

Abstract
Small-molecule antagonists disable discrete biochemical properties of protein targets. For multi-domain protein targets, the pharmacologic consequence of drug action is limited by selective disruption of one domain-specific activity. More broadly, target inhibition is kinetically limited by the durability and degree of target engagement. These features of traditional drug molecules are challenging to the development of inhibitors targeting transcription factors and chromatin-associated epigenetic proteins, which function as multi-domain biomolecular scaffolds and generally feature rapid association and dissociation kinetics. We therefore devised a chemical strategy to prompt ligand-dependent target protein degradation, via chemical conjugation with derivatized phthalimides that hijack the function of the Cereblon E3 ubiquitin ligase complex. Using this approach, we converted an acetyl-lysine competitive antagonist that displaces BET bromodomains from chromatin (JQ1) to a phthalimide-conjugated ligand that prompts immediate Cereblon-dependent BET protein degradation (dBET1). Expression proteomics confirms high specificity for BET family members BRD2, BRD3 and BRD4 among 7429 proteins detected. Degradation of BET bromodomains is associated with a more rapid and robust apoptotic response compared to bromodomain inhibition in primary human leukemic blasts, and dBET1 exhibits in vivo efficacy in a human leukemia xenograft. The reach of this approach is illustrated by a second series of probes that degrade the cytosolic signaling protein, FKBP12. Together, these findings identify a facile and general new strategy to control target protein stability, with implications for approaching previously intractable protein targets.
Phthalimide drug molecules emerged in the 1950s with thalidomide, developed initially as a sedative but infamously withdrawn from human use owing to catastrophic teratogenicity (1). Subsequently, the phthalimides have been successfully repurposed for erythema nodosum leprosum, multiple myeloma (MM) and myelodyspasia. The remarkable efficacy of the phthalimides thalidomide, lenalidomide and pomalidomide in MM (Celgene Corporation; Fig 1A), has prompted broad investigation into the mechanism-of-action of phthalimide imunomodulatory drugs (IMiDs). In 2010, Hiroshi Handa and colleagues utilized ligand-affinity chromatography to identify the cellular target of thalidomide as Cereblon (CRBN), a component of a cullin-RING ubiquitin ligase (CRL) complex (2). Recently with William Kaelin, our group and others reported that phthalimides bind CRBN without apparent target protein inhibition, rather prompting CRBN-dependent proteasomal degradation of ubiquitylated, neo-substrate transcription factors IKZF1 and IKZF3 (3, 4). Crystallographic and biochemical studies now establish that lenalidomide and pomalidomide bind CRBN to form a cryptic interface that promotes recruitment of IKZF1 and IKZF3 (5).
Figure 1. Design and characterization of dBET1.
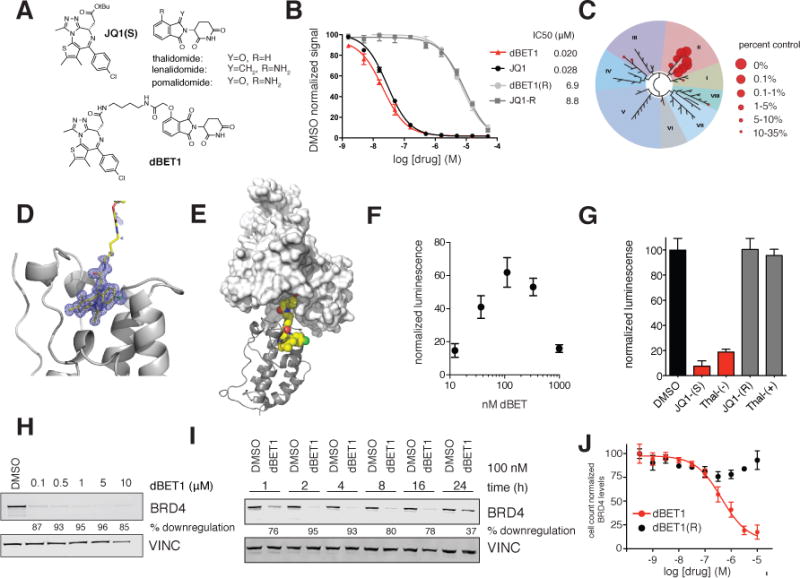
(A) Chemical structure of JQ1(S), the phthalimides and dBET1 (B) DMSO normalized BRD4 binding signal measured by AlphaScreen for the indicated compounds. Values represent mean ± stdev of triplicate analysis (C) Selectivity of dBET1 for binding to BETs over other human bromodomains, as determined by single point screening (BromoScan) (D) Crystal structure of dBET1 bound to bromodomain 1 of BRD4 (E) Docking of (D) into the published DDB1-CRBN structure (F) dimerization assay measuring dBET1 induced proximity between recombinant BRD4 bromodomain (1) and recombinant CRBN-DDB1. Values represent mean ± stdev of quadruplicate analysis and are normalized to DMSO. (G) competition of dBET1 induced proximity at 111 nM as shown in (F) in the presence of DMSO (vehicle), JQ1(S), thal-(−), JQ1(R) and thal-(+) all at a final concentration of 1 μM. Values represent mean ± stdev of quadruplicate analysis and are normalized to DMSO. (H) Immunoblot analysis for BRD4 and Vinculin after 18 h treatment of MV4;11 cells with the indicated concentrations of dBET1 (I) Immunoblot analysis for BRD4 and Vinculin after treatment of MV4;11 cells with 100 nM dBET1 at the indicated timepoints (J) Cell count normalized BRD4 levels as determined by high-content assay in SUM149 cells treated with the indicated concentrations of dBET1 and dBET1(R) for 18 h. Values represent mean ± stdev of triplicate analysis, are normalized to DMSO treated cells and baseline corrected based on immunoblots in Supplementary Figure 2C
Ligand-induced target protein destabilization has proven a desirable and efficacious therapeutic strategy, in particular for cancer as with PML degradation by arsenic trioxide in acute promyelocytic leukemia (6) and estrogen receptor degradation by fulvestrant (7). As illustrated by these compounds and others, target destabilization has theoretical advantages over traditional small-molecule antagonists including prolonged efficacy (need for compensatory protein resynthesis), increased potency (potential for repeated, catalytic ligand action), and broader spectrum activity (due to whole protein degradation). Historically, target-degrading compounds have emerged from serendipity or target-specific campaigns in medicinal chemistry.
Chemical biologists have devised elegant solutions to modulate the stability and degradation of proteins using engineered cellular systems involving the use of chemical dimerizers (8), destabilized FKBP12 chimera (9, 10) and hydrophobic tagging (11), but these approaches have been limited to targeting non-endogenous fusion proteins. Others have attempted to induce degradation of endogenous proteins through the recruitment of E3 ligases using peptidic binding ligands paired with cell-permeating peptides (12–14) and non-specific aminopetidase inhibitors (15). Regrettably, the peptidic nature of the best validated of these reagents results in low cellular potency of target protein degradation (EC50 25 – 150 μM), limiting broader utility. To date, a facile chemical technology allowing mechanism-based and target-specific protein degradation has proven elusive, and no technology has been shown to induce the degradation of a targeted protein in vivo.
RING-domain E3 ubiquitin-protein ligases lack enzymatic activity, rather functioning as adaptors to E2 ubiquitin-conjugating enzymes. Inspired by Handa’s ligand-affinity chromatography result, we hypothesized that rational design of bifunctional phthalimide-conjugated ligands could serve as further chemical adaptors to confer CRBN- and ligand-dependent target protein degradation. To test this hypothesis, we selected BRD4 as an exemplary, non-enzymatic protein target. BRD4 is a transcriptional co-activator involved in dynamic transcriptional activation and elongation. BRD4 binds to enhancer and promoter regions adjacent to target genes, via recognition of side-chain acetylated lysine on histone proteins and transcription factors (TFs) by twin acetyl-lysine binding modules or bromodomains (16). Recently, we developed a first direct-acting inhibitor of BET bromodomains (JQ1) (17), that displaces BRD4 from chromatin leading to impaired signal transduction from master regulatory TFs to RNA Polymerase II(18–20). Molecular recognition of the BRD4 bromodomains by JQ1 is stereo-specific, and only the (+)-JQ1 enantiomer (JQ1S; from here forward JQ1) is active; the (−)-JQ1 enantiomer (JQ1R) is inactive (Fig 1A). Silencing of BRD4 expression by RNA interference in murine and human models of MM and acute myeloid leukemia (AML) elicited rapid transcriptional downregulation of the MYC oncogene and a potent anti-proliferative response (19, 21). These and other studies in cancer, inflammation(22) and heart disease(23, 24), establish a desirable mechanistic and translational purpose to target BRD4 for selective degradation.
Identifying that the carboxyl group on JQ1(25) and the aryl ring of thalidomide can tolerate chemical substitution, we designed the bifunctional dBET1 (Fig 1A). As expected, this substitution did not impair potent binding to the first bromodomain of BRD4 (BRD4(1); IC50 = 20 nM), while the epimeric dBET1(R) was inactive (Fig 1B). Selectivity profiling confirmed potent and BET-specific target engagement among 32 bromodomains studied by phage display and displacement (BromoScan; Fig. 1C). A high-resolution crystal structure (1.0 Å) of dBET1 bound to BRD4(1) confirmed the mode of molecular recognition as comparable to JQ1 (Fig 1D, Fig S1). Ordered density for the dBET1 ligand was found only to the first two carbon atoms of the butane spacer, suggesting conformational flexibility of the conjugated phthalimide. Using the dBET1-BRD4(1) crystal structure and the recently reported structure of CRBN bound to thalidomide(5), we modeled the feasibility of ternary complex formation in silico. An extended conformation of dBET1 was capable of bridging ordered BRD4(1) and CRBN without destructive steric interactions (Fig 1E). Further, the known modular nature of CRL complexes biologically suggests that chemical recruitment of BRD4 may lead to CRBN-dependent degradation (26, 27). To experimentally assess the chemical adaptor function of dBET1, we developed a homogeneous proximity assay for recombinant, human CRBN-DDB1 and BRD4(1) using AlphaScreen technology. As shown in Figure 1F, luminescence arising from proximity of acceptor (CRBN-bound) and donor (BRD4-bound) beads increases in the presence of low (10 – 100 nM) concentrations of dBET1. At higher concentrations of dBET1 (e.g. 1 μM), luminescence diminishes consistent with independent occupancy of CRBN and BRD4 binding sites by excess dBET1 (the hook effect). Indeed, inhibition of chemical adaptor function may be accomplished with competitive co-incubation with free JQ1 or thalidomide, each in a stereo-specific manner (Fig. 1G).
To functionally assess the effect of dBET1 on BRD4 stability in cells, we treated a human AML cell line (MV4;11) for 18 hours with increasing concentrations of dBET1 and assayed endogenous BRD4 levels by immunoblot. Pronounced loss of BRD4 (> 85%) was observed with concentrations of dBET1 as low as 100 nM (Fig 1H). The epimeric control dBET1(R) compound that lacks BRD4 binding (> 300 fold weaker binding in homogenous assays) was inactive, demonstrating that BRD4 degradation requires target protein engagement (Fig S2A, B). The kinetics of BRD4 degradation were next determined in a time course experiment using 100 nM dBET1 in MV4;11 cells. Marked depletion of BRD4 was observed at 1 hour and complete degradation was observed at 2 hours of treatment (Fig 1I). A partial recovery in BRD4 abundance at 24 hours establishes the possibility of compound metabolic instability, a recognized liability of phthalimide drug molecules. To quantify dose-responsive effects on BRD4 protein stability, we developed a cell-count normalized, immunofluorescence-based high-content assay in an adherent human cancer cell line (SUM149 breast cancer cells; Fig 1J). Potent depletion of total BRD4 was observed for dBET1 (EC50 = 430 nM) without apparent activity for dBET1(R). These observations were confirmed by immunoblot in SUM149, which have been used for baseline normalization of the assay (Fig S2C, D). Additional cultured adherent and non-adherent human cancer cell lines showed comparable response (SUM159, MOLM13; Fig S3).
To critically assess the mechanism of dBET1-induced BRD4 degradation, we separately studied requirements on proteasome function, BRD4 binding and CRBN binding, using chemical genetic and gene editing approaches. First, we confirmed that treatment with either JQ1 or thalidomide alone was insufficient to induce BRD4 degradation in MV4;11 cells (Fig S4A). BRD4 stability was rescued by pre-treatment with the irreversible proteasome inhibitor carfilzomib (0.4 μM), establishing a requirement for proteasome function in dBET1-mediated BRD4 degradation (Fig 2A). Pre-treatment with excess JQ1 (10 μM) or thalidomide (10 μM) abolished dBET1-induced BRD4 degradation, consistent with a requirement for both BRD4 and CRBN engagement (Fig 2A). Cullin-based ubiquitin ligases require neddylation of the cullin subunit for processive E3 ligase activity and target polyubiquitination(4, 28). Pre-treatment with the selective NAE1 inhibitor MLN4924(29) rescued BRD4 stability from dBET1 exposure, further supporting dependence on active RING E3 ligase activity (Fig S4B). Finally, to definitively confirm the cellular requirement for CRBN, we employed a recently published human MM cell line (MM1.S-CRBN−/−) that features an engineered knockout of CRBN by CRISPR/Cas9 technology(4). Whereas treatment of MM1.SWT cells with dBET1 promoted rapid degradation of BRD4, exposure of MM1.S-CRBN−/− cells to dBET1 was ineffectual (Fig 2B). These data provide mechanistic evidence for CRBN-dependent proteasomal degradation of BRD4 by dBET1.
Figure 2. Chemical and genetic rescue of dBET1 and dFKBP-1 mediated degradation.
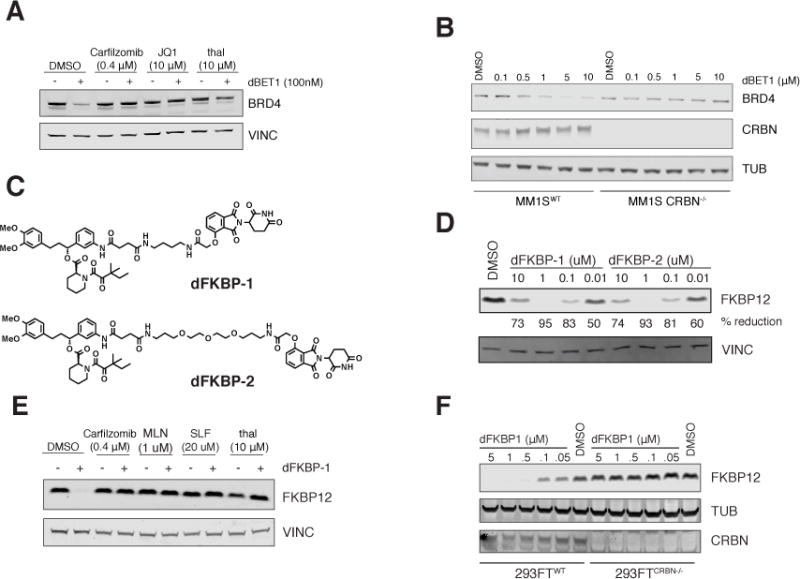
(A) Immunoblot analysis for BRD4 and Vinculin after a 4 h pre-treatment with either DMSO, Carfilzomib (400 nM), JQ1 (10 μM) or thalidomide (10 μM) followed by a 2 h dBET1 treatment at a concentration of 100 nM in MV4;11 cells. (B) Immunoblot analysis for BRD4, CRBN and tubulin after treatment of MM1SWT or MM1SCRBN−/− with dBET1 for 18 h at the indicated concentrations. (C) structures of dFKBP-1 and dFKBP-2 (D) Immunoblot analysis for FKBP12 and Vinculin after 18 h treatment with the indicated compounds (E) Immunoblot analysis for FKBP12 and Vinculin after a 4 h pre-treatment with either DMSO, Carfilzomib (400 nM), MLN4924 (1 μM), SLF (20 μM) or thalidomide (10 μM) followed by a 4 h dFKBP-1 treatment at a concentration of 1 μM in MV4;11 cells. (F) Immunoblot analysis for FKBP12, CRBN and tubulin after treatment of 293FTWT or 293FTCRBN−/− with dFKBP12 at the indicated concentrations for 18 h.
To assess the feasibility of extending this strategy to other protein targets, we designed and synthesized phthalimide-conjugated ligands to the cytosolic signaling protein, FKBP12 (FKBP1A). FKBP12 has been extensively studied in the chemical biology literature, including studies of engineered target degradation and a growing literature on chemical dimerizers. Research on FKBP12 has identified a role in cardiac development, ryanodine receptor function, and oncogenic signaling, among other biological phenotypes, rendering chemical probes of FKBP12 stability desirable to a diverse community. At a known permissive site on the FKBP12-directed ligand SLF, we positioned two chemical spacers to create the conjugated phthalimides dFKBP-1 and dFKBP-2 (Fig 2C). Both potently decreased FKBP12 abundance in MV4;11 cells (Fig 2D), leading to over 80% reduction of FKBP12 at 0.1 μM and 50% reduction at 0.01 μM. Prior reports of conjugated PROTAC ligands first demonstrate activity at 25 μM, revealing a 1000-fold improvement in potency with an all small-molecule strategy. As with dBET1, destabilization of FKBP12 by dFKBP-1 was rescued by pre-treatment with carfilzomib, MLN4924, free SLF or free thalidomide (Fig 2E). CRBN-dependent degradation was established using previously published isogenic 293FT cell lines which are wild-type (293FT-WT) or deficient (293FT-CRBN−/−) for CRBN (4). Treatment of 293FT-WT cells with dFKBP-1 induced potent, dose-dependent degradation of FKBP12, whereas 293FT-CRBN−/− were unaffected (Fig 2F).
An unbiased, proteome-wide approach was selected to assess the cellular consequences of dBET1 treatment on protein stability. Recent advances in quantitative proteomics have availed the technical feasibility of differential quantification of thousands of cellular proteins in a highly parallel format(30). Here, we compared the acute impact of dBET1 treatment (250 nM) to JQ1 (250 nM) and vehicle (DMSO 0.0025%) controls on protein stability in MV4;11 cells. A two hour incubation was selected to capture primary, immediate consequences of small-molecule action and to mitigate expected, confounding effects on suppressed transactivation of BRD4 target genes. Three biological sample replicates were prepared for each treatment condition using isobaric tagging that allowed the detection of 7,429 proteins using a lower cut-off of at least two identified spectra per protein. Following BET bromodomain inhibition with JQ1, few proteomic changes are observed (Fig 3A). Only MYC was significantly depleted by more than 2-fold after 2 hours of JQ1 treatment, confirming the reported rapid and selective effect of BET bromodomain inhibition on MYC expression in AML (Fig 3A,C)(21). JQ1 treatment also downregulated the oncoprotein PIM1 (Fig 3A,C).
Figure 3. Highly selective BET bromodomain degradation measured by expression proteomics.
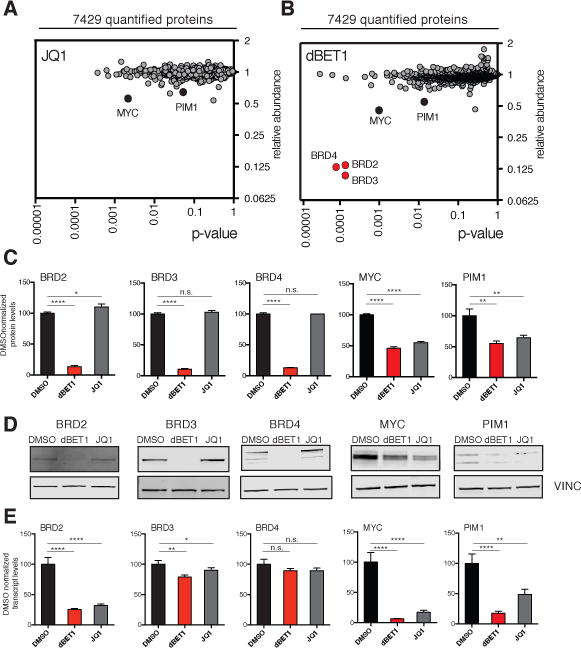
MV4;11 cells were treated for 2 hours with DMSO, 250 nM dBET1 or 250 nM JQ1. (A) depicts fold change of abundance of 7429 proteins comparing JQ1 to DMSO treatment as well as their respective p-value (t-test). Data results from triplicate analysis. (B) as for (A) but comparing 250 nM dBET1 to DMSO treatment. Data results from triplicate analysis. (C) bargraph depiction of selected proteins from (A) and (B) normalized to DMSO. Values represent mean ± stdev of triplicates. (D) Immunoblot analysis of BRD2, BRD3, BRD4, MYC, PIM1 and VINC after 2 h treatment of MV4;11 cells with either DMSO, 250 nM dBET1 or 250 nM JQ1. (E) bargraph depiction of qRT-PCR analysis of transcript levels of BRD2, BRD3, BRD4, MYC and PIM1 after 2 h treatment of MV4;11 cells with either DMSO, 250 nM dBET1 or 250 nM JQ1. Values represent mean +/− stdev of triplicates.
Treatment with dBET1 elicited a comparable, modest effect on MYC and PIM1 expression. Remarkably, only three additional proteins were identified as significantly (p < 0.001) and markedly (> 5-fold) depleted in dBET1-treated cells: BRD2, BRD3 and BRD4 (Fig 3B,C). These findings are consistent with the anticipated, BET-specific bromodomain target spectrum of the JQ1 bromodomain-biasing element on dBET1(17). The remaining testis-specific member BRDT is not detected in MV4;11 leukemia cells and was therefore not quantified. To validate these findings, we performed orthogonal detection of BRD2, BRD3, BRD4, MYC and PIM1 by immunoblot following treatment of MV4;11 leukemia cells with dBET1 or JQ1, as above. BET family members were degraded only by dBET1, whereas MYC and PIM1 abundance was decreased by both dBET1 and JQ1, and to a lesser degree (Fig 3D). No effect on Ikaros TF expression was observed in either treatment condition (Fig S5). MYC and PIM1 are often associated with massive adjacent enhancer loci by epigenomic profiling, (18, 20) suggestive of a transcriptional mechanism of downregulation. We therefore measured mRNA transcript abundance for each depleted gene product (Fig 3E). Treatment with either JQ1 or dBET1 downregulated MYC and PIM1 transcription, suggestive of secondary transcriptional effects. Transcription of BRD4 and BRD3 were unaffected, consistent with post-transcriptional effects. Interestingly, transcription of BRD2 was affected by JQ1 and dBET1, whereas protein stability of the BRD2 gene product was only influenced by dBET1, suggestive of transcriptional and post- transcriptional consequences. These data establish a highly selective effect of dBET1 on target protein stability, proteome-wide.
We next explored the differential anti-proliferative consequences of BET degradation with dBET1 to BET bromodomain inhibition with JQ1. Degradation of BRD4 by dBET1 was associated with a more potent apoptotic consequence in MV4;11 than JQ1 as measured by caspase activation (Caspase-GLO; Fig 4A), cleavage of poly(ADP-ribose) polymerase (PARP), cleavage of Caspase-3 (immunoblot; Fig 4B) and Annexin V staining (flow cytometry; SFig 6A). The enhanced apoptotic response to dBET1 was confirmed in additional cultured human cell lines including DHL4 (B-cell lymphoma; Fig 4A). Kinetic studies of apoptotic response were then performed in MV4;11 cells cultured for either 4 or 8 hours followed by drug washout. Induction of apoptosis was assessed at 24 hours. While pulsed treatment with JQ1 did not yield a pronounced apoptotic response, we observed significantly increased apoptosis after only 4 h of dBET1 treatment that was enhanced at 8 h (Fig 4C, 4D). Indeed, dBET1 induced a potent and superior inhibitory effect on MV4;11 cell proliferation at 24 hours (measured by ATP content, IC50 = 0.14 μM, compared to IC50 = 1.1 μM with JQ1; Fig 4E) consistent with the reported, pronounced inhibitory effect of RNA silencing of BRD4 in this and other models of MLL-rearranged leukemia(21).
Figure 4. Improved kinetic properties in induction of apoptosis and in vivo proof of concept.
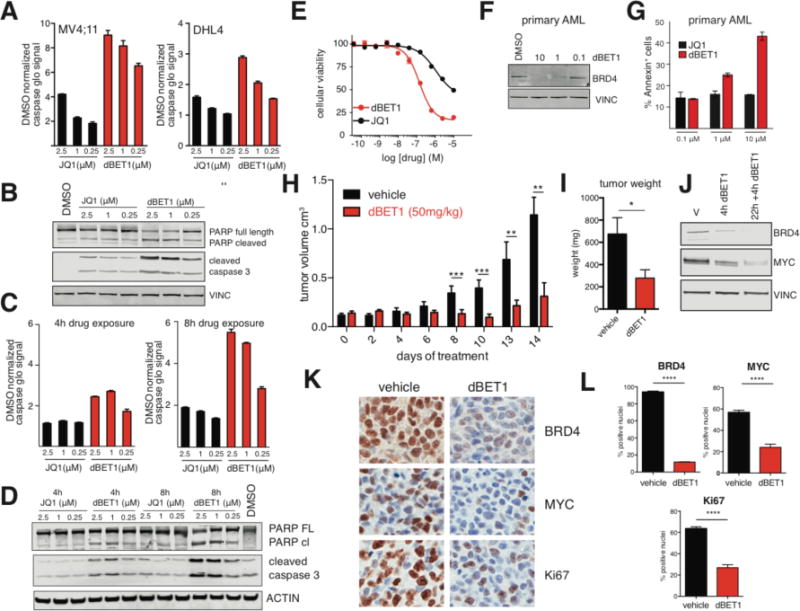
(A) Bar graph depiction of fold increase of apoptosis assessed via caspase glo assay relative to DMSO treated controls, 24 h treatment in MV4;11 or DHL4 cells. Values represent mean ± stdev of quadruplicate analysis (B) Immunoblot analysis of cleaved caspase 3, PARP cleavage and vinculin after treatment with dBET1 and JQ1 at the indicated concentrations for 24 h. (C) bar graph depiction of fold increase of apoptosis assessed via caspase glo assay relative to DMSO treated controls. MV4;11 cells were treated for 4 or 8 h with JQ1 or dBET1 at the indicated concentrations. Drug were washed out with PBS (3×) before being plated in drug-free media for a final treatment duration of 24 h. (D) Immunoblot analysis of cleaved caspase 3, PARP cleavage and vinculin after identical treatment conditions described in (C). (E) Cellular viability dose-response data of dBET1 and JQ1 after treatment of MV4;11 for 24 h as determined by ATP levels. Values represent mean ± stdev of quadruplicate analysis. (F) Immunoblot analysis for BRD4 and Vinculin after treatment of primary patient cells with the indicated concentrations of dBET1 for 24 hours. (G) bargraph depiction of fraction of annexin V positive primary patient cells after 24 h treatment with either dBET1 or JQ1 at the indicated concentrations. Values represent the average of duplicates and the range as error bars. Representative counter plots in SFig 6. (H) tumor volume of vehicle treated mice (n=5) or mice treated with dBET1 at a concentration of 50 mg/kg (n=6) over a treatment period of 14 days. (I) tumor weight after termination of the experiment shown in (H) on day 14. (J) Immunoblot analysis for BRD4, MYC and Vinculin using tumor lysates from mice that have been treated either once for 4 h or twice for 22 h and 4h compared to a vehicle treated control. (K) Immunohistochemistry staining for BRD4, MYC and Ki67 of a representative tumor of a dBET1 treated and a control treated mouse. (L) quantification of (K) based on 3 independent areas within that section. Data represents mean ± stdev of triplicate analysis and is normalized to DMSO.
The rapid biochemical activity and robust apoptotic response of cultured cell lines to dBET1 established the feasibility of assessing effects on primary human AML cells, where ex vivo proliferation is negligible in short-term cultures. Exposure of primary leukemic patient blasts to dBET1 elicited dose-proportionate depletion of BRD4 (Fig 4F) and induction of apoptosis (Annexin V/PI staining; Fig 4G, SFig 6B). Importantly, the effect of BRD4 degradation by dBET1 again elicited a significantly greater apoptotic response in primary AML cells and AML cell lines than displacement of BRD4 by JQ1 (Fig 4G). Together, these data exemplify ligand-dependent target protein degradation and support the thesis that target degradation can elicit a more pronounced biological consequence than domain-specific target inhibition.
To model the therapeutic opportunity of BRD4 degradation in vivo, we evaluated the tolerability and anti-tumor efficacy of repeat-dose dBET1 in an established murine xenograft model of human MV4;11 leukemia cells (31). Tumor-bearing mice were treated with dBET1 administered by intraperitoneal injection (50 mg/kg daily) or vehicle control. After 14 days of therapy a first tumor reached institutional limits for tumor size, and the study was terminated for comparative assessment of efficacy and modulation of BRD4 stability and function. Administration of dBET1 attenuated tumor progression as determined by serial volumetric measurement (Fig 4H), and decreased tumor weight assessed post-mortem (Fig 4I). Acute pharmacodynamic degradation of BRD4 was observed four hours after a first or second daily treatment with dBET1 (50 mg/kg IP) by immunoblot (Fig 4J), accompanied by downregulation of MYC (Fig 4J). These findings were confirmed by quantitative immunohistochemistry for BRD4 and MYC following repeat-dose exposure to dBET1 for 14 days (Fig 4K). A statistically significant destabilization of BRD4, downregulation of MYC and inhibition of proliferation (Ki67 staining) was observed with dBET1 compared to vehicle control in excised tumors (Fig 4K, 4L). Pharmacokinetic studies of dBET1 (50 mg/kg IP) corroborate adequate drug exposure in vivo (Cmax = 392 nM; SFig 7B), above the EC50 for BRD4 knock-down observed in vitro (<100 nM). Notably, two weeks of dBET1 was well tolerated by mice without a meaningful effect on weight, white blood count, hematocrit or platelet count (SFig 7C, 7D).
In summary, we present a mechanism-based, chemical strategy for endogenous target protein degradation. Facile conjugation of a novel linked phthalimide to selective small molecules produces CRBN-dependent post-translational degradation with exquisite target-specific activity, compared to established modulators of proteostasis (i.e. HSP90 inhibitors, NAE1 inhibitors). Though this approach is CRBN-dependent, CRBN is ubiquitously expressed in physiologic and pathophysiologic (e.g. neoplastic, and inflammatory) tissues, supporting broad utility in developmental and disease biology. The increased apoptotic response of primary AML cells to dBET1, even compared to the efficacious tool compound JQ1, highlights the potential superiority of BET degradation over BET bromodomain inhibition and prompts consideration of therapeutic development. Pharmacologic destabilization of BRD4 by dBET1 resulted in anti-tumor efficacy in a human leukemia xenograft, establishing phthalimide-conjugation as the first such system to induce target degradation in vivo. The facile extension of this approach to new targets (here, FKBP12), low molecular mass, high cell permeability, potent cellular activity, and synthetic simplicity addresses limitations associated with prior, pioneering systems. We expect that modulation of ligand and linker features will allow fine-tuning of biophysical properties of future phthalimide-conjugates, as needed. A more general implication of this research is the feasibility of approaching intractable protein targets (e.g. KRAS, MYC) using phthalimide-conjugation of target-binding ligands that may or may not possess target-specific inhibitory activity.
Supplementary Material
Acknowledgments
We thank W. Kaelin, R. Mazitschek and S. Orkin for engaging discussions, S.-H. Seo and S. Deangelo for assistance in the purification of BRD4, C. Ott for help with in vivo model studies, and N. Thoma for CRBN expression reagents. This research was supported by generous philanthropic gifts from Marc Cohen and Alain Cohen, the William Lawrence and Blanche Hughes Foundation, the NIH (R01-CA176745 and P01-CA066996 to J.E.B.). G.E.W is supported by an EMBO long-term fellowship. D.L.B. is a Merck Fellow of the Damon Runyon Cancer Research Foundation (DRG-2196-14). Quantitative proteomics studies were performed by Ryan Kunz of the Thermo Fisher Scientific Center for Multiplexed Proteomics at Harvard Medical School.
References
- 1.Bartlett JB, Dredge K, Dalgleish AG. Nat Rev Cancer. 2004;4:314–322. doi: 10.1038/nrc1323. [DOI] [PubMed] [Google Scholar]
- 2.Ito T, et al. Science. 2010;327:1345–1350. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 3.Krönke J, et al. Science. 2014;343:301–305. doi: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu G, et al. Science. 2014;343:305–309. doi: 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fischer ES, et al. Nature. 2014;512:49–53. doi: 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lallemand-Breitenbach V, et al. J Exp Med. 2001;193:1361–1371. doi: 10.1084/jem.193.12.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu YL, et al. Mol Cell. 2005;18:413–424. doi: 10.1016/j.molcel.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 8.Pratt MR, Schwartz EC, Muir TW. Proc Natl Acad Sci USA. 2007;104:11209–11214. doi: 10.1073/pnas.0700816104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banaszynski LA, et al. Cell. 2006;126:995–1004. doi: 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banaszynski LA, et al. Nat Med. 2008;14:1123–1127. doi: 10.1038/nm.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neklesa TK, et al. Nat Chem Biol. 2011;7:538–543. doi: 10.1038/nchembio.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakamoto KM, et al. Proc Natl Acad Sci USA. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schneekloth J, et al. J Am Chem Soc. 2004;126:3748–3754. doi: 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- 14.Hines J, et al. Proc Natl Acad Sci USA. 2013;110:8942–8947. doi: 10.1073/pnas.1217206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Itoh Y, et al. J Am Chem Soc. 2010;132:5820–5826. doi: 10.1021/ja100691p. [DOI] [PubMed] [Google Scholar]
- 16.Filippakopoulos P, Knapp S. Nat Rev Drug Discov. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- 17.Filippakopoulos P, et al. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chapuy B, et al. Cancer Cell. 2013;24:777–790. doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delmore JE, et al. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lovén J, et al. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zuber J, et al. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nicodeme E, et al. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anand P, et al. Cell. 2013;154:569–582. doi: 10.1016/j.cell.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown JD, et al. Mol Cell. 2014;56:219–231. doi: 10.1016/j.molcel.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anders L, et al. Nat Biotechnol. 2013;32:92–96. doi: 10.1038/nbt.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fischer ES, et al. Cell. 2011;147:1024–1039. doi: 10.1016/j.cell.2011.10.035. [DOI] [PubMed] [Google Scholar]
- 27.Petroski MD, Deshaies RJ. Nat Rev Mol Cell Biol. 6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 28.Sufan RI, Ohh M. Neoplasia. 2006;8:956–963. doi: 10.1593/neo.06520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soucy TA, et al. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 30.Huttlin EL, et al. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mertz A JA, et al. Proc Natl Acad Sci USA. 2011;108:16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.