

Abstract
Challenges in determining the structures of heterogeneous and dynamic protein complexes have greatly hampered past efforts to obtain a mechanistic understanding of many important biological processes. One such process is chaperone-assisted protein folding, where obtaining structural ensembles of chaperone:substrate complexes would ultimately reveal how chaperones help proteins fold into their native state. To address this problem, we devised a novel structural biology approach based on X-ray crystallography, termed Residual Electron and Anomalous Density (READ). READ enabled us to visualize even sparsely populated conformations of the substrate protein immunity protein 7 (Im7) in complex with the E. coli chaperone Spy. This study resulted in a series of snapshots depicting the various folding states of Im7 while bound to Spy. The ensemble shows that Spy-associated Im7 samples conformations ranging from unfolded to partially folded and native-like states, and reveals how a substrate can explore its folding landscape while bound to a chaperone.
High-resolution structural models of protein-protein interactions are critical for obtaining mechanistic insights into biological processes1. However, many protein-protein interactions are highly dynamic, making it difficult to obtain high-resolution data. Particularly challenging are interactions of intrinsically or conditionally disordered sections of proteins with their partner proteins. Recent advances in X-ray crystallography2 and NMR spectroscopy3,4 continue to improve our ability to analyze biomolecules that exist in multiple conformations. X-ray crystallography has historically provided valuable information on small-scale conformational changes, but observing large-amplitude heterogeneous conformational changes often falls beyond the reach of current crystallographic techniques. NMR can theoretically be used to determine heterogeneous ensembles5,6, but in practice, this proves to be very challenging.
Despite the importance of understanding how proteins fold into their native state within the cell, our knowledge about this critical process remains limited. It is clear that molecular chaperones aid in protein folding. However, exactly how they facilitate the folding process is still being debated7,8. Structural characterization of chaperone-assisted protein folding likely would help bring clarity to this question. Structural models of chaperone-substrate complexes have recently begun to provide information as to how a chaperone can recognize its substrate9–18. However, the impact that chaperones have on their substrates, and how these interactions affect the folding process remain largely unknown. For most chaperones, it is still unclear whether the chaperone actively participates in and affects the folding of the substrate proteins, or merely provides a suitable microenvironment enabling the substrate to fold on its own. This is a truly fundamental question in the chaperone field, and one that has eluded the community largely because of the highly dynamic nature of the chaperone-substrate complexes.
To address this question, we investigated the ATP-independent Escherichia coli periplasmic chaperone Spy. Spy prevents protein aggregation and aids in protein folding under various stress conditions, including treatment with tannin and butanol19. We originally discovered Spy by its ability to stabilize the protein-folding model Im719–21 in vivo and recently demonstrated that Im7 folds while associated with Spy22. The crystal structure of Spy revealed that it forms a thin α-helical homodimeric cradle19,23. Crosslinking and genetic experiments suggested that Spy interacts with substrates somewhere on its concave side19,24. By using a novel X-ray crystallography-based approach to model disorder in crystal structures, we have now determined the high-resolution ensemble of the dynamic Spy:Im7 complex. This work provides a detailed view of chaperone-mediated protein folding and shows how substrates like Im7 find their native fold while bound to their chaperones.
RESULTS
Crystallizing the Spy:Im7 complex
We reasoned that to obtain crystals of complexes between Spy (domain boundaries in Supplementary Fig. 1) and its substrate proteins, our best approach was to identify crystallization conditions that yielded Spy crystals in the presence of protein substrates but not in their absence. We therefore screened crystallization conditions for Spy with four different substrate proteins: a fragment of the largely unfolded bovine α-casein protein19,25, wild-type (WT) E. coli Im719,26, an unfolded variant of Im7 (L18A L19A L37A)27, and the N-terminal half of Im7 (Im76-45), which encompasses the entire Spy-binding portion of Im724. We found conditions in which all four substrates co-crystallized with Spy, but in which Spy alone did not yield crystals. Subsequent crystal washing and dissolution experiments confirmed the presence of the substrates in the co-crystals (Supplementary Fig. 2). The crystals diffracted to ~1.8 Å resolution. We used Spy:Im76-45 selenomethionine crystals for phasing with single-wavelength anomalous diffraction (SAD) experiments, and used this solution to build the well-ordered Spy portions of all four complexes. However, modeling of the substrate in the complex proved to be a substantial challenge, as the electron density of the substrate was discontinuous and fragmented. Even the minimal binding portion of Im7 (Im76-45) showed highly dispersed electron density (Fig. 1a). We hypothesized that the fragmented density was due to multiple, partially occupied conformations of the substrate bound within the crystal. Such residual density is typically not considered usable by traditional X-ray crystallography methods. Thus, we developed a new approach to interpret the chaperone-bound substrate in multiple conformations.
Figure 1.

Crystallographic data and ensemble selection. (a) 2mFo−DFc omit map of residual Im76-45 and flexible linker electron density contoured at 0.5 σ. This is the residual density that is used in the READ selection. (b) Composites of iodine positions detected from anomalous signals using pI-Phe substitutions, colored and numbered by sequence. Multiple iodine positions were detected for most residues. Agreement to the residual Im76-45 electron density (c) and anomalous iodine signals (d) for ensembles of varying size generated by randomly choosing from the MD pool (blue) and from the selection procedure (black). The agreement from back-calculating a subset of data excluded from the selection procedure is shown by the red curve (cross-validation). The cost function, χ2, decreases as the agreement to the experimental data increases and is defined in the Online Methods.
READ: a strategy to visualize heterogeneous and dynamic biomolecules
To determine the structure of the substrate portion of these Spy:substrate complexes, we conceived of an approach that we term READ, for Residual Electron and Anomalous Density. We split this approach into five steps: (1) By using a well-diffracting Spy:substrate co-crystal, we first determined the structure of the folded domain of Spy and obtained high quality residual electron density within the dynamic regions of the substrate. (2) We then labeled individual residues in the flexible regions of the substrate with the strong anomalous scatterer iodine, which serves to locate these residues in three-dimensional space using their anomalous density. (3) We performed molecular dynamics (MD) simulations to generate a pool of energetically reasonable conformations of the dynamic complex and (4) applied a sample-and-select algorithm to determine the minimal set of substrate conformations that fit both the residual and anomalous density. (5) Finally, we validated the ensemble using multiple statistical tests. Importantly, even though we only labeled a subset of the residues in the flexible regions of the substrate with iodine, the residual electron density can provide spatial information on many of the other flexible residues28,29. These two forms of data are therefore complementary: by labeling individual residues, one can locate them to specific points in space. The electron density then allowed us to connect the labeled residues of the substrate by confining the protein chain within regions of detectable density. In this way, the two forms of data together were able to describe multiple conformations of the substrate within the crystal. As described in detail below, we developed the READ method to uncover the ensemble of conformations that the Spy-binding domain of Im7 (i.e., Im76-4524) adopts while bound to Spy. However, we believe that READ will prove generally applicable to visualizing heterogeneous and dynamic complexes that have previously escaped detailed structural analysis.
Collecting READ data for the Spy:Im76-45 complex
To apply the READ technique to the folding mechanism employed by the chaperone Spy, we selected Im76-45 for further investigation because NMR data suggested that Im76-45 could recapitulate unfolded, partially folded, and native-like states of Im7 (Supplementary Fig. 3)30. Moreover, binding experiments indicated that Im76-45 comprises the entire Spy-binding region24. To introduce the anomalous scatterer iodine, we replaced eight Im76-45 residues with the non-canonical amino acid 4-iodophenylalanine (pI-Phe). Its strong anomalous scattering31 allowed us to track the positions of these individual Im76-45 residues one at a time, potentially even if the residue was found in several locations in the same crystal. We then co-crystallized Spy and the eight Im76-45 peptides, each of which harbored an individual pI-Phe substitution at one distinct position, and collected anomalous data for all eight Spy:Im76-45 complexes (Fig. 1B, Supplementary Table 1 Supplementary Dataset 1, and Supplementary Table 2). Consistent with our electron density map, we found that the majority of anomalous signals emerged in the cradle of Spy, implying that this is the likely Im7 substrate binding site. Consistent with the fragmented density, however, we observed multiple iodine positions for seven of the eight substituted residues. Together, these results indicated that the Im7 substrate binds Spy in multiple conformations.
READ sample-and-select procedure
To determine the structural ensemble that Im76-45 adopts while bound to Spy, we combined the residual electron density and the anomalous signals from our pI-Phe substituted Spy:Im76-45 complexes. To generate an accurate depiction of the chaperone-substrate interactions, we devised a selection protocol based on a sample-and-select procedure employed in NMR spectroscopy4. This procedure iteratively constructs structural ensembles and then compares them to the experimental data. During each round of the selection, a genetic algorithm alters the ensemble and its agreement to the experimental data is re-evaluated. If successful, the selection identifies the smallest group of specific conformations that best fits the residual electron density and anomalous signals. The READ sample-and-select algorithm is diagrammed in Fig. 2.
Figure 2.

Flowchart of the READ sample-and-select process.
Prior to performing the selection, we generated a large and diverse pool of chaperone-substrate complexes using coarse-grained MD simulations in a pseudo-crystal environment (Fig. 2 and Supplementary Fig. 4). The coarse-grained simulations are based on a single-residue resolution model for protein folding32 and were extended here to describe Spy-Im76-45 binding events (Online Methods). The initial conditions of the binding simulations are not biased toward a particular conformation of the substrate or any specific chaperone-substrate interaction (Online Methods). Im76-45 binds and unbinds to Spy throughout the simulations. This strategy allows a wide range of substrate conformations to interact with the chaperone. From the MD simulations, we extracted ~10,000 diverse Spy:Im76-45 complexes to be used by the ensuing selection. Each complex within this pool comprises one Spy dimer bound to a single Im76-45 substrate. This pool was then used by the selection algorithm to identify the minimal ensemble that best satisfies both the residual electron and anomalous crystallographic data.
The anomalous scattering portion of the selection uses our basic knowledge of pI-Phe geometry: the iodine is separated from its respective Cα atom in each coarse-grained conformer by 6.5 Å. The selection then picks ensembles that best reproduce the collection of iodine anomalous signals. Simultaneously, it uses the residual electron density to help choose ensembles. To make the electron density selection practical, we needed to develop a method to rapidly evaluate the agreement between the selected sub-ensembles and the experimental electron density on-the-fly during the selection procedure. To accomplish this task, we generated a compressed version of the experimental 2mFo−DFc electron density map for use in the selection. This process provided us with a target map that the ensuing selection tried to recapitulate. To reduce the extent of 3D space to be explored, this compressed map was created by only using density from regions of space significantly sampled by Im76-45 in the Spy:Im76-45 MD simulations. For each of the ~10,000 complexes in the coarse-grained MD pool, the electron density at the Cα positions of Im76-45 was extracted and used to construct an electron density map (Online Methods). These individual electron density maps from the separate conformers could then be combined (Fig. 2) and compared to the averaged experimental electron density map as part of the selection algorithm.
This approach allowed us to simultaneously use both the iodine anomalous signals and the residual electron density in the selection procedure. The selection resulted in small ensembles from the MD pool that best fit the READ data (Fig. 1c,d). Before analyzing the details of the Spy:Im76-45 complex, we first engaged in a series of validation tests to verify the ensemble and selection procedure (Supplementary Note 1, Figures 1c,d, Supplemental Figures 5-7). Combined, these validation tests confirmed that the selection procedure and selected six-member ensemble recapitulate the experimental data. Of note, the final six-membered ensemble was the largest ensemble that could simultaneously decrease the RFree and pass the 10-fold cross-validation test. This ensemble depicts the conformations that the substrate Im76-45 adopts while bound to the chaperone Spy (Fig. 3 Supplementary Movie 1, and Table 1).
Figure 3.

Spy:Im76-45 ensemble, arranged by RMSD to native state of Im76-45. Although the six-membered ensemble from the READ selection should be considered only as an ensemble, for clarity, the individual conformers are shown separately here. Spy is depicted as a gray surface and the Im76-45 conformer is shown as orange balls. Atoms that were either not directly selected in the READ procedure, or whose position could not be justified based on agreement with the residual electron density were removed, leading to non-contiguous sections. Dashed lines connect non-contiguous segments of the Im76-45 substrate. Residues of the Spy flexible linker region that fit the residual electron density are shown as larger gray spheres. Shown below each ensemble member is the RMSD of each conformer to the native state of Im76-45, as well as the percentage of contacts between Im76-45 and Spy that are hydrophobic.
Table 1.
Crystallography Statistics
| SeMet Spy:Im76-45 | Spy:Im76-45 | Spy:Casein 148-177, substrate not modeled | Spy H96L:Im7 L18A L19A L37A, substrate not modeled | Spy H96L:WT Im7, substrate not modeled | |
|---|---|---|---|---|---|
| PDB ID | 5INA | 5IOG | 5IOE | 5IOA | |
| Data collection | |||||
| Space group | P4122 | P4122 | P4122 | P4122 | P4122 |
| Cell dimensions | |||||
| a, b, c (Å) | 42.9, 42.9, 259.3 | 42.9, 42.9, 260.2 | 43.0, 43.0, 258.2 | 43.1, 43.1, 258.7 | 43.1, 43.14, 260.2 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 64.82–2.44(2.53–2.44) | 30.50–1.79(1.83–1.79) | 36.88–1.77 (1.80–1.77) | 30.48–1.87(1.91–1.87) | 33.21–1.87(1.91–1.87) |
| Rmerge (%) | 10.6(36) | 8.2(108) | 6.2(134) | 8.4(152) | 9.6(249) |
| I/σ(I) | 15.1(6.8) | 7.0(1.1) | 15.3(1.6) | 13.8(1.8) | 13.2(1.3) |
| Completeness (%) | 100(100) | 94.0(90.1) | 99.9(99.5) | 100(100) | 96.8(93.1) |
| Redundancy | 15.6(15.6) | 4.3(4.2) | 8.7(8.2) | 9.6(9.4) | 8.2(8.2) |
| CC1/2 | 0.998(0.689) | 0.999(0.745) | 0.999(0.676) | 0.998(0.606) | |
| Refinement | |||||
| Resolution (Å) | 1.79 | 1.77 | 1.87 | 1.87 | |
| No. of Reflections | 22583 | 25052 | 21505 | 20838 | |
| Rwork/Rfree | 0.22/0.23 | 0.21/0.24 | 0.22/0.24 | 0.21/0.25 | |
| No. of Atoms | 1765 | 1669 | 1715 | 1653 | |
| Protein | 1586 | 1493 | 1541 | 1444 | |
| Ligand/ion | 30 | 56 | 60 | 30 | |
| Water | 149 | 120 | 114 | 179 | |
| B-factors | 49.4 | 48.5 | 47.4 | 39.2 | |
| Protein | 49.0 | 47.5 | 46.3 | 38.3 | |
| Ligand/ion | 48.6 | 65.9 | 80.4 | 62.9 | |
| Water | 54.2 | 51.9 | 44.5 | 42.1 | |
| r.m.s. Deviations | |||||
| Bond lengths (Å) | 0.013 | 0.013 | 0.013 | 0.014 | |
| Bond angles (º) | 1.24 | 1.30 | 1.24 | 1.39 | |
Folding and interactions of Im7 while bound to Spy
Our results showed that by using this novel READ approach, we were able to obtain structural information about the dynamic interaction of a chaperone with its substrate protein. We were particularly interested in finding answers to one of the most fundamental questions in chaperone biology—how does chaperone binding affect substrate structure and vice versa. By analyzing the individual structures of the six-member ensemble of Im76-45 bound to Spy, we observed that Im76-45 takes on several different conformations while bound. We found these conformations to be highly heterogeneous and to include unfolded, partially folded, and native-like states (Fig. 3). The ensemble primarily encompasses Im76-45 laying diagonally within the Spy cradle in several different orientations, but some conformations traverse as far as the tips or even extend over the side of the cradle (Figs. 3,4a).
Figure 4.

Contact maps of Spy:Im76-45 complex. (a) Spy:Im76-45 contact map projected onto the bound Spy dimer (above) and Im76-45 (below) structures. For clarity, Im76-45 is represented with a single conformation. The frequency plotted is calculated as the average contact frequency from Spy to every residue of Im76-45 and vice-versa. As the residues involved in contacts are more evenly distributed in Im76-45 compared to Spy, its contact map was amplified. (b) Detailed contact maps of Spy:Im76-45. Contacts to the two Spy monomers are depicted separately. Note that the flexible linker region of Spy (residues 47–57) is not represented in the 2D contact maps.
We constructed a contact map of the complex, which shows the frequency of interactions for chaperone-substrate residue pairs (Fig. 4). We found that the primary interaction sites on Spy reside at the N and C termini (Arg122, Thr124, and Phe29) as well as on the concave face of the chaperone (Arg61, Arg43, Lys47, His96, and Met46). The Spy-contacting residues comprise a mixture of charged, polar, and hydrophobic residues. Surprisingly, we noted that in the ensemble, Im76-45 interacts with only 38% of the hydrophobic residues in the Spy cradle, but interacts with 61% of the hydrophilic residues in the cradle. This mixture suggests the importance of both electrostatic and hydrophobic components in binding the Im76-45 ensemble. With respect to the substrate, we observed that nearly every residue in Im76-45 is in contact with Spy (Fig. 4a). However, we did notice that despite this uniformity, regions of Im76-45 preferentially interact with different regions in Spy (Fig. 4b). For example, the N-terminal half of Im76-45 binds more consistently in the Spy cradle, whereas the C-terminal half predominantly binds to the outer edges of Spy’s concave surface.
Not unexpectedly, we found that as Im76-45 progresses from the unfolded to the native state, its interactions with Spy shift accordingly. Whereas the least-folded Im76-45 pose in the ensemble forms the most hydrophobic contacts with Spy (Fig. 3), the two most-folded conformations form the fewest hydrophobic contacts (Fig. 3). This shift in contacts is likely due to hydrophobic residues of Im76-45 preferentially forming intra-molecular contacts upon folding (i.e., hydrophobic collapse), effectively removing themselves from the interaction sites. The diversity of conformations and binding sites observed here emphasizes the dynamic and heterogeneous nature of the chaperone-substrate ensemble. Although we do not yet have time resolution data of these various snapshots of Im76-45, this ensemble illustrates how a substrate samples its folding landscape while bound to a chaperone.
Spy changes conformation upon substrate binding
Comparing the structure of Spy in its substrate-bound and apo19 states revealed that the Spy dimer also undergoes significant conformational changes upon substrate binding (Fig. 5a and Supplementary Movie 2). Upon substrate binding, the Spy dimer twists 9° about its center relative to its apo19 form. This twist yields asymmetry and results in substantially different interaction patterns in the two Spy monomers (Fig. 4b). It is possible that this twist serves to increase heterogeneity in Spy by providing more binding poses. Additionally, we observed that the linker region (residues 47–57) of Spy, which participates in substrate interaction, becomes mostly disordered upon binding the substrate16,19. This increased disorder might explain how Spy is able to recognize and bind different substrates and/or differing conformations of the same substrate. Importantly, we observed the same structural changes in Spy regardless of which of the four substrates was bound (Fig. 5b, Table 1). The RMSD between the well-folded sections of Spy in the four chaperone-substrate complexes was very small, less than 0.3 Å. Combined with competition experiments showing that the substrates compete in solution for Spy binding (Fig. 5c and Supplementary Fig. 8), we conclude that all the tested substrates share the same overall Spy binding site.
Figure 5.

Spy conformation changes upon substrate binding. (a) Overlay of apo Spy (PDB ID: 3O39, gray) and bound Spy (green). (b) Overlay of WT Spy bound to Im76-45 (green), H96L Spy bound to Im7 L18A L19 AL13A (blue), H96L Spy bound to WT Im7 (yellow), and WT Spy bound to casein (salmon). (c) Competition assay showing Im76-45 competes with Im7 L18A L19A L37A H40W for the same binding site on Spy (further substrate competition assays are shown in Supplementary Fig. 8). Error bars depict standard deviations of n=3 technical replicates.
DISCUSSION
To shed light on how chaperones interact with their substrates, we developed a novel structural biology method (READ) and applied it to determine a conformational ensemble of the chaperone Spy bound to substrate. As a substrate, we used Im76-45, the chaperone-interacting portion of the protein-folding model protein Im720,21. In the chaperone-bound ensemble, Im76-45 samples unfolded, partially folded, and native-like states. The ensemble provides an unprecedented description of the conformations that a substrate assumes while exploring its chaperone-associated folding landscape. This substrate-chaperone ensemble helps accomplish the longstanding goal of obtaining a detailed view of how a chaperone aids protein folding.
We recently showed that Im7 can fold while remaining continuously bound to Spy22. The high-resolution ensemble obtained here now provides insight into exactly how this occurs. The structures of our ensemble agree well with lower-resolution crosslinking data, which indicate that chaperone-substrate interactions primarily occur on the concave surface of Spy24. The ensemble suggests a model in which Spy provides an amphipathic surface that allows substrate proteins to assume different conformations while bound to the chaperone. This model is consistent with previous studies postulating that the flexible binding of chaperones allows for substrate protein folding33. The amphipathic concave surface of Spy likely facilitates this flexible binding and may be a crucial feature for Spy and potentially other chaperones12, allowing them to bind multiple conformations of many different substrates.
In contrast to Spy’s binding hotspots, Im76-45 displays substantially less specificity in its binding sites. Nearly all Im76-45 residues come in contact with Spy. Unfolded substrate conformers interact with Spy through both hydrophobic and hydrophilic interactions, whereas the binding of native-like states is mainly hydrophilic. This trend suggests that complex formation between an ATP-independent chaperone and its unfolded substrate may initially involve hydrophobic interactions, effectively shielding the exposed aggregation-sensitive hydrophobic regions in the substrate. Once the substrate begins to fold within this protected environment, it progressively buries its own hydrophobic residues, and its interactions with the chaperone shift towards becoming more electrostatic. Notably, the most frequent contacts between Spy and Im76-45 are charge-charge interactions. The negatively charged Im7 residues Glu21, Asp32, and Asp35 reside on the surface of Im7 and form interactions with Spy’s positively charged cradle in both the unfolded and native-like states. Residues Asp32 and Asp35 are close to each other in the folded state of Im7. This proximity likely causes electrostatic repulsion that destabilizes Im7’s native state. Interaction with Spy’s positively-charged residues likely relieves the charge repulsion between Asp32 and Asp35, promoting their compaction into a helical conformation. As inter-molecular hydrophobic interactions between Spy and the substrate become progressively replaced by intra-molecular interactions within the substrate, the affinity between chaperone and substrates could decrease, eventually leading to release of the folded client protein.
Recently, we employed a genetic selection system to improve the chaperone activity of Spy. This selection resulted in “Super Spy” variants that were more effective at both preventing aggregation and promoting protein folding24. In conjunction with our bound Im76-45 ensemble, these mutants now allowed us to investigate structural features important to chaperone function. Previous analysis revealed that the Super Spy variants either bound Im7 tighter than WT Spy, increased chaperone flexibility as measured via H/D exchange, or both24. Our ensemble revealed that two of the Super Spy mutations (H96L and Q100L) form part of the chaperone contact surface that binds to Im76-45 (Fig. 4a). Moreover, our co-structure suggests that the L32P substitution, which increases Spy’s flexibility24, could operate by unhinging the N-terminal helix and effectively expanding the size of the disordered linker. This possibility is supported by the Spy:substrate structures, in which the linker region becomes more flexible compared to the apo19 state (Fig. 6a). This expansion would increase the structural plasticity for substrate binding34. By sampling multiple conformations, this linker region may allow diverse substrate conformations to be accommodated.
Figure 6.
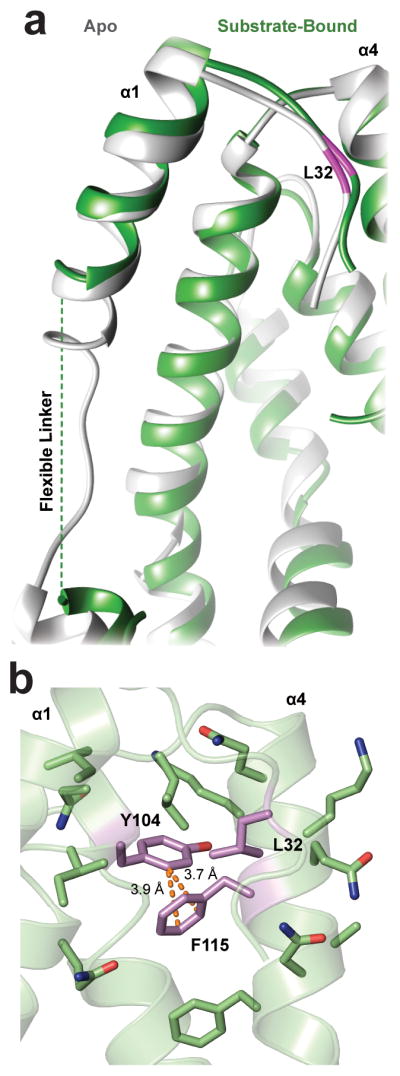
Flexibility of Spy linker region and effect of Super Spy mutants. (a) The Spy linker region adopts one dominant conformation in its apo state (PDB ID 3039, gray), but expands and adopts multiple conformations in bound states (green). (b) F115 and L32 tether Spy’s linker region to its cradle, decreasing Spy activity by limiting linker region flexibility. The Super Spy mutants F115L, F115I, and L32P are proposed to gain activity by increasing the flexibility or size of this linker region. L32, F115, and Y104 are rendered in purple to illustrate residues that are most affected by Super Spy mutations; CH⋯π hydrogen bonds are depicted by orange dashes.
Other Super Spy mutations (F115I and F115L) caused increased flexibility but not tighter substrate binding24. This residue does not directly contact Im76-45 in our READ-derived ensemble. Instead, when Spy is bound to substrate, F115 engages in close CH⋯π hydrogen bonds with Tyr104 (Fig. 6b). This interaction presumably reduces the mobility of the C-terminal helix. The F115I/L substitutions would replace these hydrogen bonds with hydrophobic interactions that have little angular dependence. As a result, the C-terminus, and possibly also the flexible linker, is likely to become more flexible and thus more accommodating of different conformations of substrates. Overall, comparison of our ensemble to the Super Spy variants provides specific examples to corroborate the importance of conformational flexibility in chaperone-substrate interactions34.
Despite extensive studies, exactly how complex chaperone machines help proteins fold remains controversial7,8. Our study indicates that the chaperone Spy employs a simple surface binding approach that allows the substrate to explore various conformations and form transiently favorable interactions while being protected from aggregation. We speculate that many other chaperones could utilize a similar strategy. ATP and co-chaperone dependencies may have emerged later through evolution to better modulate and control chaperone action.
In addition to insights into chaperone function, this work presents a new method for determining heterogeneous structural ensembles via a hybrid methodology of X-ray crystallography and computational modeling. Heterogeneous dynamic complexes or disordered regions of single proteins, once considered solely approachable by NMR spectroscopy, can now be visualized through X-ray crystallography. Consequently, this technique could enable structural characterization of many important dynamic and heterogeneous biomolecular systems.
ONLINE METHODS
For computational methods, including simulations of Spy-substrate interactions, binning the residual Im7 electron density, ensemble selection, validation tests, and contact map generation, please see Supplementary Note 1.
Spy truncation mutants’ construction and in vitro and in vivo activity measurements
To facilitate crystallization, we used Spy 29-124, a truncated Spy version that removes the unstructured N- and C-terminal tails (full length Spy is 138 amino acids). To determine if these alterations impact Spy’s chaperone activity in vitro, we performed in vitro chaperone activity assays and found that they had no significant effect; these deletions also had only a minor effect on Spy’s ability to stabilize Im7 in vivo (Supplementary Fig. 1). The in vitro activity of Spy 29-124 was assessed using the aldolase refolding assay as previously described24. Briefly, in the denaturing step, 100 μM aldolase was denatured in buffer containing 6.6 M GdmCl, 40 mM HEPES pH 7.5, and 50 mM NaCl overnight at 22 °C (room temperature). In the refolding step, denatured aldolase was diluted to 3 μM in refolding buffer (40 mM HEPES, 150 mM NaCl, 5 mM DTT pH 7.5) in the presence of 6 μM WT Spy or Spy 29-124 (Spy:aldolase = 2:1). As a control, an identical experiment without Spy added was also performed. The refolding temperature was 37 °C with continuous shaking. The refolding status was monitored at different time points (1 min, 4 min, 10 min, and 20 min) and tested by diluting the refolding sample by 15-fold into the reaction buffer (0.15 mM NADH, 2 mM F1,6-DP, 1.8 U/ml GDH/TPI, 40 mM HEPES, and 150 mM NaCl pH 7.5) at 28 °C. The absorbance was monitored for 1.5 min at 340 nm. The percentage refolding was calculated and averaged over three repeats.
To determine the in vivo activity of the Spy mutants, the quantity of the unstable Im7 variant L53A I54A expressed in the periplasm was compared during Spy variant co-expression as previously described19. Plasmid Spy (pTrc-spy)19 was used as the template for the construction of the variant plasmids of Spy for in vivo chaperone activity measurement (Supplementary Table 3). To use the native signal sequence of spy for the periplasmic export of the Spy variants, an NheI site was first introduced between the signal sequence and the mature protein coding region of Spy. The vector was then digested with NheI and BamHI, purified, and ligated with the linear fragments corresponding to truncated sequences (21–130, 24–130, 27–130, 30–130, and 33–130) of Spy.
Cells containing a strain that expressed the unstable Im7 mutant IL53A I54A (pCDFTrc-ssIm7L53A I54A)19 were transformed with plasmids that expressed either WT or one of the five truncated Spy mutants and grown to mid-log phase in LB medium at 37 °C. Im7 L53A I54A and Spy expression were induced with various concentrations of IPTG for 2 h to compare the in vivo chaperone activity of WT Spy and the truncated Spy mutants at similar expression levels. Periplasmic fractions were prepared as previously described35 and were separated on 16% Tricine gel (Life Technologies Inc.). The bands corresponding to Spy and the C-terminal His-tagged Im7 were either directly visualized on Coomassie stained gels or determined by western blot using anti-His antibody (Abcam ab1187; validation provided on manufacturer’s website).
Protein expression and purification
The gene for spy 29-124 was amplified from plasmid pET28sumo-spy19 with primer 1 (5′-CGC GGG ATC CTT CAA AGA CCT GAA CCT GAC CG-3′) and primer 2 (5′-CGC GCT CGA GTT ATG TCA GAC GCT TCT CAA AAT TAG C-3′), and was cloned into pET28sumo via BamHI and XhoI sites. The H96L variant was made by Phusion site-directed mutagenesis (New England Biolabs). WT and H96L Spy 29-124 were expressed and purified as described previously19 with the exception that Ni-HisTrap columns (GE Healthcare) were utilized instead of the Ni-NTA beads and mini-chromatography column. ULP1 cleavage occurred following elution from the Ni-HisTrap column overnight at 4 °C while dialyzing to 40 mM Tris, 300 mM NaCl, pH 8.0. After dialysis, Spy was passed over the HisTrap column to remove the cleaved SUMO tag (20 mM imidazole was left over from the dialysis). Cleavage of the SUMO tag leaves a single serine in position 28 of Spy. The flow-through was then concentrated and diluted 5 times with 20 mM Tris, pH 8 for further purification on a HiTrap Q column. Spy has an isoelectric point of 9.5 and therefore was collected in the flow-through. The flow-through containing Spy was concentrated and diluted 5-fold with 50 mM sodium phosphate at pH 6.5 and passed over a HiTrap SP column. Spy was then eluted with a gradient from 0 M to 1 M NaCl. Re-buffering to the final reaction buffer was accomplished by gel filtration, passing the pooled and concentrated fractions containing Spy over a HiLoad 75 column in 40 mM HEPES, 100 mM NaCl, pH 7.5. Fractions containing Spy were then concentrated, frozen in liquid nitrogen, and stored at −80 °C. WT Im7, Im7 L18A L19A L37A H40W, and Im7 L18A L19A L37A were purified by the same protocol as Spy, but without the SP column step. In addition to WT Im7 and these various Im7 mutants, co-crystallization experiments extensively utilized Im76-45, a minimal Spy-binding segment that encompasses the first two helices of Im7 and contains 46% of the total Im7 sequence. It displays partial helicity when free in solution (Supplementary Fig. 3)24,30. The 6-45 portion of Im7 (H2N-SISDYTEAEFVQLLKEIEKENVAATDDVLD VLLEHFVKIT-OH), 4-iodophenlyalanine variants, and a peptide corresponding to a portion of bovine alpha casein S1 148-177 (Ac-ELFRQFYQLDAYPSGAWYYVPLGTQYTDAP-amide) were obtained from New England Peptide at ≥ 95% purity. Anomalous signals for residues E12, E14, L19, and E21 substitutions were determined using a peptide containing Im7 6–26, which was also obtained from New England Peptide at ≥ 95% purity.
Protein crystallization
Co-crystals of WT Spy 29-124 and Spy H96L 29-124 in complex with Im7 variants and casein were grown by vapor diffusion. 25–130 mg/ml dimer Spy was incubated with various Im7 or casein substrates at concentrations ranging from equimolar to three-fold excess substrate in 22%–33% PEG 3000, 0.88–1.0 M imidazole pH 8.0, and 40–310 mM zinc acetate at 20 °C. Crystals were flash frozen in liquid nitrogen using 35% PEG 3000 as a cryo-protectant. It is worthwhile to note that the flash freezing could somewhat bias the conformations observed in the crystal structure36. However, we chose to freeze the crystal to provide us with the maximum capability to identify and interpret the iodine anomalous signals.
Assessing presence of substrate in crystals
Crystals were washed by sequential transfer between three to six 2 μl drops of mother liquor, incubating in each wash solution for 2–10 s in an effort to remove all surface bound and precipitated substrate protein before being dissolved for visualization by SDS-PAGE. Before loading, samples were boiled for 10 min in reducing loading buffer, and then loaded onto 16% Tricine gels. Wash samples and dissolved crystal samples were analyzed by Lumetein staining (Biotium) and Flamingo staining (Bio-Rad) per manufacturer’s instructions, and imaged using a FluorChem M Imager (ProteinSimple).
X-ray crystallography
Data were collected at the LS-CAT beamlines at the Advanced Photon Source at 100 K. SeMet and native Spy:Im76-45 crystals were collected at 12.7 keV and 9.7 keV, respectively. Spy:Casein 148-177, and Spy H96L:WT Im7 crystals were collected also collected at 12.7 keV. Data integration and scaling were performed with iMosflm37 and AIMLESS38, respectively. As molecular replacement attempts using the previously published apo Spy structures (PDB IDs: 3O39 and 3OEO) were unsuccessful, the Spy:Im76-45 complex was solved using Se-SAD phasing with SeMet-Spy, followed by density modification and initial model building by AutoSol in Phenix39. The initial model was completed and refined using the native Spy:Im76-45 complex data. The rest of the structures were built using the native Spy:Im76-45 structure as a molecular replacement search model. Refinements, including TLS refinement, were performed using COOT40 and Phenix39. All refined structures were validated using the Molprobity server41, with Clashscores ranking better than the 90th percentile for all structures. Structural figures were rendered using PyMOL42 and UCSF Chimera43, and movies generated using UCSF Chimera43. Several partially occupied zinc atoms were observed in the crystal structure. Although some of these zinc atoms could also potentially modelled as water molecules, doing so resulted in an increase in the RFree. Additionally, a section of density near His A96 that is potentially partially occupied by a combination of water, Spy linker region, and possibly zinc, was modelled as containing water molecules. Spy H96L:Im76-45 was employed for iodine anomalous scattering experiments due to increased robustness and reproducibility of the crystals.
The expected anomalous scatterers in the structures were S in methionine residues of Spy, Zn from the crystallization buffer, and I in the single pI-Phe residue of each synthetic Im76-45 peptide. Each I site is expected to be partially occupied as Im76-45 had diffuse density corresponding to multiple, partially occupied conformers; the Zn sites also may be partially occupied. To identify I, S, and Zn atomic positions using anomalous scattering, datasets were collected at 6.5 keV and 14.0 keV at 100 K using the ID-D beamline at LS-CAT31. Anomalous difference maps for initial anomalous signal screening were calculated with phases from a molecular replacement search using the native Spy:Im76-45 (with no Im76-45 built in) complex as the search model.
Anomalous difference maps calculated with the 14.0 keV data were used as controls to distinguish iodine from zinc atoms, as the iodine and zinc anomalous scattering factors are comparable at 14.0 keV, whereas at 6.5 keV, f″ is ~9-fold greater for iodine than for zinc31. Anomalous differences were also collected and analyzed for a crystal of WT Spy 29-124:Im76-45 containing no iodine. The resulting anomalous difference map was inspected for peaks corresponding to sulfur, which were then excluded when selecting iodine peaks. Also, peaks that overlapped with Spy in the crystal lattice were excluded from analysis.
As an initial screen for placing iodine atoms in the 6.5 keV anomalous difference maps, the median methionine sulfur signal was used as a cutoff for each individual map to control for varying data quality between crystals. Then, all anomalous atoms were refined in Phenix44 using anomalous group refinement. Refined B-factor of placed iodine ions was then used to estimate the positional fluctuation of the anomalous signals. This positional fluctuation was used as estimated error in the ensuing selection. A summary of all the anomalous signal heights (Supplementary Table 1) and anomalous difference maps (Supplemental Dataset 1) are displayed at varying contour levels for maximum clarity of iodine and methionine peak heights.
Substrate binding to Spy
The dissociation constant of Im76-45 was determined via a fluorescence-based competition experiment with Im7 L18A L19A L37A H40W, and its ability to compete with casein 148-177 for Spy binding was tested. Im7 L18A L19A L37A H40W was chosen for competition experiments due to its tight binding (Supplementary Fig. 8) and substantial fluorescence change upon binding. This mutant binds to Spy tighter than Im7 L18A L19A L37A. 10 μM Spy 29-124 dimer was mixed with 10 μM Im7 L18A L19A L37A H40W or casein 148-177 to form a 1:1 complex in a buffer containing 40 mM HEPES pH 7.5 and 100 mM NaCl at 22 °C. Complex formation was monitored with a QuantaMaster 400 (Photon Technology International) using the tryptophan fluorescence of Im7 L18A L19A L37A H40W. Naturally tryptophan-free Im76-45 was then titrated into the complex to compete with Im7 L18A L19A L37A H40W for Spy binding. The observed fluorescence intensity at 350 nm was plotted as a function of the logarithm of the Im76-45 or casein 148-177 concentration. The data were fit for a one-site-binding competition model (OriginLab 9.1):
where A1 and A2 are the maximum and minimum asymptotes, respectively, and x is the concentration of Im76-45. x0 is the apparent KD for Im76-45 based on its ability to compete with Im7 L18A L19A L37A H40W. Using the KD of Im7 L18A L19A L37A H40W binding to Spy 29-124, we then calculated the KD for Im76-45 binding to Spy 29-124 using the Cheng-Prusoff equation:
where L is the concentration of Im7 L18A L19A L37A H40W and KD is the dissociation constant for Im7 L18A L19A L37A H40W binding to Spy. Due to interaction between higher oligomer states of Im76-45 and casein 148-177 (Supplementary Fig. 8), the competition curve was unable to be fit for casein 148-177 competing with Im76-45.
The stoichiometry of binding of casein 148-177 and Spy was determined by tryptophan fluorescence of the casein upon Spy 29-124 addition. Increasing concentrations of Spy 29-124 were titrated to 20 μM of casein 148-177 in 40 mM HEPES (pH 7.5), 100 mM NaCl, at 22 °C. Complex formation was monitored with a QuantaMaster 400 (Photon Technology International) using the tryptophan fluorescence of casein 148-177. The observed fluorescence intensity at 339 nm was plotted as a function of the Spy 29-124 dimer concentration and fit with a quadratic equation using Origin 9.1 (OriginLab).
To determine the dissociation constant, increasing concentrations of Spy 29-124 were titrated to 0.25 μM of casein in 40 mM HEPES (pH 7.5), 100 mM NaCl, at 22 °C. Complex formation was monitored with a QuantaMaster 400 (Photon Technology International) using the tryptophan fluorescence of casein 148-177. The observed fluorescence intensity at 339 nm was corrected for dilution due to the titration and then plotted as a function of the Spy 29-124 dimer concentration. The data were fit using a square hyperbola function in Origin 9.1 (OriginLab):
where F is the recorded fluorescence signal, Fmax is the maximum fluorescence reached upon saturation of the complex, L is the concentration of free Spy in solution, KD is the dissociation constant, and C is a parameter for the offset. The calculated KD is an average of three independent repetitions. The measured dissociation constants for the different substrates ranged from 0.1 to 1 μM.
Isothermal titration calorimetry (ITC)
Spy 29-124 and Im7-L18A L19A L37A H40W were dialyzed overnight against 40 mM HEPES, 100 mM NaCl, pH 7.5. 165 μM Spy dimer was loaded into a syringe and titrated into a cell containing 15 μM Im7 L18A L19 AL37A H40W at 25 °C in an iTC200 (Malvern Instruments) with an injection interval of 120 s and an initial delay time of 60 s. The solution was stirred at 1000 rpm, and the reference power was set to 6 μcal s−1 in high feedback mode. Data analysis was conducted using a plugin for Origin 7 (OriginLab), the software provided by the manufacturer.
Analytical ultracentrifugation
Sedimentation velocity experiments for the Im76-45 and the bovine α-S1-casein peptide were performed using a Beckman Proteome Lab XL-I analytical ultracentrifuge (Beckman Coulter). Both peptides were first dialyzed against 40 mM HEPES, 100 mM NaCl, pH 7.5, then diluted to a concentration of 10 μM using the dialysis buffer. Samples were loaded into cells containing standard sector shaped 2-channel Epon centerpieces with 1.2 cm path-length (Beckman Coulter) and equilibrated to 22 °C for at least 1 h prior to sedimentation. All samples were spun at 48,000 rpm in a Beckman AN-50 Ti rotor, and the sedimentation of the protein was monitored continuously using interference optics, since the Im76-45 does not absorb strongly at 280 nm. Data analysis was conducted with SEDFIT (version 14.1)45, using the continuous c(s) distribution model. The confidence level for the maximum entropy (ME) regularization was set to 0.95. Buffer density and viscosity were calculated using SEDNTERP (http://sednterp.unh.edu/).
Supplementary Material
Acknowledgments
All maps, structure factors, crystal datasets, code, and PDB files are available upon request. The authors would like to thank J. Smith, D. Akey, U. Jakob, D. Smith, Z. Wawrzak, and F. Stull for critical comments and suggestions. Use of the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the US DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (grant 085P1000817). This work was funded by an NRSA National Institutes of Health (NIH) grant GM108298 (L.S.A.), a Boehringer Ingelheim Fonds fellowship (P.K.), a National Natural Science Foundation of China (NSFC) grant 31400664 (S.Q.) who is also sponsored by Shanghai Pujing Program, NIH grant GM102829 (J.C.A.B.), NIH grant GM107233 (C.L.B.) and NSF grant CHE1506273 (C.L.B.) J.C.A.B. is supported as a Howard Hughes Medical Institute Investigator.
Footnotes
ACCESSION CODES
Structures and datasets in this work have been deposited in the PDB under the IDs 5INA, 5IOG, 5IOE, and 5IOA.
AUTHOR CONTRIBUTIONS
Overall concept was conceived by S.H. and J.B. Experiments were designed by S.H., S.Q., J.B., R.T., H.B., and P.K. Experiments were performed by S.H., S.Q., P.K., R.M., and L.W. Analysis and computational modeling was designed by C.B., L.S., P.A., L.A., H.B., and S.H. Computational analysis was carried out by Q.X., S.H., L.S., L.A., P.A., P.K., and R.M. The manuscript was written primarily by S.H. and J.B., with assistance from L.S., L.A. and all other authors.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Keskin O, Gursoy A, Ma B, Nussinov R. Principles of protein-protein interactions: what are the preferred ways for proteins to interact? Chemical Reviews. 2008;108:1225–44. doi: 10.1021/cr040409x. [DOI] [PubMed] [Google Scholar]
- 2.Fraser JS, et al. Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:16247–16252. doi: 10.1073/pnas.1111325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kay LE. New Views of Functionally Dynamic Proteins by Solution NMR Spectroscopy. J Mol Biol. 2016;428:323–31. doi: 10.1016/j.jmb.2015.11.028. [DOI] [PubMed] [Google Scholar]
- 4.Salmon L, Blackledge M. Investigating protein conformational energy landscapes and atomic resolution dynamics from NMR dipolar couplings: a review. Rep Prog Phys. 2015;78:126601. doi: 10.1088/0034-4885/78/12/126601. [DOI] [PubMed] [Google Scholar]
- 5.Blackledge MJ, et al. Conformational Backbone Dynamics of the Cyclic Decapeptide Antamanide - Application of a New Multiconformational Search Algorithm-Based on Nmr Data. Biochemistry. 1993;32:10960–10974. doi: 10.1021/bi00092a005. [DOI] [PubMed] [Google Scholar]
- 6.Guerry P, et al. Mapping the Population of Protein Conformational Energy Sub-States from NMR Dipolar Couplings. Angewandte Chemie-International Edition. 2013;52:3181–3185. doi: 10.1002/anie.201209669. [DOI] [PubMed] [Google Scholar]
- 7.Jewett AI, Shea JE. Reconciling theories of chaperonin accelerated folding with experimental evidence. Cell Mol Life Sci. 2010;67:255–76. doi: 10.1007/s00018-009-0164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mashaghi A, et al. Reshaping of the conformational search of a protein by the chaperone trigger factor. Nature. 2013;500:98–101. doi: 10.1038/nature12293. [DOI] [PubMed] [Google Scholar]
- 9.Buckle AM, Zahn R, Fersht AR. A structural model for GroEL-polypeptide recognition. Proc Natl Acad Sci U S A. 1997;94:3571–5. doi: 10.1073/pnas.94.8.3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez-Hackert E, Hendrickson WA. Promiscuous substrate recognition in folding and assembly activities of the trigger factor chaperone. Cell. 2009;138:923–34. doi: 10.1016/j.cell.2009.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saio T, Guan X, Rossi P, Economou A, Kalodimos CG. Structural basis for protein antiaggregation activity of the trigger factor chaperone. Science. 2014;344:1250494. doi: 10.1126/science.1250494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joachimiak LA, Walzthoeni T, Liu CW, Aebersold R, Frydman J. The structural basis of substrate recognition by the eukaryotic chaperonin TRiC/CCT. Cell. 2014;159:1042–55. doi: 10.1016/j.cell.2014.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen DH, et al. Visualizing GroEL/ES in the act of encapsulating a folding protein. Cell. 2013;153:1354–65. doi: 10.1016/j.cell.2013.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karagoz GE, et al. Hsp90-Tau complex reveals molecular basis for specificity in chaperone action. Cell. 2014;156:963–74. doi: 10.1016/j.cell.2014.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dekker C, et al. The crystal structure of yeast CCT reveals intrinsic asymmetry of eukaryotic cytosolic chaperonins. EMBO J. 2011;30:3078–90. doi: 10.1038/emboj.2011.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Munoz IG, et al. Crystal structure of the open conformation of the mammalian chaperonin CCT in complex with tubulin. Nat Struct Mol Biol. 2011;18:14–9. doi: 10.1038/nsmb.1971. [DOI] [PubMed] [Google Scholar]
- 17.Elad N, et al. Topologies of a substrate protein bound to the chaperonin GroEL. Mol Cell. 2007;26:415–26. doi: 10.1016/j.molcel.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Albert A, et al. Structure of GroEL in complex with an early folding intermediate of alanine glyoxylate aminotransferase. J Biol Chem. 2010;285:6371–6. doi: 10.1074/jbc.M109.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quan S, et al. Genetic selection designed to stabilize proteins uncovers a chaperone called Spy. Nat Struct Mol Biol. 2011;18:262–9. doi: 10.1038/nsmb.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friel CT, Smith DA, Vendruscolo M, Gsponer J, Radford SE. The mechanism of folding of Im7 reveals competition between functional and kinetic evolutionary constraints. Nat Struct Mol Biol. 2009;16:318–24. doi: 10.1038/nsmb.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Figueiredo AM, Whittaker SB, Knowling SE, Radford SE, Moore GR. Conformational dynamics is more important than helical propensity for the folding of the all alpha-helical protein Im7. Protein Sci. 2013;22:1722–38. doi: 10.1002/pro.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stull F, Koldewey P, Humes JR, Radford SE, Bardwell JC. Substrate protein folds while it is bound to the ATP-independent chaperone Spy. Nat Struct Mol Biol. 2016;23:53–8. doi: 10.1038/nsmb.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwon E, Kim DY, Gross CA, Gross JD, Kim KK. The crystal structure Escherichia coli Spy. Protein Science. 2010;19:2252–2259. doi: 10.1002/pro.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quan S, et al. Super Spy variants implicate flexibility in chaperone action. Elife. 2014;3:e01584. doi: 10.7554/eLife.01584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Creamer LK, Richardson T, Parry DA. Secondary structure of bovine alpha s1- and beta-casein in solution. Arch Biochem Biophys. 1981;211:689–96. doi: 10.1016/0003-9861(81)90505-1. [DOI] [PubMed] [Google Scholar]
- 26.Chak KF, Safo MK, Ku WY, Hsieh SY, Yuan HS. The crystal structure of the immunity protein of colicin E7 suggests a possible colicin-interacting surface. Proceedings of the National Academy of Sciences. 1996;93:6437–6442. doi: 10.1073/pnas.93.13.6437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pashley CL, et al. Conformational Properties of the Unfolded State of Im7 in Nondenaturing Conditions. Journal of Molecular Biology. 2012;416:300–318. doi: 10.1016/j.jmb.2011.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burling FT, Weis WI, Flaherty KM, Brunger AT. Direct observation of protein solvation and discrete disorder with experimental crystallographic phases. Science. 1996;271:72–77. doi: 10.1126/science.271.5245.72. [DOI] [PubMed] [Google Scholar]
- 29.van den Bedem H, Dhanik A, Latombe JC, Deacon AM. Modeling discrete heterogeneity in X-ray diffraction data by fitting multi-conformers. Acta Crystallographica Section D-Biological Crystallography. 2009;65:1107–1117. doi: 10.1107/S0907444909030613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salmon L, et al. Capturing a dynamic chaperone-substrate interaction using NMR-informed molecular modeling. Journal of the American Chemical Society. 2015 doi: 10.1021/jacs.6b02382. Under Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brennan S, Cowan PL. A Suite of Programs for Calculating X-Ray Absorption, Reflection, and Diffraction Performance for a Variety of Materials at Arbitrary Wavelengths. Review of Scientific Instruments. 1992;63:850–853. [Google Scholar]
- 32.Karanicolas J, Brooks CL., III The origins of asymmetry in the folding transition states of protein L and protein G. Protein Science. 2002;11:2351–2361. doi: 10.1110/ps.0205402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jewett AI, Shea JE. Folding on the chaperone: Yield enhancement through loose binding. Journal of Molecular Biology. 2006;363:945–957. doi: 10.1016/j.jmb.2006.08.040. [DOI] [PubMed] [Google Scholar]
- 34.Bardwell JC, Jakob U. Conditional disorder in chaperone action. Trends Biochem Sci. 2012;37:517–25. doi: 10.1016/j.tibs.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quan S, Hiniker A, Collet JF, Bardwell JC. Isolation of bacteria envelope proteins. Methods Mol Biol. 2013;966:359–66. doi: 10.1007/978-1-62703-245-2_22. [DOI] [PubMed] [Google Scholar]
- 36.Fischer M, Shoichet BK, Fraser JS. One Crystal, Two Temperatures: Cryocooling Penalties Alter Ligand Binding to Transient Protein Sites. Chembiochem. 2015;16:1560–4. doi: 10.1002/cbic.201500196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr. 2011;67:271–81. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–42. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D-Biological Crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallographica Section D-Biological Crystallography. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica Section D-Biological Crystallography. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.3r1. 2010. [Google Scholar]
- 43.Pettersen EF, et al. UCSF chimera - A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 44.Afonine PV, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallographica Section D-Biological Crystallography. 2012;D68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophysical Journal. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.