

Abstract
A keloid is a benign skin tumor formed by an overgrowth of granulation tissue in affected patients. Peroxisome proliferator-activated receptor-γ (PPAR-γ) agonists were reported to be able to regulate extracellular matrix production in human dermal fibroblasts. This study explored the underlying molecular mechanism of PPAR-γ agonist troglitazone treatment for fibroblasts obtained from keloid patients. The data revealed that troglitazone treatment of keloid fibroblasts (KFs) downregulated the expression of early growth response-1 (Egr1) and collagen-1 (Col1). Level of Egr1 were closely associated with KF-induced fibrosis. The miRNA profiling data revealed that miR-543 was transcriptionally activated after troglitazone treatment. Bioinformatic analysis and experimental data showed that miR-543 was able to target Egr1. ELISA data confirmed that Col1 protein in the supernatant were modulated by the feedback regulatory axis of PPAR-γ agonist-induced miR-543 to inhibit Egr1 expression, whereas PPAR-γ antagonist treatment abolished such effect on Col1 suppression in KFs. This study demonstrated that the PPAR-γ agonist-mediated miR-543 and Egr1 signaling plays an important role in the suppression of collagen synthesis in KFs. Future in vivo studies are needed to confirm these in vitro data.
Keywords: Benign tumor, keloid, PPAR-γ agonist, troglitazone, Egr1, miR-543
Introduction
A keloid is a benign skin tumor caused by an overgrowth of granulation tissue in predisposed patients. Histologically, keloids are characterized by an excessive deposition of extracellular matrix (ECM) components, particularly collagen III and later collagen I [1]. The precise etiology of keloids remains unknown, however they are initiated after skin injury and frequently occur in genetically susceptible individuals [2]. Keloids can cause physical and psychological distress in patients [3]. Pathologically, overexpression of various growth factors and cytokines, which is considered a paradigm for skin fibrosis or uncontrolled wound healing, contribute to keloid formation [2]. While tissue fibrosis in normal scar formation is known to be a reactive process in which various factors modulate relevant pathways, it is unclear molecularly how growth factors or cytokines abnormally affect wound healing or skin repairation in keloid formation, or how the associated underlying molecular and biochemical mechanisms are affected causing dysfunction [4,5]. Current keloid treatment involves removing excess scar tissue and preventing further “growth”, by administering intralesional injections of corticosteroids or radiation therapy [3]. Therefore there is an urgent need to develop a novel strategy to effectively control keloid formation and to better understand the mechanisms underlying keloid formation predisposition and pathogenesis.
Recent studies identified the nuclear receptor peroxisome proliferator-activated receptor-γ (PPAR-γ) as an important factor in the endogenous anti-fibrotic defense mechanism [6,7]. Activation of PPAR-γ by both natural and synthetic agonists can effectively inhibit pro-fibrotic tissue reactions in many organs [8]. PPAR-γ is a nuclear receptor and prostaglandin PGJ2 or arachidonic acid metabolite 5-hydroxyicosatetraenoic acid (5-HETE) can activate PPAR-γ, which in turn forms a heterodimer with retinoid X receptor-α to transcriptionally regulate expression of the target genes [9]. PPAR-γ plays an essential role in the regulation of embryo development, cell differentiation, lipid metabolism, and tumorigenesis [10,11]. A previous study demonstrated that synthetic PPAR-γ agonist RS5444 was able to inhibit anaplastic thyroid cancer growth in vitro and in nude mice [12]. The mechanisms responsible for PPAR-γ agonist antitumor activity include growth inhibition, induction of differentiation and apoptosis, promotion of cell cycle arrest, and anti-angiogenic activity [13]. In our previous study, we demonstrated that the PPAR-γ agonist troglitazone inhibited expression of collagen-1 (Col1) in hypertrophic scar fibroblasts [14]. Indeed, fibroblasts are the major effector cells in the development of fibrosis and inappropriate fibroblast growth and activation are the fundamental pathogenic alterations underlying abnormal fibrosis [15,16]. Therefore, in the present study, we explored the molecular mechanisms underlying the anti-fibrotic properties of the PPAR-γ agonist troglitazone in keloid fibroblasts (KFs). We first determined the effect of troglitazone on the downregulation of Col1 and Egr1 proteins in KFs in vitro, and then profiled differentially expressed microRNAs (miRNAs) that may mediate the effects of troglitazone in KFs. After that, we performed gene knock-down and knock-in investigations to identify the PPAR-γ-miR-543-Egr1 signaling axis that responds to troglitazone-mediated suppression of collagen synthesis in KFs.
Materials and methods
Patients and sample collection
This study was performed according to a written study protocol that has been approved by the Ethics Committee of Xijing Hospital affiliated to Fourth Military Medical University (Shaanxi, China). All patients provided a written informed consent before inclusion in this study. Keloids (K) and paired normal skin (NS) tissues were obtained from 57 patients who were diagnosed with keloid affliction by trained clinicians and pathologists according to clinical criteria in Xijing Hospital between April 2009 and July 2014 [17]. The clinical diagnosis was based on i) keloid extension beyond the area of the original injury; ii) rounder and smoother surface of keloid; and iii) the presence of irritation, such as itching or pain. These tissues were obtained from the surgery room and used for fibroblast isolation and culture as well as for immunohistochemical analysis of protein expression (see below). All experiments involving human subjects were performed in compliance with the Declaration of Helsinki.
Fibroblast isolation, culture, and drug treatment
Human primary KFs and normal skin fibroblasts (NSFBs) were isolated from surgically excised tissues and cultured in vitro according to previous studies [4,5]. For treatment with troglitazone or PPAR-γ antagonist GW9662, KFs and NSFBs were seeded in six-well plates and cultured overnight; on the next day, the cell culture medium was refreshed with Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (control group) or DMEM with 10% FBS and 30 mmol troglitazone (ab141112, Abcam, Cambridge, MA, USA) or 1 μM GW9662 (ab141125, Abcam) and then cultured for 72 or 48 h according to our previous study [6,14].
Quantitative reverse transcription-PCR (qRT-PCR)
Total RNA was isolated from cells using the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions and subsequently reverse transcribed into cDNA using the Transcriptor First Strand cDNA synthesis kit (Roche Applied Science, Mannheim, Germany). qPCR was then performed with primers for Egr1 (5’-GGTCAGTGGCCTAGTGAGC-3’ and 5’-GTGCCGCTGAGTAAATGGGA-3’), Col1 (5’-GTTGCTGCTTGCAGTAACCTT-3’ and 5’-AGGGCCAAGTCCAACTCCTT-3’), and miR-543 (5’-AAACAUUCGCGGUGCACUUCUU-3’). The relative levels of miR-543, and Egr1 were normalized to U6, and levels of Col1 were normalized GAPDH, using the 2-ΔΔCt method using Applied Biosystems 7500 software version 2.0.1. Each experiment was performed in triplicate and repeated thrice.
Protein extraction and Western blot
Protein extraction and Western blot analysis for Egr1 expression were performed according to our previous study [14] with the following antibodies: A mouse monoclonal anti-human Egr1 (ab55160; Abcam, 1:500) or β-actin antibody (A5441; Sigma-Aldrich, MO, USA, 1:1000). Protein signals were visualized using an Odyssey Infrared Imaging System (LI-COR Biosciences, NE, USA). Protein levels were quantified using Image J software through pixel analysis of bands with normalization to β-actin as a loading control.
Immunohistochemistry
Keloids (K) and paired normal skin (NS) tissues were fixed in 4% paraformaldehyde overnight at room temperature, and then subjected to tissue processing and embedded in paraffin. The paraffin blocks were cut into 4 μm-thick sections. For immunohistochemistry, tissue sections were deparaffinized in xylene, rehydrated in a series of ethanol solutions, and then steamed in a pressure cooker containing 0.01 M citrate buffer for the antigen retrieval. Next, the sections were incubated with 1% H2O2 in phosphate buffered saline (PBS) for 30 min to block the endogenous peroxidase activity and then with 20% of normal serum for 30 min at room temperature. The sections were then incubated overnight at 4°C with a rabbit anti-Egr1 antibody (ab55160; Abcam) at a dilution of 1:100. On the next day, the sections were washed with PBS thrice and then incubated with poly-HRP IgG (PV-6000; Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd., Beijing, China) for 15 min and subjected to color reactions to visualize positive signals in 3,3’-diaminobenzidene solution (ZLI-9032; Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd.). The sections were finally counterstained with hematoxylin and mounted with a coverslip. After the immunostained sections were reviewed by a pathologist under a light microscope, the levels of Egr1 protein in tissues were semi-quantitatively analyzed using MetaMorph image analysis software (Universal Imaging Corp., CA, USA). The staining results were expressed as the mean ± standard deviation (SD) of optical density from six different digital images.
Enzyme-linked immunosorbent assay (ELISA)
The level of Col1 supernatant protein was assessed using an ELISA with a Human Collagen Type I ELISA Kit (ACEL, Kanagawa, Japan) according to the manufacturer’s protocol. Briefly, the conditioned medium from cell culture was added to an ELISA Kit plate that was precoated with an anti-Col1 antibody, then a biotinylated secondary antibody was added and the plate was incubated at room temperature for 2 h. The color development catalyzed by horseradish peroxidase was terminated with 2.5 mol sulfuric acid and the absorption was measured at 450 nm. The protein concentration was normalized to the relative absorbance rate of the standards and expressed as mean ± SD.
Locked nucleic acid (LNA)-based miRNA microarray assay
Profiling of differentially expressed miRNAs in RNA samples of treated cells was performed using locked nucleic acid (LNA) mercury microarrays (Exiqon, Vedbaek, Denmark). In brief, total RNA was isolated using the TRIzol agent (Invitrogen, Carlsbad, CA, USA) and miRNeasy mini kit (Qiagen) according to the manufacturers’ instructions and then quantified using a NanoDrop 1000 (Thermo-Fisher Scientific, Waltham, MA, USA) to determine the A260/A280 ratio. RNA samples of each pool of cells (2 μg) was labeled using the miRCURY Hy3/Hy5 Power labeling kit and hybridized with the miRCURY LNA Array (version 16.0), which contains probes to capture different miRNAs and covers all human miRNAs annotated in miRBase 18.0 (http://www.mirbase.org/) and viral miRNAs related to these species. Following the washing steps, the slides were scanned with the Axon GenePix 4000B microarray scanner and scanned images were imported into GenePix Pro 6.0 software (Axon) for grid alignment and data extraction.
Oligonucleotides, plasmid construction and transfection
The FAM-modified 2’-OMe oligonucleotides for targeting the miRNAs were chemically synthesized and purified with high-performance liquid chromatography (GenePharma, Shanghai, China). The 2’-O-me-miR mimics were composed of RNA duplexes for the following miRNAs: miR-543, 5’-AAACAUUCGCGGUGCACUUCUU-3’; miR-4439, 5’-GUGACUGAUACCUUGGAGGCAU-3’; miR-3157, 5’-UUCAGCCAGGCUAGUGCAGUCU-3’; miR-92b, 5’-UAUUGCACUCGUCCCGGCCUCC-3’; and miR-1249, 5’-ACGCCCUUCCCCCCCUUCUUCA-3’. The sequences of 2’-O-Me-miR inhibitors were: miR-543 inhibitor, 5’-AAGAAGUGCACCGCGAAUGUUU-3’; miR-4439 inhibitor, 5’-AUGCCUCCAAGGUAUCAGUCAC-3’; miR-3157 inhibitor, 5’-AGACUGCACUAGCCUGGCUGAA-3’; miR-92b inhibitor, 5’-GGAGGCCGGGACGAGUGCAAUA-3’; and miR-1249 inhibitor, 5’-UGAAGAAGGGGGGGAAGGGCGU-3’. The sequence of the 2’-O-me scramble oligonucleotide (control) was 5’-CAGUACUUUUGUGUAGUACAA-3’.
The sequences of the Egr1 siRNA were 5’-AGGACAAGAAAGCAGACAAAAGUTT-3’ and 5’-ACUUUUGTCUGCUUUCUUGUCCUTT-3’. The sequences of scrambled siRNA were 5’-GCAAACAUCCCAGAGGUAU-3’ and 5’-AUACCUCUGGGAUGUUUGC-3’. These oligonucleotides and constructed siRNA vectors were transfected into cells using Lipofectamine™ 2000 (Invitrogen) according to the manufacturer’s instructions.
Plasmid construction and transfection
For Egr1 overexpression, the full-length open reading frame of Egr1 cDNA was amplified using PCR with primers (5’-ATGGCCGCGGCCAAGGCCGAG-3’ and 5’-TTAGCAAATTTCAATTGTCCTG-3’) and then subcloned into a pcDNA3.0 vector (Invitrogen). Sequencing was performed to confirm correct cloning insertion. The vector was then transfected into cells using Lipofectamine™ 2000 according to the manufacturer’s instructions.
Luciferase-reporter activity assay
The 3’-untranslated region of Egr1 cDNA was amplified using PCR with primers (5’-GTGTGATGCGCCTTGCTGATG-3’ and 5’-AGGATATACACCACTATCC-3’), while this Egr1 cDNA with point substitutions according to the miRNA complementary sites was also amplified with appropriate primers. The amplified PCR products were cloned into the pMD-18T vector (TaKaRa, Shiga, Japan), and confirmed by DNA sequencing. These Egr1 cDNA fragments were then released from pMD-18T vectors and subcloned into pGL3 vectors (Promega, Madison, WI, USA) and confirmed with DNA sequenced. These vectors were named ‘pGL3-Egr1-UTR’ and ‘pGL3-Smad3-UTR-mut’, respectively. The luciferase reporter vectors containing the putative wild type peroxisome proliferator response element (PPRE-wt) and PPRE deletion mutant (PPRE-mut) were constructed in our previous study [14].
For the luciferase-reporter activity assay, cells were seeded into 24-well plates and grown overnight. On the next day, cells were transfected with 1 μg of luciferase-reporter plasmids per well using Lipofectamine™ 2000 Transfection Reagent. Luciferase activities were measured using the dual-luciferase reporter gene assay kit (Promega) according to the manufacturer’s instructions.
Chromatin immunoprecipitation assay (ChIP)
After the KFs were treated with 30 mM troglitazone for 72 h, the cells were cross-linked with 1% formaldehyde for 30 min and quenched prior to sonication as described previously [9]. The input fraction was corresponding to 10% chromatin solution. The sheared chromatin was immunoprecipitated with an anti-PPAR-a antibody (ab45036, Abcam, 1:75) or IgG at 4°C overnight and subsequently mixed with 40 μl of protein A/G Sepharose beads (Promega) and 2 μl of 10 mg/ml herring sperm DNA (Sigma). After that, immunoprecipitated DNA samples were amplified using PCR with primers containing the PPAR-γ binding sites (-1975 to -1806 bp, 5’-GGATGGCCTTCTGGACACG-3’ and 5’-TCCGAAGTTGCCATGTGCA-3’).
Statistical analysis
All experiments were repeated three times and the quantitative data were summarized as the mean ± SD for statistical analysis using two-sided Student’s t-tests. All statistical analyses were performed using PRISM Software, Version 4 (GraphPad Software, La Jolla, CA, USA) and a P value ≤ 0.05 was considered statistically significant.
Results
PPAR-γ agonist suppression of Egr1 and Col1 expression in KFs
To confirm the anti-fibrotic effect of PPAR-γ agonist troglitazone, we grew and treated KFs with 30 mmol troglitazone for 72 h according to a previous study [6]. We found that troglitazone significantly reduced the level of Col1 mRNA and secreted protein in KFs compared to negative control cells (Figure 1A and 1C, P = 0.026 and P = 0.031, respectively). Similarly, levels of Egr1 mRNA were also reduced by 72 h treatment with troglitazone (Figure 1A and 1B, P = 0.018). Egr1 was reported to be a profibrogenic protein and highly expressed in several fibrotic diseases [18-20]. Thus, our current data indicate that PPAR-γ agonist functions as negative regulators of collagen synthesis in KFs.
Figure 1.

Effect of 30 mM troglitazone treatment for 72 h in KFs on Col1 and Egr1 expression. A. qRT-PCR analysis reveals levels of Col1 and Egr1 mRNA were reduced by approximately 50% when treated with troglitazone (*P = 0.018 and *P = 0.026). B. Western blot analysis indicated levels of Egr1 protein after normalization to β-actin were reduced when treated with troglitazone and quantified with Image J software. C. ELISA analysis indicated a reduced Col1 supernatant protein level in the troglitazone-treated KFs (*P = 0.031).
Egr1 expression detected in keloid tissues in clinic
We next determined Egr1 expression and its clinical significance in keloids. The immunohistochemical data showed that Egr1 protein was upregulated in keloid tissues compared to normal skin tissues from 57 keloid-affected patients (Figure 2A and 2B, P = 0.006).
Figure 2.
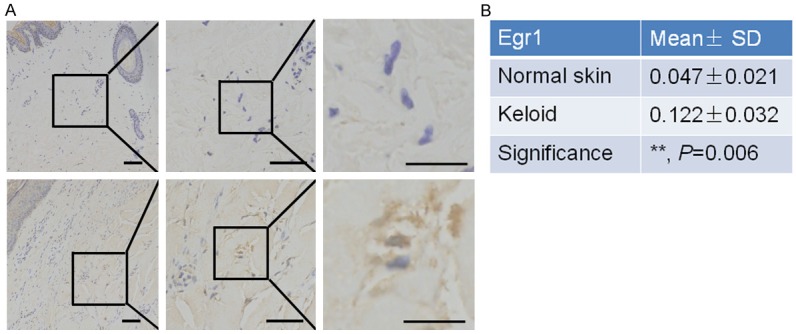
Overexpression of Egr1 protein in KFs of keloid tissues. A. Immunohistochemical analysis of Egr1 protein in keloid and paired normal skin tissues. Upper, K; lower, NS. Magnification: ×40 (left), ×200 (middle), and ×400 (right). B. Summarized data of immunostaining intensity of Egr1 protein between keloid and normal skin tissues. **P = 0.006.
Egr1 overexpression promotes collagen synthesis in KFs
We assessed whether Egr1 upregulation can induce collagen synthesis in NSFBs after Egr1 cDNA transfection into NSFBs. Our data showed that overexpression of Egr1 protein in NSFBs induced the secretion of collagen protein in the conditioned medium and Col1 mRNA in NSFBs compared to controls (Figure 3A-C). In contrast, the knockdown of Egr1 expression reduced levels of collagen secreted protein and Col1 mRNA in KFs (Figure 3D-F). These findings, together with the data of previous studies [8,14], indicated that Egr1 was involved in skin fibrosis.
Figure 3.

Regulation of Col1 synthesis by Egr1 in KFs. A. qRT-PCR analysis indicates that ectopic Egr1 expression significantly promoted synthesis of Col1 mRNA (***P < 0.001). B. Western blot expression of Egr1 in ectopic Egr1 expression KFs and quantified with Image J software. C. ELISA analysis of Col1 supernatant protein level in ectopic Egr1 expressed KFs (**P = 0.003). D. qRT-PCR showing that knockdown of Egr1 expression (**P = 0.012) significantly decreased synthesis of Col1 (*P = 0.019). E. Western blot analysis of Egr1 proteins in Egr1 knocked down KFs and quantified with Image J software. F. ELISA analysis of Col1 supernatant protein level in Egr1 knocked down KFs (*P = 0.025). Graphical data represented as the mean ± SD.
miR-543 transcriptional activation by PPAR-γ agonist inhibits Egr1 and Col1 expression in KFs
miRNAs are a class of naturally-occurring small non-coding RNAs that are 18-22 nucleotides in length and function to posttranscriptionally regulate expression of their target genes [21-24]. In this study, our analysis didn’t find a PPRE in the Egr1 gene, indicating that PPAR-γ agonist inhibition of Egr1 expression could be indirect. Therefore, we performed a miRNA microarray analysis to profile differentially expressed miRNAs in KFs after troglitazone treatment. We found five upregulated miRNAs (miR-3157, miR-1249, miR-92b, miR-4439 and miR-543) in troglitazone-treated KFs vs. control cells (Figure 4A, right panel) and we further confirmed their expression using qRT-PCR (Figure 4B).
Figure 4.

Functional analysis of a PPRE-binding site in the miR-543 promoter in KFs. A. miRNA microarray analysis of differentially expressed miRNAs in troglitazone-treated KFs. Shades of red represent increased gene expression, whereas shades of blue represent decreased expression. B. qRT-PCR analysis of mature miRNA levels in troglitazone-treated KFs. C. qRT-PCR analysis of the levels of Col1 mRNA in KFs in response to miRNA mimics. The levels of Col1 mRNA were decreased in KFs in response to treatment with miR-3157 (*P = 0.05), miR-92b (*P = 0.046), and miR-543 (*P = 0.023), whereas there was no change in Col1 mRNA upon miR-1249 and miR-4439 treatment. D. qRT-PCR analysis of Col1 mRNA in normal skin fibroblasts (NSFBs) upregulated in response to miRNA inhibitor treatment. Col1 mRNA was upregulated in NSFBs in response to treatment with miR-3157 (*P = 0.047), miR-92b (*P = 0.043), and miR-543 (*P = 0.046) inhibitors, whereas it did not change upon miR-1249 and miR-4439 inhibitor. E. qRT-PCR analysis indicating the miR-543 level is increased in KFs treated with troglitazone and suppressed by treatment with GW9662 (***P < 0.001). F. qRT-PCR analysis of endogenous miR-543 and Egr1 expression in KFs transfected with control or miR-543 mimics (***P < 0.001 and *P = 0.012). G. Schematic illustration of miR-543 promoter upstream constructs containing the putative PPRE-binding sites. The stippled box is the TK minimal promoter. Distal promoter fragments were ligated (dashed line) and aligned to their native position in the 2 kb construct for ease of comparison. H. ChIP assay using an anti-PPAR-γ antibody or IgG. The immunoprecipitated DNA fragments and input were detected using PCR with specific primers flanking the putative PPRE-binding sites (**P = 0.002). I. Luciferase reporter assay analysis of KFs transfected with equal copy numbers of the indicated promoter constructs (miR-543 2 kb wild-type promoter (miR-543 wt) versus a site 1 mutated miR-543 promoter (miR-543 mut)). Change in expression for each reporter is expressed in relation to its respective empty vector or vehicle control. The data represent the mean from three independent experiments in triplicate (**P = 0.002).
Furthermore, we associated expression of these miRNAs with collagen synthesis in KFs by transiently transfecting NSFBs with miRNA mimics and KFs with miRNA inhibitors for these five miRNAs. Our results showed that miR-543 significantly affected collagen secretion in KFs (Figure 4C, P = 0.006), whereas transient transfection of these five miRNA inhibitors in NSFBs revealed that miR-543 promotion had a significant effect on collagen secretion (Figure 4D, P = 0.046). Thus, miR-543 caught our attention for further investigations since i) miR-543 possesses the greatest effect on Col1 synthesis in our screening experiment; ii) miR-543 was a negative regulator of tumor metastasis, suggesting that its disturbance plays a role in collagenous protein barriers [25]. Nevertheless, the mechanisms underlying the function of miR-543 in keloids remains largely unknown.
We then assessed miR-543 levels in KFs using qRT-PCR and found that miR-543 levels were significantly higher in troglitazone-treated KFs than in the controls (Figure 4E, P < 0.001). Interestingly, overexpression of miR-543 significantly reduced level of Egr1 mRNA in KFs (Figure 4F, P < 0.001 and P = 0.012). To support this finding, bioinformatics analyses identified a region -1863 to -1854 bp from the transcription start site of the miR-543 promoter as a potential PPRE (Figure 4G). To ascertain the functionality of this PPRE, we cloned a 2 kb DNA fragment corresponding to the promoter region of miR-543 and performed a ChIP analysis (Figure 4H) and found troglitazone treatment induced PPAR-γ recruitment to the PPRE site upstream of the miR-543 transcription start site (Figure 4H, P = 0.002). We then performed luciferase-reporter assay and found that the transcriptional activity of the plasmid carrying the wild-type promoter increased upon troglitazone treatment in KFs, whereas there was no change in luciferase activity of the mutant plasmid, indicating that the predicted PPRE was responsible for PPAR-γ transactivation of miR-543 expression in vitro (Figure 4I, P = 0.002). These data demonstrated that PPAR-γ transcriptionally induces miR-543 expression in KFs.
PPAR-γ agonist and antagonist regulate Egr1 via miR-543
We next assessed whether miR-543 was able to inhibit Egr1 expression in KFs by transfection of a miR-543 inhibitor into KFs and found that miR-543 inhibition was able to restore expression of Egr1 mRNA in troglitazone-treated KFs (Figure 5A, P = 0.019 and P = 0.03). To further assess whether the effect of troglitazone on KFs was mediated by PPAR-γ, we treated KFs with a PPAR-γ-specific antagonist GW9662 and found that Egr1 mRNA level was increased (Figure 5B, P = 0.019) and that transfection of miR-543 mimics inhibited Egr1 mRNA levels in KFs (Figure 5B, P = 0.028). Furthermore, our bioinformatic analysis showed that the Egr1 3’-UTR contains a miR-543-binding site (Figure 5C) and excessive activation of Egr1 occurred in fibrotic diseases [18-20]. Our luciferase reporter assay showed a 65% decrease in the relative luciferase activity of the wild-type Egr1 3’-UTR vector after miR-543 mimics transfection in KFs (Figure 5D, P = 0.016), in contrast, the mutant construct completely abolished this effect (Figure 5D), suggesting that miR-543 can directly bind to the Egr1 3’-UTR and inhibit Egr1 expression in KFs, which is consistent with Egr1 expression data in Figure 5E. These findings indicated that miR-543 was able to negatively regulate Egr1 expression.
Figure 5.

Egr1 is a direct target of miR-543 in KFs. A. qRT-PCR analysis showing Egr1 expression is significantly decreased in troglitazone-treated KFs (**P = 0.03), and such inhibition is abolished by miR-543 inhibitor treatment (**P = 0.019). B. qRT-PCR analysis showing Egr1 expression is significantly increased in GW9662-treated KFs (*P = 0.019) and such upregulation is abolished by miR-543 mimics (*P = 0.028). GAPDH was used as the internal loading control. C. Prediction of miR-543 target sequences in the 3’-UTR of the Egr1 gene and its mutant containing seven mutated nucleotides. D. Luciferase reporter assay of the wild-type (WT) or mutant (MUT) reporter plasmid co-transfected with miR-543 mimics (miR-543) or the mimics control (control) into KFs. A Renilla luciferase vector was used as the internal control (*P = 0.016; ns = no significance). E. Western blot analysis of Egr1 protein levels in KFs treated with troglitazone, GW9662, miR-543 mimics or miR-543 inhibitors. Troglitazone significantly repressed Egr1 in KFs; however, Western blot results showed that miR-543 silencing did not suppress Col1 synthesis in KFs. Ectopic miR-543 expression decreased Col1 synthesis in GW9662 treated KFs. Protein expression were quantified with Image J software.
miR-543 mediates pro-collagen synthesis of PPAR-γ agonist in KFs
We determined the effect of miR-543 on Col1 synthesis through Egr1 downregulation by transfecting miR-543 inhibitors or mimics into KFs in the presence of troglitazone or GW9662. Our data showed that Col1 supernatant protein was significantly downregulated in KFs cultured with troglitazone (Figure 6A, P = 0.028), whereas transfection with a miR-543 inhibitor increased collagen synthesis compared to the control cells (Figure 6A, P = 0.024). However, level of Col1 protein in the supernatant were significantly increased after exposure of KFs to GW9662 exposure (Figure 6B, P = 0.029), whereas miR-543 overexpression suppressed Col1 protein levels in these cells (Figure 6B, P = 0.046), indicating that level of Col1 prote in the supernatant of in KFs were modulated by a feedback axis of the PPAR-γ agonist/miR-543/Egr1/Col1.
Figure 6.

ELISA analysis of the effect of miR-543 on inhibition of Col1 mRNA and protein levels in KFs. A. Troglitazone treatment of KFs decreased Col1 supernatant protein level (*P = 0.028), whereas treatment with a miR-543 inhibitor increased Col1 supernatant protein level (*P = 0.024). B. GW9662 treatment of KFs increased Col1 protein levels (*P = 0.029), whereas miR-543 mimics decreased Col1 levels (*P = 0.046).
Discussion
A potential therapy for liver fibrosis using PPAR-γ agonists was first proposed 16 years ago by Miyahara et al. [26]. Emerging evidence also indicates that PPAR-γ is involved in the control of fibrogenesis by regulating multiple signaling pathways [27]. Our previous study showed that a PPAR-γ agonist inhibited Col1 expression in hypertrophic scar fibroblasts [14]. Keloids occur in predisposed patients as a result of excessive deposition of collagens in an abnormal response to skin injury [1]. In this regard, PPAR-γ agonists could be therapeutic agents to suppress keloid formation. However, the understanding of the gene networks to regulate programs of gene expression is a major challenge in antifibrotic therapy [28]. In the current study, we demonstrated that a PPAR-γ agonist suppressed Col1 expression in KFs through transcriptional activation of miR-543 expression and in turn inhibition of Egr1 expression.
Egr1, a transcription factor also known as NGF1A and Krox-24, belongs to a family of zinc finger DNA-binding proteins [19]. Egr1 expression is generally low or undetectable in normal tissues [18], but is induced by injury and stress-associated stimuli, such as mechanical force, shear stress, growth factors, and cytokines [19]. Previous studies showed that ectopic induction of Egr1 expression was able to induce expression of collagen genes in normal fibroblasts in vitro, whereas fibroblasts lacking Egr1 exhibit a diminished response to transforming growth factor (TGF)-β stimulation, indicating that Egr1 is an important downstream mediator of TGF-β signaling in tissue fibrosis [29]. Therefore, Egr1 is a profibrogenic gene and is highly expressed in several fibrotic diseases [18-20]. Egr1 is regulated by multiple fibrogenic signals, and plays an important role in fibrosis [30]. Increased Egr1 expression was detected in synovial fibroblasts in patients with rheumatoid arthritis [31], and in the fibrous caps of atherosclerotic vascular lesions [32]. The expression of Egr1 was associated with tissue fibrosis in peritoneal adhesions [33] and in pulmonary artery fibroblasts in hypoxic animal models [34]. Previous studies report that TGF-β-induced Egr1 can bind to the COL1A2 promoter to promote expression of collagen genes [35]. Our current study further confirmed that Egr1 expression was able to induce Col1 expression and that a PPAR-γ agonist reduced Egr1 expression in KFs. However, it is necessary to point out that the research approach in our study of investigating collagen synthesis inhibition using a PPAR-γ agonist was not in agreement with previous work showing that the TGF-β1-initiated pathway affected Egr1 expression and stimulated collagen synthesis [35]. TGF-β1 is able to induce rapid and transient accumulation of Egr1 and interact with a consensus Egr1-binding element site of the human COL1A2 promoter in vitro and in vivo [35], supporting the notion that Egr1 positively regulates collagen synthesis. However, the remaining concern or question is whether other factors can also regulate Egr1 expression. In addition, TGF-β1 can regulate several gene pathways and modulate expression of various miRNAs, such as miR-543 and others [36-38]. Our study profiled differentially expressed miRNAs to identify aberrant miR-543 expression in KFs after PPAR-γ agonist treatment. Therefore, we combined miRNA levels, gene expression, PPAR-γ agonist and collagen synthesis in the picture.
Our current study linked miR-543 in the regulation of Col1 expression. Indeed, a previous study showed that aberrant expression of miRNAs was found during development and progression of fibrogenic diseases [39] and multiple lines of evidence indicated that miRNA-associated gene regulation networks play a critical role in antifibrotic progression [40]. Recent studies demonstrated that PPAR-γ could regulate different miRNAs, strongly suggesting that expression and function of profibrotic genes are regulated by antifibrotic miRNAs in response to PPAR-γ agonist treatment [41]. A previous functional study demonstrated that miRNA-mediated transcriptional or post-transcriptional regulation of multiple targeting genes could suppress development and progression of fibrosis [42]. The miR-543 mentioned in the article has also been reported to act as a tumor suppressor in endometrial and ovarian cancers [43,44], and as an oncogene in hepatocellular carcinoma and gastric cancers [45,46]. In addition, miR-543 was reported to regulate the aging process of human mesenchymal stem cells and insulin resistance of hepatocyte cells [47,48]. Our current data analyzed global miRNA expression patterns in PPAR-γ-treated KFs and identified miR-543 as a major player. Thus, further elucidation of the role of miR-543 in PPAR-γ agonist-reduced Col1 expression and fibrosis in KFs could provide mechanistic information, including its potential antifibrotic role in KFs.
The development and progression of keloids is a complicated profibrotic process [2,49] and the pro-collagen synthesis signaling pathways play a central role in its pathogenesis [50]. Our current study provides a proof-of-principle and one-sided story for the regulation of keloid development. However, it remains to be determined why only a small percentage of individuals exhibit abnormal responses to skin injury and form keloids, whereas most other individuals will undergo a normal skin repair and wound healing. Our current data may provide insightful information for developing a novel therapeutic strategy for keloid formation and help to understand the molecular mechanisms of keloid formation and progression. Further study is needed to validate our current data in vivo.
Acknowledgements
This study was supported in part by grants from National Natural Science Foundation of China (Nos. 81372069, 81171811, 81272098, 81501684 and 81201470), Shaanxi Natural Science Foundation (No. 2014JM4180).
Disclosure of conflict of interest
None.
References
- 1.Liu Y, Wang X, Yang D, Xiao Z, Chen X. MicroRNA-21 affects proliferation and apoptosis by regulating expression of PTEN in human keloid fibroblasts. Plast Reconstr Surg. 2014;134:561e–573e. doi: 10.1097/PRS.0000000000000577. [DOI] [PubMed] [Google Scholar]
- 2.Andrews JP, Marttala J, Macarak E, Rosenbloom J, Uitto J. Keloids: The paradigm of skin fibrosis - Pathomechanisms and treatment. Matrix Biol. 2016;51:37–46. doi: 10.1016/j.matbio.2016.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meaume S, Le Pillouer-Prost A, Richert B, Roseeuw D, Vadoud J. Management of scars: updated practical guidelines and use of silicones. Eur J Dermatol. 2014;24:435–443. doi: 10.1684/ejd.2014.2356. [DOI] [PubMed] [Google Scholar]
- 4.Shin JU, Kim SH, Kim H, Noh JY, Jin S, Park CO, Lee WJ, Lee DW, Lee JH, Lee KH. TSLP is a potential initiator of collagen synthesis and an activator of cxcr4/sdf-1 axis in keloid pathogenesis. J Invest Dermatol. 2016;136:507–515. doi: 10.1016/j.jid.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 5.Dong X, Zhang C, Ma S, Wen H. Mast cell chymase in keloid induces profibrotic response via transforming growth factor-β1/Smad activation in keloid fibroblasts. Int J Clin Exp Pathol. 2014;7:3596–3607. [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang GY, Cheng T, Zheng MH, Yi CG, Pan H, Li ZJ, Chen XL, Yu Q, Jiang LF, Zhou FY, Li XY, Yang JQ, Chu TG, Gao WY. Peroxisome proliferator-activated receptor-gamma (PPAR-gamma) agonist inhibits transforming growth factor-beta1 and matrix production in human dermal fibroblasts. J Plast Reconstr Aesthet Surg. 2010;63:1209–1216. doi: 10.1016/j.bjps.2009.06.032. [DOI] [PubMed] [Google Scholar]
- 7.Zambrano S, Blanca AJ, Ruiz-Armenta MV, Miguel-Carrasco JL, Arévalo M, Mate A, Vázquez CM. L-carnitine attenuates the development of kidney fibrosis in hypertensive rats by upregulating PPAR-γ. Am J Hypertens. 2014;27:460–470. doi: 10.1093/ajh/hpt268. [DOI] [PubMed] [Google Scholar]
- 8.Deng YL, Xiong XZ, Cheng NS. Organ fibrosis inhibited by blocking transforming growth factor-β signaling via peroxisome proliferator-activatedreceptor γ agonists. Hepatobiliary Pancreat Dis Int. 2012;11:467–478. doi: 10.1016/s1499-3872(12)60210-0. [DOI] [PubMed] [Google Scholar]
- 9.O’Flaherty JT, Rogers LC, Paumi CM, Hantgan RR, Thomas LR, Clay CE, High K, Chen YQ, Willingham MC, Smitherman PK, Kute TE, Rao A, Cramer SD, Morrow CS. 5-Oxo-ETE analogs and the proliferation of cancer cells. Biochim Biophys Acta. 2005;1736:228–236. doi: 10.1016/j.bbalip.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 10.Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 11.Tyagi S, Gupta P, Saini AS, Kaushal C, Sharma S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J Adv Pharm Technol Res. 2011;2:236–240. doi: 10.4103/2231-4040.90879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marlow LA, Reynolds LA, Cleland AS, Cooper SJ, Gumz ML, Kurakata S, Fujiwara K, Zhang Y, Sebo T, Grant C, McIver B, Wadsworth JT, Radisky DC, Smallridge RC, Copland JA. Reactivation of suppressed RhoB is a critical step for the inhibition of anaplastic thyroid cancer growth. Cancer Res. 2009;69:1536–1544. doi: 10.1158/0008-5472.CAN-08-3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Copland JA, Marlow LA, Kurakata S, Fujiwara K, Wong AK, Kreinest PA, Williams SF, Haugen BR, Klopper JP, Smallridge RC. Novel high-affinity PPAR-γ agonist alone and in combination with paclitaxel inhibits human anaplastic thyroid carcinoma tumor growth via p21waf1/cip1. Oncogene. 2006;25:2304–2317. doi: 10.1038/sj.onc.1209267. [DOI] [PubMed] [Google Scholar]
- 14.Zhu HY, Li C, Zheng Z, Zhou Q, Guan H, Su LL, Han JT, Zhu XX, Wang SY, Li J, Hu DH. Peroxisome proliferator-activated receptor-γ (PPAR-γ) agonist inhibits collagen synthesis in human hypertrophic scar fibroblasts by targeting Smad3 via miR-145. Biochem Biophys Res Commun. 2015;459:49–53. doi: 10.1016/j.bbrc.2015.02.061. [DOI] [PubMed] [Google Scholar]
- 15.Dantas AT, Pereira MC, de Melo Rego MJ, da Rocha LF Jr, Pitta Ida R, Marques CD, Duarte AL, Pitta MG. The Role of PPAR Gamma in Systemic Sclerosis. PPAR Res. 2015;2015:124624. doi: 10.1155/2015/124624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee WJ, Ahn HM, Roh H, Na Y, Choi IK, Lee JH, Kim YO, Lew DH, Yun CO. Decorin-expressing adenovirus decreases collagen synthesis and upregulates MMP expression inkeloid fibroblasts and keloid spheroids. Exp Dermatol. 2015;24:591–597. doi: 10.1111/exd.12719. [DOI] [PubMed] [Google Scholar]
- 17.Zhu HY, Bai WD, Li C, Zheng Z, Guan H, Liu JQ, Yang XK, Han SC, Gao JX, Wang HT, Hu DH. Knockdown of lncRNA-ATB suppresses autocrine secretion of TGF-β2 by targeting ZNF217 via miR-200c in keloid fibroblasts. Sci Rep. 2016;6:24728. doi: 10.1038/srep24728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomes PG, Donohue TM Jr. Role of Early Growth Response-1 in the Development of Alcohol-Induced Steatosis. Curr Mol Pharmacol. 2015 doi: 10.2174/1874467208666150817112529. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Xu N, Xu J, Kong B, Copple B, Guo GL, Wang L. E2F1 is a novel fibrogenic gene that regulates cholestatic liver fibrosis through the Egr-1/SHP/EID1 network. Hepatology. 2014;60:919–930. doi: 10.1002/hep.27121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Derdak Z, Villegas KA, Wands JR. Early growth response-1 transcription factor promotes hepatic fibrosis and steatosis in long-term ethanol-fed Long-Evans rats. Liver Int. 2012;32:761–770. doi: 10.1111/j.1478-3231.2012.02752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartel DP. MicroRNAs target recognition regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Debernardi S, Massat NJ, Radon TP, Sangaralingam A, Banissi A, Ennis DP, Dowe T, Chelala C, Pereira SP, Kocher HM, Young BD, Bond-Smith G, Hutchins R, Crnogorac-Jurcevic T. Noninvasive urinary miRNA biomarkers for early detection of pancreatic adenocarcinoma. Am J Cancer Res. 2015;5:3455–3466. [PMC free article] [PubMed] [Google Scholar]
- 23.Silva M, Hernandez ME, Rojas F, Li L, Subramanian S, Wilson MJ. MicroRNA miR-182 cluster mediated modulation of RECK without changes in cell surface membrane type-1 matrix metalloproteinase (MT1-MMP) Am J Cancer Res. 2015;5:2918–2928. [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu HY, Li C, Bai WD, Su LL, Liu JQ, Li Y, Shi JH, Cai WX, Bai XZ, Jia YH, Zhao B, Wu X, Li J, Hu DH. MicroRNA-21 regulates hTERT via PTEN in hypertrophic scar fibroblasts. PLoS One. 2014;9:e97114. doi: 10.1371/journal.pone.0097114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song N, Liu H, Ma X, Zhang S. Placental growth factor promotes metastases of ovarian cancer through MiR-543-regulated MMP7. Cell Physiol Biochem. 2015;37:1104–1112. doi: 10.1159/000430235. [DOI] [PubMed] [Google Scholar]
- 26.Miyahara T, Schrum L, Rippe R, Xiong S, Yee HF Jr, Motomura K, Anania FA, Willson TM, Tsukamoto H. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J Biol Chem. 2000;275:35715–35722. doi: 10.1074/jbc.M006577200. [DOI] [PubMed] [Google Scholar]
- 27.Chen M, Li H, Wang G, Shen X, Zhao S, Su W. Atorvastatin prevents advanced glycation end products (AGEs)-induced cardiac fibrosis via activating peroxisome proliferator-activated receptor gamma (PPAR-γ) Metabolism. 2016;65:441–453. doi: 10.1016/j.metabol.2015.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Liu XY, He YJ, Yang QH, Huang W, Liu ZH, Ye GR, Tang SH, Shu JC. Induction of autophagy and apoptosis by miR-148a through the sonic hedgehog signaling pathway in hepatic stellate cells. Am J Cancer Res. 2015;5:2569–2589. [PMC free article] [PubMed] [Google Scholar]
- 29.Bhattacharyya S, Fang F, Tourtellotte W, Varga J. Egr-1: new conductor for the tissue repair orchestra directs harmony (regeneration) or cacophony (fibrosis) J Pathol. 2013;229:286–297. doi: 10.1002/path.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhattacharyya S, Wu M, Fang F, Tourtellotte W, Feghali-Bostwick C, Varga J. Early growth response transcription factors: key mediators of fibrosis and novel targets for anti-fibrotic therapy. Matrix Biol. 2011;30:235–242. doi: 10.1016/j.matbio.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aicher WK, Heer AH, Trabandt A, Bridges SL Jr, Schroeder HW Jr, Stransky G, Gay RE, Eibel H, Peter HH, Siebenlist U. Overexpression of zinc-finger transcription factor Z-225/Egr-1 in synoviocytes from rheumatoid arthritis patients. J Immunol. 1994;152:5940–5948. [PubMed] [Google Scholar]
- 32.McCaffrey TA, Fu C, Du B, Eksinar S, Kent KC, Bush H Jr, Kreiger K, Rosengart T, Cybulsky MI, Silverman ES, Collins T. High-level expression of Egr-1 and Egr-1-inducible genes in mouse and human atherosclerosis. J Clin Invest. 2000;105:653–662. doi: 10.1172/JCI8592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roy S, Clark CJ, Mohebali K, Bhatt U, Wallace WA, Nahman NS, Ellison EC, Melvin WS, Sen CK. Reactive oxygen species and EGR-1 gene expression in surgical postoperative peritoneal adhesions. World J Surg. 2004;28:316–320. doi: 10.1007/s00268-003-7403-z. [DOI] [PubMed] [Google Scholar]
- 34.Banks MF, Gerasimovskaya EV, Tucker DA, Frid MG, Carpenter TC, Stenmark KR. Egr-1 antisense oligonucleotides inhibit hypoxia-induced proliferation of pulmonary artery adventitial fibroblasts. J Appl Physiol (1985) 2005;98:732–738. doi: 10.1152/japplphysiol.00821.2004. [DOI] [PubMed] [Google Scholar]
- 35.Chen SJ, Ning H, Ishida W, Sodin-Semrl S, Takagawa S, Mori Y, Varga J. The early-immediate gene EGR-1 is induced by transforming growth factor-beta and mediates stimulation of collagen gene expression. J Biol Chem. 2006;281:21183–21197. doi: 10.1074/jbc.M603270200. [DOI] [PubMed] [Google Scholar]
- 36.Kumar P, Naumann U, Aigner L, Wischhusen J, Beier CP, Beier D. Impaired TGF-β induced growth inhibition contributes to the increased proliferation rate of neural stem cells harboring mutant p53. Am J Cancer Res. 2015;5:3436–3445. [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X, Lu H, Li T, Yu L, Liu G, Peng X, Zhao J. Krüppel-like factor 8 promotes tumorigenic mammary stem cell induction by targeting miR-146a. Am J Cancer Res. 2013;3:356–373. [PMC free article] [PubMed] [Google Scholar]
- 38.Bai W, Chen Y, Yang J, Niu P, Tian L, Gao A. Aberrant miRNA profiles associated with chronic benzene poisoning. Exp Mol Pathol. 2014;96:426–430. doi: 10.1016/j.yexmp.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 39.O’Reilly S. MicroRNAs in fibrosis: opportunities and challenges. Arthritis Res Ther. 2016;18:11. doi: 10.1186/s13075-016-0929-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krishna CV, Singh J, Thangavel C, Rattan S. Role of MicroRNAs in Gastrointestinal Smooth Muscle Fibrosis and Dysfunction: Novel Molecular Perspectives on the Pathophysiology and Therapeutic Targeting. Am J Physiol Gastrointest Liver Physiol. 2016;310:G449–59. doi: 10.1152/ajpgi.00445.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dharap A, Pokrzywa C, Murali S, Kaimal B, Vemuganti R. Mutual induction of transcription factor PPARγ and microRNAs miR-145 and miR-329. J Neurochem. 2015;135:139–146. doi: 10.1111/jnc.13220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun JY, Huang Y, Li JP, Zhang X, Wang L, Meng YL, Yan B, Bian YQ, Zhao J, Wang WZ, Yang AG, Zhang R. MicroRNA-320a suppresses human colon cancer cell proliferation by directly targeting β-catenin. Biochem Biophys Res Commun. 2012;420:787–792. doi: 10.1016/j.bbrc.2012.03.075. [DOI] [PubMed] [Google Scholar]
- 43.Bing L, Hong C, Li-Xin S, Wei G. MicroRNA-543 suppresses endometrial cancer oncogenicity via targeting FAK and TWIST1 expression. Arch Gynecol Obstet. 2014;290:533–541. doi: 10.1007/s00404-014-3219-3. [DOI] [PubMed] [Google Scholar]
- 44.Song N, Liu H, Ma X, Zhang S. Placental growth factor promotes metastases of ovarian cancer through MiR-543-regulated MMP7. Cell Physiol Biochem. 2015;37:1104–1112. doi: 10.1159/000430235. [DOI] [PubMed] [Google Scholar]
- 45.Yu L, Zhou L, Cheng Y, Sun L, Fan J, Liang J, Guo M, Liu N, Zhu L. MicroRNA-543 acts as an oncogene by targeting PAQR3 in hepatocellular carcinoma. Am J Cancer Res. 2014;4:897–906. [PMC free article] [PubMed] [Google Scholar]
- 46.Li J, Dong G, Wang B, Gao W, Yang Q. miR-543 promotes gastric cancer cell proliferation by targeting SIRT1. Biochem Biophys Res Commun. 2016;469:15–21. doi: 10.1016/j.bbrc.2015.11.062. [DOI] [PubMed] [Google Scholar]
- 47.Lee S, Yu KR, Ryu YS, Oh YS, Hong IS, Kim HS, Lee JY, Kim S, Seo KW, Kang KS. miR-543 and miR-590-3p regulate human mesenchymal stem cell aging via direct targeting of AIMP3/p18. Age (Dordr) 2014;36:9724. doi: 10.1007/s11357-014-9724-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu X, Chi L, Zhang W, Bai T, Zhao W, Feng Z, Tian H. Down-regulation of the miR-543 alleviates insulin resistance through targeting the SIRT1. Biochem Biophys Res Commun. 2015;468:781–787. doi: 10.1016/j.bbrc.2015.11.032. [DOI] [PubMed] [Google Scholar]
- 49.Unahabhokha T, Sucontphunt A, Nimmannit U, Chanvorachote P, Yongsanguanchai N, Pongrakhananon V. Molecular signalings in keloid disease and current therapeutic approaches from natural based compounds. Pharm Biol. 2015;53:457–463. doi: 10.3109/13880209.2014.918157. [DOI] [PubMed] [Google Scholar]
- 50.Zhou P, Shi L, Li Q, Lu D. Overexpression of RACK1 inhibits collagen synthesis in keloid fibroblasts via inhibition of transforming growth factor-β1/Smad signaling pathway. Int J Clin Exp Med. 2015;8:15262–15268. [PMC free article] [PubMed] [Google Scholar]