

Abstract
Coarse-grained features of macromolecular assemblies are understood via a set of order parameters (OPs) constructed in terms of their all-atom configuration. OPs are shown to be slowly changing in time and capture the large-scale spatial features of macromolecular assemblies. The relationship of these variables to the classic notion of OPs based on symmetry breaking phase transitions is discussed. OPs based on space warping transformations are analyzed in detail as they naturally provide a connection between overall structure of an assembly and all-atom configuration. These OPs serve as the basis of a multiscale analysis that yields Langevin equations for OP dynamics. In this context, the characteristics of OPs and PCA modes are compared. The OPs enable efficient all-atom multiscale simulations of the dynamics of macromolecular assemblies in response to changes in microenvironmental conditions, as demonstrated on the structural transitions of cowpea chlorotic mottle virus capsid (CCMV) and RNA of the satellite tobacco mosaic virus (STMV).
Graphical abstract
I. INTRODUCTION
A hallmark of macromolecular assemblies is the emergence of collective modes from rapidly fluctuating atomistic degrees of freedom (DoF). These systems exhibit dual macroscopic/microscopic behavior thereby reflecting the interplay of equilibrium and nonequilibrium processes across multiple time and length scales. Biologically relevant examples that display such coupling include processes affecting the structure and dynamics of macromolecular assemblies like viruses, ribosomes, liposomes, and intracellular granules.1–4 These systems are typically composed of multimillion atoms. They function on length scales of nanometers involving processes that occur on time scales ranging from nanoseconds to milliseconds. While molecular dynamics (MD) has been widely used to simulate macromolecular structures at an atomistic level, the simulation time for large nanoscale assemblies has been limited to tens or sometimes few hundreds of nanoseconds.5,6 Feasibility of such simulations also depends on the extent of computing resources available. Recently, billion atom MD simulations have been accomplished.7,8 However, these simulations neglect Coulomb interactions, bonded forces, and the rapidly fluctuating hydrogen atoms. All the latter are central to biomolecular structure and dynamics. Thus, capturing bionanosystem behavior across diverse temporal and spatial scales presents great challenges in structural biology, fundamental mathematics and physics, and theoretical and computational chemistry.
Significant effort has been devoted to reduce the dimensionality of many-atom systems and accelerate their simulations by projecting the equation of motion in a low dimensional space.9–13 This is accomplished via modeling a system in terms of its collective DoF. The number of such DoF is often much lesser than the all-atom ones. Thus, macromolecular models based only on collective modes involve tracking much smaller number of dynamical variables than the all-atom description. Consequently, computational cost of implementing these reduced dimensional models is moderate. A list of relevant approaches would include bead- and shape-based coarse-graining models,14–16 rigid region decomposition,17 symmetry constrained18 and curvilinear coordinate19 models, as well as principal component analysis (PCA)20,21 and normal-mode analysis guided approaches.22,23 These models have been successful in investigating structural transitions in a very rich set of biomolecular systems including BPTI, lysozyme, ligand-binding proteins,24 trans-membrane proteins,25 RNA segments,20,26 GRoEL,27 and viral protein capsids of different symmetries.15,28 However, they suffer from one or more of the following difficulties in the context of bionanosystem simulations: (1) characteristic variables are not slowly varying in time, (2) nonlinear motions like macromolecular twist is not readily accounted for, (3) internal dynamics, and hence inelasticity of collisions is neglected, (4) symmetry-breaking processes cannot be accounted for, (5) the forces involved must be calibrated for most new applications, and (6) generating intermediate all-atom trajectories for “on-the-fly” dimensionality reduction becomes very expensive for large systems.
A coarse-grained theory of macromolecular assemblies is statistical in character since specifying the coarse-grained variables leaves great uncertainty in the detailed all-atom state. Thus, the theory should provide an algorithm for evolving the coarse-grained variables and another for coevolving the probability of the detailed all-atom states. Acknowledging this multiscale perspective, we have discovered novel theoretical techniques that probe the cross-talk across scales in time and space, yet preserve the key all-atom aspects of the assembly dynamics.29–35 The result is a set of stochastic equations for the evolution of coarse-grained variables, and those for constructing the coevolving probability of the all-atom states.
This is achieved via the introduction of a set of order parameters (OPs) that describe the overall organization of a system. These OPs capture changes in symmetry that follow large-scale structural transitions. Such transitions emerge from the interplay of long-range organization and order-destroying effects of thermal fluctuations. To account for this cross-talk between variables on different time and length scales, OPs enable generation of an ensemble of all-atom states. This ensemble, in turn, affects evolution of the overall structure through the thermal forces and diffusivities. Thus, emergence of new structures resulting from changes in conditions imposed on the macromolecular assemblies is probed. All these properties are critical for the practical implementation of a multiscale molecular dynamics/order parameter extrapolation (MD/OPX) approach32,36,37 and more recently a fully self-consistent multiscale approach and software deductive multi-scale simulator (DMS).38 These approaches have captured polyalanine folding from a linear to a globular state,39 Ostwald’s ripening in nanocomposites,31 pathways of structural transition and disassembly in virus capsids,38 counterion induced collapse in viral RNA, and stability of RNA–protein complexes.33
In this article we review several examples of OPs in the context of modeling macromolecular assemblies (section II.A). A special class of macromolecular OPs, notably the space warping ones, is shown to account for slow collective DoF relegating the high frequency fluctuations to all-atom ensembles (section II.B). An attempt is made to place these coarse-grained variables within the classic notion of OPs that indicate symmetry breaking in phase transition theory40 (section II.C). This analysis further reveals the physical significance of several of the space warping OPs (section II.D). These OPs serve as the basis of a multiscale simulation algorithm that captures the slow dynamics of macromolecular assemblies simultaneously preserving all-atom details (section II.E). In this context, the applicability of these OPs is compared to that of the PCA modes (section II.F). The OPs are used to simulate structural transitions in cowpea chlorotic mottle virus (CCMV) capsid and the free and protein bound states of RNA in satellite tobacco mosaic virus (STMV) over a range of temperature and salinity (section III). These simulations yield key insights on macromolecular structural transitions and identify a regime of physical conditions over which the OP mediated multiscale simulations are applicable.
II. METHODOLOGY
A. Types of Order Parameters
OPs are coarse-grained variables characterizing the large-scale spatial organization of a system. Several types of OPs have been identified. Examples and the phenomena they have been used to describe are as follows.
Scaled coordinates: Collective and single-particle behaviors in quantum systems,41 and scaled center-of-mass coordinates for proteins in macromolecular assemblies.42,43
Curvilinear coordinates: Macromolecular conformational dynamics.19
Density-like variables: Release of drug molecules from a nanocapsule, the dynamics of enveloped viruses,35 and liquid crystal phase transitions.44,45
Space warping parameters: Overall size, shape and state of deformation of viruses and other macromolecular assemblies.30,33,34
Subsystem OPs: The asymmetric motions of different subunits of a complex macromolecular assembly.31,35
Hierarchical OPs: Disassembly/collapse dynamics of the icosahedral or other structures of viruses.46
Other examples of OPs in the theory of macromolecular structures commonly used are system diameter, end-to-end distance, radius of gyration, solvent accessible surface area (SASA), and measure of similarity to a reference structure in molecular biophysics (e.g., root-mean-square deviation of atomic positions between simulation and reference structures). However, these do not form a complete set, facilitate the construction of all-atom configurations, or evolve much slower than the typical time scale of atomistic fluctuations. Therefore, unlike the space-warping variables, these OPs cannot underlie a multiscale methodology (section II.E). Furthermore, space-warping OPs subsume the slowly varying parameters from the aforementioned list.33 A central property of the space warping OPs is that they evolve slowly. The origins of slowness include (a) inertia associated with the coherent dynamics of many atoms evolving simultaneously, (b) migration over long distances, (c) stochastic forces that tend to cancel, and (d) presence of high free energy barriers involving collective motion. In the following, discussion on structural space warping OPs is extended.
B. Construction of Space Warping Order Parameters
We construct space warping OPs to capture coherent motions of many-atom systems as follows. Consider a macromolecular assembly described via the positions of its N constituent atoms labeled i = 1,…, N. In our approach, is related to a reference position . Deformation of space taking to is continuous and is used to introduce OP via the transformation
| (1) |
where denotes the kth OP and the factor uki is defined in terms of a basis function for reference position of atom i. Index k labeling the is a set of three integers {k1k2k3} such that is a product of Legendre polynomials of orders k1, k2, k3 for the X, Y, Z components of respectively. As the change, space is deformed, and so do the macromolecules embedded in it. Since we seek a dimensionality reduction, the number of is much less than the number N of atoms. Thus, we take a finite truncation of the k sum in (1); this necessitates introduction of a residual (denoted ) to correct the coherent deformation generated by the . With this
| (2) |
An explicit expression for the is obtained by minimizing the mass-weighted square residuals with respect to the .32 This yields
| (3) |
With the above formulation, dimensionality reduction for many-atom systems from the N-atom configuration to M OPs is achieved through
| (4) |
Next, we make use of the Liouville equation to elucidate the rate of OP dynamics. The Liouville operator is defined , where and are the momentum of and net force on atom i. Given eq 3, one may compute as . This yields
| (5) |
By definition, the basis functions uki vary smoothly across the system. Thus, near equilibrium, linear combinations of rapidly fluctuating atomic momenta tend to cancel, thereby reducing the rate of OP evolution. As a result, these OPs can be extrapolated over longer periods in time relative to individual atoms. For the various choices of k, evolution of the corresponding tracks collective (correlated) motions on different time scales. With this, eq 5 provides mathematical basis for the simulation approach implemented in MD/OPX.32
C. Role of Symmetry
The concept of OPs originated in the theory of phase transitions. In that context, they are variables that are zero in one phase (usually above the critical point), and nonzero in another.47 More generally, they change discontinuously across a first-order transition and their derivatives change across a second-order case indicating a change in the physical state of the system. For example, a magnetic system above the Curie temperature is isotropic, but anisotropy emerges below this temperature as atomic-scale magnets tend, on the average, to have a preferred direction.40 Net magnetization of the system serves as an OP capturing the emergent order of atomic-scale magnets that underlie this symmetry breaking transition. Similarly, the space-warping OP here provides a framework that captures the emergent order observed in viruses and other macromolecular assemblies under appropriate conditions in the host medium. For example, if is independent of i then the OP is proportional to the center-of-mass of the assembly.46 Some of the space warping OPs defined in this way constitutes a strain tensor accounting for compression-extension-rotation, while others describe more complex deformations like tapering, twisting and bending (section II.D). Furthermore, the effective OP masses, μk as defined in eq 3, decrease for larger values of k. This suggests that OPs with higher k probe deformations of smaller regions in space. Thus, a model based on this set of OPs captures a spatially diverse range of coherent deformations through the various choices of k. Thereby, such a model can describe the emergence of structural order and accompanying symmetry changes in macromolecular systems.
A typical example of macromolecular structural transition that resembles classical phase transition from isotropic to anisotropic states is now discussed. Consider a nanoscale assembly consisting of a spherical nanocore with a number of surface-attached viral capsid protein pentamers. In the absence of nanocore, pentamers often self-assemble into icosahedral structures of different T-numbers depending on the host medium conditions.28 However, in the presence of the nanocore, arrangement of the surface-attached proteins may differ dramatically from those of the icosahedral structures in a nanocore free solution.48 In particular, symmetry of the surface assemblies can be strongly influenced by the size and surface properties of the nanocore. If the core particle is of radius equals to the cavity of a T = 1 structure, then the pentamers would likely assemble in a T = 1 symmetry.48 However, if the core particle diameter is increased beyond the cavity size (but less than the diameter of the next T number structure) then a point is reached wherein bare spots appear on the core. Consequently, some preferred pentamer-to-neighbor-pentamer interactions are lost and these pentamers undergo Brownian motion over a range of temperatures. With this, the orientational order defining a T = 1 icosahedron can be disturbed and a symmetry breaking transition resulting in an assembly of randomly oriented pentamers is realized.
The above example can be placed more explicitly in the context of the classic relationship between symmetry breaking and emergence of OPs as follows. First, express the distribution of the pentamer density in terms of spherical harmonics and associated weights. At high temperature, the symmetric structure of pentamers is lost and hence the surface density is uniform (wherein weights of all harmonics except those for the lowest order ones are zero). Depending on the structure that emerges, at lower temperature, the weights of certain harmonics can depart from their values in the uniform state. In this case, weights of the spherical harmonics following the emergent geometry pattern (e.g., T = 1 icosahedron) serve as OPs in a manner similar to that used in the theory of ferromagnetism. When are spherical harmonics, the in eq 3 provides a way to capture this transition. From this example it is seen, the space warping OPs are directly analogous to those appearing in the classical theory of phase transition for cases where simple symmetry can be identified.
The classical phase transition theories, like that for magnetization, are built on the properties of infinite systems, e.g., renormalization group concepts.49 In contrast, macromolecular assemblies are finite, in fact small in extent and hence cannot completely follow the theory of macroscopic phase transitions. Furthermore, macromolecular assemblies can be in conformational states without a simple, readily identifiable symmetry, e.g., ribosomes. Nonetheless, as pH and other conditions in the host medium change, the system can switch to a different conformation.50 Such a system experiences structural transition between two states, neither of which has a readily identifiable symmetry. This suggests that often OPs in macromolecules cannot be readily associated with the breaking of symmetry even if they signify a dramatic change of order. Therefore, other metrics are required to signal the emergence of new order in macromolecular systems when there are no readily identifiable symmetries involved.
For the space warping OP formulation of eq 3 such metrics include
The onset of long-time tails in correlation functions of OP momenta indicating the coupling of a proposed set of OPs to emergent ones.46
The systematic growth of residuals in eq 2 during structural transitions indicating the emergence of new types of organization from rapidly fluctuating atomistic processes.46
Numerical procedures for constructing the emergent OPs are provided in section III. With this, the space warping OPs can be used for the classical symmetry breaking type transitions in macromolecular systems and also provide an approach in cases where symmetries are not readily identifiable.
D. Structural Characterization of Macromolecular Systems
Macromolecular assemblies display a rich array of structural organizations. For example, simple nonenveloped viruses often show an icosahedral arrangement of pentameric or hexameric protein subunits. However, under certain conditions they may exist in capsular, twisted and conical shapes.28 Transition between these structures occurs via symmetry breaking pathways that account for changes in overall assembly organization. As shown below, the set of space warping OPs is rich enough that many types of symmetry are embedded in them. As in nature, observed symmetries for a given system emerge due to the underlying interatomic forces. This feature of the present approach enhances the capability to discover pathways of structural transition or self-assembly.29,31
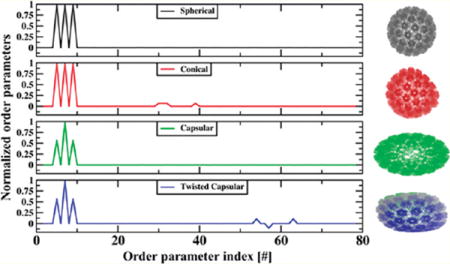
Considering a T = 7 virus capsid as shown in Figure 1, it is demonstrated that specific combinations of space warping OPs capture the deformation of the symmetric T = 7 virus capsid into conical, capsular and twisted capsular forms. Transformations captured via these OPs are understood below in terms of a set of fundamental global and local deformations.51 Consider the example of a tapering deformation (Figure 1). Take Uk, k = 100, 110, 101 to be x0, x0y0, and x0z0, respectively. Neglecting residuals, eq 1 becomes , and similarly for yi and zi (where xi,yi,zi are the three Cartesian components of r⇀ vector and ϕkα is the α-th component of OP ). This relationship can be written in the tensorial form
| (6) |
For the choice of ϕk as specified in Figure 1 (i.e., ϕ100x, ϕ100y, ϕ110x, ϕ110z = 0 and ϕ100x, ϕ110y, ϕ101z, ϕ101y, ϕ101x ≠ 0), J is the Jacobian matrix for tapering along the X axis.51 Thus, these OPs capture structures that are tapered with respect to the reference configuration . Similar matrix transformations can be constructed using other combinations of OPs to explain the twisting and bending transitions of Figure 1. In this context, examples of OPs that enable motions like extension-compression-rotation were provided earlier.39,46 Thus, the space warping variables are coarse-grained in character and, in addition, are rich enough to capture the emergence of overall order and symmetry breaking in macromolecular assemblies. With this, they enable a multiscale methodology as follows.
Figure 1.
Normalized space warping OPs with specified k-indices are shown to deform a T = 7 capsid into conical, capsular, and twisted capsular forms. Cartesian components of only 33 OPs are chosen for this analysis as they are sufficient to capture these transformations.
E. Deductive Multiscale Analysis
Equation 3 implies that for a given set of atomic positions the corresponding OPs are uniquely defined. However, the converse is not true, i.e., there exists multiple all-atom configurations consistent with a given set of OPs . Thus, the OP construction scheme constitutes a many-to-one mapping from the all-atom to the coarse-grained description. As a consequence, the OP description retains overall structural information, losing all-atom details. Atomistic structures are reconstructed via a procedure called deductive multiscale analysis (DMA) that evolves the OPs with an ensemble of all-atom configurations as follows.
The description adapted starts with the probability density ρ of the N atomic positions and momenta Γ. However, this formulation masks the underlying hierarchical organization of a macromolecular assembly. To address this, in DMA ρ is hypothesized to depend on Γ both directly, and via a set of OPs, indirectly. With this ansatz, a multiscale analysis of the Liouville equation yields sets of coupled Langevin equations for the OPs31,52
| (7) |
where τ = ɛ2t and ɛ is a smallness parameter, e.g., ratio of typical atomic mass to that of the entire assembly. The variance of noise is bound by diffusivity . The factors are related to the ensemble average of correlation functions between hierarchical OP momenta via
| (8) |
where μk is the effective mass associated with ϕk as defined in eq 3 and is the lowest order Liouville operator. The thermal-average force is given by
| (9) |
for OP-constrained Helmholtz free-energy F, where
| (10) |
Q(ϕ,β) is the partition function constructed from configurations consistent with the set of (denoted ϕ collectively).
In the above formalism, thermal average forces are constructed at each Langevin time step via Monte Carlo integration of atomic forces obtained from interatomic force fields (e.g., CHARMM53). Note that this does not require any assumption on the form of the dependence of the thermal average forces on the OPs; this is automatically incorporated because the all-atom ensembles used to carry out the thermal averaging are constructed for the values of the OPs at the given Langevin time step. Furthermore, this formalism accounts for the full impact of fluctuations as the random forces in the Langevin equations are constructed to be consistent with the diffusivity factors .
The thermodynamic forces involved are small so that the state is captured by the slowly varying OPs. As the system evolves toward equilibrium the thermodynamic forces vanish and the system is fluctuation dominated. Both these cases are accounted for in our multiscale OP approach since the intensity of fluctuations is chosen to be consistent with the diffusion factors. Finally, if the thermodynamic forces are extremely large then the time scale of OP evolution approaches that of atomistic fluctuations. With this, the time scale separation between fast and slow variables is violated and the theory does not hold.
Inherent in our DMA approach is the capability to reconstruct atomistically resolved states given the evolving coarse-grained dynamics. Any coarse-grained theory carries an inherent uncertainty in the fine scale states.54 DMA addresses this by providing the conditional probability density for the atomistic configurations given the instantaneous values of the OPs. These configurations are generated by a procedure denoted hybrid sampling in which the all-atom structures are reconstructed from the coarse-grained description via randomly varying the residuals at constant values of OPs (eq 2).34 Therefore, the OPs constrain the ensemble of atomic states, while the latter determine the thermal average forces and diffusivity factors that control OP evolution (eq 7). Thus, the OPs imply a multiscale simulation algorithm that accounts for cross-talk between coarse-grained and atomic DoF. All these ideas are implemented as the DMS software.
F. Use of PCA and Legendre Basis Functions for Order Parameter Construction
Bionanosystems undergo large-scale conformational changes involving collective motions of strongly interacting clusters of atoms. Such structural changes often correspond to the functionally relevant motions of macromolecular systems.4,21 Collective DoF can be observed through calculating the autocorrelations of the normalized 3N-dimensional atomic displacement vectors from consecutive time windows as
| (11) |
where denotes the normalized 3N-dimensional atomic displacement vector during the l-th time window of size δt for l = 1,…, L.
There is a variety of coarse-grained variables in the literature that are used to analyze coherent motions in biomolecular systems (section I). Here, we test the suitability of using the basis vectors obtained from PCA for constructing the slow variables used in the multiscale analysis of section II.E. To address this issue, consider using the PCA modes (or eigenvectors) as the basis functions for constructing OPs (see the Appendix). Correlations between the PCA derived basis functions Uk are compared to those of the Legendre polynomial ones uki at selected time intervals. Strong correlation in the long time behavior of these variables implies slow evolution of the functions used to construct the basis vector which, in turn, underlies coherent OP behavior. With this, define a correlation matrix C for the basis vectors via
| (12) |
where denotes the kth PCA or Legendre basis vector (k = 1,…, M) from the l-th time window (l = 1,…, L). In analogy with the PCA modes, the Legendre polynomials are constructed here using atomic displacements of a given time interval tl (and not absolute positions as in eq 1, Appendix). The average structure required to calculate these PCA modes is changed with each time window, and similarly for the reference coordinates needed to construct OPs. This treatment of the Legendre polynomials enables a fair comparison with PCA modes in the context of analyzing basis function behavior as both variables are then expressed in terms of atomic displacements for a given time interval. This particular construction of uki is used for PCA comparisons only.
III. RESULTS AND DISCUSSION
A. Suitability of Coarse-Grained variables for Deductive Multiscaling
PCA has been applied to reduce the dimensionality of MD trajectories for analyzing large-scale structural changes.21,55 Their behavior is compared with that of the space warping OPs for dimensionality reduction and deductive multiscaling. For demonstration, we choose the CCMV capsid as our model system because of its extensively studied structural transition phenomena.56,57 The crystal structure of wild-type CCMV is solved at 3.2 Å resolution by X-ray crystallography.57 Its capsid consists of 180 chemically identical protein subunits that form a 286-Å-diameter icosahedral shell with a T = 3 quasi-symmetry.
Macromolecular assemblies (e.g., virus capsids) evolve in stochastic fashion, and therefore simulating their long-time behavior should account for an ensemble of atomic positions and momenta. Thus, an analysis aimed at extracting low-frequency modes (e.g., space warping OPs or PCA) from MD should in principle be performed with an ensemble of trajectories. Here, 30 replica NAMD58 runs with different random atomic velocity initializations are executed starting with the swollen CCMV capsid. Since the characteristic time of OP evolution is expected to be ∼O(N) times longer than that of atomistic fluctuations (see eqs5 and 7), each MD trajectory is run for 105 fs (100 ps) for investigating the low-frequency modes.
While it is ideal to study the time evolution of all atoms in the system, it is impractical to perform PCA diagonalization of the large 3N × 3N atomic positional covariance matrix for the 400 000 atom capsid. Thus, only data on 10% of Cα atoms (every tenth residue, 2940 in total) in the CCMV capsid backbone is extracted for our PCA study. We divide the 100 ps ensemble-averaged trajectory of swollen CCMV capsid into 10 time windows, each of which is input to the MD analysis program carma59 for PCA. Every calculation yields 8,820 eigenvectors and their eigenvalues. The first 20 PCA eigenfuctions are found to capture about 95% of structural changes in swollen CCMV capsid. We then choose these 20 modes from each time window and calculate their correlation matrix C from consecutive time windows using eq 12 to compare their similarities and thereby monitor coherence.
Figure 2a shows dot products of atomic displacement vectors , i.e., the Δ matrix expressed via eq 11 using 10 ps time windows. The overlap of from neighboring time windows (the upper and lower diagonal entries) are found to be significant (about 0.6). As the distance between time windows increases, the autocorrelations decrease gradually to about 0.2. These correlations imply that the capsid undergoes slowly evolving collective motions over time intervals of 10 ps or longer. Figure 2b shows the correlation matrix C of PCA basis vectors from the first two consecutive time windows calculated using eq 12. These PCA vectors are found to display small overlapping with their dot products distributed between −0.02 and +0.02. Basis vectors from other sets of consecutive time windows also give similar results. In contrast, Legendre polynomials of atomic displacements display significantly larger correlation between the basis vectors of the two consecutive time windows (similar values as the correlations of Δ) compared with that of the PCA (see Figure 2c). The antidiagonal entries have switching positive and negative values because of the odd and even properties of Legendre polynomials used.
Figure 2.
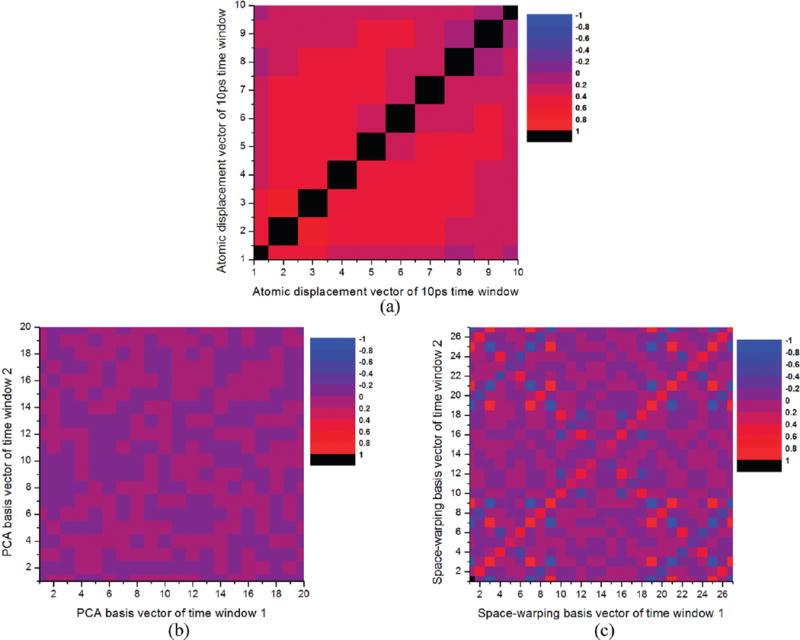
Correlation matrices of (a) atomic displacement vectors of the ten consecutive time windows of 100 ps ensemble-averaged trajectory and (b) PCA and (c) space warping basis vectors from the first two consecutive time windows. Twenty PCA and 27 Legendre basis vectors are used as they keep the residuals σi in eqs A2 and A3 small.
Space warping basis vectors constructed with Legendre polynomials of atomic displacements at consecutive time intervals display large similarities implying coherence of the basis vectors uki as shown in Figure 2c, which, in turn, reflects the slowly evolving nature of the collective motions illustrated in Figure 2a. This suggest that the OPs constructed from these basis vectors are suitable to project system configuration over long time (10 ps or longer). Therefore, they can serve as a basis for the multiscale analysis introduced in section II.E. The comparison of Legendre with PCA derived basis vectors implies that the latter capture correlated motions on time scales lesser than 10 ps, not suitable to project system configuration over long time, and do not manifest the coherence in evolution required to construct OPs for deductive multiscaling and related simulations. Thus, temporal scales captured through these PCA modes are shorter than those described by OPs constructed from the Legendre polynomials of Figure 2. In the deductive multiscale formalism however, such local motions are captured through the construction of all-atom ensembles that probe events on time scales much smaller than that of OP evolution (section II.E). Alternatively, incorporation of more OPs in the coarse-grained description enables capturing short scale collective motions32 (as discussed in sections III.B and III.C). Thus, the space warping OPs when embedded in our multiscale approach together with the implied quasi-equilibrium ensemble of OP constrained atomic configurations capture dynamics on shorter, similar, as well as longer scales than PCA modes. As a result, the OPs provide an ideal and versatile reduced description that enables the deductive multiscaling of N-atom macromolecular assemblies.
B. Symmetry Breaking and CCMV Capsid Structural Transition
The slowly evolving nature of OPs suggests a multiscale MD/OPX approach to simulate bionanosystem dynamics.32,36,37 In this approach, a set of replica MD runs with different velocity initializations is used to estimate the rate of OP change. Since OPs evolve coherently, they are readily extrapolated over long time periods using this rate through eq 5. Thus, the slow overall dynamics of the system is simulated. At every time step, the replica MD runs are repeated to reassess the rate of OP change that enables further extrapolation in time.
Biomolecular systems of nanoscale size provide ideal examples for illustrating the effectiveness of an OP based simulation methodology. MD/OPX was used to simulate the shrinkage of a swollen CCMV capsid in vacuum for 200 ns.36 33 OPs were constructed by using Legendre polynomials of atomic coordinates over order (0, 1, 2) in X, Y, and Z directions. Low-order Legendre polynomials were selected because they vary smoothly in space and thus can capture the overall nanoscale deformation of the virus capsid. Thirty 500 fs replica MDs were used for calculating the rate of OP evolution and extrapolating them to evolve the system. As the OPs are slowly varying in time, the length of MD runs used to compute the rate of their change are typically much smaller than the extrapolation time step. With this, adaptive timesteps in the range of 50–60 ps are achieved to extrapolate the OPs. These are 4 orders of magnitude larger than the typical MD timesteps (e.g., 1 fs). As shown in Figure 3a, the CCMV shrinkage was found to be an energy-driven, symmetry-breaking process that involves large-scale translation and rotation of pentamers and hexamers in the capsid. The capsomeres undergo cooperative motions through strongly coupled allosteric interactions during shrinkage. As a result, this viral structural transition starts locally and then propagates across the capsid, i.e., they proceed via intermediate states that are not constrained to the icosahedral symmetry of the initial and final states (i.e., T = 3).
Figure 3.
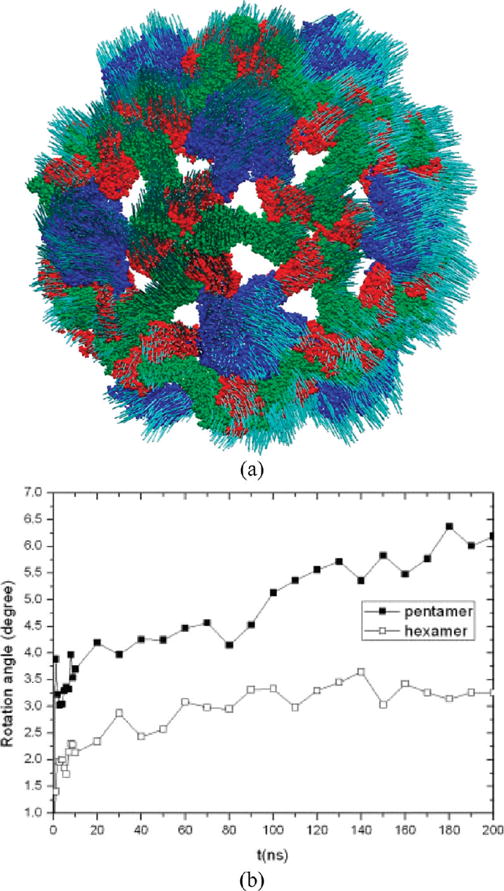
200 ns MD/OPX simulation of the shrinkage of swollen CCMV capsid: (a) CPK representation of Cα atoms of the resulting structure with arrows indicating the atomic displacements from their original positions in the starting PDB structure to their final positions and (b) time courses of the average rotation angle for pentamers and hexamers calculated via superimposing their structures to the initial configurations.
Variables that capture the symmetry breaking nature of this CCMV capsid structural transition are the hexamer and pentamer orientation angles (Figure 3b). Their values change gradually during capsid shrinkage, indicating change in orientational order of the capsid subsystems. Insight into the nature of transitions involving these variables can be gained via analogy with the ferromagnetic phase transitions. For example, magnetic orientational order changes as temperature changes across the Curie value. Similarly, the capsid structural transition involves capsomere rotation as the pH and thus the protonation state of protein residues in the system changes.37,56 However, unlike traditional examples in ferromagnetic transitions where the crystal structure remains intact, the orientational reordering of capsomeres accompanies shrinkage of the T = 3 capsid. This behavior, in turn, is similar to ferromagnetic phase transitions in alloys that simultaneously involve negative expansion of lattice parameters.60 Thus, transitions involving change in capsomere orientations for a fixed T-number could occur as analogues to the change in lattice structure accompanying some ferromagnetic transitions.61 Such structural transitions are also observed in molecular solids and liquid crystals44 where eularian angles are used as orientational OPs.
C. Structural Transition between States of Uncertain Symmetry
Traditionally, OPs are used to characterize macroscopic phase transitions wherein symmetry breaking is usually apparent since the symmetries of the states on either side of the transition are readily identified. While transitions in macromolecular systems often demonstrate large-scale structural reorganization, the symmetries involved can be less apparent (section II). Here, we use DMS38 simulations to illustrate new OP related metrics that signal the change of order in macromolecular systems without readily identifiable symmetry. These simulations are based on the two-way transfer of structural information between OPs (coarse-grained description) and atomistic configurations (characterized by quasi-equilibrium probability densities) as described in section II.E. The demonstration system, RNA of STMV, is chosen because it (a) has interesting complexity, containing 949 nucleotides arranged in 30 double stranded helical stems joined via single-stranded loops, (b) is highly flexible, (c) expresses nonlinear motions and, (d) unlike CCMV, does not have a readily identifiable initial symmetry.62 With this, applicability of the space warping OPs of section II as coarse-grained variables that efficiently probe highly complex motions is tested.
The initial state of the STMV RNA is taken to be when it resides inside the capsid with associated protein subunits. The simulated evolution follows after the capsid is removed instantaneously. In a 1:1 electrolyte like NaCl, the RNA initially expands, then shrinks due to electrostatic shielding by the diffusive counterion cloud, and finally fluctuates among atomistic states of similar energy.33 All-atom configurations resulting from this simulation imply that the RNA tertiary structure is highly disrupted, although secondary structures remain after 50 ns.33 In contrast, in a 2:1 electrolyte like MgCl2, Mg2+ ions tightly bind to the RNA. Thus, secondary and tertiary structure is preserved during the 25 ns simulation (see Figure S1). This DMS predicted RNA stability in 0.3 M 2:1 electrolyte is in agreement with previous experimental and theoretical predictions6,63 and reconfirmed here with NAMD simulation results at 310 K (Figure S2a).
To further test the robustness of DMS, the RNA simulation in 0.3 M MgCl2 is repeated over a range of temperatures between 310 and 425 K. At each temperature 25 ns all-atom trajectories are obtained. Details of conditions and parameters used for these simulations are listed in Tables S1 and S2 in the Supporting Information. Increase in temperature results in the emergence of new collective motions. To capture these motions, OPs that were not included to the initial set of collective variables are added to the reduced description and subsequently evolved via Langevin equations (see section SI2 of the Supporting Information for details). Such OPs, denoted corresponds to values of k for which nonlinear local motions like tapering, twisting and bending are probed (Figure 4b). This illustrates that new space warping OPs can be readily added to capture complex motions as they emerge in response to change changes in the microenvironment.
Figure 4.
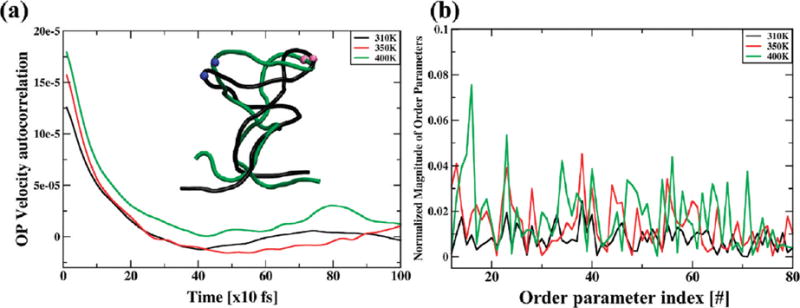
(a) Velocity autocorrelation functions for a typical OP (Φ100X). Absence of a long-time tail indicates lack of coupling to other slow variables not included in the set of OPs. As temperature increases, so does the area under a curve, implying greater OP diffusion. (b) Appearance of new OPs (k = 2 and 3 here) indicates emergence of collective motions as temperature increases. Some of these OPs correspond to those for twisting and tapering (Figure 1), thereby capturing the emergence of nonlinear deformation about thermo-labile centers of the RNA helix (inset). These emergent OPs are obtained from the population of growing residuals as described in Figure S3 in the Supporting Information.
Tracking the decay characteristic of OP velocity autocorrelation functions calculated by using eq 8 provides a criterion of completeness for a given set of OPs. In particular, appearance of long time tails in these correlation functions typically suggests there are additional OPs that couple to the set considered originally.33,46 As these “missing” modes are accounted for via the introduction of additional OPs ( ), the corresponding OP velocity autocorrelation functions rapidly decay indicating completeness of the reduced description (Figure 4a). With this, the growth of residuals (Figure S3) and emergence of long tails in correlation functions (manifested during the construction of all-atom ensembles via hybrid sampling as described in section II.E) indicate the necessity of additional OPs that capture a shift in structural organization. Therefore, these quantities serve as OP related metrics that signal the change of order in macromolecular systems even if symmetry breaking is not readily identifiable.
The DMS results at 310 K and 425 K are benchmarked with those of 5 ns NAMD simulations. The OP trajectories obtained from these simulations are found to be in agreement (Figure S2); this validates the applicability of DMS simulations over a biologically relevant range of temperatures. However, as temperature increases the time scale of OP evolution decreases. This limits the size of Langevin OP timesteps and, subsequently, the applicability of the noninertial Langevin equations that underlie DMS simulations. Thus, simulations in the 310–425 K range are carried out using DMS; higher temperature simulations are performed using NAMD to capture the inertial regime that restricts DMS applicability (section SI2, in the Supporting Information). Comparison with MD at multiple temperatures ensures that DMS predicted results are free from artifacts. This also confirms that the predicted temperature behavior of the RNA is not affected by the switch in simulation methodology (DMS to MD) at 450 K.
The present investigation of RNA dynamics over a range of temperatures provides insights into a macromolecular phase transition. The essence of this transition is seen in Figure 5 where ensemble average values of three selected OPs are plotted versus temperature. The temperature-dependent behavior of average OP values suggests a phase transition type phenomenon occurs between 475 and 500 K. OPs undergo a drastic change in magnitude across this transition region (Figure 5). Observed increase in OPs implies expansion of the RNA structure. Thus, space warping OPs indicate a substantial shift in order between states of RNA in which symmetries are not distinct. Since these OPs capture overall RNA structure, analogous transition behavior is observed for other coarse-grained descriptors like the number of hydrogen bonds (Figure 5), radius of gyration (Figure S4), and end-to-end distance.
Figure 5.
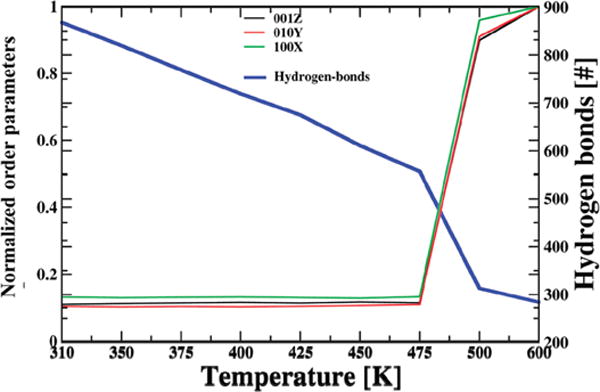
Phase transition behavior of STMV RNA captured via the quasi-discontinuous change of space warping OPs between 475 and 500 K suggesting that these OPs are appropriate indictors of a change in organization even if the nature of symmetry breaking is not apparent. The change in OPs indicates thermal expansion of the RNA. Similar behavior is reflected in the average number of intranucleic acid hydrogen bonds that rapidly decreases across this region.
Most RNAs have a degree of heterogeneity in the nucleotide sequence. With this, one might expect heterogeneous nucleation sites for the transition. i.e., nucleotides are not all equally responsible for the structural changes in RNA. In the present simulations, the 14th and 19th nucleotide (ADE and URA, respectively) in each of the 30 RNA helices were found to be more thermo-labile, i.e., sensitive to changes in temperature. As shown in Figure 6a, enhanced motion of these nucleotides is reflected in significant shifts in their dihedral angles as the temperature changes. A Poisson–Boltzmann evaluation of electrostatic energies64 shows that these two nucleotides are the most stable when RNA is embedded inside the virus capsid (Figure S5). Strong protein-nucleic acid interactions hold dihedral angles neighboring these nucleotides unchanged. However, when the capsid is removed, setting the RNA free as simulated here, the associated dihedrals change appreciably. Consequently, these dihedrals sample a range of values that evolve away from those of the protein-bound RNA (Figure 6a). As the transition zone is entered the motion of these thermo-labile centers increase until a RNA-wide transition is expressed. For example, at these temperatures (475–500 K), the internucleotide hydrogen bond breaks providing additional DoF to the RNA helices (Figure 5). This, together with enhanced mobility of the thermo-labile centers, results in correlated nucleotide motions in their vicinity. Emergence of such motions like twisting or bending (Figure 4) disrupts secondary structure of the double stranded RNA stem and mediates the propagation of an instability front across the entire macromolecule (Figure S6) that transforms all the helices to coils (Figure 6c). Resulting shift in order of the overall RNA structure is indicated by the discontinuous temperature behavior of space warping OPs across the transition region (Figure 4). Thus, the thermo-labile nucleotides provide centers of nucleation that initiate the observed first-order like phase transition between the encapsidated and coiled states of STMV RNA. Root mean square (rms) fluctuations about the mean dihedrals for nucleotides of the RNA helices are presented (Figure 6b). Such fluctuations increase near the transition zone as expected if the analogy to macroscopic phase transitions is to hold.
Figure 6.
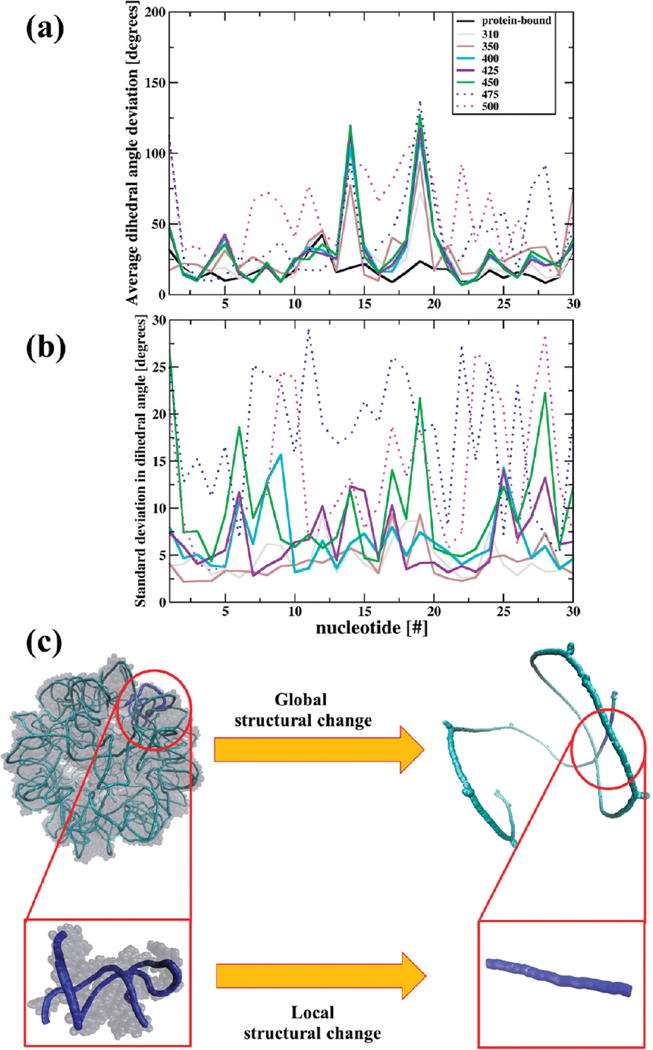
(a) Average deviation of nucleotide dihedral angles from their initial values at t = 0 for 30 nucleotides comprising of the RNA helix for temperatures between 310 and 500 K. At temperatures lower than the transition temperatures, nucleotides 14 and 19 show maximum deviation from their room temperature values. At temperatures in the transition zone, a more RNA-wide motion occurs, as indicated by the increase in dihedral deviation of neighboring nucleotides. In contrast, the protein bound RNA helix is the least mobile. (b) rms fluctuations about mean dihedral angles showing distinct increase with temperature. Dihedral angles are calculated using the AMIGOS73 program. Only η-type dihedrals73 are plotted for illustrative purposes. At each temperature, the ensemble average dihedrals are averaged over all the thirty RNA helices to obtain data for this plot. (c) Global and local changes in the structure of viral RNA accompanying the transition between states of uncertain symmetry.
In the above, the temperature-dependent behavior of STMV RNA is investigated from the space warping OPs, nucleation and fluctuation perspectives. Results suggest a first-order like phase transition occurs between 475 and 500 K. Such behavior reflects the interplay between local and overall structures of RNA. Although there is no experimental data available for the transition temperature of STMV RNA, earlier simulations on smaller RNA loops suggest a melting point between 420 to 430 K.65 Given that the present system is much larger (i.e., 949 versus 14 nucleotides and an order or 2-fold greater number of dinucleotide bonds), the high transition temperature suggested here is not surprising. Besides, significant discrepancies between experimental and theoretical results on macromolecular structure and dynamics are well-known.66 These differences can be attributed to factors such as the finite size of all-atom ensembles constructed at every Langevin time step for computing the thermal forces and diffusions (section II.E), possible incompatibility of force-fields with high temperature simulation,65 discrepancies in thermal expansion coefficient of TIP3P water model,67 and problems with simulating the counterion environment as realized in the experiments.68 Nonetheless, RNA simulations in aqueous medium have been performed for temperatures in the range 300–700 K.65,69 These simulations have provided insights into the thermorigidity and functionality of multiple RNA structures. Even though the above simulation inconsistencies might affect computing exact transition temperatures, the behavior of OPs across transition regions as reported here qualitatively follows previously observed trends.66,69,70 With this, the space warping OPs used for macromolecular assemblies have properties analogous to those used in the theory of macroscopic phase transitions.
Our results also suggest that proteins on the inner surface of STMV capsid greatly enhance the stability of the thermo-labile nucleotides, i.e., suppress motion of the nucleation centers (see Figures 6a and S7). Consequently, viruses can survive in high temperatures (∼400 K) without losing the RNA tertiary structure.71 Thus, the free-energy minimizing structure of a virus can enable it to withstand severe temperatures, as commonly observed. At very high temperatures however, these protein–RNA interactions are lost and so the virus ultimately loses stability.
D. Performance of Multiscale Techniques
Structural transitions in CCMV and RNA of STMV provide examples of phenomenon in which accounting for the cross-talk among multiple time and length scales becomes critical. Since both DMS and MD/OPX are based on the interplay of all-atom and coarse-grained variables, such multiscale methodologies naturally account for this cross-talk. Furthermore, application of these techniques leads to efficient computation of slow processes. For example, MD/OPX simulation with adaptive OP extrapolation timestepping is about 6 times faster than direct MD. Similarly, DMS is found to be about 11 times faster than NAMD at 310 K. This efficiency reduces by 4 fold at 425 K. This is expected as the OPs loose coherence with increase in temperature. However, a direct comparison with conventional MD run is not appropriate. This is because at each OP time step 200–300 all-atom structures consistent with the instantaneous OP values are obtained. Thus, DMS or MD/OPX simulation corresponds to an ensemble of 200–300 conventional MD runs, for which the timecourses of the spacing warping OPs are essentially the same, while the detailed atomistic configuration varies among members of the ensemble. Finally, a single MD run may not be representative of an ensemble of possible time courses, which, in contrast is automatically overcome in the all-atom multiscale approach.
IV. CONCLUSIONS
Space warping OPs capture key elements of symmetry breaking manifested in structural transitions of macromolecular assemblies. Their generality allows them to capture a variety of symmetries that emerge as a consequence of the underlying interatomic forces and conditions to which the system is subjected. The space warping OPs enable a multiscale analysis which accounts for the cross-talk between the coarse-grained and all-atom DoF, and characterizes an ensemble of atomic configurations coevolving with the OPs. In this context, spacing warping OPs together with the multiscale analysis capture a more diverse range of coherent motions than do PCA modes. These ideas are demonstrated via simulations of symmetry breaking accompanying CCMV capsid shrinkage, and a first order structural transition in RNA of STMV. The multiscale methodology presented here is robust to a range of salinity and biologically relevant temperatures. However, high temperature regimes reduce the multiscale simulation performance due to the loss of time scale separation between coarse- and atomic-scale processes. Depending upon conditions, OP-mediated multiscale simulations are much more efficient than conventional MD simulations.
Supplementary Material
Acknowledgments
This research was supported in part by National Science Foundation through the Collaborative Research in Chemistry Program (Grant No. CHE-082651), NIH National Institute of Biomedical Imaging and Bioengineering (Grant No. R21 EB008951), the METAcyt project, and Indiana University College of Arts and Sciences through the Center for Cell and Virus Theory.
APPENDIX
Principal Component Analysis for Dimensionality Reduction
Principal component analysis (PCA) involves diagonalization of the positional covariance matrix for selected atoms (like Cα in protein backbone)
| (A1) |
where xi and xj are atomic coordinates and the 〈⋯〉 denote trajectory averages over a selected time window tl with l = 1,…, L. This generates an orthogonal set of eigenvectors (i.e., basis vectors or modes), denoted U, each associated with an eigenvalue that indicates the amplitude of fluctuations along that eigenvector. Eigenvalues divided by their sum describe relative contributions of the associated eigenvectors to major conformational changes observed in the trajectory. For reduced representation of the system, a subset of PCA eigenvectors with largest eigenvalues is chosen and the number of these eigenvectors M is normally much smaller than the total number of atoms N,21,72 i.e.,U ≡ {Uk, k = 1,…, M} for M ≪ N.
Simple changes in the definition of space warping OPs (eq 1) enable its construction using the PCA basis vectors. To achieve this, space warping OPs are recast in terms of displacements from a mean configuration. This yields
| (A2) |
where are atomic coordinates of the system reference configuration which can be starting structure of a time window and ϕk are the OPs tracking displacement from this structure. Similarly, the Legendre polynomials constructing uki are defined as functions of atomic displacements, (i = 1,…, 3N) from the selected time window tl (section II.B). Introducing these uki into eq 1 yields analogues of A2 in the Legendre basis
| (A3) |
where the factor uki is constructed using functions of the form . This treatment of the Legendre polynomials enables a fair comparison with PCA modes in the context of constructing basis vectors as functions of atomic displacements.
Footnotes
Special Issue: B: Macromolecular Systems Understood through Multiscale and Enhanced Sampling Techniques
Supporting Information
Comparison of DMS and NAMD simulation results with tables listing the simulation parameters, temperature dependence of OP evolution, and the dynamics of STMV RNA at high temperatures. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.Xie A, van der Meer AF, Austin RH. Phys Rev Lett. 2002;88(1):018102. doi: 10.1103/PhysRevLett.88.018102. [DOI] [PubMed] [Google Scholar]
- 2.Donko Z, Kalman GJ, Hartmann P, Golden KI, Kutasi K. Phys Rev Lett. 2003;90(22):226804. doi: 10.1103/PhysRevLett.90.226804. [DOI] [PubMed] [Google Scholar]
- 3.Delarue M, Dumas P. Proc Natl Acad Sci USA. 2004;101(18):6957–62. doi: 10.1073/pnas.0400301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Canady MA, Tsuruta H, Johnson JE. J Mol Biol. 2001;311:803–814. doi: 10.1006/jmbi.2001.4896. [DOI] [PubMed] [Google Scholar]
- 5.Klepeis JL, Lindorff-Larsen K, Dror RO, Shaw DE. Curr Opin Struct Biol. 2009;19(2):120–7. doi: 10.1016/j.sbi.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 6.Freddolino PL, Arkhipov AS, Larson SB, McPherson A, Schulten K. Structure. 2006;14(3):437–449. doi: 10.1016/j.str.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 7.Abraham FF, Walkup R, Gao H, Duchaineau M, Diaz De La Rubia T, Seager M. Proc Natl Acad Sci USA. 2002;99:5777–5782. doi: 10.1073/pnas.062012699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmed S, Islam S, Mohammed S. IEEE Trans Electron Devices. 2010;57:164–173. [Google Scholar]
- 9.Lange OF, Grubmuller H. J Chem Phys. 2006;124(21):214903. doi: 10.1063/1.2199530. [DOI] [PubMed] [Google Scholar]
- 10.Dey BK, Rabitz H, Askar A. J Chem Phys. 2003;119(11):5379–5387. [Google Scholar]
- 11.Kitao A, Go N. Curr Opin Struct Biol. 1999;9(2):164–169. doi: 10.1016/S0959-440X(99)80023-2. [DOI] [PubMed] [Google Scholar]
- 12.Elezgaray J, Sanejouand YH. Biopolymers. 1998;46(7):493–501. doi: 10.1002/(SICI)1097-0282(199812)46:7<493::AID-BIP7>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 13.Amadei A, Linssen ABM, Berendsen HJC. Proteins: Struct, Funct Bioinf. 1993;17(4):412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- 14.Bellesia G, Jewett AI, Shea JE. Protein Sci. 2011;20(5):818–826. doi: 10.1002/pro.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arkhipov A, Freddolino PL, Schulten K. Structure. 2006;14(12):1767–77. doi: 10.1016/j.str.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 16.Freddolino PL, Arkhipov A, Shih AY, Yin Y, Chen Z, Schulten K. Application of residue-based and shape-based coarse graining to biomolecular simulations. Chapman and Hall/CRC Press, Taylor and Francis Group; London: 2008. pp. 299–315. Chapter 20. [Google Scholar]
- 17.Gohlke H, Thorpe MF. Biophys J. 2006;91:2115–2120. doi: 10.1529/biophysj.106.083568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maher M, Puget JF, Backofen R. Proceedings of the 4th International Conference on Principles and Practice of Constraint Programming. Vol. 1520. Springer-Verlag; Weinheim, Germany: 1998. Constraint techniques forsolving the protein structure prediction problem; pp. 72–86. [Google Scholar]
- 19.Shreif Z, Ortoleva P. J Stat Phys. 2008;130:669–685. [Google Scholar]
- 20.Riccardi L, Nguyen PH, Stock G. J Phys Chem B. 2009;113:16660. doi: 10.1021/jp9076036. [DOI] [PubMed] [Google Scholar]
- 21.Hayward S, Kitao A, Go N. Proteins: Struct, Funct Genet. 1995;23(2):177–186. doi: 10.1002/prot.340230207. [DOI] [PubMed] [Google Scholar]
- 22.Sweet CR, Petrone P, Pande VS, Izaguirre JA. J Chem Phys. 2008;128:145101. doi: 10.1063/1.2883966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bahar I, Rader AJ. Curr Opin Struct Biol. 2005;15(5):586–592. doi: 10.1016/j.sbi.2005.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang C-E, Shen T, Trylska J, Tozzini V, McCammon JA. Biophys J. 2006;90:3880–3885. doi: 10.1529/biophysj.105.074575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ayton GS, Voth GA. J Struct Biol. 2007;157:570–578. doi: 10.1016/j.jsb.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 26.Pasquali S, Derreumaux P. J Phys Chem B. 2010;114(37):11957–11966. doi: 10.1021/jp102497y. [DOI] [PubMed] [Google Scholar]
- 27.Groot BLd, Vriend G, Berendsen HJC. J Mol Biol. 1999;286:1241–1249. doi: 10.1006/jmbi.1998.2568. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen HD, Reddy VS, Brooks CL. J Am Chem Soc. 2009;131:2606–2614. doi: 10.1021/ja807730x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miao Y, Ortoleva P. J Chem Phys. 2006;125(4):44901. doi: 10.1063/1.2218838. [DOI] [PubMed] [Google Scholar]
- 30.Miao Y, Ortoleva PJ. J Chem Phys. 2006;125(21):214901. doi: 10.1063/1.2400858. [DOI] [PubMed] [Google Scholar]
- 31.Pankavich S, Shreif Z, Miao Y, Ortoleva P. J Chem Phys. 2009;130:194115. doi: 10.1063/1.3134683. [DOI] [PubMed] [Google Scholar]
- 32.Miao Y, Ortoleva PJ. J Comput Chem. 2008;30(3):423–437. doi: 10.1002/jcc.21071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singharoy A, Cheluvaraja S, Ortoleva PJ. J Chem Phys. 2011;134(4):044104. doi: 10.1063/1.3524532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheluvaraja S, Ortoleva P. J Chem Phys. 2010;132(7):075102. doi: 10.1063/1.3316793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shreif Z, Adhangale P, Cheluvaraja S, Perera R, Kuhn R, Ortoleva P. Sci Model Sim. 2008;15(1–3):363–380. [Google Scholar]
- 36.Miao Y, Ortoleva PJ. Biopolymers. 2009;93(1):61–73. doi: 10.1002/bip.21299. [DOI] [PubMed] [Google Scholar]
- 37.Miao Y, Johnson JE, Ortoleva PJ. J Phys Chem B. 2010;114(34):11181–11195. doi: 10.1021/jp102314e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joshi H, Singharoy A, Sereda YV, Cheluvaraja SC, Ortoleva PJ. Prog Biophys Mol Biol. 2011;107(1):200–17. doi: 10.1016/j.pbiomolbio.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaqaman K, Ortoleva PJ. J Comput Chem. 2002;23:484–491. doi: 10.1002/jcc.10040. [DOI] [PubMed] [Google Scholar]
- 40.Hill TL. Statistical Mechanics: Principles and Selected Applications. Dover Publications; Mineola, NY: 1987. [Google Scholar]
- 41.Iyengar SS, Ortoleva P. J Chem Phys. 2008;128(16):164716. doi: 10.1063/1.2877226. [DOI] [PubMed] [Google Scholar]
- 42.Shea JE, Oppenheim I. J Phys Chem B. 1996;100:19035e19042. [Google Scholar]
- 43.Cukier RI, Deutch JM. J Chem Phys. 1969;50:36e41. [Google Scholar]
- 44.Shreif Z, Pankavich S, Ortoleva PJ. Phys Rev E. 2009;80(3 Pt 1):031703. doi: 10.1103/PhysRevE.80.031703. [DOI] [PubMed] [Google Scholar]
- 45.Rothman DH, Zaleski S. Rev Mod Phys. 1994;66:1417. [Google Scholar]
- 46.Singharoy A, Sereda YV, Ortoleva PJ. J Chem Theor Comput. 2012 doi: 10.1021/ct200574x. in review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McQuarrie DA. Statistical Mechanics. Harper and Row; New York: 1976. [Google Scholar]
- 48.Sun J, DuFort C, Daniel M-C, Murali A, Chen C, Gopinath K, Stein B, De M, Rotello VM, Holzenburg A, Kao CC, Dragnea B. Proc Natl Acad Sci. 2007;104(4):1354–1359. doi: 10.1073/pnas.0610542104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma SK. Modern Theory of Critical Phenomena (Frontiers in Physics) Perseus Books (Sd); New York: 1976. [Google Scholar]
- 50.Whitelam S, Feng EH, Hagan MF, Geissler PL. Soft Matter. 2009;5:1251–1262. doi: 10.1039/b810031d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barr AH. Proceedings of the 11th annual conference on Computer graphics and interactive techniques. ACM; New York: 1984. Global and local deformations of solid primitives; pp. 21–30. [Google Scholar]
- 52.Pankavich S, Miao Y, Ortoleva J, Shreif Z, Ortoleva P. J Chem Phys. 2008;128(23):234908. doi: 10.1063/1.2931572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.MacKerell AD. Abstr Papers Am Chem Soc. 1998;216:U696–U696. [Google Scholar]
- 54.Ayton GS, Noid WG, Voth GA. Curr Opin Struct Biol. 2007;17:192–198. doi: 10.1016/j.sbi.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 55.Levy RM, Srinivasan AR, Olson WK, McCammon JA. Biopolymers. 1984;23(6):1099–1112. doi: 10.1002/bip.360230610. [DOI] [PubMed] [Google Scholar]
- 56.Liu HJ, Qu CX, Johnson JE, Case DA. J Struct Biol. 2003;142(3):356–363. doi: 10.1016/s1047-8477(03)00028-5. [DOI] [PubMed] [Google Scholar]
- 57.Speir JA, Munshi S, Wang GJ, Baker TS, Johnson JE. Structure. 1995;3(1):63–78. doi: 10.1016/s0969-2126(01)00135-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nicholas MG. J Comput Chem. 2006;27(14):1765–1768. doi: 10.1002/jcc.20482. [DOI] [PubMed] [Google Scholar]
- 60.Yousuf M, Sahu PC, Jajoo HK, Rajagopalan S, Rajan KG. J Phys F: Met Phys. 1986;16(3):373. [Google Scholar]
- 61.Morellon L, Blasco J, Algarabel PA, Ibarra MR. Phys Rev B. 2000;62:1022–1026. [Google Scholar]
- 62.Schneemann A. Annu Rev Microbiol. 2006;60:51. doi: 10.1146/annurev.micro.60.080805.142304. [DOI] [PubMed] [Google Scholar]
- 63.Day J, Kuznetsov YG, Larson SB, Greenwood A, McPherson A. Biophys J. 2001;80:2364. doi: 10.1016/S0006-3495(01)76206-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singharoy A, Yesnik A, Ortoleva P. J Chem Phys. 2010;132:1741120. doi: 10.1063/1.3424771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Villa A, Widjajakusuma E, Stock G. J Phys Chem B. 2008;112(1):134–42. doi: 10.1021/jp0764337. [DOI] [PubMed] [Google Scholar]
- 66.García A, Onuchic J. Proc Natl Acad Sci US A. 2003;100(24):13898–903. doi: 10.1073/pnas.2335541100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hess B, van der Vegt NFA. J Phys Chem B. 2006;110(35):17616–17626. doi: 10.1021/jp0641029. [DOI] [PubMed] [Google Scholar]
- 68.Antao VP, Lai SY, Tinoco IJ. Nucleic Acids Res. 1991;19(21):5901–5905. doi: 10.1093/nar/19.21.5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin MM, Meinhold L, Shorokhov D, Zewail AH. Phys Chem Chem Phys. 2008;10(29):4227–4239. doi: 10.1039/b804675c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sorin EJ, Rhee YM, Nakatani BJ, Pande VS. Biophys J. 2003;85(2):790–803. doi: 10.1016/S0006-3495(03)74520-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Flint JS, Racaniello VR, Krug R, Flint SJ, Enquist LW, Skalka AM, Racaniello VR, Krug RM. Principles of Virology: Molecular Biology, Pathogenesis, and Control. American Society Microbiology; Washington, DC: 1999. [Google Scholar]
- 72.Bradley MJ, Chivers PT, Baker NA. J Mol Biol. 2008;378(5):1155–1173. doi: 10.1016/j.jmb.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Duarte C, Wadley L, Pyle A. Nucleic Acids Res. 2003;31:4755–4761. doi: 10.1093/nar/gkg682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.