

Abstract
The proteasome inhibitor MLN9708 is an orally administered drug that is hydrolyzed into its active form, MLN2238 (ixazomib). Compared with Bortezomib, MLN2238 has a shorter proteasome dissociation half-life and a lower incidence and severity of peripheral neuropathy, which makes it an attractive candidate for colorectal cancer treatment. In the present study, we observed that MLN2238 induced autophagy, as evidenced by conversion of the autophagosomal marker LC3 from LC3I to LC3II, in colorectal cancer cell lines. Mcl-1, an anti-apoptotic Bcl-2 family protein, was markedly elevated after treating a colorectal cancer cell line with MLN2238. We proved that inhibiting Mcl-1 expression enhances MLN2238 induced apoptosis and negatively regulates autophagy. Co-administration of BH3 mimetic ABT-737 with MLN2238 synergistically kills colorectal cancer cells through MCL-1 neutralization and autophagy inhibition. Furthermore, the synergistic killing effect of the combination therapy is correlated with P53 status in colorectal cancer. These data highlight that the combination of ABT-737 with MLN9708 is a promising therapeutic strategy for human colorectal cancer.
Keywords: Colorectal cancer, MLN9708, ABT-737, autophagy, MCL-1
Introduction
The latest cancer statistics according to GLOBOCAN reveal that colorectal cancer is the third most commonly diagnosed cancer in males and the second most common in females, with over 1.3 million new cancer cases and nearly 700,000 deaths in 2012 [1]. Despite the introduction of several new drugs for the treatment of colorectal cancer over the last decade that have prolonged survival of advanced stage patients, the disease remains lethal and new treatments with milder side-effects are urgently needed.
Proteasome inhibitors have been shown to inhibit proliferation and induce apoptosis in a variety of cell types. Hematologic cancer derived cells are among the most sensitive to proteasome inhibition [2]. However, the efficacy of proteasome inhibitors as single agent using in vitro and in vivo colorectal cancer models is limited and often requires high doses [3,4]. These drugs also do not show obvious activity in patients with metastatic colorectal cancer in clinical trials [5]. Autophagy has been reported as a mechanism of drug resistance that may reverse or retard therapeutic efficiency [6,7]. Autophagy is known as a catabolic process that targets cellular organelles and cytoplasmic constituents to the lysosomes for degradation [8]. The role of autophagy in colorectal cancer and other cancers remains controversial, it can cause both cell death and survival [9-11]. Several anticancer drugs have been shown to induce autophagy as well as apoptosis [12]. Proteasome inhibitors can induce ER stress in cancer cells [13] and ER stress response is known to be involved in both apoptosis and autophagy [14,15]. Autophagy appears to be up regulated in colorectal cancer and generally associated with poor prognosis and drug resistance [16]. Experimental studies have indicated that in the case of colorectal cancer cell lines that were resistant to nutrient deprivation culture conditions, autophagy might provide an alternative source of energy through degradation of its organelles [17]. Inhibition of autophagy substantially increased apoptosis and significantly attenuated senescence in colorectal cancer cells by blocking p53/p21 pathway [18]. The inhibition also reduced cancer stem-like cell tumorigenicity in vivo [19]. Recent studies also suggest that autophagy inhibitors given in combination with pro-apoptotic agents may enhance chemosensitization in human cancer cells [20].
In the present study, we investigated the effectiveness of combining BH3 mimetic, ABT-737, with proteasome inhibitor, MLN2238 (ixazomib), on colorectal cancer cell lines in vitro. We observed that sensitivity of colorectal cancer cell lines to proteasome inhibition was autophagy-related. In addition, we found that BCL-2 family proteins were involved in the proteasome inhibitor efficacy. In this study, we show that the combination of the two drugs synergistically kill colon cancer cells. Further, we found that autophagy induced by MLN2238 can be negatively regulated by the BH3 mimetic ABT-737. Mechanistic analysis suggests that this combination increases cell apoptosis and cell necrosis by negatively regulating Mcl-1. Therefore, combination treatment with MLN2238 and ABT-737 down regulates autophagy by reducing Mcl-1 expression and offers an attractive strategy to enhance the antitumor efficacy of MLN2238.
Materials and methods
Cells and cell culture
Human colon cancer cell lines HT29, LOVO, SW480, and HCT116 were purchased from the American Type Culture Collection (Manassas, VA) and were authenticated by STR assay two months before experiments. RKO and HCT116 P53-/- cell lines were kindly provided by Dr. Eric Wickstrom at Thomas Jefferson University (Philadelphia, PA). HCT116 Mcl-1 overexpression cell line was generated following the protocol as reported previously [50]. MC38 cell line was kindly provided by Dr. Scott Waldman at Thomas Jefferson University (Philadelphia, PA). The cells were maintained in RPMI 1640 or EMEM or DMEM supplemented with 10% (v/v) fetal bovine serum (Invitrogen, Carlsbad, CA, USA). All cells were cultured at 37°C in a humidified incubator containing 5% CO2.
Chemicals
Ixazomib was provided by Millennium Pharmaceuticals (Cambridge, MA, USA) and ABT-737 were purchased from selleckchem (Houston, TX, USA). Inhibitors were diluted in DMSO at stock concentrations of 10 and 20 mM for in vitro use, respectively. DMSO was purchased from Sigma (St Louis, MO, USA). Mcl-1 siRNA smart-pool and non-targeting siRNA control were purchased from Dharmacon. The transfection of siRNA was performed using Lipofectamine® RNAiMAX (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Cells were then cultured for a 48 h recovery period before being collected or treated as indicated.
Cell viability assay
MTS assays were performed using tetrazolium based CellTiter 96® AQueous One Solution Cell Proliferation assay (Promega; Fitchburg, WI). HCT116, RKO, SW480, LOVO and HT-29 cells were seeded in 96 well plates at 3,000 cells/well. Cells were treated with various concentrations of MLN2238 and/or ABT737 one day after plating. MTS assay was performed at 24 h after treatment with MLN2238 and/or ABT737.
Calculation of combination index
The effect of combination between MLN2238 and ABT-737 at a fixed ratio (1:10) was analyzed using Calcusyn software (Biosoft). Determination of synergy or antagonism was quantified by the combination index (CI). CI=1 indicates an additive effect; < 1, synergy; > 1, antagonism.
Flow cytometry assay
Apoptosis was detected using an FITC Annexin-V Apoptosis Detection Kit (BD Pharmingen, San Diego, CA, USA) according to the manufacturer’s instructions. Cells were seeded in 60-mm culture plates for a final population of 70% confluence. After treatments, cells were collected with 0.15% trypsin at the indicated times, pelleted and washed twice with ice-cold PBS. Cells were resuspended in binding buffer (1 × 106 cells/ml). Then 100 μl of the cell suspension (1 × 105 cells) was incubated with 5 μl of Annexin-V FITC and 5 μl of propidium iodide (PI) for 15 min at room temperature in the dark. The population of apoptosis cells was analyzed using an LSR-II flow cytometer (BD FACSCalibur, Becton Dickinson, San Jose, CA, USA).
Western blot analysis
Cells were washed twice with ice-cold PBS and then lysed in M-Per mammalian lysis buffer (Thermo Scientific; Waltham, MA) plus protease and phosphatase inhibitors (2 μg/ml leupeptin, 2 μg/ml aprotinin, 1 mM PMSF and 0.1 mM Na3VO4). The protein concentration of the lysates was determined with the Bradford reagent (Bio Rad; Hercules, CA) and equal amounts of protein were subjected to SDS-PAGE on a 10% or 15% gel. Separated proteins were transferred to a nitrocellulose membrane, which was then exposed to 5% non-fat dried milk in TBS containing 0.1% Tween 20 (0.1% TBST) for 1 h at room temperature and incubated overnight at 4°C with antibodies against Mcl-1, LC-3, Atg5, P62, phospho-S6 (p-S6), phospho-ERK (p-ERK), Bcl-2, Bcl-xl, Bim (Cell Signaling Technology; Danvers, MA), Beclin-1, Noxa, PARP, total S-6 (T-S6), total ERK (T-ERK) (Santa Cruz Biotechnology; Santa Cruz, CA), actin (Sigma-Aldrich; St. Louis, MO). The membranes were then washed with 0.1% TBST before incubation with horseradish peroxidase-conjugated goat antibodies to rabbit or mouse (Santa Cruz Biotechnology). Immune complexes were detected with chemiluminescence reagents (Pierce ECL Substrate; Thermo Scientific). The gels were then quantitatively analyzed comparing the expression levels of the experimental group with a control of actin. Images show a representative experiment out of 3 with nearly identical results.
In vivo studies
Animal experiments were approved by Experimental Animal Ethics Committee of Fudan University Shanghai medical college. A total of 5 × 105 MC38 murine colorectal cancer cells were inoculated subcutaneously in the right flank of female C57BL/6 mice (6 weeks old) using Matrigel (BD, diluted to 4 mg/ml in PBS). Tumors were measured every other day, volume was calculated with the formula ([length] × [width]2)/2. When the tumors reached a volume of ~100 mm3, Mice were randomly assigned into four groups receiving: vehicle, MLN2238, ABT-737 and combo drugs. MLN2238 was prepared in a 5% solution of 2-hydroxypropyl-β-cyclodextrin (Sigma-Aldrich). Mice were injected intraperitoneally with 30 mg/kg of ABT-737 formulated daily (in 2.5% DMSO, 62.5% H2O containing 5% dextrose, 5% Tween-80, 30% propylene glycol, final pH 4.5) everyday, or MLN2238 (11 mg/kg, i.v) twice weekly for 3 weeks and both. Animals were euthanized when their tumors reached 2 cm3.
Statistical analyses
The values shown represent the mean ± SD for triplicate experiments. The statistical significance of the differences between experimental variables was determined using the Student’s t test. P < 0.05 was considered statistically significant. Student’s t test and linear regression were performed using Excel (Microsoft) and Graphpad Prism (Graphpad Software Inc.).
Results
ABT-737 sensitizes colorectal cancer cells to MLN2238
To assess whether ABT-737 could augment the efficacy of the MLN2238, we initially treated colorectal cancer cell lines (HCT116, LOVO, SW480 and HT29) with MLN2238, ABT-737 or the combination of both agents. Quantitative assessment of viable cells was performed at 24 hours using a standard MTS assay. In all cell lines, MLN2238 (200-500 nM) alone produced a modest reduction in cell viability, however, as dose increased the efficacy had no obvious improvement (Figure 1A). Since ABT-737 (2 μM) alone did not have cytotoxicity in these cells (Figure 1B), we co-administrated MLN2238 (200 nM) with ABT-737 (2 μM). This combination reduced cell viability to a greater extent than MLN2238 alone (Figure 1C). We also noticed that not all four cell lines responded the same to the combination treatment. In HCT116 and LOVO, the combination of MLN2238 with ABT-737 significantly decreased cell viability (Figure 1C *p < 0.005), while in SW480 and HT29, the combination showed less efficacy.
Figure 1.
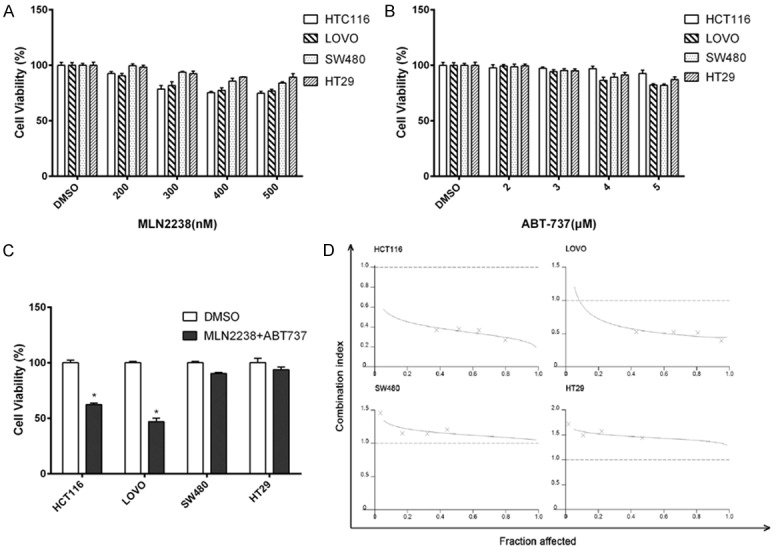
ABT-737 sensitizes colorectal cancer cells to MLN2238. Cultured HCT116, LOVO, SW480, HT-29 human colorectal cancer cell lines were incubated with (A) MLN2238 or (B) ABT-737 for 24 h at indicated doses. (C) Cell lines were treated with MLN2238 (200 nM) and ABT-737 (2 μM). Cell viability was determined using the MTS reduction assay. Experiments were conducted in triplicate. Mean ± SD. *, P < 0.005, relative to DMSO. (D) Cell lines were treated with MLN2238 and ABT-737 at a fixed ratio (1:10), cell viability was measured, and the CI was calculated. Isobologram showing CI < 1 indicates synergy.
To determine whether the cytotoxic effect of the drug combination was synergistic, we performed an analysis with the combination index (CI). The cell lines were treated with different concentrations of MLN2238 and ABT-737 at a fixed ratio (1:10) and cell viability was determined. The CI value was then calculated using Calcusyn software. As shown in the isobologram, the CI values also showed differences between cell lines. A synergistic killing effect was observed in HCT116 and LOVO, but not in SW480 and HT29 (Figure 1D).
MLN2238 induces significant apoptosis after ABT-737 co-treatment
MTS data showed that MLN2238 and ABT-737 combination treatments displayed strong synergistic killing effects for HCT116 and LOVO cells. We thus investigated the type of cell death induced by treatment of colon cancer cells with MLN2238 alone or in combination with ABT-737. Utilization of propidium iodide (PI) and fluorescein isothiocyanate (FITC)-conjugated Annexin V (Annexin V-FITC) is a standard procedure to monitor the progression of apoptosis. Early apoptotic cells are Annexin V-positive and PI-negative (Annexin V+/PI−), whereas late apoptotic cells are Annexin V/PI double positive (Annexin V+/PI+) [21]. The results of Annexin V/PI staining showed a minimal cell death rate was detected when exposed to MLN2238 alone (approximately 10-15% cell death in HCT116 and LOVO). When cells were co-treated with MLN2238 and ABT-737, there was a significant increase in death rate (up to 45-50% in HCT116, nearly 60% in LOVO) (Figure 2A). There was a noticeable increase of early apoptotic cell in HCT116 and LOVO after combo treatment, but not in SW480 and HT29. The findings were similar to the MTS results. ABT-737 significantly enhanced MLN2238 induced cell death in HCT116 and LOVO (Figure 2B MLN2238 vs COMBO, P < 0.005), while no significantly increased cell death was observed in SW480 and HT-29 (Figure 2B MLN2238 vs COMBO, P > 0.1). Since the efficacy difference between cell lines may be related to different gene status, we compared the results with genomic features of these cancer cell lines and found the synergistic effect may be related to differing p53 status (HCT116 and LOVO, p53 wild-type; SW480 and HT29, p53 mutant type).
Figure 2.
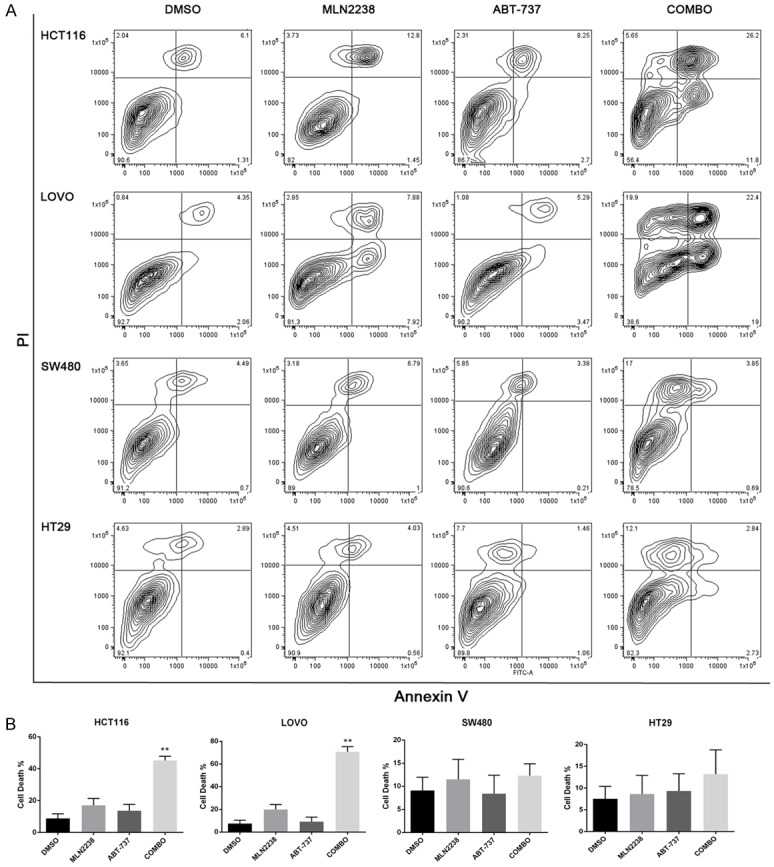
MLN2238 increases cell apoptosis after ABT-737 co-treatment. Cell lines were treated with MLN2238 (200 nM), ABT737 (2 μM) or the combination for 24 h. A. Flow cytometry analysis of annexin-V and propidium iodide (PI) staining of apoptotic cells following treatment. B. Quantitation of flow cytometry (n=5/group). Mean ± SD. *, P < 0.005, relative to MLN2238.
MLN2238 induced autophagy is inhibited by ABT-737 with degradation of the anti-apoptotic protein Mcl-1
Several anticancer drugs have been shown to induce both apoptosis and autophagy [16]. We explored whether MLN2238 can induce autophagy, by detecting expression of the light chain 3 (LC3) protein that is associated with autophagosomal membranes [22]. We found MLN2238 can induce autophagy in colon cancer cell lines HCT116, LOVO, SW480, HT-29 with the conversion of cytoplasmic LC3I to membrane-bound LC3II as detected by immunoblotting. This conversion can be inhibited by co-administering the BH3 mimetic ABT-737 in cell lines HCT116 and LOVO but not in cell lines HT-29 or SW480. We also observed decreased Beclin-1 when treated with both drugs in cell lines HCT116 and LOVO, but not in cell lines HT-29 or SW480, and there is no change in ATG5 in these cell lines (Figure 3). Additionally, MLN2238-induced cleavage of poly (ADP-ribose) polymerase (PARP) was dramatically enhanced in HCT116 and LOVO cells after ABT-737 treatment. Interestingly, RKO cells did not show a reduction of LC3I to LC3II conversion induced by MLN2238 in the combination group, but cleavage of PARP still increased. This result was in accord with Annexin V/PI staining data (Figure S1). It has been reported that Bcl-2 negatively regulates proteasome induced cell death and contributes to the proteasome resistance [23]. By checking Bcl-2 family protein status in these colon cancer cell lines, we demonstrated that Mcl-1, but not Bcl-XL or Bcl-2, accumulation was found in all of these cell lines after MLN2238 treatment. In subsequent experiments we examined whether the accumulation of Mcl-1 induced by MLN2238 in human colon cancer lines can be inhibited by co-administration of the BH3 mimetic ABT-737.
Figure 3.
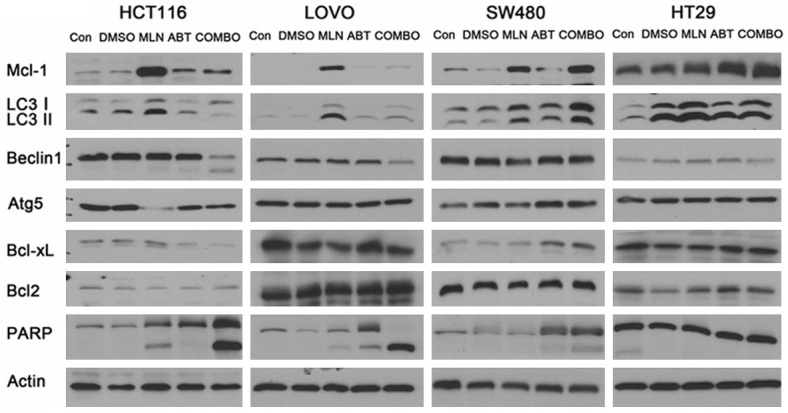
MLN2238 induced autophagy is inhibited by ABT-737 with degradation of the anti-apoptotic protein Mcl-1. Cell lines were treated with doses of MLN2238 (200 nM), ABT737 (2 μM),or the combination for 24 h.Protein lysates were harvested and western blots performed using antibodies for Mcl-1, LC3, Beclin1, Atg5, Bcl-xl, Bcl2, PARP and Actin.
MLN2238 and ABT-737 synergy is altered by regulation of Mcl-1 expression
In HCT116 and LOVO cells, we observed a significant reduction in Mcl-1 expression in the combination treatment group along with a concomitant decrease in LC3II. To determine the connection between the Mcl-1 and LC3II decrease, we overexpressed Mcl-1 in the HCT116 cell line and treated with drugs as described above. We found that in the combination group Mcl-1 decreased slightly but the conversion of LC3I to LC3II was not blocked by the co-treatment (Figure 4A). Annexin V/PI staining proved Mcl-1 overexpression in the HCT116 cell line did not show a synergistic effect in the combination treatment (Figure 4C, 4D MLN2238 vs COMBO, P > 0.1). Interestingly, Mcl-1 does not appear to be a direct activator of autophagy. Overexpression of Mcl-1 does not increase baseline or MLN2238 induced autophagy in HCT116 (Figure 4A). When HCT116 cells were treated with MLN2238 and Mcl-1 siRNA, we found an increased killing effect in the Mcl-1 knockdown group, accompanied with decreased LC3II (Figure 4B). Taken together, our data suggest that the combination of MLN22-38 with BH3 mimetic ABT-737 enhances the MLN2238 killing effect by decreasing the Mcl-1 expression and blocks autophagy by reducing the conversion of LC3I to LC3II.
Figure 4.

MLN2238 and ABT-737 synergy is altered by regulation of Mcl-1 expression. A. HCT116 cells overexpressing Mcl-1 were treated with doses of MLN2238 (200 nM), ABT737 (2 μM) or the combination for 24 h. Protein lysates were harvested and western blots performed using antibodies for Mcl-1 and LC3. B. Mcl-1 siRNA treated HCT116 cells were given dose of MLN2238 (200 nM). Protein lysates were harvested and Mcl-1 and LC3 levels were detected by western blots. C. HCT116 Mcl-1 overexpression cells were treated with doses of MLN2238 (200 nM), ABT737 (2 μM), or the combination for 24 h. Flow cytometry analysis of annexin-V and propidium iodide (PI) staining of apoptotic cells following the treatment. D. Quantitation of flow cytometry (n=5/group). Mean ± SD.
P53 related synergistic effect and LC3 expression
In order to further explore whether or not MLN2238 and ABT-737 synergism is related to P53 status in the cell lines, we treated HCT-116 P53-/- cells with drugs as indicated previously. Annexin V/PI staining of HCT116 P53-/- cells not only showed no synergy between MLN2238 and ABT-737, but HCT116 P53-/- cells also appear resistant to these two drugs individually (Figure 5A, 5B MLN2238 vs COMBO, P > 0.1). Western-blot showed the conversion of LC3I to LC3II could not be inhibited by co-administration of ABT-737 in HCT116 P53-/- cells (Figure 5C). We showed HCT116 P53-/- cells are resistant to the combination treatment with high expression of Mcl-1 and LC3II. This result shows that the sensitivity of colon cancer cell lines to combination therapy is correlated with P53 status.
Figure 5.
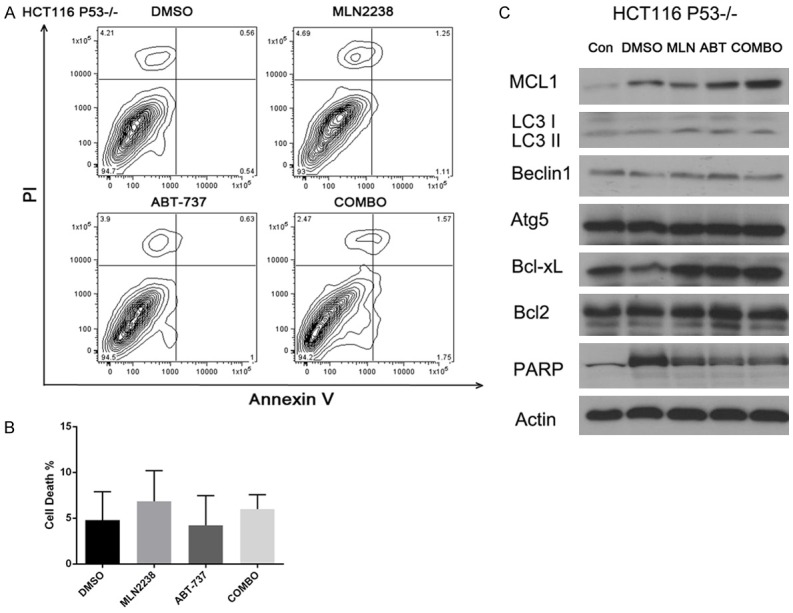
Synergistic effect of MLN2238 and ABT 737 is P53 related. HCT116 P53-/- cells were treated with doses of MLN2238 (200 nM), ABT737 (2 μM), or the combination for 24 h. A. Flow cytometry analysis of annexin-V and propidium iodide (PI) staining of apoptotic cells following the treatment. B. Quantitation of flow cytometry (n=5/group). Mean ± SD. C. Protein lysates were harvested and western blots performed using antibodies for Mcl-1, LC3, Beclin1, Atg5, Bcl-xL, Bcl2, PARP and Actin.
Combination of MLN2238 and ABT-737 inhibits tumor growth in colon cancer allograft model
To test whether the combined effects of MLN2238 and ABT-737 observed in vitro would translate to an in vivo model, we examined effects of MLN2238 or ABT-737 and combo group in MC38 mouse allografts. In our in vivo model, MLN2238 alone inhibited tumor growth compared to control, but ABT-737 alone showed no potent alone in inhibiting tumor growth. Combination of MLN2238 and ABT-737 produced greater inhibition of tumor growth than either agent did alone (Figure 6A, 6B). Mice body weights were monitored to assess the tolerability of combination therapy (Figure 6C). Body weight changes over the course of the experiment were minimal in all treatment groups, suggesting that combined MLN2238 with ABT-737 is well tolerated. These results provided a rationale for combined MLN2238 and ABT-737 therapy as a strategy to inhibit tumor growth in vivo.
Figure 6.
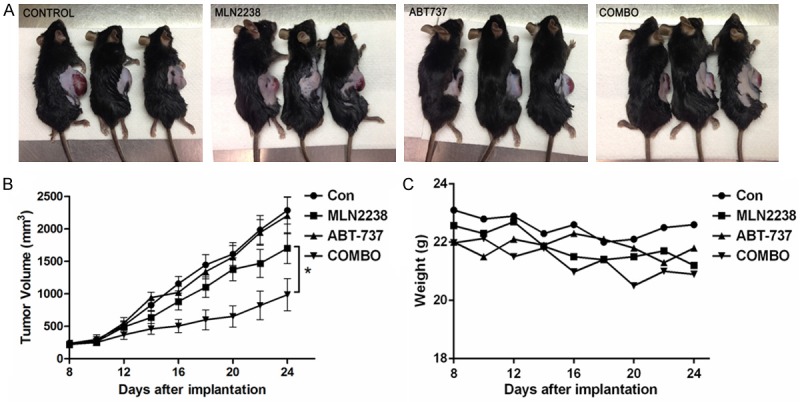
Combination of MLN2238 and ABT-737 inhibits tumor growth in colon cancer allograft model. A total of 5 × 105 MC38 murine colorectal cancer cells were inoculated subcutaneously in the right flank of female C57BL/6 mice. A. Representative tumor growth in mice treated with vehicle, MLN2238, ABT-737 and combo drugs. B. Tumor volume was calculated from day 8 to day 24 (n=6/group). Mean ± SD. *, P < 0.05. C. The average weight of the mice was monitored after treatments.
Discussion
Colorectal cancer is the second leading cause of cancer related mortality in the United States [24]. Despite the introduction of several new drugs for colorectal cancer treatment over the last decade, the disease remains lethal and new treatments with less off target toxicities are urgently needed. Proteasome inhibitors have documented efficacy in a number of hematologic malignancies, especially in multiple myeloma and mantle cell lymphoma [25,26]. However, proteasome inhibitor studies in solid tumors have yielded disappointing results [27-29]. Bortezomib’s toxicity, especially Bortezomib induced peripheral neuropathy (BIPN), may be a major obstacle in the clinical development of BTZ chemotherapy-based regimens in solid tumors. Therefore, it is imperative that a new generation proteasome inhibitors be developed with better therapeutic efficacy and fewer side effects.
MLN9708 (ixazomib citrate) is an orally administered drug that is hydrolyzed into its active form, MLN2238 (ixazomib). Compared with BTZ, MLN2238 has a shorter proteasome dissociation half-life and improved antitumor activity in human cancer xenograft models and a lower incidence and severity of peripheral neuropathy [30,31]. Recently, continuous administration of low-dose cytotoxic agents without extended intervals, also known as “low-dose chemotherapy”, has been considered as an attractive option in colorectal cancer treatment due to its significant reduction in toxic side effects and the risk of multidrug resistance development in tumor cells [32].
Given its favorable toxicity profile observed in clinical trials, MLN9708 is an attractive therapeutic agent to evaluate in human colon cancer. To the best of our knowledge, this is the first preclinical evaluation of its hydrolyzed form, MLN2238, in colon cancer.
Our data show that colon cancer cell death is about 10%-15% at 24 h in the different colon cancer cell lines, when treated with MLN2238 alone. However, there was a significant increase in death rate (up to 50-65%) when MLN2238 was combined with BH3 mimetic ABT-737. Colony assay data also show that combination of MLN2238 and ABT-737 inhibit the colony formation in colon cancer cell lines (Figure S3). Recent evidence indicates that anticancer drugs can trigger apoptosis and/or autophagy, both of which are regulated by the Bcl-2 protein family [6,33]. The Bcl-2 family proteins that regulate apoptotic cell death mediate drug-sensitivity in various cancers [34], and they inhibit apoptosis by binding to Bax or Bak. Bcl-2 and Bcl-xL are also well known for their anti-autophagy abilities [35]. BH3 domain peptide competed for binding to different antiapoptotic proteins Bcl-xL [36] and led to Noxa selectively binding to Mcl-1.
In this study, we explored the effect of MLN2238 combined with or without BH3 mimetic ABT-737 upon apoptosis and autophagy, as well as Bcl-2 family protein expression levels, in human colon cancer cell lines. We found a significant upregulation of Mcl-1 in colon cancer cell lines treated with MLN2238 alone. This result is similar to previous studies about proteasome inhibitors which showed that Bortezomib upregulated Mcl-1 expression in melanoma and breast cancer [37,38]. We reported that MLN2238 induced autophagy as evidenced by conversion of the autophagosomal marker LC3 from LC3I to LC3II. In our colon cancer cell lines, HCT116 and LOVO, the combination of BH3 mimetic and MLN2238 potently downregulated Mcl-1 but not Bcl-2/Bcl-xl proteins, increased PARP cleavage and reduced LC3 II expression. We proved that inhibiting Mcl-1 expression enhances MLN2238 induced apoptosis and negatively regulates autophagy.
Autophagy is an evolutionarily conserved catabolic pathway that targets cellular organelles and cytoplasmic constituents to the lysosomes for degradation and has multiple roles in carcinogenesis and cancer therapy [12]. Autophagy may serve as a cell survival mechanism that occurs in response to cellular stress induced by nutrient deprivation or chemotherapy. However, the molecular mechanisms that govern the interplay between autophagy and apoptosis are poorly understood. Apoptosis induced cell death accounted for 20% in programmed cell death. It is likely that mitochondrial related autophagy blocked mitochondria dependent apoptotic pathway to delay apoptosis [39,40]. The finding that MLN2238 induced both apoptosis and autophagy may be related to its ability to trigger endoplasmic reticulum (ER) stress. BH3 mimetic induces autophagy via blockage of the Bcl-2-Beclin-1 interaction at the endoplasmic reticulum [41]. Since both MLN2238 and ABT-737 can induce autophagy to antagonize the apoptotic effect, we hypothesized increased autophagy would occur when these two drugs were combined. In fact, we found decreased expression of Mcl-1 and LC3II in the combination treatment group in HCT116 and LOVO cells, which is different from our hypothesis, but not in SW480, RKO or HT-29 cell lines. Immunoblots of cleaved PARP corroborate the fact that the combination treatment induced apoptosis synergistically in cell line in HCT116 and LOVO, but not in SW480 and HT-29. We attempted to determine whether the expression of other anti-apoptotic Bcl-2 family members would change after combination therapy. We found Bcl-2 and Bcl-xl expression did not change in any of the colon cancer cell lines, while Beclin-1 decreased in HCT116 and LOVO cells but not in SW480 or HT-29 cells. When we compared the result with genomic features of these colon cancer cell lines [42], we found those with WT P53 benefit the most from the combination treatment, and cell lines with P53 mutations were resistant to the combination of drugs. To confirm efficacy of combination therapy in colon cancer cell lines correlated with P53 status, we performed the treatment as indicated previously using the HCT116 P53-/- and normal HCT116 cell lines. We found the HCT116 P53-/- cell line was resistant to the combination of MLN2238 plus ABT-737 and showed increased Mcl-1 and LC3II expression as compared to control HCT116 cells. Immunoblot analyses also demonstrated that MLN2238 plus ABT-737 decreased ERK phosphorylation in HCT116 cells but not in the HCT116 P53-/- cell line (Figure S2). Our result is consistent with a previous finding that blocking LC3 lipidation or ATG5-ATG12 conjugation decreases ERK phosphorylation, independent of changes in MEK phosphorylation [43]. It has been reported that transcriptional regulation of Mcl-1 by Sp1 is repressed by p53 [44]. Transcriptional activity of STAT3 and HIF-1a which activate transcription of Mcl-1, is also inhibited by p53 [45,46]. Together, P53 status can be used as an indicator for predicting sensitivity of this combination treatment. Interestingly, RKO cell lines with WT P53 did not show a significant reduction of LC3I to LC3II conversion (Figure S1), but we still found decreased Mcl-1 and increased cleavage of PARP after the combination treatment. The reason why the RKO cell line is different from other WT P53 colon cancer cell lines may be because RKO cells possess the BRAF V600E mutation, and tumor cells with BRAF V600E mutations display high rates of induced autophagy [47]. Together, these data indicate that autophagy is serving a prosurvival function in MLN2238 treated colon cancer cells. Consistent with these results, we demonstrated that co-administration of ABT-737 enhances the anti-cancer effects of MLN2238 through decreased Mcl-1 expression and autophagy inhibition.
MLN9708 is a boronic acid analogue that selectively and reversibly inhibits the b5 subunit sites of the 20S proteasome. Structurally, MLN9708 (ixazomib citrate) is an orally administered drug that is hydrolyzed into its active form, MLN2238 (ixazomib). Compared with Bortezomib, data from clinical trials show that MLN9708 has a lower incidence of peripheral neuropathy [48,49]. This makes MLN9708 potentially more suitable for using in combination therapy regimens in human colorectal cancer.
In summary, our findings demonstrate that MLN2238 can induce both apoptosis and autophagy in human colorectal cancer cell lines, with increased Mcl-1 expression. ABT-737 was shown to potentiate MLN2238 mediated apoptosis by inhibiting autophagy, resulting in a synergistic cytotoxic effect. Furthermore, this synergistic effect correlates with P53 status. Inhibition of autophagy by MLN2238 plus ABT-737 was shown to drive more colon cancer cells into apoptosis and induce more cell death, indicating that autophagy serves a prosurvival role in these colon cancer cells subjected to cellular stress. Our in vivo data proved combination of MLN2238 and ABT-737 produced greater inhibition of tumor growth. Together, our data provide in vitro and in vivo evidence that MLN9708 plus ABT-737 has the potential to be a new therapeutic strategy for the treatment of human colorectal cancer.
Acknowledgements
The authors would like to thank Dr. Eric Wickstrom and Dr. Scott Waldman for the cell lines. This work was supported by the grants from Natural Science Foundation of China (81372432). LY, JW participated in manuscript drafting, table creation, and manuscript revision. SX, GL and DB participated the study design and revised the manuscript. JZ revised the manuscript and performed statistical analyses of data. BL and ZZ participated in the study design and were responsible for final approval of the manuscript. All authors have read and approved the final manuscript. The authors declare that they have no conflicts of interests.
Supporting Information
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Drexler HC. Activation of the cell death program by inhibition of proteasome function. Proc Natl Acad Sci U S A. 1997;94:855–860. doi: 10.1073/pnas.94.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pitts TM, Morrow M, Kaufman SA, Tentler JJ, Eckhardt SG. Vorinostat and bortezomib exert synergistic antiproliferative and proapoptotic effects in colon cancer cell models. Mol Cancer Ther. 2009;8:342–349. doi: 10.1158/1535-7163.MCT-08-0534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bychkov ML, Gasparian ME, Dolgikh DA, Kirpichnikov MP. Combination of TRAIL with Bortezomib Shifted Apoptotic Signaling from DR4 to DR5 Death Receptor by Selective Internalization and Degradation of DR4. PLoS One. 2014;9:e109756. doi: 10.1371/journal.pone.0109756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mackay H, Hedley D, Major P, Townsley C, Mackenzie M, Vincent M, Degendorfer P, Tsao MS, Nicklee T, Birle D, Wright J, Siu L, Moore M, Oza A. A phase II trial with pharmacodynamic endpoints of the proteasome inhibitor bortezomib in patients with metastatic colorectal cancer. Clin Cancer Res. 2005;11:5526–5533. doi: 10.1158/1078-0432.CCR-05-0081. [DOI] [PubMed] [Google Scholar]
- 6.Jin SV, White E. Tumor suppression by autophagy through the management of metabolic stress. Autophagy. 2008;4:563–566. doi: 10.4161/auto.5830. [DOI] [PubMed] [Google Scholar]
- 7.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaachouay H, Ohneseit P, Toulany M, Kehlbach R, Multhoff G, Rodemann HP. Autophagy contributes to resistance of tumor cells to ionizing radiation. Radiother Oncol. 2011;99:287–292. doi: 10.1016/j.radonc.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 10.Li H, Wang P, Sun Q, Ding WX, Yin XM, Sobol RW, Stolz DB, Yu J, Zhang L. Following cytochrome c release, autophagy is inhibited during chemotherapy-induced apoptosis by caspase 8-mediated cleavage of Beclin 1. Cancer Res. 2011;71:3625–3634. doi: 10.1158/0008-5472.CAN-10-4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao W, Shen Z, Shang L, Wang X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ. 2011;18:1598–1607. doi: 10.1038/cdd.2011.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amaravadi RK, Thompson CB. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin Cancer Res. 2007;13:7271–7279. doi: 10.1158/1078-0432.CCR-07-1595. [DOI] [PubMed] [Google Scholar]
- 13.Ashok BT, Kim E, Mittelman A, Tiwari RK. Proteasome inhibitors differentially affect heat shock protein response in cancer cells. Int J Mol Med. 2001;8:385–390. doi: 10.3892/ijmm.8.4.385. [DOI] [PubMed] [Google Scholar]
- 14.Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007;14:500–510. doi: 10.1038/sj.cdd.4402039. [DOI] [PubMed] [Google Scholar]
- 15.Heath-Engel HM, Chang NC, Shore GC. The endoplasmic reticulum in apoptosis and autophagy: role of the BCL-2 protein family. Oncogene. 2008;27:6419–6433. doi: 10.1038/onc.2008.309. [DOI] [PubMed] [Google Scholar]
- 16.Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, Han W, Lou F, Yang J, Zhang Q, Wang X, He C, Pan H. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4:e838. doi: 10.1038/cddis.2013.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato K, Tsuchihara K, Fujii S, Sugiyama M, Goya T, Atomi Y, Ueno T, Ochiai A, Esumi H. Autophagy is activated in colorectal cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res. 2007;67:9677–9684. doi: 10.1158/0008-5472.CAN-07-1462. [DOI] [PubMed] [Google Scholar]
- 18.Zhang JW, Zhang SS, Song JR, Sun K, Zong C, Zhao QD, Liu WT, Li R, Wu MC, Wei LX. Autophagy inhibition switches low-dose camptothecin-induced premature senescence to apoptosis in human colorectal cancer cells. Biochem Pharmacol. 2014;90:265–275. doi: 10.1016/j.bcp.2014.05.009. [DOI] [PubMed] [Google Scholar]
- 19.Wei MF, Chen MW, Chen KC, Lou PJ, Lin SY, Hung SC, Hsiao M, Yao CJ, Shieh MJ. Autophagy promotes resistance to photodynamic therapy-induced apoptosis selectively in colorectal cancer stem-like cells. Autophagy. 2014;10:1179–1192. doi: 10.4161/auto.28679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79:1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wlodkowic D, Telford W, Skommer J, Darzynkiewicz Z. Apoptosis and beyond: cytometry in studies of programmed cell death. Methods Cell Biol. 2011;103:55–98. doi: 10.1016/B978-0-12-385493-3.00004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith AJ, Dai H, Correia C, Takahashi R, Lee SH, Schmitz I, Kaufmann SH. Noxa/Bcl-2 protein interactions contribute to bortezomib resistance in human lymphoid cells. J Biol Chem. 2011;286:17682–17692. doi: 10.1074/jbc.M110.189092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 25.San Miguel JF, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, Spicka I, Petrucci MT, Palumbo A, Samoilova OS, Dmoszynska A, Abdulkadyrov KM, Schots R, Jiang B, Mateos MV, Anderson KC, Esseltine DL, Liu K, Cakana A, van de Velde H, Richardson PG. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359:906–917. doi: 10.1056/NEJMoa0801479. [DOI] [PubMed] [Google Scholar]
- 26.Kane RC, Dagher R, Farrell A, Ko CW, Sridhara R, Justice R, Pazdur R. Bortezomib for the treatment of mantle cell lymphoma. Clin Cancer Res. 2007;13:5291–5294. doi: 10.1158/1078-0432.CCR-07-0871. [DOI] [PubMed] [Google Scholar]
- 27.Markovic SN, Geyer SM, Dawkins F, Sharfman W, Albertini M, Maples W, Fracasso PM, Fitch T, Lorusso P, Adjei AA, Erlichman C. A phase II study of bortezomib in the treatment of metastatic malignant melanoma. Cancer. 2005;103:2584–2589. doi: 10.1002/cncr.21108. [DOI] [PubMed] [Google Scholar]
- 28.Kondagunta GV, Drucker B, Schwartz L, Bacik J, Marion S, Russo P, Mazumdar M, Motzer RJ. Phase II trial of bortezomib for patients with advanced renal cell carcinoma. J. Clin. Oncol. 2004;22:3720–3725. doi: 10.1200/JCO.2004.10.155. [DOI] [PubMed] [Google Scholar]
- 29.Kozuch PS, Rocha-Lima CM, Dragovich T, Hochster H, O’Neil BH, Atiq OT, Pipas JM, Ryan DP, Lenz HJ. Bortezomib with or without irinotecan in relapsed or refractory colorectal cancer: results from a randomized phase II study. J. Clin. Oncol. 2008;26:2320–2326. doi: 10.1200/JCO.2007.14.0152. [DOI] [PubMed] [Google Scholar]
- 30.Lee EC, Fitzgerald M, Bannerman B, Donelan J, Bano K, Terkelsen J, Bradley DP, Subakan O, Silva MD, Liu R, Pickard M, Li Z, Tayber O, Li P, Hales P, Carsillo M, Neppalli VT, Berger AJ, Kupperman E, Manfredi M, Bolen JB, Van Ness B, Janz S. Antitumor activity of the investigational proteasome inhibitor MLN9708 in mouse models of B-cell and plasma cell malignancies. Clin Cancer Res. 2011;17:7313–7323. doi: 10.1158/1078-0432.CCR-11-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, Yu J, Yang Y, Hales P, Bruzzese F, Liu J, Blank J, Garcia K, Tsu C, Dick L, Fleming P, Yu L, Manfredi M, Rolfe M, Bolen J. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010;70:1970–1980. doi: 10.1158/0008-5472.CAN-09-2766. [DOI] [PubMed] [Google Scholar]
- 32.Loven D, Hasnis E, Bertolini F, Shaked Y. Low-dose metronomic chemotherapy: from past experience to new paradigms in the treatment of cancer. Drug Discov Today. 2013;18:193–201. doi: 10.1016/j.drudis.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 33.Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4:600–606. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelly PN, Strasser A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ. 2011;18:1414–1424. doi: 10.1038/cdd.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 36.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 37.Qin JZ, Xin H, Sitailo LA, Denning MF, Nickoloff BJ. Enhanced killing of melanoma cells by simultaneously targeting Mcl-1 and NOXA. Cancer Res. 2006;66:9636–9645. doi: 10.1158/0008-5472.CAN-06-0747. [DOI] [PubMed] [Google Scholar]
- 38.Zhou W, Hu J, Tang H, Wang D, Huang X, He C, Zhu H. Small interfering RNA targeting mcl-1 enhances proteasome inhibitor-induced apoptosis in various solid malignant tumors. BMC Cancer. 2011;11:485. doi: 10.1186/1471-2407-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calgarotto AK, da Silva Pereira GJ, Bechara A, Paredes-Gamero EJ, Barbosa CM, Hirata H, de Souza Queiroz ML, Smaili SS, Bincoletto C. Autophagy inhibited Ehrlich ascitic tumor cells apoptosis induced by the nitrostyrene derivative compounds: relationship with cytosolic calcium mobilization. Eur J Pharmacol. 2012;678:6–14. doi: 10.1016/j.ejphar.2011.12.031. [DOI] [PubMed] [Google Scholar]
- 40.Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lian J, Wu X, He F, Karnak D, Tang W, Meng Y, Xiang D, Ji M, Lawrence TS, Xu L. A natural BH3 mimetic induces autophagy in apoptosis-resistant prostate cancer via modulating Bcl-2-Beclin1 interaction at endoplasmic reticulum. Cell Death Differ. 2011;18:60–71. doi: 10.1038/cdd.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahmed D, Eide PW, Eilertsen IA, Danielsen SA, Eknaes M, Hektoen M, Lind GE, Lothe RA. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis. 2013;2:e71. doi: 10.1038/oncsis.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martinez-Lopez N, Athonvarangkul D, Mishall P, Sahu S, Singh R. Autophagy proteins regulate ERK phosphorylation. Nat Commun. 2013;4:2799. doi: 10.1038/ncomms3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pietrzak M, Puzianowska-Kuznicka M. p53-dependent repression of the human MCL-1 gene encoding an anti-apoptotic member of the BCL-2 family: the role of Sp1 and of basic transcription factor binding sites in the MCL-1 promoter. Biol Chem. 2008;389:383–393. doi: 10.1515/BC.2008.039. [DOI] [PubMed] [Google Scholar]
- 45.Lin J, Tang H, Jin X, Jia G, Hsieh JT. p53 regulates Stat3 phosphorylation and DNA binding activity in human prostate cancer cells expressing constitutively active Stat3. Oncogene. 2002;21:3082–3088. doi: 10.1038/sj.onc.1205426. [DOI] [PubMed] [Google Scholar]
- 46.Sermeus A, Michiels C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2011;2:e164. doi: 10.1038/cddis.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mulcahy Levy JM, Foreman NK, Thorburn A. Using BRAF[V600E] as a marker of autophagy dependence in pediatric brain tumors. Autophagy. 2014;10:2077–2078. doi: 10.4161/auto.36138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumar SK, Bensinger WI, Zimmerman TM, Reeder CB, Berenson JR, Berg D, Hui AM, Gupta N, Di Bacco A, Yu J, Shou Y, Niesvizky R. Phase 1 study of weekly dosing with the investigational oral proteasome inhibitor ixazomib in relapsed/refractory multiple myeloma. Blood. 2014;124:1047–1055. doi: 10.1182/blood-2014-01-548941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Richardson PG, Baz R, Wang M, Jakubowiak AJ, Laubach JP, Harvey RD, Talpaz M, Berg D, Liu G, Yu J, Gupta N, Di Bacco A, Hui AM, Lonial S. Phase 1 study of twice-weekly ixazomib, an oral proteasome inhibitor, in relapsed/refractory multiple myeloma patients. Blood. 2014;124:1038–1046. doi: 10.1182/blood-2014-01-548826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han J, Goldstein LA, Gastman BR, Rabinowich H. Interrelated roles for Mcl-1 and BIM in regulation of TRAIL-mediated mitochondrial apoptosis. J Biol Chem. 2006;281:10153–10163. doi: 10.1074/jbc.M510349200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.