix array analyses (2log values). ATP13A3 and CAV1 expression values for each cell line are visualized with red circles and black rectangles, respectively. Dark and light colors denote samples with significant (“present call”), and absent expression, respectively. The correlations between ATP13A3 and CAV1 mRNA expression were inverse and statistically significant (calculated with a 2log Pearson test, see Materials and Methods).
Figure 9.
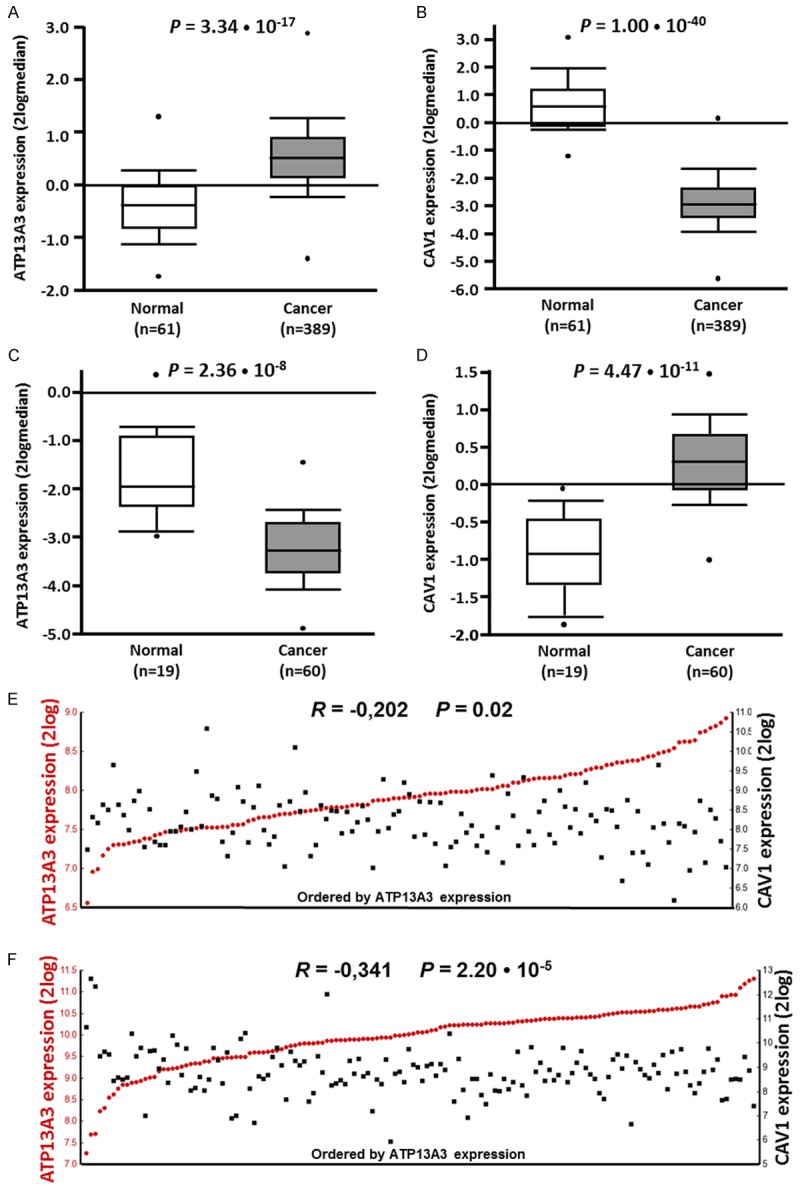
Significant ATP13A3 over- and Cav-1 under-expression and inverse correlations in other human cancer tissues. (A-D) Visual representation of ATP13A3 (A) and Cav-1 (B) mRNA expression in invasive ductal carcinoma samples in the TCGA-593 breast cancer dataset, and of ATP13A3 (C) and Cav-1 (D) mRNA expression in rectal adenocarcinoma samples in the TCGA-237 colon cancer dataset, as analyzed using Oncomine. Values are represented as 2log-median centered. ATP13A3 and CAV1 show mRNA over-and under-expression in tumor versus normal samples, respectively. The P values are calculated with a Student’s t-test. More details can be found in Table S3 in the Supporting information. (E, F) Visual representation of ATP13A3 and Cav-1 mRNA expression correlations calculated over (E) all 123 samples of the Chin-123 breast cancer dataset or (F) all 148 samples of the Sugihara colon cancer dataset as analyzed using R2. The tumors are ranked horizontally from left to right according to their ATP13A3 mRNA expression as determined by Affymetrix array analyses (2log values). ATP13A3 and Cav-1 expression values for each tumor are visualized with red circles and black rectangles, respectively. The correlations between ATP13A3 and Cav-1 expression were inverse and highly significant (calculated with a 2log Pearson test, see Materials and Methods). More details can be found in Table S3.
Results and discussion
Due to the broad context of this study, which incorporates the interplay between polyamine metabolism, oncogenes and transport activity, a brief overview is warranted.
Polyamine homeostasis via biosynthesis and transport
Polyamine homeostasis requires that polyamine biosynthesis and transport be intimately linked and balanced. The polyamine biosynthesis pathway is well understood [1] and relies on S-adenosylmethionine (SAM) and ornithine resources, which are derived from the aminoacids methionine and arginine, respectively. A detailed description is shown in Figure 1.
Figure 1.
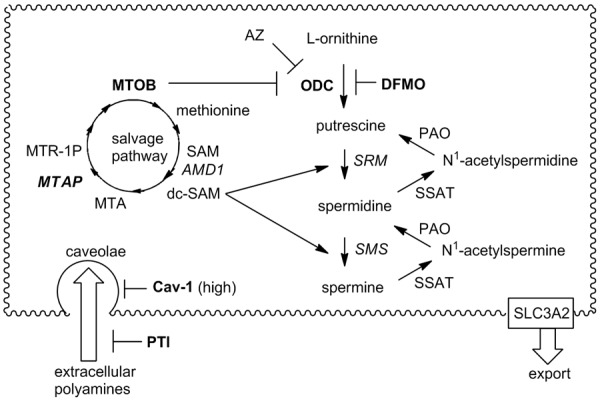
Human polyamine biosynthesis, metabolism and transport and the methionine salvage pathway. Ornithine decarboxylase (ODC) converts ornithine to putrescine and then spermidine synthase (SRM) appends an aminopropyl fragment derived from decarboxylated S-adenosylmethionine (dc-SAM) to putrescine to form Spd. The dc-SAM is generated by the action of adenosylmethionine decarboxylase (AMD1) on SAM. Spermine synthase (SMS) performs a similar task with dc-SAM to convert Spd to spermine (Spm). Catabolic enzymes like spermidine/spermine acetyl transferase (SSAT) and polyamine oxidase (PAO) allow for back-conversion from the higher polyamines (Spd and Spm) to putrescine (Put) as well as the formation of N-acetylated polyamines for cell export (presumably via SLC3A2 as seen in colon cancer cells). Abbreviations: AMD1 (adenosylmethionine decarboxylase), AZ (antizyme), Cav-1 (caveolin-1), dc-SAM (decarboxy-S-adenosylmethionine), DFMO (difluoromethylornithine), MTA (5’-methylthioadenosine), MTAP (S-methyl-5’-thioadenosine phosphorylase), MTOB (methylthiooxobutyrate), MTR-1P (methylthioribulose phosphate), ODC (ornithine decarboxylase), PAO (polyamine oxidase), PTI (polyamine transport inhibitor), SAM (S-adenosyl-L-methionine), SLC3A2 (diamine exporter), SMS (spermine synthase), SRM (spermidine synthase), SSAT (spermidine/spermine acetyltransferase). Note: a direct back-conversion from Spm to Spd is possible via spermine oxidase (not shown).
Intricate intracellular control mechanisms maintain polyamine levels via regulation of biosynthesis and transport. For example, antizyme 1 (AZ) is considered a dual regulator of polyamine biosynthesis and transport [1,24,25]. High intracellular polyamine levels cause a +1 translational frameshift which aligns two open reading frames and produces a full length AZ protein. AZ then binds to ornithine decarboxylase (ODC) to form an inactive ODC:AZ heterodimer and facilitates its degradation via the proteasome, thereby inhibiting polyamine biosynthesis [26,27]. AZ induction also inhibits polyamine transport by an unknown mechanism [28-30]. In another example, inhibition of ODC with DFMO, results in a concomitant increase in polyamine import activity [13,14,31,32] in an attempt to maintain cellular polyamine homeostasis. In summary, while there is evidence linking polyamine biosynthesis and transport, the actual biomolecules involved in this connection are largely unknown.
Biomarkers of polyamine transport and oncogenes
A handful of candidate proteins involved in polyamine import have been reviewed [31], but no comprehensive molecular explanation of how they work in concert to maintain polyamine homeostasis is yet available. These important gaps in our knowledge preclude a full understanding of polyamine homeostasis and have delayed the identification of valid biomarkers for polyamine transport in human cancers. These biomarkers are needed to stratify cancer patients with tumors which will best respond to DFMO or which may require DFMO+PTI therapy.
While it is widely known that cancer cells have increased intracellular polyamine levels, it is less clear whether these levels are achieved through increased biosynthesis or a combination of biosynthetic and import processes. Polyamine transport biomarkers would help identify where along the continuum (between Case A and Case B) specific cancer types lie. As cross-talk exists between the synthetic route and the PTS (e.g., via AZ induction), cells can shift their sources of polyamines to avoid a particular pharmacologic intervention, e.g., DFMO. Biomarkers which track this shift over time could inform drug dosing and the effectiveness of combination therapies to address this escape response. A first step in identifying these biomarkers is to understand the relationships between key oncogenic signaling pathways and polyamine metabolism.
Oncogenes and polyamine transport
The interplay of oncogenes and polyamine metabolism has been recently reviewed [1]. The Ras signaling pathway was of interest for our study because K-Ras-activating oncogenic mutations causing uncontrolled cell growth are found in the vast majority (> 90%) of pancreatic ductal adenocarcinomas (PDACs) and there is a critical need to develop new interventions for this deadly form of cancer [33]. The canonical Ras signaling pathway involves the Raf, MEK1/2, Erk1/2 and c-Myc proteins, which play critical roles in cell growth and metabolism. However, the relationships between these oncogenic proteins and putative polyamine transport proteins, like caveolin-1 (Cav-1), in pancreatic cancers are poorly understood. Due to our limited understanding of polyamine transport, there are few reports detailing the connection between polyamine transport proteins and oncogenes.
In colorectal cancers, Cav-1 has been shown to be a negative regulator of polyamine import [34]. In addition, activated K-Ras was shown to decrease the expression of the putative putrescine export protein SLC3A2 in HCT116 colon cancer cells [35]. Therefore, at least in HCT116 cells, activated K-Ras increases polyamine import and slows polyamine export, leading to elevated intracellular polyamine levels. Together these results suggest that Cav-1 expression may provide insight into a tumor’s dependence on polyamine import processes, where low Cav-1 expression is associated with high polyamine transport activity.
Bergeron and Porter first explored the effect of specific oncogene expression on polyamine metabolic enzymes and transport. Specifically, Rat-1 cells were stably transfected with Ras or N-myc oncogenes [36]. ODC activity was shown to be approximately 12-times higher in Ras-transfected cells, than in the parent or N-myc-transfected cell lines [36]. In contrast, polyamine transport was significantly increased in N-myc-transfected cells [36]. These authors concluded that the “associations between N-myc and Ras expression and critical aspects of polyamine metabolism suggest a possible role for the latter in facilitating the growth promoting properties of these oncogenes” [36].
The connection between the Myc family of proteins and polyamine transport activity is important because the Myc transcription factors affect polyamine biosynthesis by up-regulation of ODC transcription [1]. Specifically, Myc dimerizes with a partner protein, Max, and the Myc-Max complex binds to E-box motifs (CACGTG) in the ODC promoter activating ODC transcription [1]. ODC expression enhances putrescine biosynthesis and subsequent biosynthesis of the higher polyamines, Spd and Spm, via additional biosynthetic steps involving dc-SAM (Figure 1). ODC activity is sufficient for tumor promotion and ODC is considered a proto-oncogene [37]. As expected, cells which overexpress Myc have elevated levels of polyamines [1]. In this regard, Myc is key to increased intracellular polyamine levels because its up-regulation is known to increase polyamine biosynthesis and presumably import [36]. Lastly, a feedback mechanism has been proposed where the polyamines themselves regulate c-Myc translation through Chk2-dependent HuR phosphorylation [38]. Therefore, we surmised that Myc-driven tumors are likely ‘addicted’ to polyamines and may be especially sensitive to combination therapies which target polyamine biosynthesis and transport.
Since K-Ras mutations are prevalent in > 90% of PDACs and the Myc family genes are activated in nearly 70% of human cancers [1], we were interested in whether pancreatic cell lines with K-Ras mutations and high c-Myc expression also had high polyamine transport activity. We also investigated whether other genes associated with polyamine import in the literature (e.g., Cav-1), tracked with the polyamine import properties of pancreatic cancers (e.g., Vmax of 3H-Spd import).
To gain a better understanding of the relationships between specific protein expression and polyamine transport properties of pancreatic cancer cells, several human pancreatic cancer cell lines were characterized in terms of their sensitivity to DFMO, Km and Vmax values for 3H-Spd import, and the ability of Spd to rescue the cells from a DFMO IC50 dose challenge. This data was then correlated with the relative expression patterns of proteins thought to play a role in polyamine transport. In addition, three known polyamine transport inhibitors (PTIs) [13,14,39] and DFMO were evaluated in the treatment of human pancreatic cancer cell lines in vitro to better understand how their relative protein expression patterns and basal Vmax properties affected the efficacy of DFMO-only and DFMO+PTI therapies.
Cell line selection and biomarker rationale
Six human pancreatic cancer cell lines (AsPC-1, BxPC-3, Capan-1, L3.6pl, PanC-1, and Su86.86) and one murine pancreatic cancer line (PanO2) were studied along with immortalized normal human pancreatic duct cells (HPNE) as a control. These cell lines were selected based upon their expected differential expression of key proteins (e.g., ATP13A3, Cav-1, c-Myc, MTAP, and p16). All human pancreatic cell lines tested here have known K-Ras mutations [40], except for BxPC-3 cells [41]. HPNE cells were used as a baseline control and represent intermediary cells formed during acinar to ductal metaplasia. PanO2 mouse pancreatic cancer cells were also evaluated to support future in vivo comparisons in immune-competent and immune-compromised mice [42]. Chinese hamster ovary cells with active (CHO-K1) and deficient polyamine transport (CHO-MG) were used as additional controls.
The expression of each target protein was re-determined in this study for confirmation. MTAP is a key protein involved in recycling of methylthioadenosine (MTA, a byproduct of polyamine biosynthesis) to methionine (Figure 1) [43]. The pancreatic cell lines BxPC-3, CAPAN-1, PanC-1, and Su86.86 were confirmed to be MTAP-deficient, while AsPC-1 and L3.6pl were found to be MTAP positive. The Vmax and Km values for 3H-Spd import and DFMO IC50 values were determined for each cell line (Table 1) along with the relative expression of putative polyamine transport proteins ATP13A3, Cav-1, c-Myc, and MTAP, as well as the tumor suppressor p16 protein in each cell line during logarithmic growth. These data then allowed for a better description of how relative protein expression patterns correlated to polyamine import processes.
The rationale for the selected protein markers was as follows:
MTAP and p16
The S-methyl-5’-thioadenosine phosphorylase (MTAP) gene is deleted in over 47% of pancreatic cancers [44] and this deletion effectively removes a downstream metabolite (2-keto-4-methyl-thiobutyrate, MTOB), which is an inhibitor of ODC activity (Figure 1) [44]. MTAP and MTOB are important because ODC activity is known to promote tumor formation [37,45]. We hypothesized that because MTAP and the p16 tumor suppressor gene are in such close proximity at 9p21, deletion in this region can create dual loss of both p16 and MTAP, which may promote tumor progression by removing a tumor suppressor gene and the ODC inhibitor, MTOB [44].
Cav-1
Polyamines have been shown to enter human cells via a caveolin-dependent mechanism (Figure 1), where low caveolin-1 (Cav-1) expression correlated with increased polyamine transport activity [34]. Interestingly, caveolin-1 expression becomes virtually undetectable in many metastatic cancer cells, which should increase their polyamine transport activity [46]. There are, however, conflicting reports in the literature regarding caveolin-1 levels in pancreatic cancers [46,47] and a definitive study was needed to address this knowledge gap by defining the relationship between caveolin-1 expression and polyamine transport in pancreatic cancers because non-caveolin dependent pathways are also possible [31].
We note that recent studies further support the negative regulation of the PTS by Cav-1 in other tissues. Indeed, recent Cav-1 knockout (Cav-1 KO) experiments in vascular smooth muscle cells (VSMCs) demonstrated that the Cav-1 KO VSMCs had increased polyamine uptake relative to wild-type (wt) cells and that the Cav-1 KO cells were hyper-proliferative in the presence, but not the absence of exogenous polyamines [48]. In summary, Cav-1 was shown to negatively regulate polyamine uptake in VSMCs and the proliferative advantage of Cav-1 KO cells was critically dependent upon polyamine uptake [48,49].
c-Myc
c-Myc was chosen due to the previously described correlations between Myc expression and polyamine metabolism [1,36]. The potential of c-Myc to modulate both polyamine biosynthesis and transport in pancreatic cancers was an intriguing hypothesis to pursue. A strong correlation between c-Myc protein expression and Vmax of 3H-Spd uptake was observed in the current study further validating our selection of this protein.
ATP13A3
The selection of ATP13A3 as a potential biomarker of polyamine transport was based upon our previous work on polyamine transport ligands. We developed a series of fluorescent polyamine probes and optimized their structure for import via the polyamine transport system (PTS) in mammalian cells [9,50]. These novel probes ranged from having one-, two- or three-appended polyamine chains on an aryl platform and are shown in Figure 2. Interestingly the one- and two-arm designs were shown to be PTS ligands that could enter and kill cells via the PTS [51]. The three-arm motifs were shown to be efficient polyamine transport inhibitors (PTIs) [14]. In 2010, one of the synthetic probes (1a in Figure 2) was used in C. elegans to identify a novel P5-type ATPase (CATP-5), which was shown to play a role in polyamine transport [52]. The human homologue of this worm gene is ATP13A3 and the findings with CATP-5 led us to investigate ATP13A3 as a potential marker for human polyamine transport.
Figure 2.
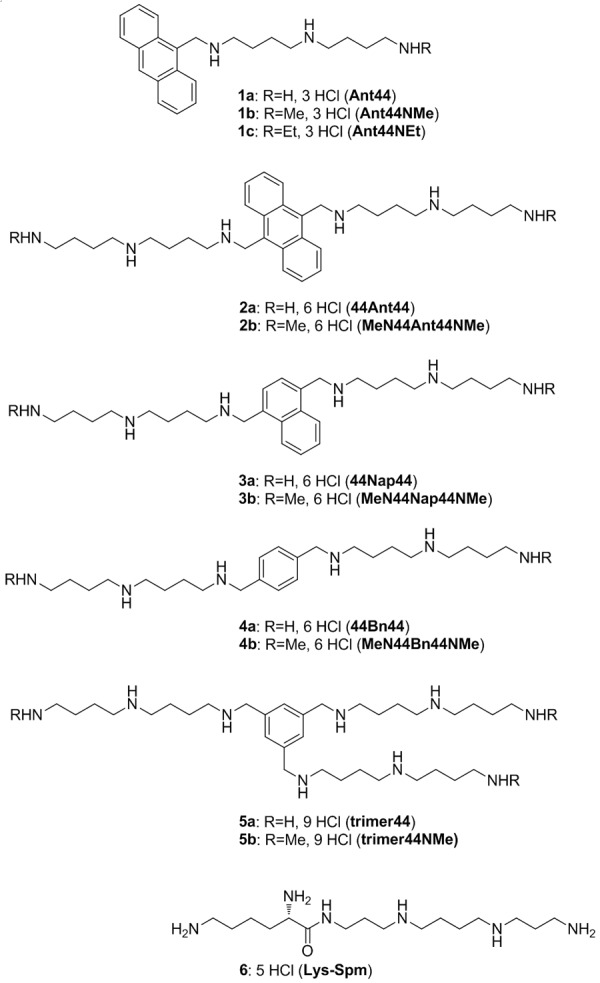
Structures of polyamine derivatives 1-6.
Our study validated three putative biomarkers for polyamine transport (Cav-1, c-Myc and ATP13A3). In contrast, MTAP and p16 protein expression patterns did not correlate with Vmax of 3H-Spd import. We note that additional genes that could also be involved (e.g., ATP13A2 [53], glypican-1 [54-56], NOS-2, and SLC3A2) [31], were not part of this study.
PTI compounds
While compounds 1-4 in Figure 2 were cytotoxic ligands for the PTS, compounds 5a, 5b and 6 were relatively non-toxic polyamine transport inhibitors (PTIs) [19,57] and were compared in this study for their activity in combination with DFMO in human and murine pancreatic cancer cell lines. We note that all of these compounds have polyamines within their structure and were expected to be competitive inhibitors of polyamine transport.
Kinetic studies
The Km for Spd and Vmax experiments using radiolabeled Spd (3H-Spd) were performed in the selected pancreatic cell lines as well as in two Chinese hamster ovary cell lines (CHO-K1 and CHO-MG cells). CHO-MG is a mutant of CHO-K1, which has no measurable polyamine uptake activity [33]. The CHO cell lines were included as controls to illustrate the effect in cell lines known to have high and low polyamine transport activity, respectively [14,51,58]. The results are shown in Table 1.
The experiments in Table 1 identified L3.6pl as the cell line with the highest Vmax and highest Km. The L3.6pl cell line was isolated after six selection rounds for metastatic capacity using orthotopic transplantation [59]. Specifically, the L3.6pl cells had over three fold higher polyamine uptake (Vmax = 24 nmol/mg protein/min) than the control HPNE cells (Vmax = 1.8 nmol/mg protein/min) and utilized a transport system with more than three-fold lower binding affinity, as evidenced by the higher Km value determined in this cell line (Km = 0.67 µM in L3.6pl vs 0.19 µM for HPNE). We speculate that this low affinity pathway provides these cells with a scavenging pathway to harvest polyamine metabolites from their surroundings resulting in a greater net import flux as seen in the Vmax determinations.
The two CHO cell lines gave the expected striking differences in 3H-Spd uptake. CHO-K1 cells had a Km = 0.24 µM and Vmax of 3 nmol/mg protein/min, while the CHO-MG line showed no measurable uptake (Vmax = 0 nmol/mg protein/min) confirming its deficient uptake pathway.
We next determined the sensitivity of these cells to DFMO and the ability of exogenous Spd (1 µM) to rescue cells when treated at their IC50 dose of DFMO (Table 2). We assigned a ‘spermidine rescue rank’ based upon the ability of Spd (at 1 µM) to rescue these cells past certain viability thresholds. The data in Table 2 were then compared to the Vmax of untreated cells in Table 1. We hypothesized that DFMO-treated cell lines with high basal polyamine transport activity would import Spd more readily and retain high cell viability due to the ability of cells to interconvert Spd to either Put or Spm as needed (See Figure 1) [1].
Table 2.
Cell line sensitivity to DFMO (72 h IC50) and spermidine rescue challengea
| Cell line | 72 h DFMO IC50 (mM) | % viability after 72 h incubation with Spd (1 µM) and the 72 h DFMO IC50 dose |
|---|---|---|
| HPNE | 17 ± 0.6 | 60-62 (Very low) |
| L3.6pl | 4.2 ± 0.11 | 89-93 (High) |
| Panc-1 | 3.8 ± 0.1 | 80-81 (Med) |
| Su 86.86 | 10.4 ± 0.44 | 80-85 (Med) |
| BxPC-3 | 14.4 ± 0.25 | 70-72 (Low) |
| AsPC-1 | 11.8 ± 0.44 | 50-60 (Not rescuable) |
| Capan-1 | 13.2 ± 0.5 | 26-43 (Not rescuable) |
| PanO2 | 0.5 ± 0.03 | 85-93 (High) |
| CHO-K1 | 4.2 ± 0.37 | 100-105 (High) |
| CHOMG | 0.05 ± 0.005 | 50-55 (Not rescuable) |
‘Spermidine Rescue Rank’ as measured via the % viability observed when each cell line was incubated 72 h at 37°C in the presence of an IC50 dose of DFMO (listed above) and Spd (1 µM) was assigned as: High (≥ 90%), Medium (≥ 80-89%), Low (≥ 70-79%), Very low (> 60-69%) and not rescuable (≤ 60%).
The rank is shown in parentheses (e.g., High, Med, Low).
As shown in Table 2, the cell lines demonstrated varying sensitivity to DFMO and some couldnot be rescued from DFMO action by exogenous Spd addition (AsPC-1, Capan-1, and CHO-MG). Interestingly, the L3.6pl cell line, which had the highest Vmax, was one of the most sensitive human cell lines to DFMO (72 h IC50 = 4.2 mM). Panc-1 (human), PanO2 (murine) and CHO (hamster) cells also showed significant rescue with exogenous Spd and had lower DFMO IC50 values (< 4.2 mM). As expected, the CHO-MG line was very sensitive to DFMO and was not rescuable with exogenous Spd due to the lack of a functional PTS. In general, cell lines with low DFMO 72 h IC50 values could be significantly rescued to higher viability with Spd in the DFMO/Spd rescue experiment. The correlation between DFMO 72 h IC50 value and % viability in the DFMO+Spd rescue experiment was good (r2 = 0.53) suggesting that the sensitivity of the cell line to DFMO alone was predictive of its rescue by Spd. Indeed, cells with DFMO IC50 values ≤ 4.2 mM were rescued to > 80% viability by exogenous Spd (1 µM).
Our interpretation is that the cell lines that rely on polyamine import more than polyamine biosynthesis would give a higher Vmax value for 3H Spd import, higher Spd rescue rank, and lower DFMO IC50 value (e.g., L3.6pl cells). In contrast, cells with a heavy reliance upon polyamine biosynthesis and lower commitment to polyamine transport should give a lower Vmax values, lowerSpd rescue rank, and a higher DFMO IC50 value (e.g., AsPC-1).
We were interested in how the dual blockade of polyamine biosynthesis and polyamine transport via the DFMO+PTI combination therapy affected the viability of cell lines with different DFMO sensitivities and Vmax values. Earlier work had identified three PTIs (5a, 5b and 6) and these were screened in the L3.6pl cell line for this property [14]. A Spd rescue assay (Figure 3) was employed to rank the potency of each PTI as an EC50 value. The PTI toxicity profile (IC50 and IC10 values) were also determined in the different cell lines.
Figure 3.

Example 48 h EC50 determination experiment in L3.6pl cells. The Control was set to 100% relative viability and was determined via the MTS reagent after 48 h growth period at 37°C. The DFMO-only cells were exposed to the 48 h DFMO IC50 dose (and gave the expected 50% viability); the DFMO+Spd cells were exposed to the 48 h IC50 DFMO dose and Spd (1 µM) (and shows increased viability due to Spd rescue); the remaining samples received a fixed DFMO IC50 dose, a fixed Spd rescuing dose (1 µM), and increasing concentrations of the PTI agent 5a. As the PTI concentration is increased from 1 to 10 µM 5a, less Spd enters the cells and lower relative viability is observed. The PTI 5a was found to be non-toxic to L3.6pl cells at 2.5 µM during 48 h exposure. As seen in the graph above, the non-toxic PTI was able to block the rescuing effect of the added Spd. Note: PTI 5b gave a similar result.The EC50 is shown graphically as the midpoint (in % viability) between the green and red lines, i.e. the DFMO+Spd and DFMO only controls.
EC50 studies
As shown in Figure 3, cells were treated with the 72 h IC50 DFMO dose, a rescuing dose of Spd (1 µM), and increasing concentrations of the PTI compound to determine the EC50 value. In an ideal case, where DFMO gave 50% viability and Spd gave 100% rescue, the EC50 value would be halfway between these two outcomes, i.e., the concentration of the PTI needed to give 75% viability. Figure 3 provides an illustration with PTI 5a. The EC50 dose of the PTI is the concentration of the PTI needed to attain the % viability halfway between the DFMO only and DFMO+Spd controls. The lower the EC50 value, the more potent the PTI is at blocking Spd import. Note: at the EC50 dose the PTI presumably blocks 50% of the entering Spd. Since reduced viability is the readout from this assay, it was critical that the PTI agent was not toxic at the concentrations tested. This was confirmed by separate control experiments via determination of the PTI IC10 value (i.e., the PTI concentration which gives 10% inhibition of cell viability). At the compound’s IC10 value, the cells are ≥ 90% viable. Rewardingly, each PTI had EC50 values well below the respective PTI’s IC10 value in each cell line.
The three PTI compounds (5a, 5b and 6 [19]) all have polyamine motifs within their structure and were expected to be competitive inhibitors of polyamine transport. Indeed, Lys-Spm 6 has been established as a competitive inhibitor of Spd transport [19]. To demonstrate this with the tri-substituted motifs, we performed 3H-Spd kinetic experiments with 5b [14] and found that 5b displayed competitive inhibition characteristics as expected (see Supporting information).
The three PTIs were then screened for their toxicity profiles (IC10 and IC50) as well as for blockade of Spd import (EC50) in several cell lines (see Table 3A and 3B). Their respective Ki values are listed in the legend of Table 1. Understanding the cytotoxicity of these inhibitors via IC10 and IC50 measurements was critical because the EC50 assay relies upon a reduced viability endpoint which should not be derived from the intrinsic toxicity of the PTI compound itself, but from the ability of the PTI to out compete Spd for the putative extracellular receptor.
Table 3A.
PTI 72 h IC10 and IC50 values (in µM) across cell linesa
| Cell Line | Trimer44 (5a) IC50 (IC10) | Trimer44NMe (5b) IC50 (IC10) | Lys-Spm (6) IC50 (IC10) |
|---|---|---|---|
| HPNE | 17.2 ± 0.7 (4) | 20.0 ± 0.1 (6) | > 200 (> 200) |
| L3.6pl | 57.1 ± 3.3 (2.5) | 75.0 ± 1.0 (5) | > 100 (> 100) |
| Panc-1 | > 200 (20) | > 200 (20) | > 200 (>200) |
| Su86.86 | 39.2 ± 1.2 (4) | 51.2 ± 1.3 (6) | > 200 (60) |
| BxPC-3 | > 100 (50) | >100 (>100) | > 100 (> 100) |
| AsPC-1 | > 100 (> 50) | >100 (>100) | > 100 (> 100) |
| Capan 1 | ND | ND | ND |
| PanO2 | 46 ± 4.9 (8) | 65.3 ± 5.6 (8) | > 200 (> 200) |
| CHOK1 | > 100 (10) | > 100 (10) | > 100 (> 100) |
| CHOMG | 66.9 ± 6.2 (10) | 121.7 ± 3.9 (10) | > 100 (> 100) |
The IC50 and IC10 values represent the concentration of PTI needed to inhibit 50% and 10% of the relative cell viability, respectively after 72 h of incubation at 37°C.
The IC10 value for these cells is the maximum amount of PTI compound that can be tolerated with minimal toxic effects to the cell (≤ 10% reduction in cell viability compared to untreated controls) after the 72 h incubation period. This represents the maximal PTI dose which would not bias assays by inducing significant toxic effects. Units are all in µM; ND = not determined.
Table 3B.
PTI 72 h EC50 values (µM)
| Cell Line | Trimer44 (5a) EC50 | Trimer44NMe (5b) EC50 | Lys-Spm (6) EC50 |
|---|---|---|---|
| HPNE | ND | ND | ND |
| L3.6pl | 1.5 ± 0.1 | 3.2 ± 0.1 | 2.7 ± 0.2 |
| Panc-1 | 2.9 ± 0.15 | 3.11 ± 0.18 | 2.06 ± 0.14 |
| Su86.86 | 0.82 ± 0.03 | 1.96 ± 0.08 | 1.08 ± 0.01 |
| BxPC-3 | 0.34 ± 0.02 | 1.03 ± 0.09 | 0.25 ± 0.02 |
| AsPC-1 | ND | ND | ND |
| Capan 1 | ND | ND | ND |
| PanO2 | 0.61 ± 0.01 | 0.90 ± 0.07 | 6.00 ± 0.57 |
| CHOK1 | 0.34 ± 0.02 | 0.90 ± 0.07 | 1.37 ± 0.05 |
| CHOMG | ND | ND | ND |
aAll EC50 values were determined after 72 h incubation and represent the concentration of PTI (in µM) needed to provide a viability value halfway between the DFMO-only and the DFMO+Spd (1 µM) viability values. For example, when the DFMO-only control (added at its IC50 value) gave 50% viability and the DFMO+Spd control gave 100% viability, the EC50 value is the concentration of PTI needed to attain 75% viability in the presence of the IC50 dose of DFMO and the rescuing dose of Spd (1 µM) (Note: see Figure 3 for an illustration). Importantly, all EC50 values were well below the IC10 values noted in Table 3A.
An ideal PTI should have a high 72 h IC50 value, a high 72 h IC10 value and a low 72 h EC50 value. Comparison between Table 3A (IC10) and 3B (EC50) entries provides insight into the therapeutic window available for each PTI in each respective cell line. The Lys-Spm conjugate 6 gave low overall toxicity and good performance across multiple cell lines. The N-methyl derivative 5b was consistently less toxic than 5a as shown by its higher IC50 values in Table 3A. In contrast, 5a was more potent than 5b as shown by its uniformly lower EC50 values (Table 3B). All three PTIs (5a, 5b, and 6) were individually effective in blocking Spd import in each of the rescuable cell lines at ≤ 6.5 µM. For most of the cell lines tested, the three PTIs had similar potencies. An exception was found in the PanO2 murine cell line, where 5a was nearly ten-fold more potent than Lys-Spm 6.
We noted that the human pancreatic cancer cell lines with the highest EC50 values (L3.6pl and Panc-1) also had the highest Vmax values in Table 1. This suggested that cells with high basal polyamine uptake rates require more PTI to block their Spd import in the presence of DFMO. We speculated that this could be due to DFMO-induced increased expression of cell surface receptors associated with the PTS or activation of adifferent PTS.
The AsPC-1 and Capan-1 lines, were not studied in the EC50 assay as they were not rescuable with exogenous Spd. Pancreatic cancers with low to no cellular polyamine import are not expected to show effects from PTI treatment.However, these two cell lines were very interesting in terms of their sensitivity to the native polyamines. Neither AsPC-1 nor Capan-1 cells could be rescued from their respective 72 h IC50 dose of DFMO, even with 10 µM Spd. Capan-1 was not rescued from the 72 h IC50 dose of DFMO with either Put or Spd (at 5 µM), but was partially rescued up to 87% viability by Spm (5 µM). In contrast, AsPC-1 cells were not rescued with any of three native polyamines even at 10 µM.
Protein expression
DFMO was recently found to be a potential chemopreventative for K-ras-driven pancreatic cancers. Rao et al. found that changes in ODC signaling occur at early stages of pancreatic precursor lesions and increases as the tumor progresses in K-ras activated p48 Cre/+-LSL-K-rasG12D/+ mice [60]. DFMO treatment reduced the progression of pancreatic intraepithelial neoplasms (PanINs) and their progression to pancreatic ductal adenocarcinoma (PDAC) [60]. Interestingly, although DFMO inhibited PDAC in the K-ras mice, some tumor outgrowth was still observed [60]. The authors suggested that exogenous polyamines from dietary sources may explain the observed tumor escape [60]. This speculation is consistent with our observations, that exogenous Spd rescues pancreatic cancer cells from a DFMO challenge via the PTS. However, as seen in our DFMO study of various pancreatic cell lines, not all cell lines respond to exogenous Spd (e.g., AsPC-1) when exposed to an IC50 concentration of DFMO. The question remains: which proteins are responsible for this differential response?
There is significant evidence in the literature that DFMO can alter specific protein levels. For example, DFMO was shown to decrease Cav-1 mRNA expression in a dose-dependent manner and to increase the mRNA expression of the p21, p27 and p53 tumor suppressor genes [60]. Overexpression of Cav-1 in the pancreatic cancer cell line, Panc 10.05 that does not normally express Cav-1, induced an epithelial phenotype with increased expression of membranous E-cadherin and β-catenin [61]. Other studies have reported a decrease in c-Myc expression upon DFMO treatment [62-67]. Therefore, we looked at both the basal level of the target proteins and their relative changes upon DFMO treatment to better understand their roles in Spd rescue from DFMO.Western blots were used to determine relative protein expression in untreated and treated pancreatic cells lines.
No correlations between Vmax and MTAP or p16 expression were observed (Figure 4) [5]. Cav-1 showed variable expression across these cell lines (Table 4). Based upon work by Gerner, Cav-1 is a negative regulator of polyamine transport and low caveolin-1 (Cav-1) expression should be associated with high polyamine import [6,7]. In our study, Cav-1 alone was found to give inconsistent results as a predictor of Vmax, consistent with the varied literature findings with this protein in pancreatic cancer [46,47].
Figure 4.
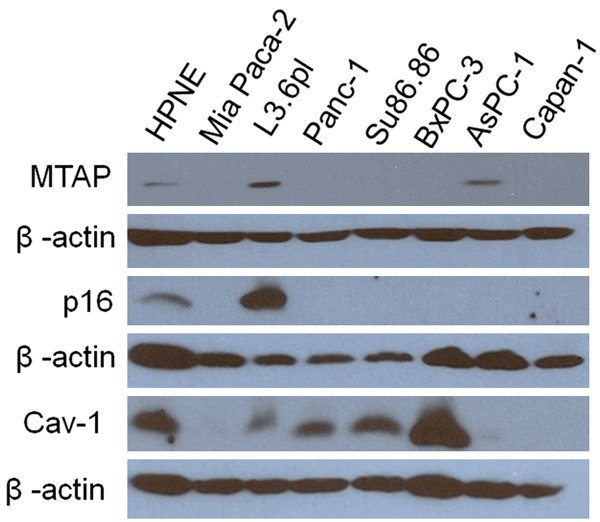
MTAP, p16, and caveolin-1 (Cav-1) protein expression versus β-actin expression in each cell line. MTAP expression was only detected in HPNE, L3.6pl, and AsPC-1 cell lines. HPNE and L3.6pl were p16 positive. MiaPaca-2 cells were included as a negative control.
Table 4.
Relative Protein expression of Cav-1, ATP13A3 and c-Myc in untreated human pancreatic cancer cell lines along with the respective Vmax values for 3H Spd import and relative Spd rescue indexa
| Cell line | Cav-1 | ATP13A3 | c-Myc | Vmax of untreated cells | Spd rescue index |
|---|---|---|---|---|---|
| HPNE | 98.8 | 13.45 | 1 | 1.8 | Very low |
| L3.6pl | 100 | 100 | 100 | 24 | High |
| Panc-1 | 126.4 | 5.7 | 75.3 | 13 | Med |
| SU86.86 | 118.3 | 9.5 | 30.2 | 7 | Med |
| BxPC-3 | 358.5 | 26.9 | 18.1 | 9 | Low |
| AspC-1 | 16.6 | 1.8 | 4.6 | 2.3 | Not rescuable |
| Capan-1 | 3.3 | 0.0 | 9.0 | 0.8 | Not rescuable |
The blots were quantified by Image J software (NIH) and normalized by dividing by the β-actin expression level for each cell line.
Entries represent protein expression and expressed in relative %, with the L3.6pl cell expression levels set to 100%. To illustrate how the relative protein expression patterns relate to the polyamine transport properties of the cell lines, the Vmax value for 3H-Spd import (nmoles Spd/mg protein/min) and the Spd rescue index observed with DFMO-treated cells are listed again for comparisons.
As shown in Figure 5 and Table 4, basal c-Myclevels show a strong correlation (r2 = 0.90) with Vmax in untreated cells. This direct relationship was consistent with Bergeron’s related experiments which showed increased polyamine uptake in Rat-1 cells upon N-myc transfection [36]. Thus, relative c-Myc expression correlates very well with basal polyamine transport activity. Basal c-Myc expression in untreated cells also had a good correlation (r2 = 0.86) with Km value. We were interested to see how c-Myc levels were modulated in the presence of DFMO. The level of c-Myc was shown to decrease upon DFMO treatment in L3.6pl cells (see Supporting information), contrary to what one might expect if c-Myc was involved in the Spd rescue process. We hypothesized that upon DFMO treatment the reduced c-Myc levels may trigger compensatory uptake pathways, possibly involving ATP13A3.
Figure 5.
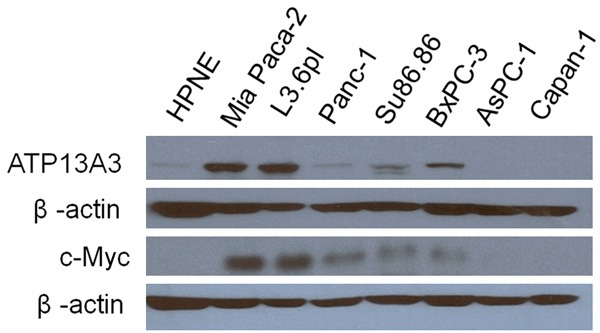
ATP13A3 and c-Myc protein expression versus β-actin expression in each cell line. ATP13A3 and c-Myc protein expression was barely detectable in the AsPC-1 and Capan-1 cell lines, which were not rescuable with Spd in previous DFMO experiments. MiaPaca-2 cells were included as a positive control for c-Myc and ATP13A3.
As shown in Table 4, basal ATP13A3 protein expression showed good correlation with Vmax (r2 = 0.75) and Km (r2 = 0.82). Cell lines with significant basal expression of ATP13A3 gave medium to high Spd rescue indices. Moreover, as shown in Figure 6, ATP13A3 was significantly upregulated (3 fold increase) in L3.6pl cells in the presence of DFMO alone or in combination with Spd (72 h incubation). This was also observed with BxPC-3 cells which showed a > 1.7 fold increase of ATP13A3 protein expression upon treatment with DFMO with or without Spd (Figure 6). Since ATP13A3 protein expression was increased in the presence of DFMO, we further investigated its role in DFMO-stimulated polyamine import using siRNA experiments.
Figure 6.
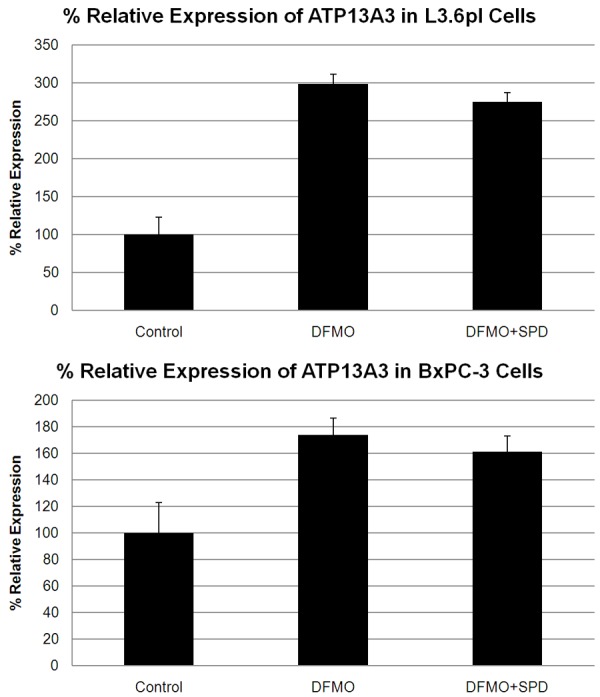
ATP13A3 protein levels in L3.6pl (top) and Bx-PC3 cells (bottom) in the presence of DFMO or DFMO+Spd as analyzed by Western blot. Expression was normalized using β-actin levels. Both cell lines showed a significant increase in relative ATP13A3 expression in the presence of the 48 h IC50 DFMO dose (8 mM) or DFMO + (1 µM Spd).
siRNA experiments
siRNA experiments confirmed the role of ATP13A3 in Spd rescue of DFMO-treated L3.6pl cells. As shown in Figure 7, L3.6pl cells were treated for 48 h with either scrambled siRNA or ATP13A3 targeting siRNA (at 75 nM). Note: Parallel experiments confirmed the knockdown of ATP13A3 protein by Western blot (Figure 7A). The media was then removed, the cells were washed and fresh media added. A 48 h IC50 DFMO dose (8 mM) was then added along with Spd (1 µM). As expected, DFMO and the DFMO+Spd controls showed 50% and 90% viability, respectively. ATP13A3 siRNA caused a significant lower viability (62%) in the presence of DFMO+Spd. In contrast, the scrambled siRNA control showed the expected high viability (89%) in the presence of the same DFMO+Spd challenge.
Figure 7.
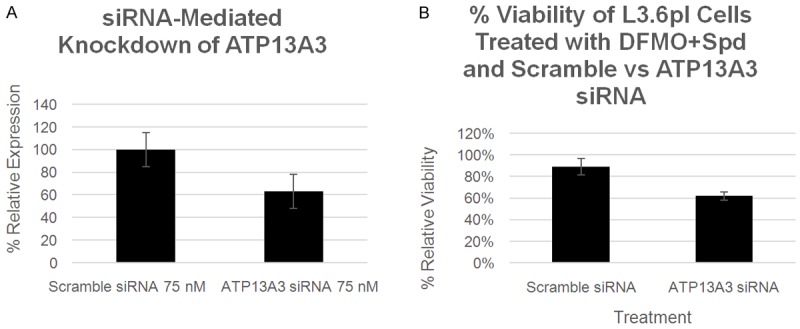
A: ATP13A3 protein levels in L3.6pl cells as analyzed by Western blot. Expression was normalized using β-actin protein levels. Decreased ATP13A3 expression was observed in the presence of the ATP13A3 siRNA versus the scrambled siRNA (p < 0.05). B: The effect of scrambled siRNA versus ATP13A3 siRNA on relative L3.6pl cell viability % when challenged with a 48 h IC50 DFMO dose (8 mM) and Spd (1 µM). Reduced ATP13A3 expression results in reduced rescue by Spd (p < 0.01). Other control experiments are shown in Figure S1 in the Supporting information.
Therefore, at first glance c-Myc and ATP13A3 showed strong correlations to polyamine transport activity, while Cav-1 did not. The mixed findings with Cav-1 could be explained, however, if one takes into account the relative ATP13A3 expression in cells. Indeed, low Cav-1 and high ATP13A3 expression may facilitate the cell’s ability to escape DFMO therapy via caveolin-dependent endocytosis. This inverse relationship can be used to explain the rest of the results in Table 4. For example, Capan-1 cells, which had the lowest Cav-1 expression and were expected to have high polyamine transport activity, actually had the lowest Vmax value and no detectable ATP13A3 to facilitate polyamine import via the putative DFMO-stimulated polyamine import process. A similar pattern was observed with AspC-1 cells.
We speculated that Cav-1 protein levels may also explain why BxPC-3 cells, which had the second highest ATP13A3 level measured, gave an unexpected low Spd rescue index. Indeed, as seen in Table 4, BxPC-3 had 3.6 fold higher Cav-1 levels than any other cell line tested in our study and these high Cav-1 levels did not significantly change in the presence of DFMO or DFMO+Spd (See Supporting information). This suggests that the high Cav-1 levels of BxPC-3 may limit its ability to scavenge polyamines via a caveolin-dependent process [68]. In contrast, L3.6pl cells had ~3.6-fold lower Cav-1 expression than BxPC-3, very high ATP13A3 expression and a high Spd rescue index.
Based upon these results, we propose that the relative ATP13A3 protein expression level provides an indicator of the cell’s ability to escape DFMO, especially when viewed in the context of the cell’s Cav-1 expression level. One interpretation of this apparent Cav-1 dependence is that cells with high ATP13A3 expression may escape DFMO using a caveolin-dependent polyamine uptake mechanism.
Biomarkers
The potential connection between basal expression of ATP13A3 and Cav-1 proteins was followed up using a bioinformatics approach to delineate the potential correlation between these two protein markers. We specifically focused on how the ATP13A3 and Cav-1 ratios vary in specific cancer types. Due to the lack of comprehensive protein expression profiles in the public databases for both markers, we looked at the available datasets for their relative mRNA levels.
First, we were interested to know whether theinverse expression pattern of ATP13A3 and Cav-1 existed in other human pancreatic cell lines. Analysis of mRNA expression profiles in the public domain, indeed, showed significant opposite expression patterns for ATP13A3 and Cav-1 in two independent pancreatic cell line collections; the general GSK-950 cell line set (27 pancreatic cell lines; Figure 8A), and the Sadandam-47 pancreas set (20 pancreatic cell lines; Figure 8B). This mRNA analysis provided supportive evidence that an inverse relationship does exist in specific human pancreatic cancer subtypes.
Next, we looked for similar inverse expression patterns in public human cancer patient samples. Our first analysis of public human sample data (Table S1) showed that both genes have good expression levels (500-1,000 after MAS5.0 normalization). ATP13A3 is almost always significantly expressed in human cancer samples, at stable, medium levels (99.6% of samples). CAV1 is less widely expressed (76.8% of samples), and shows a higher variance in expression level. We found invariant, significant ATP13A3 over-expression in pancreas cancer versus normal tissue samples. However, we could not establish a robust inverse expression pattern with Cav-1, due to the paucity of public human pancreas cancer sets (Table S3).
We then widened our analysis to other cancer types for which more datasets were available. These include the most frequent and lethal human tumor types [69,70]. The results show that ATP13A3 is often significantly over-expressed in tumor tissue in several different tumor sub-types. In these cancers, accompanying significantly lower Cav-1 tumor expression can be present (Tables S2 and S3). There was no evidence for ATP13A3 DNA copy gain versus Cav-1 copy loss (not shown). Remarkably, this high ATP13A3/low CAV1 tumor mRNA expression pattern appears to be specific for certain cancer types. With one exception (mantle cell lymphoma), it was not found in hematopoietic tumors. This inverse expression pattern was present much more often in solid tumor types and was especially frequent in epithelial tumors (carcinomas), more so thanin sarcomas and blastomas. Figure 9A-D shows examples for aggressive breast ductal carcinomas (high ATP13A3/low Cav-1) and rectal adenocarcinomas (low ATP13A3/high Cav-1). We also noted that several of the most common carcinomas, responsible for the large majority of the worldwide cancer deaths, like bladder, breast, cervix, colon, lung, ovary, and prostate cancer [69,70] have high ATP13A3/low Cav-1 tumor expression. Finally, when we compared ATP13A3/Cav-1 expression in individual samples of datasets representing the carcinomas described above (and in Table S2), we found frequent, significant inverse correlations for ATP13A3 versus CAV1 expression for the whole dataset (Table S4). Figure 9E and 9F show examples of these inverse correlations in large breast and colon datasets.
Together, these data suggest that inverse regulation of ATP13A3 and Cav-1 expression is present in aggressive, solid human cancers, and that high ATP13A3 versus low Cav-1 expression could be involved in tumor progression. These results provide for the first time testable predictions into which cancers will best respond to DFMO. For example, cancers with low ATP13A3 and high Cav-1 (as seen with rectal adenocarcinomas) will likely respond to DFMO only, while other cancers with high ATP13A3 and low Cav-1 expression (as seen with invasive ductal carcinomas of the breast) may respond better to DFMO+PTI combination therapy.
Conclusions
Three PTIs (5a, 5b, and 6) were evaluated for their efficacy in treating pancreatic cancer cell lines in combination with DFMO. The results were rationalized in terms of the cells’ relative commitment to polyamine biosynthesis and/or polyamine transport for growth. Cell lines with low Vmax values, low Spd rescue rankings as well as low c-Myc and nearly undetectable ATP13A3 levels were considered to be heavily committed to polyamine biosynthesis and required high concentrations of DFMO to inhibit their growth (e.g., AsPC-1 and Capan-1). In contrast, cell lines with high Vmax, high Spd rescue rankings, high relative c-Myc and ATP13A3 protein expression (e.g., L3.6pl) demonstrated significant commitment to polyamine transport and required lower DFMO concentrations to inhibit their growth. Interestingly, the human pancreatic cancer cell lines which were most sensitive to DFMO (i.e., gave the lowest DFMO IC50 values (Panc-1 and L3.6pl) also gave the highest EC50 values for the PTI agents indicating that more PTI was needed to competitively block the active polyamine transport systems of these cell lines. In addition, these cells had a Spd Km that was significantly higher than the HPNE control (Km = 0.19 µM) and ‘non-rescuable’ AsPC-1 and Capan-1 cell lines (Km = 0.18 and 0.12 µM, respectively; Table 2) suggesting that a lower affinity PTS facilitates uptake of extracellular polyamines and may play a role in the ability of these cells to escape DFMO challenge.
Tumors which behave like AsPC-1 and Capan-1 cells should be sensitive to a DFMO-only therapy due to their significant commitment to polyamine biosynthesis but may require high doses of DFMO to affect their growth. In contrast, metastatic tumor cells like the L3.6pl cell line which demonstrate enhanced polyamine scavenging abilities should be more sensitive to the DFMO+PTI combination therapy (which blocks both polyamine biosynthesis and import). Most promising is that the combination therapy of DFMO+PTI should work with both tumor types, with the PTI showing low efficacy in tumors with poor transport activity, but high efficacy in tumors demonstrating high polyamine transport activity. In this regard, the PTI is an important adjuvant to DFMO.
Together the experiments in this study provide novel insights into the regulation of polyamine homeostasis in pancreatic cancers. The fact that the inverse relationship between high ATP13A3 and low Cav-1 mRNA expression exists in many specific human cancers is exciting and provides multiple opportunities for future experiments to test this hypothesis and to test these potential biomarker combinations and the DFMO+PTI combination therapy.
In summary, this paper demonstrates for the first time the role of ATP13A3 in polyamine transport in human pancreatic cancers and provides potential biomarkers to select tumors most susceptible to DFMO. This discovery provides a new approach to potentially stratify cancer patients in current and future clinical trials with DFMO [71,72] and provides a potential ‘catch-all’ strategy in the form of a DFMO+PTI combination therapy for tumors which may escape.
Acknowledgements
The authors would like to thank Dr. Laurence von Kalm (UCF) for his helpful discussions regarding this project, financial support by the 2011 Department of Defense Congressionally Directed Medical Research Program Peer Review Cancer Research Program (PRCRP) Discovery Award (#CA110724), Dr. Isaiah Fidler at the University of Texas M. D. Anderson Cancer Center for the generous gift of L3.6pl cells, Dr. Cheryl Baker at BioCurity Holdings, Inc. in Orlando, FL for the gift of hTERT-HPNE control cells and Dr. Wayne Flintoff at the University of Western Ontario, Canada for the gift of CHO-MG cells.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Casero RA Jr, Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat Rev Drug Discov. 2007;6:373–390. doi: 10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
- 2.Igarashi K, Sakamoto I, Goto N, Kashiwagi K, Honma R, Hirose S. Interaction between polyamines and nucleic acids or phospholipids. Arch Biochem Biophys. 1982;219:438–443. doi: 10.1016/0003-9861(82)90175-8. [DOI] [PubMed] [Google Scholar]
- 3.Gerner EW, Meyskens FL. Polyamines and cancer: old molecules, new understanding. Nat Rev Cancer. 2004;4:781–792. doi: 10.1038/nrc1454. [DOI] [PubMed] [Google Scholar]
- 4.Gerner EW. Cancer chemoprevention locks onto a new polyamine metabolic target. Cancer Prev Res (Phila) 2010;3:125–127. doi: 10.1158/1940-6207.CAPR-09-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soda K. The mechanisms by which polyamines accelerate tumor spread. J Exp Clin Cancer Res. 2011;30:95–104. doi: 10.1186/1756-9966-30-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meyskens FL Jr, Gerner EW. Development of Difluoromethylornithine (DFMO) as a chemoprevention agent. Clin Cancer Res. 1999;5:945–951. [PubMed] [Google Scholar]
- 7.Samal K, Zhao P, Kendzicky A, Yco LP, McClung H, Gerner E, Burns M, Bachmann AS, Sholler G. AMXT-1501, a novel polyamine transport inhibitor, synergizes with DFMO in inhibiting neuroblastoma cell proliferation by targeting both ornithine decarboxylase and polyamine transport. Int J Cancer. 2013;133:1323–1333. doi: 10.1002/ijc.28139. [DOI] [PubMed] [Google Scholar]
- 8.Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134:703–707. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 9.Gardner RA, Delcros JG, Konate F, Breitbeil F, Martin B, Sigman M, Phanstiel O. N1-Substituent effects in the selective delivery of polyamine-conjugates into cells containing active polyamine transporters. J Med Chem. 2004;47:6055–6069. doi: 10.1021/jm0497040. [DOI] [PubMed] [Google Scholar]
- 10.Phanstiel O, Kaur N, Delcros JG. Structure-activity investigations of polyamine-anthracene conjugates and their uptake via the polyamine transporter. Amino Acids. 2007;33:305–313. doi: 10.1007/s00726-007-0527-y. [DOI] [PubMed] [Google Scholar]
- 11.Muth A, Kamel J, Kaur N, Shicora AC, Ayene IS, Gilmour SK, Phanstiel O 4th. Development of Polyamine Transport Ligands with Improved Metabolic Stability and Selectivity against Specific Human Cancers. J Med Chem. 2013;56:5819–5828. doi: 10.1021/jm400496a. [DOI] [PubMed] [Google Scholar]
- 12.Calvaresi EC, Hergenrother PJ. Glucose conjugation for the specific targeting and treatment of cancer. Chem Sci. 2013;4:2319–2333. doi: 10.1039/C3SC22205E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Phanstiel O, Archer JJ. Design of polyamine transport inhibitors as therapeutics. In: Casero RA, Woster PM, editors. Polyamine Drug Discovery. Royal Society of Chemistry; 2012. pp. 162–190. [Google Scholar]
- 14.Muth A, Madan M, Archer JJ, Ocampo N, Rodriguez L, Phanstiel O 4th. Polyamine transport inhibitors: design, synthesis, and combination therapies with difluoromethylornithine. J Med Chem. 2014;57:348–363. doi: 10.1021/jm401174a. [DOI] [PubMed] [Google Scholar]
- 15.Gerner EW, Meyskens FL Jr, Goldschmid S, Lance P, Pelot D. Rationale for, and design of, a clinical trial targeting polyamine metabolism for colon cancer chemoprevention. Amino Acids. 2007;33:189–195. doi: 10.1007/s00726-007-0515-2. [DOI] [PubMed] [Google Scholar]
- 16.Svensson KJ, Welch JE, Kucharzewska P, Bengtson P, Bjurberg M, Pahlman S, Ten Dam GB, Persson L, Belting M. Hypoxia-mediated induction of the polyamine system provides opportunities for tumor growth inhibition by combined targeting of vascular endothelial growth factor and ornithine decarboxylase. Cancer Res. 2008;68:9291–9301. doi: 10.1158/0008-5472.CAN-08-2340. [DOI] [PubMed] [Google Scholar]
- 17.Samal K, Zhao P, Kendzicky A, Yco LP, McClung H, Gerner E, Burns M, Bachmann AS, Sholler G. AMXT-1501, a novel polyamine transport inhibitor, synergizes with DFMO in inhibiting neuroblastoma cell proliferation by targeting both ornithine decarboxylase and polyamine transport. Int J Cancer. 2013;133:1323–1333. doi: 10.1002/ijc.28139. [DOI] [PubMed] [Google Scholar]
- 18.Skorupski KA, O’Brien TG, Guerrero T, Rodriguez CO, Burns MR. Phase I/II clinical trial of 2-difluoromethyl-ornithine (DFMO) and a novel polyamine transport inhibitor (MQT 1426) for feline oral squamous cell carcinoma. Vet Comp Oncol. 2011;9:275–282. doi: 10.1111/j.1476-5829.2011.00264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weeks RS, Vanderwerf SM, Carlson CL, Burns MR, O’Day CL, Cai F, Devens BH, Webb HK. Novel lysine-spermine conjugate inhibits polyamine transport and inhibits cell growth when given with DFMO. Exp Cell Res. 2000;261:293–302. doi: 10.1006/excr.2000.5033. [DOI] [PubMed] [Google Scholar]
- 20.Penchev VR, Rasheed ZA, Maitra A, Matsui W. Heterogeneity and targeting of pancreatic cancer stem cells. Clin Cancer Res. 2012;18:4277–4284. doi: 10.1158/1078-0432.CCR-11-3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohammed A, Janakiram NB, Madka V, Ritchie RL, Brewer M, Biddick L, Patlolla JM, Sadeghi M, Lightfoot S, Steele VE, Rao CV. Eflornithine (DFMO) prevents progression of pancreatic cancer by modulating ornithine decarboxylase signaling. Cancer Prev Res (Phila) 2014;7:1198–1209. doi: 10.1158/1940-6207.CAPR-14-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Muertter RN, Edgar R. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37:D885–890. doi: 10.1093/nar/gkn764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Revet I, Huizenga G, Chan A, Koster J, Volckmann R, van Sluis P, Ora I, Versteeg R, Geerts D. The MSX1 homeobox transcription factor is a downstream target of PHOX2B and activates the Delta-Notch pathway in neuroblastoma. Exp Cell Res. 2008;314:707–719. doi: 10.1016/j.yexcr.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 24.Pegg AE. Regulation of ornithine decarboxylase. J Biol Chem. 2006;281:14529–14532. doi: 10.1074/jbc.R500031200. [DOI] [PubMed] [Google Scholar]
- 25.Pegg AE. Mammalian polyamine metabolism and function. IUBMB Life. 2009;61:880–894. doi: 10.1002/iub.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coffino P. Regulation of cellular polyamines by antizyme. Nat Rev Mol Cell Biol. 2001;2:188–194. doi: 10.1038/35056508. [DOI] [PubMed] [Google Scholar]
- 27.Zhu C, Lang DW, Coffino P. Antizyme2 is a negative regulator of ornithine decarboxylase and polyamine transport. J Biol Chem. 1999;274:26425–26430. doi: 10.1074/jbc.274.37.26425. [DOI] [PubMed] [Google Scholar]
- 28.Mitchell JL, Leyser A, Holtorff MS, Bates JS, Frydman B, Valasinas AL, Reddy VK, Marton LJ. Antizyme induction by polyamine analogues as a factor of cell growth inhibition. Biochem J. 2002;366:663–671. doi: 10.1042/BJ20011612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitchell JL, Simkus CL, Thane TK, Tokarz P, Bonar MM, Frydman B, Valasinas AL, Reddy VK, Marton LJ. Antizyme induction mediates feedback limitation of the incorporation of specific polyamine analogues in tissue culture. Biochem J. 2004;384:271–279. doi: 10.1042/BJ20040972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mitchell JL, Thane TK, Sequeira JM, Marton LJ, Thokala R. Antizyme and antizyme inhibitor activities influence cellular responses to polyamine analogs. Amino Acids. 2007;33:291–297. doi: 10.1007/s00726-007-0523-2. [DOI] [PubMed] [Google Scholar]
- 31.Poulin R, Casero RA, Soulet D. Recent advances in the molecular biology of metazoan polyamine transport. Amino Acids. 2012;42:711–723. doi: 10.1007/s00726-011-0987-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heston WD, Kadmon D, Covey DF, Fair WR. Differential effect of alpha-difluoromethylornithine on the in vivo uptake of 14C-labeled polyamines and methylglyoxal bis(guanylhydrazone) by a rat prostate-derived tumor. Cancer Res. 1984;44:1034–1040. [PubMed] [Google Scholar]
- 33.Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer. 2014;111:817–822. doi: 10.1038/bjc.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roy UK, Rial NS, Kachel KL, Gerner EW. Activated K-RAS increases polyamine uptake in human colon cancer cells through modulation of caveolar endocytosis. Mol Carcinog. 2008;47:538–553. doi: 10.1002/mc.20414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uemura T, Yerushalmi HF, Tsaprailis G, Stringer DE, Pastorian KE, Hawel L 3rd, Byus CV, Gerner EW. Identification and characterization of a diamine exporter in colon epithelial cells. J Biol Chem. 2008;283:26428–26435. doi: 10.1074/jbc.M804714200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang BK, Libby PR, Bergeron RJ, Porter CW. Modulation of polyamine biosynthesis and transport by oncogene transfection. Biochem Biophys Res Commun. 1988;157:264–270. doi: 10.1016/s0006-291x(88)80042-1. [DOI] [PubMed] [Google Scholar]
- 37.Megosh L, Gilmour SK, Rosson D, Soler AP, Blessing M, Sawicki JA, O’Brien TG. Increased frequency of spontaneous skin tumors in transgenic mice which overexpress ornithine decarboxylase. Cancer Res. 1995;55:4205–4209. [PubMed] [Google Scholar]
- 38.Liu L, Rao JN, Zou T, Xiao L, Wang PY, Turner DJ, Gorospe M, Wang JY. Polyamines regulate c-Myc translation through Chk2-dependent HuR phosphorylation. Mol Biol Cell. 2009;20:4885–4898. doi: 10.1091/mbc.E09-07-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weeks RS, Vanderwerf SM, Carlson CL, Burns MR, O’Day CL, Cai F, Devens BH, Webb HK. Novel lysine-spermine conjugate inhibits polyamine transport and inhibits cell growth when given with DFMO. Exp Cell Res. 2000;261:293–302. doi: 10.1006/excr.2000.5033. [DOI] [PubMed] [Google Scholar]
- 40.Gysin S, Rickert P, Kastury K, McMahon M. Analysis of genomic DNA alterations and mRNA expression patterns in a panel of human pancreatic cancer cell lines. Genes Chromosomes Cancer. 2005;44:37–51. doi: 10.1002/gcc.20216. [DOI] [PubMed] [Google Scholar]
- 41.Hamidi H, Lu M, Chau K, Anderson L, Fejzo M, Ginther C, Linnartz R, Zubel A, Slamon DJ, Finn RS. KRAS mutational subtype and copy number predict in vitro response of human pancreatic cancer cell lines to MEK inhibition. Br J Cancer. 2014;111:1788–1801. doi: 10.1038/bjc.2014.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hayes CS, Burns MR, Gilmour SK. Polyamine blockade promotes antitumor immunity. Oncoimmunology. 2014;3:e27360. doi: 10.4161/onci.27360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bertino JR, Waud WR, Parker WB, Lubin M. Targeting tumors that lack methylthioadenosine phosphorylase (MTAP) activity: current strategies. Cancer Biol Ther. 2011;11:627–632. doi: 10.4161/cbt.11.7.14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Subhi AL, Tang B, Balsara BR, Altomare DA, Testa JR, Cooper HS, Hoffman JP, Meropol NJ, Kruger WD. Loss of methylthioadenosine phosphorylase and elevated ornithine decarboxylase is common in pancreatic cancer. Clin Cancer Res. 2004;10:7290–7296. doi: 10.1158/1078-0432.CCR-04-0972. [DOI] [PubMed] [Google Scholar]
- 45.Gilmour SK, Birchler M, Smith MK, Rayca K, Mostochuk J. Effect of elevated levels of ornithine decarboxylase on cell cycle progression in skin. Cell Growth Differ. 1999;10:739–748. [PubMed] [Google Scholar]
- 46.Lin M, DiVito MM, Merajver SD, Boyanapalli M, van Golen KL. Regulation of pancreatic cancer cell migration and invasion by RhoC GTPase and caveolin-1. Mol Cancer. 2005;4:21–34. doi: 10.1186/1476-4598-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanase CP, Dima S, Mihai M, Raducan E, Nicolescu MI, Albulescu L, Voiculescu B, Dumitrascu T, Cruceru LM, Leabu M, Popescu I, Hinescu ME. Caveolin-1 overexpression correlates with tumour progression markers in pancreatic ductal adenocarcinoma. J Mol Histol. 2009;40:23–29. doi: 10.1007/s10735-008-9209-7. [DOI] [PubMed] [Google Scholar]
- 48.Grossi M, Rippe C, Sathanoori R, Sward K, Forte A, Erlinge D, Persson L, Hellstrand P, Nilsson BO. Vascular smooth muscle cell proliferation depends on caveolin-1-regulated polyamine uptake. Biosci Rep. 2014;34:729–740. doi: 10.1042/BSR20140140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grossi M, Phanstiel O, Rippe C, Sward K, Alajbegovic A, Albinsson S, Forte A, Persson L, Hellstrand P, Nilsson BO. Inhibition of polyamine uptake potentiates the anti-proliferative effect of polyamine synthesis inhibition and preserves the contractile phenotype of vascular smooth muscle cells. J Cell Physiol. 2016;231:1334–1342. doi: 10.1002/jcp.25236. [DOI] [PubMed] [Google Scholar]
- 50.Phanstiel O 4th, Kaur N, Delcros JG. Structure-activity investigations of polyamine-anthracene conjugates and their uptake via the polyamine transporter. Amino Acids. 2007;33:305–313. doi: 10.1007/s00726-007-0527-y. [DOI] [PubMed] [Google Scholar]
- 51.Muth A, Kamel J, Kaur N, Shicora AC, Ayene IS, Gilmour SK, Phanstiel O 4th. Development of polyamine transport ligands with improved metabolic stability and selectivity against specific human cancers. J Med Chem. 2013;56:5819–5828. doi: 10.1021/jm400496a. [DOI] [PubMed] [Google Scholar]
- 52.Heinick A, Urban K, Roth S, Spies D, Nunes F, Phanstiel O 4th, Liebau E, Luersen K. Caenorhabditis elegans P5B-type ATPase CATP-5 operates in polyamine transport and is crucial for norspermidine-mediated suppression of RNA interference. FASEB J. 2010;24:206–217. doi: 10.1096/fj.09-135889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pinto Fde T, Corradi GR, Hera DP, Adamo HP. CHO cells expressing the human P(5)-ATPase ATP13A2 are more sensitive to the toxic effects of herbicide paraquat. Neurochem Int. 2012;60:243–248. doi: 10.1016/j.neuint.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 54.Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, LeBleu VS, Mittendorf EA, Weitz J, Rahbari N, Reissfelder C, Pilarsky C, Fraga MF, Piwnica-Worms D, Kalluri R. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523:177–182. doi: 10.1038/nature14581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Diamandis EP, Plebani M. Glypican-1 as a highly sensitive and specific pancreatic cancer biomarker. Clin Chem Lab Med. 2016;54:e1–2. doi: 10.1515/cclm-2015-0773. [DOI] [PubMed] [Google Scholar]
- 56.Belting M, Mani K, Jonsson M, Cheng F, Sandgren S, Jonsson S, Ding K, Delcros JG, Fransson LA. Glypican-1 is a vehicle for polyamine uptake in mammalian cells: a pivital role for nitrosothiol-derived nitric oxide. J Biol Chem. 2003;278:47181–47189. doi: 10.1074/jbc.M308325200. [DOI] [PubMed] [Google Scholar]
- 57.Muth A, Madan M, Archer JJ, Ocampo N, Rodriguez L, Phanstiel O. Polyamine transport inhibitors: design, synthesis, and combination therapies with difluoromethylornithine. J Med Chem. 2014;57:348–363. doi: 10.1021/jm401174a. [DOI] [PubMed] [Google Scholar]
- 58.Mandel JL, Flintoff WF. Isolation of mutant mammalian cells altered in polyamine transport. J Cell Physiol. 1978;97:335–344. doi: 10.1002/jcp.1040970308. [DOI] [PubMed] [Google Scholar]
- 59.Bruns CJ, Harbison MT, Kuniyasu H, Eue I, Fidler IJ. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia. 1999;1:50–62. doi: 10.1038/sj.neo.7900005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mohammed A, Janakiram NB, Madka V, Ritchie RL, Brewer M, Biddick L, Patlolla JM, Sadeghi M, Lightfoot S, Steele VE, Rao CV. Eflornithine (DFMO) Prevents Progression of Pancreatic Cancer by Modulating Ornithine Decarboxylase Signaling. Cancer Prev Res (Phila) 2014;7:1198–1209. doi: 10.1158/1940-6207.CAPR-14-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Salem AF, Bonuccelli G, Bevilacqua G, Arafat H, Pestell RG, Sotgia F, Lisanti MP. Caveolin-1 promotes pancreatic cancer cell differentiation and restores membranous E-cadherin via suppression of the epithelial-mesenchymal transition. Cell Cycle. 2011;10:3692–3700. doi: 10.4161/cc.10.21.17895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Celano P, Baylin SB, Casero RA Jr. Polyamines differentially modulate the transcription of growth-associated genes in human colon carcinoma cells. J Biol Chem. 1989;264:8922–8927. [PubMed] [Google Scholar]
- 63.Celano P, Berchtold CM, Giardiello FM, Casero RA Jr. Modulation of growth gene expression by selective alteration of polyamines in human colon carcinoma cells. Biochem Biophys Res Commun. 1989;165:384–390. doi: 10.1016/0006-291x(89)91082-6. [DOI] [PubMed] [Google Scholar]
- 64.Klinken SP, Holmes KL, Morse HC 3rd, Thorgeirsson SS. Transcriptional and post-transcriptional regulation of c-myc, c-myb, and p53 during proliferation and differentiation of murine erythroleukemia cells treated with DFMO and DMSO. Exp Cell Res. 1988;178:185–198. doi: 10.1016/0014-4827(88)90390-4. [DOI] [PubMed] [Google Scholar]
- 65.Tao L, Kramer PM, Wang W, Yang S, Lubet RA, Steele VE, Pereira MA. Altered expression of c-myc, p16 and p27 in rat colon tumors and its reversal by short-term treatment with chemopreventive agents. Carcinogenesis. 2002;23:1447–1454. doi: 10.1093/carcin/23.9.1447. [DOI] [PubMed] [Google Scholar]
- 66.Raul F. Revival of 2-(difluoromethyl)ornithine (DFMO), an inhibitor of polyamine biosynthesis, as a cancer chemopreventive agent. Biochem Soc Trans. 2007;35:353–355. doi: 10.1042/BST0350353. [DOI] [PubMed] [Google Scholar]
- 67.Liu L, Li L, Rao JN, Zou T, Zhang HM, Boneva D, Bernard MS, Wang JY. Polyamine-modulated expression of c-myc plays a critical role in stimulation of normal intestinal epithelial cell proliferation. Am J Physiol Cell Physiol. 2005;288:C89–99. doi: 10.1152/ajpcell.00326.2004. [DOI] [PubMed] [Google Scholar]
- 68.Uemura T, Gerner EW. Polyamine transport systems in mammalian cells and tissues. Methods Mol Biol. 2011;720:339–348. doi: 10.1007/978-1-61779-034-8_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 70.International WCRF. http://www.wcrf.org/cancer_statistics/world_cancer_statistics.php.
- 71.Saulnier Sholler GL, Gerner EW, Bergendahl G, MacArthur RB, VanderWerff A, Ashikaga T, Bond JP, Ferguson W, Roberts W, Wada RK, Eslin D, Kraveka JM, Kaplan J, Mitchell D, Parikh NS, Neville K, Sender L, Higgins T, Kawakita M, Hiramatsu K, Moriya SS, Bachmann AS. A Phase I Trial of DFMO Targeting Polyamine Addiction in Patients with Relapsed/Refractory Neuroblastoma. PLoS One. 2015;10:e0127246. doi: 10.1371/journal.pone.0127246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Laukaitis CM, Gerner EW. DFMO: targeted risk reduction therapy for colorectal neoplasia. Best Pract Res Clin Gastroenterol. 2011;25:495–506. doi: 10.1016/j.bpg.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.